
Abstract
Bipolar disorder, characterized by extreme manic and depressive moods, is a prevalent debilitating disease of unknown etiology. Because mood stabilizers, antipsychotics, antidepressants, and mood-regulating neuromodulators increase the inhibitory serine-phosphorylation of glycogen synthase kinase-3 (GSK3), we hypothesized that deficient GSK3 serine-phosphorylation may increase vulnerability to mood-related behavioral disturbances. This was tested by measuring behavioral characteristics of GSK3α/β21A/21A/9A/9A knockin mice with serine-to-alanine mutations to block inhibitory serine-phosphorylation of GSK3. GSK3 knockin mice displayed increased susceptibility to amphetamine-induced hyperactivity and to stress-induced depressive-like behaviors. Furthermore, serine-phosphorylation of GSK3 was reduced during both mood-related behavioral responses in wild-type mouse brain and in blood cells from patients with bipolar disorder. Therefore, proper control of GSK3 by serine-phosphorylation, which is targeted by agents therapeutic for bipolar disorder, is an important mechanism that regulates mood stabilization, and mice with disabled GSK3 serine-phosphorylation may provide a valuable model to study bipolar disorder.
Similar content being viewed by others

INTRODUCTION
Bipolar disorder afflicts approximately 2% of the population and is debilitating and life-threatening if untreated. The defining characteristic of bipolar disorder is uncontrollable extreme shifts in mood between mania and depression, usually triggered by environmental factors such as stress (Feder et al, 2009; Martinowich et al, 2009). The pathophysiological mechanisms underlying bipolar disorder are unknown, but may include genetic variations, abnormal development of neural networks, and dysregulation of neurotransmitter, neuroendocrine, and signaling activities (Gold and Chrousos, 2002; Martinowich et al, 2009; Pittenger and Duman, 2008). Therefore, mechanistic insights gained by identifying key mood-regulating molecules and animal models that present heightened sensitivity to both manic-like and depressive-like behaviors are critical for deciphering the pathophysiology of bipolar disorder and developing new therapeutic interventions (Gould and Einat, 2007a).
Glycogen synthase kinase-3 (GSK3) is a highly conserved serine/threonine protein kinase. Two isoforms of GSK3, GSK3α and GSK3β, share 84% sequence homology but are encoded by different genes (Woodgett, 1990). GSK3 is constitutively partially active, and is predominantly regulated in an inhibitory manner by phosphorylation of serines in the N-terminal regions of its two isoforms, serine-9 in GSK3β and serine-21 in GSK3α (Woodgett, 1990). This phosphorylation transforms the N terminus into a pseudo-substrate that blocks access of substrates to GSK3 (Frame et al, 2001). The inhibitory mechanism of GSK3 serine phosphorylation is used by many neuromodulators and neurotransmitter receptors to regulate GSK3 (Beaulieu et al, 2004; Li et al, 2004; Mai et al, 2002).
GSK3 has been suspected of contributing to bipolar disorder since the discovery that lithium, an effective mood stabilizer used therapeutically for bipolar disorder, is a selective inhibitor of GSK3 (Klein and Melton, 1996; Stambolic et al, 1996). Besides directly inhibiting GSK3, lithium also increases the inhibitory serine-phosphorylation of GSK3 (De Sarno et al, 2002), as do other neuromodulators implicated in mood disorders, such as serotonin (Li et al, 2004) and brain-derived neurotrophic factor (BDNF) (Mai et al, 2002), raising the possibility that this action is critical for regulating mood (Beaulieu et al, 2009; Phiel and Klein, 2001; Rowe et al, 2007). Several studies provided links between GSK3 and manic- or depressive-like behaviors in animals or humans. Evidence that GSK3 contributes to manic-like behavior include the findings that GSK3β overexpressing transgenic mice show hyperactivity (Prickaerts et al, 2006), amphetamine-induced hyperactivity concurs with reduced serine-dephosphorylation of GSK3 (Beaulieu et al, 2004), and both lithium and selective GSK3 inhibitors reduce hyperactivity in animals (Beaulieu et al, 2004; Gould et al, 2004a). Besides lithium, the anti-manic agents valproate and antipsychotic drugs increase GSK3 serine phosphorylation or directly inhibit its activity (Chen et al, 1999; De Sarno et al, 2002; Kozlovsky et al, 2006; Li et al, 2007b). In addition, studies of GSK3 in depressive-like behaviors have shown that mice with GSK3β haploinsufficiency, lacking one copy of the gene encoding GSK3β, show an anti-depressant-like behavioral phenotype (O'Brien et al, 2004), which effectively normalizes depressive behaviors induced by serotonin deficiency (Beaulieu et al, 2008). Similarly, an anti-depressant-like behavioral phenotype also occurred in GSK3α knockout mice (Kaidanovich-Beilin et al, 2009). Substantial evidence also indicates that GSK3 is a potential therapeutic target in the treatment of depression, as administration of selective GSK3 inhibitors elicit antidepressant-like behavioral effects in animals (Gould et al, 2004a; Kaidanovich-Beilin et al, 2004; Rosa et al, 2008), established antidepressants (fluoxetine and imipramine) increase the inhibitory serine phosphorylation of GSK3 in mouse brain (Li et al, 2004), and antidepressant-induced neurogenesis (Malberg et al, 2000) is blocked in mice expressing GSK3 with disabled serine-phosphorylation (Eom and Jope, 2009). In accordance with animal studies, a postmortem examination of brain samples revealed an increase in GSK3 activity in suicide subjects with major depression (Karege et al, 2007), and GSK3β polymorphisms are associated with brain region structural changes observed in patients with major depression (Inkster et al, 2009), suggesting that abnormal GSK3 activity occurs in humans with depression.
Because GSK3 is linked to both manic-like and depressive-like behaviors, and serine phosphorylation of GSK3 is the major regulatory mechanism controlling GSK3 and is targeted by lithium and other bipolar disorder treatments, we tested the hypothesis that increased susceptibility to behavioral changes relevant to bipolar disorder may be caused by deficient serine phosphorylation of GSK3. Here, we report that disabling the inhibitory serine-phosphorylation of GSK3 promotes both manic-like and depressive-like behavioral disturbances in mice, and serine-phosphorylation of GSK3 is reduced during each of these behavioral disturbances in both mice and bipolar disorder patients, establishing this mode of GSK3 regulation as a key modulator of both polarities of mood.
MATERIALS AND METHODS
Animals
The Institutional Animal Care and Use Committee at the University of Alabama at Birmingham approved the experimental protocol using mice. Mice were housed 3–4 per cage with free access to food and water in a 12 h light–dark cycle animal facility. Male homozygous GSK3α21A/21A/β9A/9A knockin (GSK3 KI) mice (12–20 weeks of age) were used along with strain-, age-, and generation-matched wild-type (WT) mice (both were kindly provided by DR Alessi, Dundee, Scotland) (McManus et al, 2005) for all experiments. Where indicated, mice were injected intraperitoneally with the indicated dose of d-amphetamine in saline, 0.83 mg/kg LPS (PBS, 0.1% DMSO), or vehicle. Lithium carbonate was administered at 0.2% in mouse chow for 4 weeks, supplemented with water and saline. Serum corticosterone levels were measured by ELISA according to the manufacturer's instructions (Cayman Chemical).
Behavior Tests
All behavior tests were conducted between 1000–1500 hours.
Home cage activity
Each mouse was individually placed in a standard micro-isolator cage (29 cm L × 19 cm W × 13 cm H) with sufficient food and water for 6 days. The spontaneous horizontal and vertical locomotor activities of mice were continuously monitored by an infrared beam activity monitor system, in which a total of eight beams spanned the width of each cage. Six lower beams measuring the horizontal activity are evenly spaced (4.5 cm apart, 2.5 cm from cage floor), and would be broken by mice normally walking the length of the cage. Two upper beams measuring the vertical activity are located near the cage ends (1.8 cm from each cage wall and 7.0 cm above cage floor), and could be broken by rearing or climbing upside-down on the cage rack, but not by food or water intake. Data were recorded by a computer programmed to store data at 2 min intervals for each of the beams. Data from the first day and night of accommodation were discarded to allow a 24 h acclimation to the fresh cage. Data are expressed as the beam breaks during 12 h light and dark cycles over a 5-day period.
