
Abstract
Prenatal nicotine exposure (PNE) is linked to numerous psychiatric disorders including attention deficit hyperactivity disorder (ADHD). Current literature suggests that core deficits observed in ADHD reflect abnormal inhibitory control governed by the prefrontal cortex. Yet, it is unclear how neural activity in the medial prefrontal cortex (mPFC) is modulated during tasks that assess response inhibition or if these neural correlates, along with behavior, are affected by PNE. To address this issue, we recorded from single mPFC neurons in control and PNE rats as they performed a stop-signal task. We found that PNE rats were faster for all trial-types, made more premature responses, and were less likely to inhibit behavior on ‘STOP’ trials during which rats had to inhibit an already initiated response. Activity in mPFC was modulated by response direction and was positively correlated with accuracy and movement time in control but not PNE rats. Although the number of single neurons correlated with response direction was significantly reduced by PNE, neural activity observed on general STOP trials was largely unaffected. However, dramatic behavioral deficits on STOP trials immediately following non-conflicting (GO) trials in the PNE group appear to be mediated by the loss of conflict monitoring signals in mPFC. We conclude that prenatal nicotine exposure makes rats impulsive and disrupts firing of mPFC neurons that carry signals related to response direction and conflict monitoring.
Similar content being viewed by others


INTRODUCTION
Prenatal nicotine exposure (PNE) has been shown to increase the incidence of psychiatric disorders in offspring, including but not limited to, attention deficit hyperactivity disorder (ADHD), conduct disorder, and addiction (Cornelius and Day, 2009; Ernst et al, 2001; Milberger et al, 1996, 1998; Fried and Watkinson, 2001; Wickstrom, 2007), all of which are characterized by diminished executive control (Mick et al, 2002; Milberger et al, 1996, 1998; Morgan and Lilienfeld, 2000). In rodents, the behavioral disturbances described after PNE, and the benefits observed after methylphenidate treatment, have pinpointed PNE as a valuable animal model to investigate mechanisms that underlie poor impulse control as defined by the inability to inhibit prepotent movement (Schneider et al, 2011; Zhu et al, 2012).
Although it is clear that PNE disrupts many brain systems involved in executive control, it is unknown how or what neural correlates related to the control of behavior are disrupted. We know that dopaminergic and noradrenergic functions are affected by PNE (Navarro et al, 1990; Seidler et al, 1992; Slotkin, 1998), and that PNE alters morphology, volume, and dopamine turnover in medial prefrontal cortex (mPFC) (Muhammad et al, 2012; Muneoka et al, 1997; Mychasiuk et al, 2013; Schneider et al, 2011; Zhu et al, 2012), but we still do not understand how neural signals related to executive control mechanisms are affected. Although neighboring structures have been explored (Emeric et al, 2008; Hanes et al, 1998; Schall and Boucher, 2007), it is still unclear how firing in mPFC is normally modulated during tasks that probe response inhibition. This is surprising considering the number of studies that have implicated mPFC in inhibitory control. For instance, perturbation of the rat prelimbic prefrontal cortex reveals stop-signal reaction time deficits (Bari et al, 2011) and increased premature responses during performance of reaction time tasks (Pezze et al, 2014; Risterucci et al, 2003). However, similar approaches have yielded conflicting results (Christakou et al, 2001; Eagle et al, 2008; Eagle and Robbins, 2003). Elucidating the relationship between mPFC activity and response selection during the need for elevated executive control would help us to better understand dysfunctions observed in psychiatric disorders that impair inhibitory restraint.
To address this issue we recorded single mPFC cells from control and PNE rats in our rodent variant of the stop-signal task (Bryden et al, 2012). It has been shown that mPFC is both affected by PNE and is critical for normal performance on stop-signal paradigms (Bari et al, 2011; Schneider et al, 2011; Zhu et al, 2012). Here, we show that PNE makes rats more impulsive and attenuates neural selectivity related to the direction of the response and monitoring of conflict in mPFC.
MATERIALS AND METHODS
Subjects
Ten male and 10 female Long Evans rats were obtained at 175–200 g from Charles River Labs. Rats used for recording were bred in our animal facility as described below. Due to unexpected loss, eight additional male control rats were obtained from Charles River Labs and were combined into a single control group due to homogeneity in behavior and neurophysiology. All experiments were approved by the University of Maryland College Park under university and NIH guidelines.
Prenatal Nicotine Exposure
Procedures were similar to those described by Schneider et al (2010). In a subset (n=5) of nulliparous female rats, nicotine was added to their only source of drinking water while the control mothers (n=5) were provided with unadulterated drinking water. Nicotine bitartrate (Sigma, St Louis, MO) was dissolved in water. To acclimate the nicotine-exposed dams to the taste of nicotine, the dosage was increased weekly over the course of 3 weeks (0.02 mg/ml, 0.04 mg/ml, and 0.06 mg/ml). The range of nicotine between 1 and 6 mg/kg/day has been shown to produce plasma nicotine levels in the range of 10–50 ng/ml in habitual smokers (Benowitz and Jacob, 1987). In addition, experiments that have administered nicotine via drinking water at doses comparable to ours have found plasma nicotine levels between 21 and 60 ng/ml (Paz et al, 2007; Schneider et al, 2010). Our mothers consumed an average of 5.93 mg/kg/day of nicotine during pregnancy, which is within the range shown to produce behavioral deficits in offspring without causing physical impairments (Paz et al, 2007; Schneider et al, 2010). Nicotine-exposed mothers in the present experiment consumed significantly less water than controls during pregnancy (98.89 ml/kg/day; 131 ml/kg/day; t-test; P<0.01) and gained weight at a slower rate prior to pregnancy (0.21% gain per day; 0.68% gain per day; t-test; P<0.01), characteristics that have been observed before by Schneider et al (2010). Pregnancy duration and fluid consumption comparisons are detailed in Supplementary Table 1.
