
Abstract
RNA targets of multitargeted RNA-binding proteins (RBPs) can be studied by various methods including mobility shift assays, iterative in vitro selection techniques and computational approaches. These techniques, however, cannot be used to identify the cellular context within which mRNAs associate, nor can they be used to elucidate the dynamic composition of RNAs in ribonucleoprotein (RNP) complexes in response to physiological stimuli. But by combining biochemical and genomics procedures to isolate and identify RNAs associated with RNA-binding proteins, information regarding RNA–protein and RNA–RNA interactions can be examined more directly within a cellular context. Several protocols — including the yeast three-hybrid system and immunoprecipitations that use physical or chemical cross-linking — have been developed to address this issue. Cross-linking procedures in general, however, are limited by inefficiency and sequence biases. The approach outlined here, termed RNP immunoprecipitation–microarray (RIP-Chip), allows the identification of discrete subsets of RNAs associated with multi-targeted RNA-binding proteins and provides information regarding changes in the intracellular composition of mRNPs in response to physical, chemical or developmental inducements of living systems. Thus, RIP-Chip can be used to identify subsets of RNAs that have related functions and are potentially co-regulated, as well as proteins that are associated with them in RNP complexes. Using RIP-Chip, the identification and/or quantification of RNAs in RNP complexes can be accomplished within a few hours or days depending on the RNA detection method used.
*Note: In the version of the article originally published, in the last sentence of the ANTICIPATED RESULTS section, the callout should be to reference 19 instead of 18. The error has been corrected in the HTML and PDF versions of the article.
Similar content being viewed by others

Introduction
Global gene expression analysis has received much attention in recent years owing to the growing availability of microarray and other high-throughput sequencing technologies. Most studies have focused on profiling the steady-state levels of mRNA of various cellular, developmental and disease responses using transcriptomics. It is believed, however, that the steady-state levels of mRNAs in a cell do not always directly correlate with the steady-state protein levels1,2. Much of this discrepancy may be accounted for by RNA processing and translational events that occur post-transcriptionally.
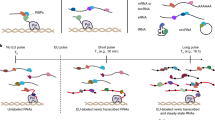
Post-transcriptional regulation of gene expression is a ribonucleoprotein-driven process, which involves RBPs and noncoding RNAs (e.g., microRNAs) that affect splicing, nuclear export, subcellular localization, mRNA decay and translation3,4,5,6,7. One of the challenges for understanding post-transcriptional control of gene expression is identifying the RNAs associated with RBPs in a cellular context. Traditionally, mRNA targets have been identified individually using in vitro techniques such as cross-linking with ultraviolet light, nitrocellulose filter binding and RNA electromobility shift assays (REMSAs)8. Although these methods have provided abundant biochemical information, they are limited in their ability to identify de novo targets when starting with an RBP and its unknown RNA targets. Additionally, bioinformatic algorithms have been developed to search for previously uncharacterized mRNA targets of particular RBPs, but such approaches are plagued by the fact that singular RNA binding sites tend to be very small (4–12 nucleotides) and therefore appear more frequently among mRNAs than expected. Biochemical methods for identifying de novo targets en masse were initially addressed by applying iterative in vitro selection against genomic RNA libraries9. This led to the elucidation of over 100 mRNAs derived from human brain cells that were able to bind to the HuB (Hel-N1) RNA-binding protein. But because this was an in vitro binding procedure, there was no certainty that the RNAs were ever in the same RNP context or associated directly with the RBP. Thus, although these in vitro technologies can identify RNA components and may suggest global binding sites for RBPs, they cannot be used to determine RNA species within the context of cell extracts or the dynamics of RNP interactions after physiological perturbations. One solution to finding in vivo RNA substrates of RBPs has been the yeast three-hybrid system10,11,12. Although this creative adaptation of the yeast two-hybrid system has been used to identify some cognate targets of RBPs, it is limited in that it requires an artificial target reporter, and it has not been used to simultaneously identify multiple targets en masse. Moreover, there are limitations to the procedure, which concern the sequence preferences for transcription by yeast RNA polymerase and structural constraints on potential RNA-binding elements. For example, RNAs that are uridylate-rich tend to cause premature termination by the yeast transcription apparatus, making them unavailable to interact with RBPs that prefer single-stranded U-rich RNA binding sites. Also, secondary structures in RNAs can significantly influence the outcome of a three-hybrid screen. Indeed, important examples of this type of mRNA target discovery have emerged using this procedure12.