Open field test
Mice were placed in a plexiglas open field (Med Associates, St Albans, VT, USA) outfitted with photobeam detectors, and activity was monitored using the activity monitoring software (Med Associates). Mice were allowed to habituate in the open field for 15 min, and activity during this period was recorded as baseline activity, including ambulatory distance and center ambulation. Following this 15 min time, mice were given a single intraperitoneal injection of d-amphetamine (Sigma, St Louis, MO, USA) and immediately returned to the open field to be monitored for an additional 30 min. Travel distance during each 5 min block and total distance travelled during the 15 min pre-amphetamine and the 30 min post-amphetamine time periods were recorded.
Learned helplessness
Mice were placed in one side of a Gemini Avoidance system shuttle box (San Diego Instruments, San Diego, CA, USA) with the gate between chambers closed, and 180 inescapable foot shocks were delivered at an amplitude of 0.3 mA, a duration of 3–5 s per shock, and a randomized inter-shock interval of 5–45 s (Duman et al, 2007). In a modified inescapable shock protocol, referred to as the reduced inescapable shock protocol, mice were given 180 foot shocks with amplitude of 0.3 mA and fixed 3 s shock duration, and a randomized inter-shock interval of 5–25 s (Supplementary Table 1). Mice exposed to the same chamber for 1 h without shocks were used as controls. Twenty-four hours after inescapable shocks, mice were returned to the shuttle box and the escape task was tested by giving 30 escape trials with each trial preceded by a foot shock at amplitude of 0.3 mA for a maximum duration of 24 s. The door of the chamber opens at the beginning of the foot shock administration to allow mice to escape. Latency to escape the shock was recorded using Gemini software, and trials in which the animal did not escape within the 24 s time limit were counted as escape failures. Mice with greater than 15 escape failures were defined as helpless.
Shock response and pain response threshold
Mice were placed in an operant chamber (Med Associates) and administered a series of 10 foot shocks with consecutive increases in amplitude from 0.1 to 1 mA at 0.1 mA increments and with a 20 s interval between shocks. At the onset of each shock, mice were observed for flinching, jumping, and vocalization. The minimum shock intensity required to elicit these responses was recorded for each mouse. As a measure of sensitivity to pain, mice were placed on a hot plate maintained at 52°C. Latency of mice to lift or lick a paw was recorded manually.
Forced swim test
Mice were placed in an automated apparatus consisting of clear Plexiglas cylinders containing distilled water (23–25°C) and outfitted with two rings of photo-beam detectors (Kinder Scientific, Poway, CA, USA). Movements were continuously monitored by computer for 6 min. Data were recorded using Motor Monitor Software (Kinder Scientific) and transferred into Microsoft Excel. The immobility time was represented by recording the resting time (seconds without beam breaks) during the last 4 min of testing (Kurtuncu et al, 2005).
Tail suspension test
Mice were tested using an automated testing system (Med Associates) consisting of an open-fronted testing chamber with a hanging vertical metal bar attached to a strain gauge that detects any movements by a mouse. The mouse tail is attached to the apparatus and movement is measured for 6 min with Med Associates software, and the immobility time was calculated as the time the force of movement was below a preset threshold during the last 4 min of testing (Crowley et al, 2004).
Elevated plus maze
Mice were placed on the central platform of an elevated plus maze that consists of a platform of 40 cm in height and four arms of 31 × 5 cm in length, with 15 cm high sides of opaque material in the two closed arms (San Diego Instruments). Entries of mice to each arm were monitored with an Ethovision camera-driven tracking system (Noldus, The Netherlands). The number of entries and time spent in each arm during the 4 min test were recorded via computer and data were analyzed using Ethovision software.
Fear conditioning
Mice were placed in an operant chamber inside an isolation box to allow exploring the operant chamber for 2 min. After this acclimation, they were administered three series of a 15 s, 75 decibel white noise tone each followed immediately by a 0.5 mA foot shock. Twenty-four hours after training, mice were tested for contextual memory by placing them in the same operant chamber for 5 min. Freezing was monitored by Med Associates Video Freeze software. Three hours after contextual testing, the cued memory was tested by placing mice in a novel context for 3 min (pretone), followed by a 75 decibel white noise tone. Freezing was monitored using Video Freeze software.
Electrophysiology
Hippocampal slices for electrophysiology were prepared from 8–12-week old mice using standard methods (Scheiderer et al, 2004). Mice were rapidly decapitated, and brains were removed and briefly rinsed in artificial cerebrospinal fluid (aCSF) containing 119 mM NaCl, 2.5 mM KCl, 2.5 mM CaCl2, 1.3 mM MgSO4, 1 mM NaH2PO4, 26 mM NaHCO3, and 10 mM glucose. Coronal slices (400 μm) were cut from the ventral pole of the hippocampus using a Vibratome (Vibratome, St Louis, MO, USA) in aCSF with continuous 95% O2/5% CO2. Slices were allowed to rest for 1 h in oxygenated aCSF before electrophysiological recording. For all experiments, slices were placed in a submersion recording chamber and continuously bathed in oxygenated aCSF at 28–30°C. A bipolar tungsten-stimulating electrode (FHC, Bowdoinham, ME, USA) was placed in the stratum radiatum of CA3 to stimulate the axons of the Schaeffer collaterals. Extracellular dendritic field excitatory post-synaptic potentials (fEPSPs) were recorded from stratum radiatum of CA1 with a glass microelectrode filled with aCSF using AxoPatch 2B (Molecular Devices, Sunnyvale, CA, USA). Baseline synaptic transmission was elicited using paired pulses of electrical stimuli (0.1 Hz, 100 μs duration, 20 ms apart). Stimulation intensity was adjusted to elicit fEPSPs between 0.6 and 0.8 mV in amplitude. At least 20 min of a stable baseline was acquired before induction of long-term potentiation (LTP) or long-term depression (LTD). To induce LTP, high frequency stimulation was applied to slices with four trains of 50 pulses at 100 Hz, separated by 20 s. Stimulation intensity was escalated to 1.5 × baseline stimulation during tetanus and returned to baseline immediately after the course of stimulation. To induce NMDA receptor-dependent LTD by low frequency stimulation, slices were stimulated with single pulses at a frequency of 1 Hz for 15 min. Stimulus-response curves and paired pulse data were collected after a stable 20 min baseline. Slices were stimulated at a range of intensities from 0 to 30 μA for 10 sweeps and the fEPSP slope was recorded. Two stimulus-response curves were collected and averaged from each slice, and only slices in which the two curves differed by <10% were included in the final average. The paired pulse ratio was tested over a range of 10–160 ms for 10 sets of pulses, and the ratio of the second fEPSP to the first was recorded. Data were filtered at 3 kHz and acquired using National Instruments acquisition software. Data from each experiment was normalized to its own baseline. The normalized slope of the fEPSP was plotted against time, with each point representing an average of five data points.