All pups were cross-fostered to control mothers to isolate the effects of nicotine exposure prenatally and minimize unique rearing practices by nicotine-exposed mothers. Pups were not exposed to nicotine in any manner after birth. Cross-fostering was performed on postnatal day 3 to ensure that any handling of pups by experimenters did not cause maternal rejection (Zhu et al, 2012). Pups from the same litter were cross-fostered to the same control dam. As a result, all cross-fostering was successful and we obtained 39 PNE pups (mean litter size=13.0; sex ratio=56.6) and 45 control pups (mean litter size=12.3; sex ratio=49.0) from 3 nicotine-exposed mothers and 4 control mothers. The pups were weaned on postnatal day 21. We used male pups in all recording experiments because PNE has been shown to have more dramatic behavioral effects on males than females and ADHD-like diagnoses are more prevalent in males (Pauly et al, 2004; Peters and Tang, 1982; Romero and Chen, 2004; Vaglenova et al, 2004). Control and PNE pup weights were not significantly different from each other during first day of training (postnatal day 49; PNE=271 g; control=259 g; t-test; P=0.56). Eight male pups per group were randomly selected from three control (dam C1, n=3; dam C3, n=2; dam C5, n=3) and three nicotine-exposed mothers (dam N3, n=3; dam N4, n=3; dam N5, n=2) to undergo training and electrode surgery (see below).
Behavioral Task
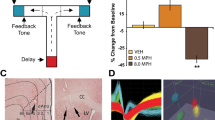
Each trial began by illumination of house lights that instructed the rat to nose-poke into the central port (Figure 1a) (Bryden et al, 2012). Nose-poking initiated a 1000 ms pre-cue delay period. At the end of this delay, a directional light to the animal’s left or right flashed for 100 ms. The trial was aborted if the rat exited the port at any time prior to offset of the directional cue light. On 80% of trials, presentation of either the left or right light signaled the direction in which the animal could respond to obtain reward in the corresponding fluid well below. On 20% of trials, the trial began in the same manner but, simultaneous with the rat exiting the nose-port, the light opposite to the location of the originally cued direction turned on and remained illuminated until the behavioral response was made. The trial was aborted if the rat took longer than 3 s to respond to the reward well after unpoke regardless of trial-type. On a fraction of sessions (28%), a variable 0 to 100-ms delay between unpoke and stop-cue onset (stop-signal delay) was implemented to further induce prepotency toward the GO cue. STOP trials were pseudorandomly interleaved with GO trials. Rats were required to inhibit the movement signaled by the first light and respond in the direction of the second light. After correct responses, rats were required to remain in the well 800–1000 ms before reward delivery (10% sucrose solution). The time necessary to stop and redirect behavior (stop change reaction time) on STOP trials was computed by calculating the difference between average correct STOP trial movement time and average correct GO trial movement time. The inter-trial intervals were 3 s and 4 s for correct and incorrect trials, respectively.

(a and b) An overview of the stop-signal task procedure (a) and trial-types (b) (Bryden et al, 2012). There were a total of four different trial-types: GO-left, GO-right, STOP-left–GO-right and STOP-right–GO-left; however, response direction (ie left and right) will be referenced to the directional preferences of individual neurons (preferred or nonpreferred). (c and d) Coronal slices ~3.3 mm anterior to bregma. Each dot represents the final position of the recording electrode for each animal based on histology. Dashed lines reflect the estimated track that the electrode traversed through the brain, and gray boxes represent estimated recorded area.
Surgery and Single-unit Recording
Procedures were the same as described previously (Bryden et al, 2011). A drivable bundle of 10 25 μm diameter FeNiCr wires (California Fine Wire) was chronically implanted in mPFC after training (+3.3 mm anterior to bregma, +0.6 mm lateral to bregma, 2 mm ventral to brain). Electrodes were driven 40 or 80 μm daily.
Data Analysis
Average movement times (calculated as the time from port exit to well entry), percent correct, and proportion of premature responses were calculated in each session then averaged across sessions. Differences between groups and/or trial types were compared via Wilcoxon (P<0.05). Significance levels were adjusted via Bonferroni correction.
Baseline firing was taken during a 1 s epoch starting 2 s prior to trial initiation (nose-poke). Task related activity was examined during the period between nose-poke exit and well entry (‘response epoch’), while the movement was being made and/or cancelled. Cells were excluded if both average baseline and response epoch firing rates deviated >5SD from the mean. To capture post-response activity, the 1 s epoch beginning at nose-poke exit was used in an analysis that examined firing on STOP and GO trials after either STOP or GO trials. This epoch was used to examine trial sequence effects. Abbreviations for trials that are differentiated by the trial-type preceding it are labeled as lowercase (GO (g) or STOP (s)) which indicates the trial-type before the trial marked by the uppercase letter (GO (G) or STOP (S)).
Least-squares multiple regression analysis was used to determine the number of cells where firing rate was significantly correlated with either the trial-type, movement time, and/or response direction parameters when variance for the two remaining factors was accounted for (Bryden, Roesch, 2015).
Y=β0+β1MovementTime+β2Direction+β3Trial-Type
where Y=log-transformed firing rate (spikes/s) during the response epoch, MovementTime=latency between unpoke and well entry, Direction=coded as: −1=ipsilateral and 1=contralateral, and Trial-Type=coded as: −1=GO and 1=STOP. A constant of 1 was added to each value of Y where the transform of zero equals zero.
Correlations between firing rate and behavioral measures (percent correct and movement time) were calculated using Pearson’s r after averaging values within each session. Correlation coefficients were determined to be statistically different via Student’s t-test after Fisher’s z-transformation for correlation coefficients.