Immunoblotting and ELISA
Mice were decapitated, and brain regions were rapidly dissected and homogenized in ice-cold lysis buffer containing 20 mM Tris-HCl, pH 7.4, 150 mM NaCl, 2 mM EDTA, 1% triton-X-100, 10% glycerol, 10 μg/ml leupeptin, 10 μg/ml aprotinin, 5 μg/ml pepstatin, 0.1 mM β-glycerophosphate, 1 mM phenylmethanesulfonyl fluoride, 1 mM sodium vanadate, 50 mM NaF, and 100 nM okadaic acid. The lysates were centrifuged at 20 800 g for 15 min. Protein concentrations were determined by the Bradford method (BioRad). Samples were mixed with Laemmli sample buffer (2% SDS), placed in a boiling water bath for 5 min, proteins were resolved in SDS-polyacrylamide gels, transferred to nitrocellulose, and incubated with antibodies to phospho-Ser9-GSK3β, phospho-Ser21-GSK3α, phospho-Ser473-Akt, phospho-Thr308-Akt, Akt, phospho-Thr202/Tyr204-ERK1/2, ERK1/2, phospho-Ser380-PTEN, PTEN, phospho-Thr183/Tyr185-JNK, JNK, phospho-Thr180/Tyr182-p38, p38 (Cell Signaling, Danvers, MA, USA), total GSK3α/β (Upstate Biotechnology, Lake Placid, NY, USA), phospho-Tyr279/216GSK3α/β, GSK3α/β (Millipore), and β-actin (Sigma). Immunoblots were developed using horseradish peroxidase-conjugated goat anti-mouse, or goat anti-rabbit IgG, followed by detection with enhanced chemiluminescence, and the protein bands were quantitated with a densitometer. IL-6 levels were measured by ELISA from 100 μg protein of hippocampal extracts according to the manufacturer's instructions (eBioscience).
Human Subjects, Demographic and Clinical Evaluation
Study with human subjects was approved by the University of Alabama at Birmingham Institutional Review Board and was conducted at the University of Alabama at Birmingham (UAB). Written informed consent was obtained from all subjects after receiving a complete description of the study.
Men and women, aged 19–65 years, from any racial and ethnic group were eligible for study screen. Bipolar disorder patients were recruited from the UAB psychiatric outpatient clinic or the Center for Psychiatric Medicine in-patient service. The inclusion criteria include: (1) a primary DSM-IV diagnosis of bipolar disorder, type I or type II; (2) currently in a symptomatic episode, either manic/hypomanic with a minimal score of ⩾20 on Young Manic Rating Scale (YMRS), or depressive with a minimal score of ⩾25 on Montgomery-Asberg Depression Rating Scale (MADRS); (3) not taking psychotropic medications for at least 3 days before blood collection; and (4) no major unstable medical illnesses for 3 months before blood collection. Subjects were excluded if they concomitantly met criteria for any other major DSM-IV Axis I diagnosis, such as psychotic disorder, major depressive disorder, anxiety disorder, or alcohol and/or other substance dependence. A total of eighteen bipolar disorder patients met inclusion criteria and donated blood, among them 12 were in a manic/hypomanic episode and six were in a depressive episode. Additionally, eleven healthy control subjects were also enrolled for comparative studies (Supplementary Table 2).
Psychiatric and medical histories along with vital signs, a physical examination, and a neurological examination, were obtained before blood collection. The most recent (within 2 weeks) laboratory data on fluid balance, blood glucose, CBC with differential, thyroid function, and liver function, were reviewed to assure that these parameters were within normal range. Clinical symptoms were assessed using the YMRS for mania, the MADRS for depression, and the Clinical Global Impression for Bipolar Disorder-severity (BD-CGI-S). All assessments were completed on the day of blood collection.
Human PBMC Preparation and GSK3 Measurements
Once inclusion criteria were confirmed at screen visit, approximately 30 ml blood from each subject was immediately collected by venipuncture. PBMCs were extracted from whole blood as described previously (Li et al, 2007a), and protein lysate from PBMCs was prepared and frozen. For immunoblotting, each group of up to seven protein samples, from at least one bipolar disorder patient and one healthy control subject that were collected within a 3-month period were processed. An aliquot of the same protein lysate from SH-SY5Y cells was included in each immunoblot to adjust the intensity of immunoblot exposure. Protein bands were quantified with a densitometer.
Statistical Analysis
All data are presented as mean±SEM. Statistical analyses were conducted using SigmaStat 10.0. All data were checked for assumptions of normal distribution and homogeneity of variances in study samples, and any unexplained outlier value greater than ±2 SD from the mean of the group was excluded. For a two-group comparison, statistical analysis was performed using unpaired Student's t-test. For comparison of more than three experimental groups, one- or two-way analysis of variance (ANOVA) was used to test for significant differences (P⩽0.05) between groups. Any significant difference detected by ANOVA was followed by post hoc or inter-group comparisons using Tukey's test, Holm-Sidak comparison, or Tamhane's test. Correlation of two sets of data from the same group of samples was tested using Pearson's correlation analysis. No attempt was made for multiple comparisons due to the number of parameters that were examined. The results presented are those that may be clinically worth investigating in future studies.
RESULTS
To evaluate the role of GSK3 serine-phosphorylation in mood regulation, we used GSK3 KI mice expressing GSK3α and GSK3β with the regulatory N-terminal serines mutated to alanines (McManus et al, 2005). GSK3 KI mice reproduce and develop normally, display no overt physical or behavioral phenotype (McManus et al, 2005), and express both isoforms of GSK3 in brain at levels identical to WT mice, but without GSK3 serine-phosphorylation (Supplementary Figure 1).
GSK3 KI mice displayed increased susceptibility to hyperactivity, a behavior often used to evaluate manic-like disturbances. When home cage activity was assessed, there were no significant differences between GSK3 KI and WT mice during the light or dark cycles, suggesting normal baseline activity (Figure 1a). However, when placed in an open field, GSK3 KI mice initially displayed modest hyperactivity (Figure 1b), but they habituated to WT levels of activity upon repeated testing in the same open field (Figure 1c), suggesting that GSK3 KI mice have a heightened response to a novel environment. After habituation in the open field, administration of amphetamine (2 mg/kg), often used to model manic-like hyperactivity (Hasler et al, 2006), caused over 2.5-fold greater hyperactivity in GSK3 KI mice than WT mice during a 30-min testing period, with a total ambulatory activity of 16 798±1720 cm in GSK3 KI mice compared with 6037±839 cm in WT mice (Figure 2a). Examination of the dose-response to amphetamine showed a lower threshold for hyperactivity in GSK3 KI mice, as administration of 1 mg/kg amphetamine caused significantly greater activity of GSK3 KI mice than WT mice (Figure 2b). This heightened response of GSK3 KI mice to amphetamine shows that the serine-phosphorylation state of GSK3 directly affects the magnitude of hyperactivity, which extends a previous report that amphetamine reduced serine-phosphorylation of GSK3 (Beaulieu et al, 2004). In addition, GSK3 KI mice displayed 19 and 59% increases in hyperactivity after two and three weekly treatments with 1 mg/kg amphetamine, whereas WT mice did not display sensitization to this once-a-week low-dose repeated amphetamine administration (Figure 2c). These results indicate that serine-dephosphorylation of GSK3 not only mediates hyperactivity caused by amphetamine (Beaulieu et al, 2004), but also increases the sensitivity for the response. Similar to its effect on amphetamine-induced hyperactivity in WT mice (Gould et al, 2007b), chronic lithium administration to GSK3 KI mice partially reduced amphetamine-induced hyperactivity to the level equivalent to WT mice receiving amphetamine treatment (Figure 2d). The partial effect of lithium is likely due to its direct inhibition of GSK3 (Klein and Melton, 1996; Stambolic et al, 1996), because its additional inhibitory effect on GSK3 by increasing serine-phosphorylation is eliminated in GSK3 KI mice.

GSK3 KI mice display hyperactivity in novel, but not familiar, settings. (a) Home cage activity over a 5-day period recorded from the light cycle of day-2 (L2) to the dark cycle of day-6 (D6) was not significantly different for GSK3 KI and WT mice (n=10). (b) GSK3 KI mice traveled a significant 49% longer distance than WT mice (2735±205 cm in GSK3 KI mice vs 1838±183 cm in WT mice) during a 15-min testing period (n=40 WT, 61 KI mice, *p<0.05, Student's t-test). (c) Distance traveled on each weekly exposure (week 1, 2, and 3) to the open field. On the second and third exposures to the open field, there were no significant differences in activity between GSK3 KI and WT mice (n=14 mice/group, *p<0.05 compared to WT (Fgenotype=23.95, Fexposure=22.91, Finteraction=4.91, two way ANOVA).