RESULTS
Prenatal Nicotine Exposure Impairs Inhibitory Control
Rats were trained on the stop-signal task described in the Materials and Methods and Figure 1. Briefly, in the majority of trials (80%), rats were instructed to respond (GO) to a directional cue-light for reward. On the remaining 20% of trials, rats had to inhibit their initiated response and redirect movement in the opposite direction to achieve reward (STOP trials).
Rats in both control and PNE groups exhibited significantly slower movement speeds (latency from port exit to well entry; Figure 2a) and reduced accuracy on STOP trials (Figure 2b). Slower movement times resulted in better task performance consistent with a speed-accuracy tradeoff in both groups (correlation between movement time and percent correct; control; r=0.14; P<0.01; PNE; r=0.10; P<0.05). Consistent with this finding, incorrect STOP trial movement times were significantly faster than movement times on correctly performed STOP trials (Figure 2a; Wilcoxon; P<0.01). These results demonstrate that rats were generating a movement prior to illumination of the STOP signal, in response to illumination of the first cue light, and that inhibition and redirection of the behavioral response was necessary to correctly perform STOP trials.

(a and b) Session averaged movement times (ms) (a) and percent of correct responses (b). Error bars indicate SEM. Asterisks indicate group comparisons (Wilcoxon; P<0.05). (c) Histogram represents the proportion of premature responses (withdraw from the nose-poke prior to GO light offset) for control and PNE animals per session. All behavior was taken from neural recording sessions. Inset represents the average proportion of premature responses for control and PNE rats. Asterisk indicates significant mean difference (Wilcoxon; P<0.05). (d) Percentage of cells that showed significantly greater (increasing-type) or less (decreasing-type) activity during the response epoch (unpoke to well entry) relative to baseline (1 s epoch beginning 2 s prior to nose-poke). There were a total of 636 control cells and 558 PNE cells. (e) Percentage of cells per rat that were characterized as either increasing- or decreasing-type. Error bars indicate SEM. (f and g) Average activity (spikes/s) across GO trials, STOP trials, and STOP errors±SEM aligned on port exit for every increasing- (f) and decreasing-type (g) cell. As a temporally sensitive statistical comparison, the P-value for the t-test (black line) between control and PNE firing rates was taken from 100 ms epochs that slid every 10 ms and plotted in the graph below. The gray horizontal line refers to the P-value 0.05. PNE, prenatal nicotine exposure.
When comparing control and PNE groups, we found that PNE rats were significantly faster over all trial-types (Figure 2a; black vs gray; Wilcoxon; P<0.05). Although the two groups did not differ significantly in accurate performance of GO trials, PNE rats made significantly more errors on STOP trials than controls (Figure 2b black vs gray; Wilcoxon; P<0.05). In addition, PNE rats exhibited a greater proportion of premature responses (defined as leaving the nose-port before offset of the first cue light; Figure 2c; Wilcoxon; P<0.01). Note, these differences were significant when comparing the PNE group with the combined control group and with individual control groups (Wilcoxon; ps<0.01). We conclude that prenatal nicotine exposure limits the capacity for successful inhibitory control during performance of our task.
Impact of Prenatal Nicotine Exposure on Activity in mPFC
We recorded 636 and 558 mPFC neurons from control and PNE rats, respectively. The recording locations are illustrated in Figure 1c and d. Use of this task in the context of behavioral neurophysiology allows us to examine activity related to response inhibition and redirection of behavior. Trials during which the movement had to be stopped and redirected will be directly compared with responses on GO trials made in the same direction, which cannot be done with more typical stop-signal tasks.
We first determined how many neurons in each group exhibited activity that was significantly different during the response epoch (port exit to well entry) relative to baseline (1 s epoch beginning 2 s prior to trial initiation; Wilcoxon; P<0.05). In the control group, 19% (n=121) and 39% (n=249) of neurons significantly increased or decreased firing during the response epoch relative to baseline, respectively. In the PNE group, 22% (n=125) and 41% (n=228) of neurons exhibited significant increases or decreases during the response epoch, respectively. The proportion of increasing- and decreasing-type cells did not differ between groups (Figure 2d; increasing type: χ2, P=0.17; decreasing type: χ2, P=0.59). To provide evidence for homogeneity between rats within a group, we plotted the percentage of increasing- and decreasing-type neurons per rat. The control group did not differ from the PNE group (Figure 2e; increasing-type: Wilcoxon, P=0.43; decreasing-type: Wilcoxon, P=0.49). Even though the proportions of task-related neurons did not differ between groups, average activity in the PNE group was reduced in numerous task epochs across the population of both increasing- and decreasing-type neurons. This is illustrated by the mean firing rates (±SEM ribbons with sliding comparisons) in Figure 2f and g, which plots average firing (spikes/s) over time (aligned on port exit and averaged across trial-types).
Both increasing- and decreasing-type cells tended to be modulated by the direction of the response and/or type of trial. For example, the single neuron in Figure 3a exhibited increased firing during STOP trials relative to GO trials. Other neurons increased firing for responses made in a specific direction (Figure 3b), while other cells were modulated by both response direction and trial-type (Figure 3c). Average firing across all neurons is plotted in Supplementary Figures S1 and S2 and is described in the Supplementary Material.

(a–c) Neural firing of characteristic increasing-type cells on correct trials. The histogram below each raster plot is the average of the spikes during each individual time point across all iterations of that trial-type during the session. (a) This neuron was selective for trial-type in that activity was stronger under STOP trials relative to GO trials. (b) This neuron was selective for direction where activity was stronger when the animal responded to the right well. (c) This neuron was selective for direction and trial-type where activity was stronger for leftward responses, particularly during STOP trials. In rasters, blue diamonds and green triangles represent time of nose-poke at trial start and fluid well entry, respectively. (d) Results of the regression analysis (detailed in Materials and Methods) for every increasing- and decreasing-type cell. Each bar represents the percentage of cells where firing rate during the response epoch is correlated with a given parameter significantly and uniquely relative to the remaining parameters (partial r2; P<0.05). Many single neurons were selective for more than one parameter in this procedure. Details of this overlap in Supplementary Figure S3. Asterisk reveals significantly different proportions (χ2; P<0.05).
To determine if the counts of neurons exhibiting task-related effects were different between the two groups, we performed the least-squares multiple regression approach (see Materials and Methods) for all task-modulated neurons (ie, increasing- and decreasing-type neurons). This analysis determines the number of cells where firing rate was significantly correlated with either the trial-type (STOP/GO), movement time, and/or response direction parameters. The counts of neurons in control rats that were significantly modulated by response direction outnumbered those observed in PNE rats (Figure 3d; χ2; P<0.01). Counts of neurons modulated by movement time and trial-type were not significantly different between groups (χ2, ps>0.62). Though these results are presented as discrete categories, it was common that a neuron was significantly modulated by more than one parameter. For breakdowns of selective neurons within each category see Supplementary Figure 3. This regression analysis demonstrates that the recruitment of mPFC neurons associated with the direction of the response was affected by prenatal nicotine exposure, but overall encoding of movement time and signaling directly related to inhibitory control (ie, differential firing based on trial-type; STOP vs GO) were not different between the two groups.
Activity in mPFC was Correlated with Behavioral Performance
The data described above demonstrate that both neural activity and performance were reduced in PNE rats. Here we ask if the two were correlated. Specifically, we determine via correlation whether average firing rates were correlated with behavioral measures of accuracy and movement time between sessions.
For increasing-type cells, firing during the response epoch was positively correlated with percent correct. The regression was significant for the control (Figure 4a; r=0.37; P<0.01) but not the PNE group (Figure 4b; r=0.17; P=0.07). Further, these correlations were significantly different from one another (Figure 4a vs B; t-test; P<0.05). The correlation between movement time and firing rate was significant for increasing-type cells only in the PNE group (Figure 4d; r=0.25; P<0.01) but this correlation did not significantly differ from the control group (Figure 4c vs D; t-test; P=0.42). However, for decreasing-type neurons, there was a positive correlation between movement time and firing rate, which was only significant in the control group (Figure 4g; r=0.29; P<0.01) and was significantly different from the PNE group (Figure 4g vs Figure 4h; t-test; P<0.01). Finally, the correlation between firing rate and percent correct in decreasing-type cells was not significant in the control group (Figure 4e; r=0.07, P=0.24), but was significantly negatively correlated in the PNE group (Figure 4f; r=−0.20; P<0.01). These correlations differed significantly (Figure 4e vs Figure 4f; t-test; P<0.01). Thus overall, when activity was high for increasing- and decreasing-type neurons, rats were better and slower, respectively. These correlations were not present in PNE rats. Furthermore, in sessions where decreasing-type neuronal activity was high, PNE rats tended to perform the task poorly. Importantly, the correlation results above are not simply a product of mPFC hypoactivation as these effects are maintained in a sub-selected population of firing-rate-matched neurons between groups (Supplementary Material).

(a and b) Plots of firing rate on correct trials during the response epoch versus percent correct for all trial-types averaged for each session are shown for increasing- (a and b) and decreasing-type (e and f) cells in the control (a, e) and PNE (b, f) groups. Plots of firing rate on correct trials versus movement time averaged on each session for increasing- (c and d) and decreasing-type (g and h) cells in control (c, g) and PNE (d, h) groups. Each dot represents a single neuron. PNE, prenatal nicotine exposure.
Increased mPFC Activity During Heightened Response Conflict
The rationale behind using STOP trials on only 20% of trials is to induce a tendency for the animal to follow the initial cue light on every trial. Given the random interleaving of our trial-types and the fluidity with which rats perform the task, it is unsurprising that we observe behavioral effects contingent on the identity of the previous trial. Both groups of animals were less accurate on STOP trials following GO trials (gS) compared with STOP trials following STOP trials (sS) (Figure 5e; Wilcoxon; P<0.05), however, this effect was amplified in PNE rats (Figure 5e; Wilcoxon; P<0.05). This effect is presumably due to the heightened prepotency to respond to the first cue light induced by the previous GO trial, which made it more difficult for the rats to suppress the initial response on STOP trials. This demonstrates that the ‘competition’ or ‘conflict’ between the two opposing responses (GO vs STOP) is highest on STOP trials when the previous trial was a GO trial, and it is during these trials that PNE rats performed the worst. Frontal areas are thought to be important for monitoring conflict under such situations (Botvinick et al, 2001; Carter and van Veen, 2007; Mayr et al, 2003; Oualian and Gisquet-Verrier, 2010), therefore we next asked whether mPFC was modulated by the added conflict induced by the identity of the previous trial, and if this encoding might be disrupted by PNE.

(a and b) Population histograms of all mPFC neurons that increased significantly above baseline in control (a) and PNE (b) groups. Activity is aligned to port exit. Blue lines refer to all GO trials. Red lines represent STOP trials preceded by GO trials (‘gS’). Orange lines indicate trials where a STOP trial is preceded by a STOP trial (‘sS’). Direction preference was determined in each cell by calculating the direction that elicited the greatest firing rate during the epoch from port exit to well entry (‘response epoch’). Therefore, as defined by our analysis, the preferred direction (thick lines) is always higher than the nonpreferred direction (thin lines) during the response epoch. The direction of STOP trials is always referred to as the ultimate response the animal made. (c and d) Indices compare the difference in firing between the three trial-types presented in (a and b) for control (c) and PNE (d) groups. Leftmost distribution calculates the differences between gS and GO trials divided by the sum for each cell. The middle distribution marks the difference between sS and GO trials divided by the sum. Rightmost distribution computes the difference between gS and sS trials divided by the sum. Activity for all distributions taken during the 1 s epoch beginning at port exit to capture post decision activity and significant shifts from zero are determined via Wilcoxon (P<0.05). Distributions between groups are compared via Wilcoxon (P<0.05). (e and f) Percent of correct responses (e) and movement latencies (f) per session for sS and gS trials. Movement times are calculated as the latency from port exit to well entry. Asterisks indicate significant mean differences (Wilcoxon; P<0.05). mPFC, medial prefrontal cortex; PNE, prenatal nicotine exposure.