GSK3 KI mice display heightened manic-like hyperactivity after d-amphetamine administration. (a) Time course of response to 2 mg/kg d-amphetamine (n=20 WT, 21 KI mice, *p<0.05 compared with WT, Fgenotype=5.775, Fblock=25.450, Finteraction=2.295, two-way repeated measure ANOVA, in which factors include genotypes=WT+GSK3 KI, block=5-trial testing blocks, and interaction=genotypes × blocks). (b) Dose-response to d-amphetamine during a 30-min test (0.5, 1, and 2 mg/kg) (n=15–21 mice/group, *p<0.05 compared with WT, Fgenotype=23.95, Fexposure=22.91, Finteraction=4.91, two-way ANOVA). (c) Mice received repeated d-amphetamine injection (1 mg/kg) once a week for 3 weeks. Distance traveled in the open field after each d-amphetamine treatment was normalized to the initial response in the same mouse, data are represented as group averages of normalized responses (n=14/group, *p<0.05 compared with WT, Fgenotype=7.365, Fdose number=0.234, Finteraction=4.635, two way ANOVA). (d) Lithium was administered to GSK3 KI and WT mice for 4 weeks, followed by d-amphetamine (2 mg/kg) administration. Ambulatory distance traveled by GSK3 KI mice is plotted against each 5-trial blocks (left panel) (n=10 without lithium (control), 13 with lithium, *p<0.05 compared with lithium-free mice at the same time block, Ftreatment=3.395, Fblock=13.448, Finteraction=2.59, two-way repeated measure ANOVA). Total distance traveled by GSK3 KI mice is also compared with wild-type mice treated with d-amphetamine (n=8) (right panel). *p<0.05 compared with d-amphetamine-treated GSK3 KI mice without lithium treatment, one way ANOVA.
Besides being sensitive to hyperactivity, GSK3 KI mice were highly susceptible to stress-induced depressive-like behavior. The ‘learned helplessness’ model of depression was examined by conditioning mice with random, mild, inescapable shocks. Measurements of responses to escapable foot shocks the next day using a standard protocol (Duman et al, 2007) (Supplementary Table 1) showed that both WT and GSK3 KI mice failed to escape (Figure 3a and b), a learned helplessness behavior that models stress-induced depression. To test if GSK3 KI mice have increased vulnerability to stress-induced depressive-like behavior, the intensity of the inescapable foot shock paradigm was reduced (Supplementary Table 1) to a level that did not cause failure to escape (defined as greater than 15 escape failures in 30 trials) in most WT mice (Figure 3b). However, with this lower level of aversive stimuli, 91% of GSK3 KI mice still displayed failure to escape. Analysis of the escape latency over each five-trial block showed that WT mice had an average of 18±2 s escape latency during the first five trials, but this helpless behavior gradually diminished over repeated trials, with only 8±3 s average escape latency during the last five trials (Figure 3c). In contrast, GSK3 KI mice displayed an average escape latency of 23±1 s and the helpless behavior persisted throughout the 30 trials. Remarkably, even without previous conditioning by inescapable foot shocks, 32% (7 out of 22) of the GSK3 KI mice failed to escape, whereas no WT mice displayed this behavior (Figure 3b). GSK3 KI mice that showed significant helpless behavior during escape trials displayed increasing escape latency to more than 21 s by the third five-trial block, whereas those without this behavior had escape latencies similar to WT mice (Figure 3d). Failure of GSK3 KI mice to escape was not due to a sensory deficit since both WT and GSK3 KI mice displayed equivalent sensitivities to foot shock and pain (Supplementary Figure 2). These results indicate that blocking serine-phosphorylation of GSK3 strongly increases susceptibility to learned helpless behavior.

GSK3 KI mice display increased sensitivity to stress-related learned helplessness. (a) WT and GSK3 KI mice received standard inescapable foot shocks (IES), and average escape latency during the entire escape trial period was calculated (n=9–10/group, *p<0.05 in one way ANOVA). (b) Escape failures (defined as escape failure >15 of 30 trials) after standard IES (IES), reduced IES (rIES), and no IES, tested by escapable foot shocks (ES). With rIES only 36% of WT mice showed learned helplessness defined as greater than 15 escape failures, whereas 91% of GSK3 KI mice displayed learned helplessness. Each diamond represents total escape failures for each individual mouse, bars represent group means (n=9–15 mice/group, *p<0.05 compared with WT mice that received the same shock treatment. (c) Average escape latency during 5-trial blocks after reduced IES. WT mice had an average of 18±2 s escape latency during the first 5 trials, but the helpless behavior was gradually reduced over repeated trials, with only 8±3 s average escape latency during the last 5 trials. GSK3 KI mice displayed an average escape latency of 23±1 s and the helpless behavior persisted throughout the 30 trials. (n=15/group, *p<0.05 compared with WT control, Fgroup=31.361, Fblock=7.096, Finteraction=5.322 two-way repeated measure ANOVA). (d) Average escape latency over the 5-trial blocks in WT and GSK3 KI mice without IES, with ‘escaped’ defined as 0–15 escape failures over 30 trials, and ‘failed to escape’ defined as >15 escape failures (n=9–15 mice/group, *p<0.05 compared with WT, **p<0.05 compared with both WT and escaped GSK3 KI, Fgroup=23.581, Fblock=79.048, Finteraction=6.782, two-way repeated measure ANOVA).
The heightened susceptibility to stress-induced depressive-like behavior was also evident in two despair-based behavioral tests, the forced swim test (Cryan et al, 2002) and the tail suspension test (Cryan et al, 2005). In both tests, GSK3 KI mice spent more time immobile than WT mice (29 and 27% increases in the forced swim test and tail suspension test, respectively) (Figure 4a and b), confirming that the serine phosphorylation of GSK3 sets the threshold for depressive-like behaviors in response to stressful conditions. To further test if serine phosphorylation of GSK3 affects susceptibility to depressive-like behavior, a combination of aversive stimuli was applied before the tail suspension test. In some mouse strains, administration of a very low dose of lipopolysaccharide (LPS), a gram-negative bacteria cell wall component that induces an inflammatory response, can cause subtle expression of depressive-like behaviors (Dantzer et al, 2008). Administration of 0.83 mg/kg LPS did not change the already heightened immobility time of GSK3 KI mice compared with WT mice (Figure 4b). The immobility time after inescapable foot shocks was not different between GSK3 KI and WT mice, but applying LPS 24 h after this aversive stress caused an additional increase in the immobility time in GSK3 KI mice. The combined aversive stimuli-induced depressive-like behavior in GSK3 KI mice was accompanied by increased serum levels of IL-6 (Figure 4c), one of many inflammatory mediators that can influence mood-relevant behaviors (Dantzer et al, 2008). These findings showed that serine-phosphorylation of GSK3 is an important determinant of the susceptibility of mice to acquire depressive-like behavior when exposed to aversive stimuli.

Depressive-like and anxiety-like behaviors in GSK3 KI mice. (a) In the forced swim test, over the last 4 min of a 6 min test, GSK3 KI mice spent longer times immobile than matched WT mice (135±8 s and 104±9 s, respectively), which was a significant 29% increase in immobility (n=20/group). (b) In the tail suspension test the average immobility time of GSK3 KI mice was 27% higher than the matched WT mice (148±5 s and 117±11 s, respectively) (n=18/group). WT and GSK3 KI mice were also treated with LPS (0.83 mg/kg) or vehicle (PBS, 0.1% DMSO), the standard inescapable shock protocol (IES), or IES followed 24 h later by treatment with LPS (0.83 mg/kg) or vehicle, and 24 h later immobility time in the tail suspension test was measured. n=4/group; Means±SEM; *p<0.05 Student's t-test compared with equivalently treated WT mice. (c) Hippocampal IL-6 levels were measured in WT and GSK3 KI mice that were subjected to IES followed 24 h later by treatment with LPS (0.83 mg/kg) or vehicle. Basal IL-6 levels in untreated WT and GSK3 KI mice were 0.58±0.17 and 0.57±0.20 ng/ml, respectively (n=7 per group). (d) In the elevated plus maze during a 4-min testing period, GSK3 KI mice spent 44% less time in the open arms of the maze than matched WT mice (24±3 s vs 42±6 s), and showed a significant increase in time spent in the closed arms (194±6 s in GSK3 KI vs 170±6 s in WT mice) (n=18/group). (e) When the anxiety-like behavior was tested in the open field for thigmotaxis for 15 min, GSK3 KI mice showed a trend towards increased anxiety only during the first 5 min of testing, whereas the overall difference between GSK3 KI and WT mice throughout the 15-min test was not significantly different. Data are means±SEM, *p<0.05, **p<0.01 by Student's t-test.