Figure 5a and b plots average activity on STOP trials when the previous trial was either a GO trial-type (gS, red) or a STOP trial-type (sS, orange) for the control (Figure 5a) and PNE (Figure 5b) groups. For reference, GO (low conflict) trials during these sessions are plotted in blue. Remarkably, when conflict was the highest (gS trials; red), activity in mPFC was the most pronounced in control rats. Differences in firing between the three trials are quantified in Figure 5c and d, which compares the difference between higher and lower conflict trial-types (ie, gS-GO, sS-GO, and gS-sS) for each neuron. In the control group, the distribution comparing gS with GO (ie, gS-GO) was significantly shifted in the positive direction (Figure 5c left; Wilcoxon; P<0.01), whereas this distribution in the PNE group was not significantly shifted (Figure 5d left; Wilcoxon; P=0.87). When comparing activity on sS trials to GO trials, neither group exhibited a significantly shifted distribution (sS-GO; Figure 5c and d middle; Wilcoxon; ps>0.32). Importantly, direct comparison of gS and sS trials revealed a statistically shifted distribution in the control group (Figure 5c right; Wilcoxon; P<0.01), which differed from the equivalent distribution in the PNE group (Figure 5c right vs Figure 5d right; Wilcoxon; P<0.01). Thus, when conflict was high, firing was significantly stronger in the mPFC of control but not PNE rats. Importantly, this group difference is not simply a product of the identity of the previous trial because these differences were not observed between gG and sG trials (Supplementary Figure S4), but instead reflects the added difficulty of inhibiting the initial GO response on STOP trials. These results demonstrate that the mPFC in control animals was able to differentiate STOP trials based on the conflict induced by the preceding trial type and this neural correlate of conflict monitoring was disrupted in animals prenatally exposed to nicotine. This could explain the marked behavioral deficits on gS trials relative to sS trials in the PNE group (Figure 5e). Importantly, this conflict monitoring effect is not simply due to mPFC hypoactivation in PNE rats. In a group of sub-selected neurons matched for firing rate across groups, control neurons still capably differentiated between gS and sS trials (details in Supplementary Material).
DISCUSSION
In summary, we show that PNE makes rats impulsive, disrupts neural signals related to response encoding and conflict monitoring, and reduces overall firing in mPFC. Further, we demonstrated that correlations between neural firing and performance (accuracy and movement speed) were altered after PNE.
An underactive prefrontal cortex (ie, ‘hypofrontality’) is commonly found in addiction, ADHD, and schizophrenia (Andreasen et al, 1992; Berman et al, 1992; Carter et al, 1998; Dickstein et al, 2006; Rubia et al, 1999; Volkow et al, 2004), all of which are psychiatric disorders characterized by diminished executive function. Low firing rates might contribute to deficits in cognitive control; however, it is unlikely to be the sole source of dysfunction in our data set because the activity of rate-matched controls still reflected response conflict (see Supplementary Material). Other experimental work has reported that interference of mPFC impairs performance on response inhibition tasks as measured by stop trial accuracy and premature responding during reaction time tasks (Bari et al, 2011; Risterucci et al, 2003). Taken together, this work points to mPFC as a critical player during response inhibition and suggests that reduced prefrontal activation/function in disorders such as ADHD drive behavioral impairments (Euston et al, 2012).
Consistent with this hypothesis, patients diagnosed with ADHD have been successfully treated with the noradrenaline and dopamine (eg, methylphenidate and atomoxetine) reuptake inhibitors (Aron et al, 2003; Bari et al, 2009; Bedard et al, 2003; DeVito et al, 2009; Robinson et al, 2008; Tannock et al, 1989) that have been shown to impact prefrontal cortex in both humans and rats. For instance, in humans, atomoxetine administration increases inferior frontal activity in human participants (Chamberlain et al, 2009) and methylphenidate reverses ADHD-associated hypofrontality (Vaidya et al, 1998). In rats, atomoxetine administration increases the immediate early gene c-Fos in mPFC (Bymaster et al, 2002). This all points to decreased prefrontal firing as a root of these and other psychiatric disorders that impair executive function. Consistent with this hypothesis, we show that firing in mPFC is reduced in PNE rats. We further demonstrate reduced selectivity related to response encoding and conflict monitoring, and an absence of correlations between firing and behavior output. Our results provide a mechanism by which these drugs might ameliorate behavior deficits. Specifically, these drugs are likely to improve function by repairing signals related to response generation and detection of heightened conflict.
Maternal smoking is a risk factor for many psychiatric disorders (Blood-Siegfried and Rende, 2010; Button et al, 2007; Ernst et al, 2001; Fried and Watkinson, 2001; Jacobsen et al, 2007; Milberger et al, 1996, 1998; Pauly and Slotkin, 2008; Schmitz et al, 2006; Thapar et al, 2003; Tong and McMichael, 1992) and is still a common practice according to Substance Abuse & Mental Health Services which reported in 2012 that one in five women smoke during pregnancy. In addition to being an important issue in its own right, prenatal nicotine exposure has gained considerable traction as a suitable model for impulsive behavior as seen in ADHD. Exposing pregnant rodents to nicotine via drinking water produces offspring that bear striking resemblance to human ADHD both symptomatically and in treatment efficacy (Pauly et al, 2004; Peters and Tang, 1982; Peters et al, 1979; Schneider et al, 2010). This exposure has also been shown to have a genetic component in that pups of prenatally exposed pups also show behavioral impairments (Zhu et al, 2014). Previous studies have shown that prenatal nicotine exposure via drinking water at the same dose used in our study (0.06 mg/ml) produces increased anticipatory responses on the five-choice serial reaction time task in rats (Schneider et al, 2011). Similar results have been obtained in mice, where prenatal nicotine exposure via drinking water reduces cingulate cortex volume, reduces prefrontal dopamine turnover, and induces hyperactivity, which was diminished by oral methylphenidate treatment (Zhu et al, 2012). Thus, there is a substantial and meaningful overlap between human ADHD research and the rodent prenatal nicotine model.
It is highly unlikely that our results can be explained by impairments outside the realm of disrupted brain areas involved in executive control. As mentioned above, others have used the same method of nicotine administration, and shown impairment in the five-choice serial reaction time task (Schneider et al, 2010). Importantly, in that study, rats also performed a battery of sensorimotor tasks to assess different developmental milestones. PNE rats did exhibit lower birth weights and delayed sensorimotor development, but differences were not apparent prior to testing in the five-choice task that occurred around 3 months of age. There were no significant differences in weight between PNE and control rats on the first day of our study (postnatal day 49). In addition, PNE rats were actually faster over all trial-types and performed similarly on GO trials compared with controls. Thus, it is unlikely that developmental problems beyond those related to executive control can account for the behavioral differences described here.
We conclude that PNE reduces activity in mPFC, an area known to be critical for executive control including response inhibition. Reduced activity in mPFC after PNE is correlated with poor impulse control and is likely to be directly related to elevated levels of drug seeking observed in ADHD and in rats that have chronically self-administered cocaine. Like prenatal nicotine, prolonged cocaine self-administration leads to mPFC hypoactivation and increased drug seeking, both of which are rescued through optogenetic stimulation of prelimbic prefrontal cortex (Chen et al, 2013). Together these results suggest that reduced firing in mPFC after exposure to prenatal nicotine might not only impair normal everyday executive control functions but increase one’s predisposition to addiction (Dalley et al, 2011; Jentsch and Taylor, 1999). Based on these findings and the existence of a positive correlation between activity and behavioral performance, this work implies that global increases in mPFC firing may improve performance in animals during tasks that assess executive control and response inhibition.
FUNDING AND DISCLOSURE
This work was supported by grants from the NIDA (R01DA031695, MRR), Gemstone Honors Program, and HHMI. The authors declare no conflicts of interest.
References
Andreasen NC, Rezai K, Alliger R, Swayze VW 2nd, Flaum M, Kirchner P et al (1992). Hypofrontality in neuroleptic-naive patients and in patients with chronic schizophrenia. Assessment with xenon 133 single-photon emission computed tomography and the Tower of London. Arch Gen Psychiatry 49: 943–958.
Aron AR, Dowson JH, Sahakian BJ, Robbins TW (2003). Methylphenidate improves response inhibition in adults with attention-deficit/hyperactivity disorder. Biol Psychiatry 54: 1465–1468.
Bari A, Eagle DM, Mar AC, Robinson ES, Robbins TW (2009). Dissociable effects of noradrenaline, dopamine, and serotonin uptake blockade on stop task performance in rats. Psychopharmacology (Berl) 205: 273–283.
Bari A, Mar AC, Theobald DE, Elands SA, Oganya KC, Eagle DM et al (2011). Prefrontal and monoaminergic contributions to stop-signal task performance in rats. J Neurosci 31: 9254–9263.
Bedard AC, Ickowicz A, Logan GD, Hogg-Johnson S, Schachar R, Tannock R (2003). Selective inhibition in children with attention-deficit hyperactivity disorder off and on stimulant medication. J Abnorm Child Psychol 31: 315–327.
Benowitz N, Jacob P III (1987) Metabolism, pharmacokinetics and pharmacodynamics of nicotine in Man. In: Martin W, Van Loon G, Iwamoto E, Davis L (eds), Tobacco Smoking and Nicotine, Vol 31. Springer: New York, USA, pp 357–373.
Berman KF, Torrey EF, Daniel DG, Weinberger DR (1992). Regional cerebral blood flow in monozygotic twins discordant and concordant for schizophrenia. Arch Gen Psychiatry 49: 927–934.
Blood-Siegfried J, Rende EK (2010). The long-term effects of prenatal nicotine exposure on neurologic development. J Midwifery Womens Health 55: 143–152.
Botvinick MM, Braver TS, Barch DM, Carter CS, Cohen JD (2001). Conflict monitoring and cognitive control. Psychol Rev 108: 624–652.
Bryden DW, Burton AC, Kashtelyan V, Barnett BR, Roesch MR (2012). Response inhibition signals and miscoding of direction in dorsomedial striatum. Front Integr Neurosci 6: 69.
Bryden DW, Johnson EE, Diao X, Roesch MR (2011). Impact of expected value on neural activity in rat substantia nigra pars reticulata. Eur J Neurosci 33: 2308–2317.
Bryden DW, Roesch MR (2015). Executive control signals in orbitofrontal cortex during response inhibition. J Neurosci 35: 3903–3914.
Button TM, Maughan B, McGuffin P (2007). The relationship of maternal smoking to psychological problems in the offspring. Early Hum Dev 83: 727–732.
Bymaster FP, Katner JS, Nelson DL, Hemrick-Luecke SK, Threlkeld PG, Heiligenstein JH et al (2002). Atomoxetine increases extracellular levels of norepinephrine and dopamine in prefrontal cortex of rat: a potential mechanism for efficacy in attention deficit/hyperactivity disorder. Neuropsychopharmacology 27: 699–711.
Carter CS, Perlstein W, Ganguli R, Brar J, Mintun M, Cohen JD (1998). Functional hypofrontality and working memory dysfunction in schizophrenia. Am J Psychiatry 155: 1285–1287.