This absence of adaptability of the GSK3 KI mice to stress may involve anxiety, which often co-exists with depression. GSK3 KI mice displayed mild-anxious behavior in the elevated plus maze, spending 44% less time than WT mice in the open arms of the maze, and a significantly longer time in the closed arms (Figure 4d). However, when the anxiety-like behavior of thigmotaxis was tested for 15 min, GSK3 KI mice were slightly more anxious only during the first 5 min of testing, whereas the overall difference between GSK3 KI and WT mice throughout the 15 min test was not significant (Figure 4e), suggesting that GSK3 KI mice may only display mild anxiety-like behavior when exposed to a new environment.
To test if increased sensitivity of GSK3 KI mice to mild stress was associated with altered hippocampal function, a stress-vulnerable mood regulating region of the brain (Duman et al, 1997), fear conditioning was examined. In the fear conditioning paradigm, baseline freezing times for WT and GSK3 KI mice were equivalent during training to associate both a context and a cue (tone) with a mild aversive stimuli (foot shocks) (Figure 5a). The freezing time of GSK3 KI mice was significantly 28% higher than WT mice upon subjection 24 h later to the contextual test, a function of both hippocampus and amygdale, suggesting that GSK3 KI mice display an enhanced emotional association with a stressful stimulus that likely involves either hippocampal or amygdalar function. Upon exposure to a novel context (pretone cue test), GSK3 KI mice initially showed 59% less freezing than WT mice, which is consistent with increased activity by GSK3 KI mice when exposed to a novel environment (Figure 1). However, when exposed to a tone paired with the new context, a function primarily dependent on the amygdala (LeDoux, 2000), both GSK3 KI and WT mice displayed equivalent high freezing times, suggesting that amygdalar function is likely intact.

Altered hippocampal responses to stress in GSK3 KI mice. (a) Contextual and cued fear conditioning. Twenty-four hours after training, GSK3 KI mice spent significantly more time freezing (79±5% and 61±4% in GSK3 KI and WT mice, respectively) in the contextual test. When exposed to novel context (pre-tone), GSK3 KI mice displayed significantly less freezing (41±4% and 17±4% in WT and GSK3 KI mice, respectively), but response to tone was equivalent in both WT and GSK3 KI mice. n=14 mice/group, *p<0.05 compared with WT, contextual: Fgenotype=5.137, Fcondition=36.233, Finteraction=5.319, cued: Fgenotype=4.593, Fcondition=108.935, Finteraction=8.202, two-way ANOVA. (b) LTP. Plasticity induced by high frequency stimulation was recorded in the CA1 region of ventral hippocampal slices from WT and GSK3 KI mice. Waveforms are representative averages of 10 fEPSPs from 5 min before (solid line) and 30 min after (dotted line) tetanus. Bar graph represents potentiation 30 min after tetanus. (c) LTD. Plasticity induced by low-frequency stimulation. Waveforms are representative averages of 10 fEPSPs from 5 min before (solid line) and 30 min after (dotted line) low frequency stimulation. Bar graph represents changes from baseline (dotted line) 30 min after stimulation. *p<0.05, Student's t-test, n=6–7 per group.
To further test if neuronal activity in the hippocampus is abnormal in GSK3 KI mice, synaptic plasticity at the Schaffer collateral CA3-CA1 synapse in the ventral hippocampus was measured. The baseline synaptic transmission and paired pulse ratio revealed no significant differences between WT and GSK3 KI mice (Supplementary Figure 3). High frequency stimulation robustly induced LTP in the CA1 to a similar magnitude in WT and GSK3 KI mice (Figure 5b). However, NMDA receptor-dependent LTD induced by low frequency stimulation was converted to a slow onset LTP-like response in GSK3 KI mice (Figure 5c), a conversion previously noted to be induced by stress in WT rats (Maggio and Segal, 2009). These findings indicate that impaired serine-phosphorylation of GSK3 in GSK3 KI mice establishes a stressed state that alters responses to aversive stimuli.
As GSK3 KI mice display heightened responses to stress, we tested if they produce higher levels of corticosterone, a leading candidate to mediate stress-induced episodes of mood disorders. Basal levels of serum corticosterone were similar in GSK3 KI and WT mice, but immediately following a session of inescapable foot shocks, the serum corticosterone was greater in GSK3 KI mice than in WT mice, which was reduced to WT levels by lithium pretreatment (Supplementary Figure 4A). Immediately after exposure of GSK3 KI mice to escapable foot shocks the serum corticosterone levels were increased above basal levels, but not as much as after exposure to inescapable foot shocks (Supplementary Figure 4B), and there was a tendency (p=0.06) for the corticosterone levels to be modestly higher in helpless than in non-helpless GSK3 KI mice after the escapable foot shock treatment.
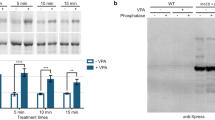
Because the absence of serine-phosphorylation of GSK3 is associated with increased susceptibility to hyperactivity and depressive-like behaviors, we examined if dephosphorylation of the inhibitory serines in GSK3 occurs in WT mice that display amphetamine-induced hyperactivity or develop learned helplessness after inescapable foot shocks. As previously reported (Beaulieu et al, 2004), amphetamine (2 mg/kg) treatment reduced the inhibitory serine phosphorylation of GSK3 in the striatum of WT mice (Supplementary Figure 5). There were also dramatic decreases in the inhibitory serine-phosphorylation of both GSK3 isoforms in the hippocampus and cerebral cortex of WT mice in the learned helpless state 24 h after inescapable foot shocks (Figure 6a). Foot shock treatments not causing learned helplessness did not change serine-phosphorylation of GSK3, showing that the treatment did not alter GSK3 unless it was associated with learned helplessness. Learned helplessness-induced decreases in serine-phosphorylated GSK3 were associated with decreased phosphorylation of Akt, while there were no changes in the activity-regulating phosphorylation of several other intracellular signaling kinases (Figure 6b; Supplementary Figure 6), indicating selective reduction in the activity of Akt, a predominant kinase that phosphorylates the regulatory serines of GSK3. Basal phosphorylation of Akt was not different between GSK3 KI and WT mice (Figure 6c), but when assessed after escapable foot shocks, lower Akt phosphorylation was found in hippocampal extracts of helpless GSK3 KI mice than in WT mice (Figure 6d). Akt phosphorylation was also significantly lower in hippocampal extracts of helpless GSK3 KI mice than in non-helpless GSK3 KI mice when the two groups were differentiated by responses to escapable foot shocks (Figure 6e), showing that the decreased Akt phosphorylation was associated with depressive-like behavior, but not the shock response in GSK3 KI mice. Thus, reduced Akt activation occurs in conjunction with decreased inhibitory serine-phosphorylation of GSK3 during depressive-like states.