Carter CS, van Veen V (2007). Anterior cingulate cortex and conflict detection: an update of theory and data. Cogn Affect Behav Neurosci 7: 367–379.
Chamberlain SR, Hampshire A, Muller U, Rubia K, Del Campo N, Craig K et al (2009). Atomoxetine modulates right inferior frontal activation during inhibitory control: a pharmacological functional magnetic resonance imaging study. Biol Psychiatry 65: 550–555.
Chen BT, Yau HJ, Hatch C, Kusumoto-Yoshida I, Cho SL, Hopf FW et al (2013). Rescuing cocaine-induced prefrontal cortex hypoactivity prevents compulsive cocaine seeking. Nature 496: 359–362.
Christakou A, Robbins TW, Everitt BJ (2001). Functional disconnection of a prefrontal cortical-dorsal striatal system disrupts choice reaction time performance: implications for attentional function. Behav Neurosci 115: 812–825.
Cornelius MD, Day NL (2009). Developmental consequences of prenatal tobacco exposure. Curr Opin Neurol 22: 121–125.
Dalley JW, Everitt BJ, Robbins TW (2011). Impulsivity, compulsivity, and top-down cognitive control. Neuron 69: 680–694.
DeVito EE, Blackwell AD, Clark L, Kent L, Dezsery AM, Turner DC et al (2009). Methylphenidate improves response inhibition but not reflection-impulsivity in children with attention deficit hyperactivity disorder (ADHD). Psychopharmacology (Berl) 202: 531–539.
Dickstein SG, Bannon K, Castellanos FX, Milham MP (2006). The neural correlates of attention deficit hyperactivity disorder: an ALE meta-analysis. J Child Psychol Psychiatry 47: 1051–1062.
Eagle DM, Baunez C, Hutcheson DM, Lehmann O, Shah AP, Robbins TW (2008). Stop-signal reaction-time task performance: role of prefrontal cortex and subthalamic nucleus. Cereb Cortex 18: 178–188.
Eagle DM, Robbins TW (2003). Lesions of the medial prefrontal cortex or nucleus accumbens core do not impair inhibitory control in rats performing a stop-signal reaction time task. Behav Brain Res 146: 131–144.
Emeric EE, Brown JW, Leslie M, Pouget P, Stuphorn V, Schall JD (2008). Performance monitoring local field potentials in the medial frontal cortex of primates: anterior cingulate cortex. J Neurophysiol 99: 759–772.
Ernst M, Moolchan ET, Robinson ML (2001). Behavioral and neural consequences of prenatal exposure to nicotine. J Am Acad Child Adolesc Psychiatry 40: 630–641.
Euston DR, Gruber AJ, McNaughton BL (2012). The role of medial prefrontal cortex in memory and decision making. Neuron 76: 1057–1070.
Fried PA, Watkinson B (2001). Differential effects on facets of attention in adolescents prenatally exposed to cigarettes and marihuana. Neurotoxicol Teratol 23: 421–430.
Hanes DP, Patterson WF 2nd, Schall JD (1998). Role of frontal eye fields in countermanding saccades: visual, movement, and fixation activity. J Neurophysiol 79: 817–834.
Jacobsen LK, Slotkin TA, Mencl WE, Frost SJ, Pugh KR (2007). Gender-specific effects of prenatal and adolescent exposure to tobacco smoke on auditory and visual attention. Neuropsychopharmacology 32: 2453–2464.
Jentsch JD, Taylor JR (1999). Impulsivity resulting from frontostriatal dysfunction in drug abuse: implications for the control of behavior by reward-related stimuli. Psychopharmacology (Berl) 146: 373–390.
Mayr U, Awh E, Laurey P (2003). Conflict adaptation effects in the absence of executive control. Nat Neurosci 6: 450–452.
Mick E, Biederman J, Faraone SV, Sayer J, Kleinman S (2002). Case-control study of attention-deficit hyperactivity disorder and maternal smoking, alcohol use, and drug use during pregnancy. J Am Acad Child Adolesc Psychiatry 41: 378–385.
Milberger S, Biederman J, Faraone SV, Chen L, Jones J (1996). Is maternal smoking during pregnancy a risk factor for attention deficit hyperactivity disorder in children? Am J Psychiatry 153: 1138–1142.
Milberger S, Biederman J, Faraone SV, Jones J (1998). Further evidence of an association between maternal smoking during pregnancy and attention deficit hyperactivity disorder: findings from a high-risk sample of siblings. J Clin Child Psychol 27: 352–358.
Morgan AB, Lilienfeld SO (2000). A meta-analytic review of the relation between antisocial behavior and neuropsychological measures of executive function. Clin Psychol Rev 20: 113–136.
Muhammad A, Mychasiuk R, Nakahashi A, Hossain SR, Gibb R, Kolb B (2012). Prenatal nicotine exposure alters neuroanatomical organization of the developing brain. Synapse 66: 950–954.
Muneoka K, Ogawa T, Kamei K, Muraoka S, Tomiyoshi R, Mimura Y et al (1997). Prenatal nicotine exposure affects the development of the central serotonergic system as well as the dopaminergic system in rat offspring: involvement of route of drug administrations. Brain Res Dev Brain Res 102: 117–126.
Mychasiuk R, Muhammad A, Gibb R, Kolb B (2013). Long-term alterations to dendritic morphology and spine density associated with prenatal exposure to nicotine. Brain Res 1499: 53–60.
Navarro HA, Mills E, Seidler FJ, Baker FE, Lappi SE, Tayyeb MI et al (1990). Prenatal nicotine exposure impairs beta-adrenergic function: persistent chronotropic subsensitivity despite recovery from deficits in receptor binding. Brain Res Bull 25: 233–237.
Oualian C, Gisquet-Verrier P (2010). The differential involvement of the prelimbic and infralimbic cortices in response conflict affects behavioral flexibility in rats trained in a new automated strategy-switching task. Learn Mem 17: 654–668.
Pauly JR, Slotkin TA (2008). Maternal tobacco smoking, nicotine replacement and neurobehavioural development. Acta Paediatr 97: 1331–1337.
Pauly JR, Sparks JA, Hauser KF, Pauly TH (2004). In utero nicotine exposure causes persistent, gender-dependant changes in locomotor activity and sensitivity to nicotine in C57Bl/6 mice. Int J Dev Neurosci 22: 329–337.
Paz R, Barsness B, Martenson T, Tanner D, Allan AM (2007). Behavioral teratogenicity induced by nonforced maternal nicotine consumption. Neuropsychopharmacology 32: 693–699.
Peters DA, Tang S (1982). Sex-dependent biological changes following prenatal nicotine exposure in the rat. Pharmacol Biochem Behav 17: 1077–1082.
Peters DA, Taub H, Tang S (1979). Postnatal effects of maternal nicotine exposure. Neurobehav Toxicol 1: 221–225.
Pezze M, McGarrity S, Mason R, Fone KC, Bast T (2014). Too little and too much: hypoactivation and disinhibition of medial prefrontal cortex cause attentional deficits. J Neurosci 34: 7931–7946.
Risterucci C, Terramorsi D, Nieoullon A, Amalric M (2003). Excitotoxic lesions of the prelimbic-infralimbic areas of the rodent prefrontal cortex disrupt motor preparatory processes. Eur J Neurosci 17: 1498–1508.
Robinson ES, Eagle DM, Mar AC, Bari A, Banerjee G, Jiang X et al (2008). Similar effects of the selective noradrenaline reuptake inhibitor atomoxetine on three distinct forms of impulsivity in the rat. Neuropsychopharmacology 33: 1028–1037.
Romero RD, Chen WJ (2004). Gender-related response in open-field activity following developmental nicotine exposure in rats. Pharmacol Biochem Behav 78: 675–681.
Rubia K, Overmeyer S, Taylor E, Brammer M, Williams SC, Simmons A et al (1999). Hypofrontality in attention deficit hyperactivity disorder during higher-order motor control: a study with functional MRI. Am J Psychiatry 156: 891–896.
Schall JD, Boucher L (2007). Executive control of gaze by the frontal lobes. Cogn Affect Behav Neurosci 7: 396–412.
Schmitz M, Denardin D, Laufer Silva T, Pianca T, Hutz MH, Faraone S et al (2006). Smoking during pregnancy and attention-deficit/hyperactivity disorder, predominantly inattentive type: a case-control study. J Am Acad Child Adolesc Psychiatry 45: 1338–1345.
Schneider T, Bizarro L, Asherson PJ, Stolerman IP (2010). Gestational exposure to nicotine in drinking water: teratogenic effects and methodological issues. Behav Pharmacol 21: 206–216.
Schneider T, Ilott N, Brolese G, Bizarro L, Asherson PJ, Stolerman IP (2011). Prenatal exposure to nicotine impairs performance of the 5-choice serial reaction time task in adult rats. Neuropsychopharmacology 36: 1114–1125.
Seidler FJ, Levin ED, Lappi SE, Slotkin TA (1992). Fetal nicotine exposure ablates the ability of postnatal nicotine challenge to release norepinephrine from rat brain regions. Brain Res Dev Brain Res 69: 288–291.
Slotkin TA (1998). Fetal nicotine or cocaine exposure: which one is worse? J Pharmacol Exp Ther 285: 931–945.
Tannock R, Schachar RJ, Carr RP, Chajczyk D, Logan GD (1989). Effects of methylphenidate on inhibitory control in hyperactive children. J Abnorm Child Psychol 17: 473–491.
Thapar A, Fowler T, Rice F, Scourfield J, van den Bree M, Thomas H et al (2003). Maternal smoking during pregnancy and attention deficit hyperactivity disorder symptoms in offspring. Am J Psychiatry 160: 1985–1989.
Tong S, McMichael AJ (1992). Maternal smoking and neuropsychological development in childhood: a review of the evidence. Dev Med Child Neurol 34: 191–197.
Vaglenova J, Birru S, Pandiella NM, Breese CR (2004). An assessment of the long-term developmental and behavioral teratogenicity of prenatal nicotine exposure. Behav Brain Res 150: 159–170.
Vaidya CJ, Austin G, Kirkorian G, Ridlehuber HW, Desmond JE, Glover GH et al (1998). Selective effects of methylphenidate in attention deficit hyperactivity disorder: a functional magnetic resonance study. Proc Natl Acad Sci USA 95: 14494–14499.
Volkow ND, Fowler JS, Wang GJ, Swanson JM (2004). Dopamine in drug abuse and addiction: results from imaging studies and treatment implications. Mol Psychiatry 9: 557–569.
Wickstrom R (2007). Effects of nicotine during pregnancy: human and experimental evidence. Curr Neuropharmacol 5: 213–222.
Zhu J, Lee KP, Spencer TJ, Biederman J, Bhide PG (2014). Transgenerational transmission of hyperactivity in a mouse model of ADHD. J Neurosci 34: 2768–2773.
Zhu J, Zhang X, Xu Y, Spencer TJ, Biederman J, Bhide PG (2012). Prenatal nicotine exposure mouse model showing hyperactivity, reduced cingulate cortex volume, reduced dopamine turnover, and responsiveness to oral methylphenidate treatment. J Neurosci 32: 9410–9418.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Supplementary Information accompanies the paper on the Neuropsychopharmacology website
Rights and permissions
About this article

Cite this article
Bryden, D., Burton, A., Barnett, B. et al. Prenatal Nicotine Exposure Impairs Executive Control Signals in Medial Prefrontal Cortex. Neuropsychopharmacol 41, 716–725 (2016). https://doi.org/10.1038/npp.2015.197
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2015.197

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}