GSK3 serine-phosphorylation is low in brains of learned helpless mice. (a) Cerebral cortical and hippocampal extracts from WT mice were immunoblotted for the indicated proteins. Abbreviations for treatments are: NN=No inescapable shock, No escapable shock (control condition); S0=inescapable shock, kill immediately (0 min); NS=No inescapable shock, followed by escapable shock, kill immediately; SS=learned helplessness paradigm with inescapable shock followed by escapable Shock. Means±SEM, n=4–7, *p<0.05 vs NN. (b) Hippocampal extracts from WT mice were immunoblotted for phospho-Thr308-Akt, phospho-Ser473-Akt, total Akt, phospho-Thr202/Tyr204-ERK1/2 (P-ERK1/2), total ERK1/2, phospho-Thr183/Tyr185-JNK (P-JNK), total JNK, phospho-Thr180/Tyr182-p38 (P-p38), total p38, phospho-Ser380-PTEN (P-PTEN), total PTEN, and β-actin. (c) Hippocampal extracts from untreated WT and GSK3 KI mice, (d) after exposing WT and GSK3 KI mice to escapable shock, and (e) with the GSK3 KI mice differentiated into non-helpless (NH) or helpless (HL) groups, were immunoblotted for the indicated proteins. n=4–7; Means±SEM; *p<0.05 by Student's t-test.
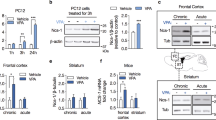
To examine if the diminished serine-phosphorylation of GSK3 observed in mice with manic-like and depressive-like behaviors is evident in patients with bipolar disorder, serine-phosphorylated GSK3 was measured in peripheral blood mononuclear cells (PBMCs) from symptomatic, drug-free bipolar disorder patients and matched healthy controls (Supplementary Table 2). Phospho-Ser21-GSK3α and phospho-Ser9-GSK3β were each approximately 35% lower in PBMCs from bipolar disorder patients than healthy controls (Figure 7a–c). Total levels of each GSK3 isoform in PBMCs were equivalent in bipolar disorder patients and healthy controls (Figure 7a), indicating that lower levels of serine-phosphorylated GSK3 in patients was not due to reduced expression of GSK3. Remarkably, reductions in phospho-Ser21-GSK3α and phospho-Ser9-GSK3β significantly correlated with severity of manic symptoms evaluated by the Young Mania Rating Scale (Figure 7d), and reduced phospho-Ser21-GSK3α also significantly correlated with severity of depressive symptoms evaluated by the Montgomery-Asberg Depression Rating Scale (Figure 7e). Therefore, decreased inhibitory serine phosphorylation of GSK3 also occurs in symptomatic bipolar disorder patients. Along with the findings from hyperactive and stressed mice, this result provides additional evidence that dysregulation of serine phosphorylation of GSK3 contributes to the etiology of mood disturbances in bipolar disorder.

GSK3 serine phosphorylation in PBMCs of bipolar disorder patients. (a) Representative immunoblots of phospho-Ser21-GSK3α, phospho-Ser9-GSK3β, and total GSK3α/β in PBMCs from healthy controls (HC) and bipolar disorder (BD) patients. (b,c) quantification of phospho-Ser21-GSK3α and phospho-Ser9-GSK3β. Values are phospho-Ser-GSK3 as % of total GSK3 in each individual subject. n=10 (HC) and 17 (BD), Means±SEM; *p<0.05 by Student's t-test. (d) Pearson correlation analysis between PBMC phospho-Ser21-GSK3α or phospho-Ser9-GSK3β and total Young Mania Rating Scale (YMRS) scores in manic/hypomanic patients. (e) Pearson correlation analysis between PBMC phospho-Ser21-GSK3α or phospho-Ser9-GSK3β and total Montgomery-Asberg Depression Rating Scale (MADRS) scores in depressed patients. n=11 in mania; n=6 in depression.
DISCUSSION
This study identified a single molecular mechanism, reduced serine-phosphorylation of GSK3, that causes behavioral alterations in mice resembling the mood symptoms in human bipolar disorder. Not only does blocking serine-phosphorylation of GSK3 increase susceptibility to both stimulus-induced hyperactivity and stress-induced depressive-like behaviors in mice, but as might be predicted if GSK3 serine-phosphorylation is critical in regulating mood-related behaviors, serine-phosphorylation of GSK3 also is reduced during both hyperactive and depressive-like states in mice. Importantly, in agreement with findings in mice, GSK3 serine-phosphorylation also is reduced in PBMCs from symptomatic bipolar disorder patients and the reduction is correlated with severity of symptoms. Taken together, all of these results indicate that insufficient serine-phosphorylation of GSK3 may have a critical pathological role in behavioral disturbances that occur in bipolar disorder.
Mice expressing normal levels of GSK3 but with the inhibitory serine-phosphorylation ablated were studied because serine-phosphorylation of GSK3 is a target of the mood stabilizer lithium and several other mood disorder treatments, but the role of this regulatory mechanism had not been studied in behavioral responses that may be related to mood disturbances. We hypothesized that ablation of the inhibitory serines in GSK3α/β would cause behavioral changes opposite to the effects of bipolar disorder treatments. This was confirmed, as prominent features of GSK3 KI mice are heightened sensitivity to amphetamine-induced hyperactivity and increased vulnerability to stress-induced depressive-like behavior. This extends previous studies of animal models that show either hyperactivity (Gould et al, 2004b; Roybal et al, 2007) or depressive-like behavior (Kalueff et al, 2007; Pittenger and Duman, 2008), and should facilitate identification of factors that contribute to bipolar disorder and that can be used to test new therapeutic strategies, such as targeting pathways converging on the serine phosphorylation of GSK3 or abnormally regulated substrates of GSK3. This study used the GSK3α and GSK3β double KI mice because both GSK3 isoforms may contribute to regulating mood-related behaviors (Kaidanovich-Beilin et al, 2009; O'Brien et al, 2004; Prickaerts et al, 2006), and all chemical inhibitors of GSK3 inhibit both GSK3α and GSK3β equally. Although some studies have identified GSK3 isoform-selective actions (Force and Woodgett, 2009; Hoeflich et al, 2000; Liang and Chuang, 2006, 2007; Phiel et al, 2003; Wang et al, 1994), there is no published information delineating a predominant role of either isoform of GSK3 on mood-related behaviors.
Importantly, decreased serine-phosphorylation of GSK3 during both manic-like and depressive-like states were also found in PMBCs from bipolar disorder patients, providing further support that abnormal regulation of GSK3 serine-phosphorylation is critically altered during mood disturbances in bipolar disorder. We used PBMCs as a surrogate system as lymphocytes in PBMCs express receptors, intracellular signaling proteins, and enzymes that are regulated by many of the same genetic and environmental influences as neurons in the brain (Gladkevich et al, 2004; Tsuang et al, 2005). The expression levels of many classes of biologically relevant processes are similar in PBMCs and brain (Sullivan et al, 2006), as may be the case with GSK3 and its regulatory signaling pathways (Castri et al, 2007; Li et al, 2007a), making PBMCs a valuable human tissue to study signaling systems that also occur in human brain. However, it is clear that further studies are needed to clarify the relationships between the activities of signaling systems in PBMCs and the brain. Although our data provide clinical evidence validating the findings of the animal behavioral studies and in mouse brain that dysregulation of GSK3 serine-phosphorylation facilitates behavioral disturbances, a larger sample size of bipolar disorder patients is required to confirm the clinical relevance. Nevertheless, the significant reduction of serine-phosphorylation of GSK3 in mice during hyperactivity or depressive-like behavioral disturbances and in symptomatic bipolar patients, as well as a significant relationship between the severity of mood symptoms in bipolar patients and dysregulated GSK3 in PBMCs, suggest that a mood state-dependent change of GSK3 serine-phosphorylation exists during mood-related behavioral disturbances.
Ablation of the regulatory serines in GSK3 sensitized GSK3 KI mice to stress, which was evident in their behavioral responses to all types of stressful conditions that were studied. The increased sensitivity to both extremes of behavioral disturbances in GSK3 KI mice could be indicative of a lack of proper regulation of GSK3 serine phosphorylation in bipolar disorder, which could result from altered signaling to GSK3 by abnormalities in mood-regulating neuromodulators. Increased dopaminergic activity is associated with hyperactivity, a behavior that often occurs in bipolar disorder, and enhanced dopamine D2 receptor activity has been reported to dephosphorylate the regulatory serines in GSK3 (Beaulieu et al, 2004). Since one effect of amphetamine is to increase dopamine release, the increased vulnerability of GSK3 KI mice to amphetamine-induced hyperactivity suggests that the dephosphorylation of GSK3 contributes to setting the threshold for hyperactivity induced by dopaminergic neurotransmission. Similarly, serotonin- and BDNF-induced signaling normally increase serine-phosphorylation of GSK3 (Beaulieu et al, 2004; Li et al, 2004; Mai et al, 2002), thus lacking regulation in GSK3 KI mice by these modulators likely models the states of serotonin and BDNF deficiency that may be associated with susceptibility to depression. Consistent with altered responses to stress, we found an abnormal LTD in GSK3 KI mice in the ventral hippocampus, the septotemporal portion of the hippocampus that is primarily associated with emotional memories (Bannerman et al, 2003). This finding may be related to a recent report (Maggio and Segal, 2009) in which an almost identical conversion of LTD to a low amplitude LTP in the ventral hippocampus occurred in WT rats exposed to acute swim stress. The induction of depressive-like learned helplessness in GSK3 KI mice is also associated with an upstream reduced phosphorylation of Akt. This modulation of Akt suggests a feedback loop of Akt regulation by GSK3, which could involve regulation of signaling of receptors to Akt or other regulators that respond to stress.
Altogether, these findings show a critical role of the serine phosphorylation of GSK3 in the signaling pathways associated with manic-like and depressive-like behaviors and indicate that impaired serine-phosphorylation of GSK3 establishes a state of increased susceptibility to stress-induced behavioral disturbances. The targets affected by deletion of serine-phosphorylation of GSK3 that mediate increased vulnerability to mood disturbances may include changes in neurotransmission, neurotrophins, activity of the HPA axis, stress-induced inflammatory responses, or other mediators.
References
Bannerman DM, Grubb M, Deacon RM, Yee BK, Feldon J, Rawlins JN (2003). Ventral hippocampal lesions affect anxiety but not spatial learning. Behav Brain Res 139: 197–213.
Beaulieu JM, Gainetdinov RR, Caron MG (2009). Akt/GSK3 signaling in the action of psychotropic drugs. Annu Rev Pharmacol Toxicol 49: 327–347.
Beaulieu JM, Sotnikova TD, Yao WD, Kockeritz L, Woodgett JR, Gainetdinov RR et al (2004). Lithium antagonizes dopamine-dependent behaviors mediated by an AKT/glycogen synthase kinase 3 signaling cascade. Proc Natl Acad Sci USA 101: 5099–5104.
Beaulieu JM, Zhang X, Rodriguiz RM, Sotnikova TD, Cools MJ, Wetsel WC et al (2008). Role of GSK3 beta in behavioral abnormalities induced by serotonin deficiency. Proc Natl Acad Sci USA 105: 1333–1338.
Castri P, Iacovelli L, De Blasi A, Giubilei F, Moretti A, Capone FT et al (2007). Reduced insulin-induced phosphatidylinositol-3-kinase activation in peripheral blood mononuclear leucocytes from patients with Alzheimer's disease. Eur J Neurosci 26: 2469–2472.
Chen G, Huang LD, Jiang YM, Manji HK (1999). The mood-stabilizing agent valproate inhibits the activity of glycogen synthase kinase-3. J Neurochem 72: 1327–1330.
Crowley JJ, Jones MD, O'Leary OF, Lucki I (2004). Automated tests for measuring the effects of antidepressants in mice. Pharmacol Biochem Behav 78: 269–274.
Cryan JF, Markou A, Lucki I (2002). Assessing antidepressant activity in rodents: recent developments and future needs. Trends Pharmacol Sci 23: 238–245.
Cryan JF, Mombereau C, Vassout A (2005). The tail suspension test as a model for assessing antidepressant activity: review of pharmacological and genetic studies in mice. Neurosci Biobehav Rev 29: 571–625.
Dantzer R, O'Connor JC, Freund GG, Johnson RW, Kelley KW (2008). From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci 9: 46–56.
De Sarno P, Li X, Jope RS (2002). Regulation of Akt and glycogen synthase kinase-3 beta phosphorylation by sodium valproate and lithium. Neuropharmacology 43: 1158–1164.
Duman CH, Schlesinger L, Kodama M, Russell DS, Duman RS (2007). A role for MAP kinase signaling in behavioral models of depression and antidepressant treatment. Biol Psychiatry 61: 661–670.
Duman RS, Heninger GR, Nestler EJ (1997). A molecular and cellular theory of depression. Arch Gen Psychiatry 54: 597–606.
Eom TY, Jope RS (2009). Blocked inhibitory serine-phosphorylation of glycogen synthase kinase-3alpha/beta impairs in vivo neural precursor cell proliferation. Biol Psychiatry 66: 494–502.
Feder A, Nestler EJ, Charney DS (2009). Psychobiology and molecular genetics of resilience. Nat Rev Neurosci 10: 446–457.
Force T, Woodgett JR (2009). Unique and overlapping functions of GSK-3 isoforms in cell differentiation and proliferation and cardiovascular development. J Biol Chem 284: 9643–9647.
Frame S, Cohen P, Biondi RM (2001). A common phosphate binding site explains the unique substrate specificity of GSK3 and its inactivation by phosphorylation. Mol Cell 7: 1321–1327.
Gladkevich A, Kauffman HF, Korf J (2004). Lymphocytes as a neural probe: potential for studying psychiatric disorders. Prog Neuropsychopharmacol Biol Psychiatry 28: 559–576.
Gold PW, Chrousos GP (2002). Organization of the stress system and its dysregulation in melancholic and atypical depression: high vs low CRH/NE states. Mol Psychiatry 7: 254–275.
Gould TD, Einat H (2007a). Animal models of bipolar disorder and mood stabilizer efficacy: a critical need for improvement. Neurosci Biobehav Rev 31: 825–831.
Gould TD, Einat H, Bhat R, Manji HK (2004a). AR-A014418, a selective GSK-3 inhibitor, produces antidepressant-like effects in the forced swim test. Int J Neuropsychopharmacol 7: 387–390.
Gould TD, O'Donnell KC, Picchini AM, Manji HK (2007b). Strain differences in lithium attenuation of d-amphetamine-induced hyperlocomotion: a mouse model for the genetics of clinical response to lithium. Neuropsychopharmacology 32: 1321–1333.
Gould TD, Zarate CA, Manji HK (2004b). Glycogen synthase kinase-3: a target for novel bipolar disorder treatments. J Clin Psychiatry 65: 10–21.
Hasler G, Drevets WC, Gould TD, Gottesman II, Manji HK (2006). Toward constructing an endophenotype strategy for bipolar disorders. Biol Psychiatry 60: 93–105.
Hoeflich KP, Luo J, Rubie EA, Tsao MS, Jin O, Woodgett JR (2000). Requirement for glycogen synthase kinase-3beta in cell survival and NF-kappaB activation. Nature 406: 86–90.
Inkster B, Nichols TE, Saemann PG, Auer DP, Holsboer F, Muglia P et al (2009). Association of GSK3beta polymorphisms with brain structural changes in major depressive disorder. Arch Gen Psychiatry 66: 721–728.
Kaidanovich-Beilin O, Lipina TV, Takao K, van Eede M, Hattori S, Laliberte C et al (2009). Abnormalities in brain structure and behavior in GSK-3alpha mutant mice. Mol Brain 2: 35.
Kaidanovich-Beilin O, Milman A, Weizman A, Pick CG, Eldar-Finkelman H (2004). Rapid antidepressive-like activity of specific glycogen synthase kinase-3 inhibitor and its effect on beta-catenin in mouse hippocampus. Biol Psychiatry 55: 781–784.
Kalueff AV, Wheaton M, Murphy DL (2007). What's wrong with my mouse model? Advances and strategies in animal modeling of anxiety and depression. Behav Brain Res 179: 1–18.
Karege F, Perroud N, Burkhardt S, Schwald M, Ballmann E, La Harpe R et al (2007). Alteration in kinase activity but not in protein levels of protein kinase B and glycogen synthase kinase-3beta in ventral prefrontal cortex of depressed suicide victims. Biol Psychiatry 61: 240–245.
Klein PS, Melton DA (1996). A molecular mechanism for the effect of lithium on development. Proc Natl Acad Sci USA 93: 8455–8459.
Kozlovsky N, Amar S, Belmaker RH, Agam G (2006). Psychotropic drugs affect Ser9-phosphorylated GSK-3beta protein levels in rodent frontal cortex. Int J Neuropsychopharmacol 9: 337–342.
Kurtuncu M, Luka LJ, Dimitrijevic N, Uz T, Manev H (2005). Reliability assessment of an automated forced swim test device using two mouse strains. J Neurosci Methods 149: 26–30.
LeDoux JE (2000). Emotion circuits in the brain. Annu Rev Neurosci 23: 155–184.
Li X, Friedman AB, Zhu W, Wang L, Boswell S, May RS et al (2007a). Lithium regulates glycogen synthase kinase-3beta in human peripheral blood mononuclear cells: implication in the treatment of bipolar disorder. Biol Psychiatry 61: 216–222.
Li X, Rosborough KM, Friedman AB, Zhu W, Roth KA (2007b). Regulation of mouse brain glycogen synthase kinase-3 by atypical antipsychotics. Int J Neuropsychopharmacol 10: 7–19.
Li X, Zhu W, Roh MS, Friedman AB, Rosborough K, Jope RS (2004). In vivo regulation of glycogen synthase kinase-3beta (GSK3beta) by serotonergic activity in mouse brain. Neuropsychopharmacology 29: 1426–1431.
Liang MH, Chuang DM (2006). Differential roles of glycogen synthase kinase-3 isoforms in the regulation of transcriptional activation. J Biol Chem 281: 30479–30484.
Liang MH, Chuang DM (2007). Regulation and function of glycogen synthase kinase-3 isoforms in neuronal survival. J Biol Chem 282: 3904–3917.
Maggio N, Segal M (2009). Differential modulation of long-term depression by acute stress in the rat dorsal and ventral hippocampus. J Neurosci 29: 8633–8638.
Mai L, Jope RS, Li X (2002). BDNF-mediated signal transduction is modulated by GSK3beta and mood stabilizing agents. J Neurochem 82: 75–83.
Malberg JE, Eisch AJ, Nestler EJ, Duman RS (2000). Chronic antidepressant treatment increases neurogenesis in adult rat hippocampus. J Neurosci 20: 9104–9110.
Martinowich K, Schloesser RJ, Manji HK (2009). Bipolar disorder: from genes to behavior pathways. J Clin Invest 119: 726–736.
McManus EJ, Sakamoto K, Armit LJ, Ronaldson L, Shpiro N, Marquez R et al (2005). Role that phosphorylation of GSK3 plays in insulin and Wnt signalling defined by knockin analysis. EMBO J 24: 1571–1583.
O'Brien WT, Harper AD, Jove F, Woodgett JR, Maretto S, Piccolo S et al (2004). Glycogen synthase kinase-3beta haploinsufficiency mimics the behavioral and molecular effects of lithium. J Neurosci 24: 6791–6798.
Phiel CJ, Klein PS (2001). Molecular targets of lithium action. Annu Rev Pharmacol Toxicol 41: 789–813.
Phiel CJ, Wilson CA, Lee VM, Klein PS (2003). GSK-3alpha regulates production of Alzheimer's disease amyloid-beta peptides. Nature 423: 435–439.
Pittenger C, Duman RS (2008). Stress, depression, and neuroplasticity: a convergence of mechanisms. Neuropsychopharmacology 33: 88–109.
Prickaerts J, Moechars D, Cryns K, Lenaerts I, van Craenendonck H, Goris I et al (2006). Transgenic mice overexpressing glycogen synthase kinase 3beta: a putative model of hyperactivity and mania. J Neurosci 26: 9022–9029.
Rosa AO, Kaster MP, Binfare RW, Morales S, Martin-Aparicio E, Navarro-Rico ML et al (2008). Antidepressant-like effect of the novel thiadiazolidinone NP031115 in mice. Prog Neuropsychopharmacol Biol Psychiatry 32: 1549–1556.
Rowe MK, Wiest C, Chuang DM (2007). GSK-3 is a viable potential target for therapeutic intervention in bipolar disorder. Neurosci Biobehav Rev 31: 920–931.
Roybal K, Theobold D, Graham A, DiNieri JA, Russo SJ, Krishnan V et al (2007). Mania-like behavior induced by disruption of CLOCK. Proc Natl Acad Sci USA 104: 6406–6411.
Scheiderer CL, Dobrunz LE, McMahon LL (2004). Novel form of long-term synaptic depression in rat hippocampus induced by activation of alpha 1 adrenergic receptors. J Neurophysiol 91: 1071–1077.
Stambolic V, Ruel L, Woodgett JR (1996). Lithium inhibits glycogen synthase kinase-3 activity and mimics wingless signalling in intact cells. Curr Biol 6: 1664–1668.
Sullivan PF, Fan C, Perou CM (2006). Evaluating the comparability of gene expression in blood and brain. Am J Med Genet B Neuropsychiatr Genet 141: 261–268.
Tsuang MT, Nossova N, Yager T, Tsuang MM, Guo SC, Shyu KG et al (2005). Assessing the validity of blood-based gene expression profiles for the classification of schizophrenia and bipolar disorder: a preliminary report. Am J Med Genet B Neuropsychiatr Genet 133: 1–5.
Wang QM, Park IK, Fiol CJ, Roach PJ, DePaoli-Roach AA (1994). Isoform differences in substrate recognition by glycogen synthase kinases 3 alpha and 3 beta in the phosphorylation of phosphatase inhibitor 2. Biochemistry 33: 143–147.
Woodgett JR (1990). Molecular cloning and expression of glycogen synthase kinase-3/factor A. EMBO J 9: 2431–2438.
Acknowledgements
We thank Dr Dario Alessi for generously providing the GSK3 knockin mice to RSJ, Liqin Liu, Min Liu, and Xiaohong Li for technical support and data management, Dr Mathieu Lesort and Dr Thomas Van Groen for behavioral technical advice, and clinical recruitment and assessment by the UAB Psychiatric Research Office and the Center for Psychiatric Medicine. This work was supported by NIH Grants MH64555, MH73723, MH86622 (XL), MH38752 (RSJ), MH57014 (JDS), MH82304 (LLM), NS47466 (UAB Behavioral Assessment Core), NS61788 (Training Program in the Neurobiology of Cognition and Cognitive Disorders, PI, JDS, awardee AP), NARSAD independent research award (XL), NARSAD Young Investigator award (EB), and the Evelyn F McKnight Brain Research Foundation.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Neuropsychopharmacology website
Supplementary information
Rights and permissions
About this article
Cite this article
Polter, A., Beurel, E., Yang, S. et al. Deficiency in the Inhibitory Serine-Phosphorylation of Glycogen Synthase Kinase-3 Increases Sensitivity to Mood Disturbances. Neuropsychopharmacol 35, 1761–1774 (2010). https://doi.org/10.1038/npp.2010.43
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2010.43
Keywords
This article is cited by
-
Differential diagnosis of unipolar versus bipolar depression by GSK3 levels in peripheral blood: a pilot experimental study
International Journal of Bipolar Disorders (2023)
-
Preclinical models of treatment-resistant depression: challenges and perspectives
Pharmacological Reports (2023)
-
AgRP neurons trigger long-term potentiation and facilitate food seeking
Translational Psychiatry (2021)
-
Advances toward precision medicine for bipolar disorder: mechanisms & molecules
Molecular Psychiatry (2021)
-
GSK-3β activation is required for ZIP-induced disruption of learned fear
Scientific Reports (2020)