The RIP-Chip (sometimes called RIP-on-Chip or RIP-SEQ) protocol described here allows the identification of multiple RNA targets of RBPs globally and within the context of a cell extract that may or may not have been subjected to physical or chemical cross-linking. To accomplish these goals with cytoplasmic RNAs, we use a mild lysis buffer and optimized conditions, which leave nuclei essentially intact and minimize inappropriate exchange of mRNAs during subsequent immunoprecipitation13,14,15,16. Other conditions that use nuclear extracts have also been adapted for this procedure17. For standard RIP-Chip, extracts are immunoprecipitated and the pellets washed extensively, the RNP is then released and dissociated into RNA and protein as the RNA is extracted. Once the RNA is purified, it can be detected by various methods including microarray analysis or high-throughput sequencing. More recently, Mili and Steitz demonstrated the ability of c-fos mRNA, a known target of HuR, to migrate to HuR within an extract after incubation of extracts from two different cell types18. Indeed, it has been known for many years that exogenous RNAs can bind to RBPs when added to cell extracts. Thus it is likely that small pools of free RNAs and RBPs are present in all cells, and are available for post-lysis interactions. Our RIP-Chip conditions are optimized to minimize inappropriate interactions13,14,15,19.
During the development of RIP-Chip we experimented with chemical cross-linking using reversible formaldehyde (potentially a hazardous compound), and found the procedures cumbersome and yielding high background binding20. After the advent of RIP-Chip several related protocols were reported, which use RNA–protein cross-linking to identify RNAs bound to RBPs17,21,22,23,24,25,26. But we have not found cross-linking to be necessary to identify RNAs associated with RBPs using cell extracts, and prefer to avoid any potential artifacts that may be introduced because these reagents have the potential to severely reduce cell lysis efficiency, to introduce sequence biases, increase background and to be incompletely reversible19,24. More recently, Kaneko and Manley found that using 0.1% formaldehyde rather than 1% formaldehyde for cross-linking RNA to RNA polymerase II significantly improves the quality and recovery of bound RNA17.
Several factors need to be considered when optimizing a RIP-Chip protocol. First, extracts are kept cold and immediately frozen after lysis and centrifugation. Upon thawing of cell lysates, the extract is diluted tenfold. Next it should be noted that the immunoprecipitation and washing conditions may vary depending on the specific mRNP being investigated. It may be necessary to use more stringent wash conditions such as adding from 0.5 M to 3 M urea, 0.1% or less SDS, or deoxycholate to the wash buffer to reduce background. An additional consideration is the accessibility of the epitope (or tag) to which the immunoprecipitating antibody is directed. Some RBPs may be obstructed to some degree by other components of the RNP complex or to be shielded in a cellular subcompartment. We have found it useful in some instances to use an epitope-tagged RBP rather than endogenous proteins for mRNP isolation. Whereas exogenously expressing a tagged RBP has the potential to affect function, exceedingly high levels of expression are not necessary. For example, the PAB RBP is autoregulated by adenylates in the 5′ UTR of its mRNA, and this can balance exogenous and endogenous levels of the protein19. It may also be possible to further reduce background in a RIP-Chip assay by the use of tandem affinity purified (TAP) tags27,28. Biotinylated tags are also useful in reducing background because they allow for harsher washing conditions because of their high affinity for the strepavidin ligand29. Furthermore, as discussed below, the introduction of EDTA to the immunoprecipitation reaction can be useful for reducing background and gaining access to epitopes, presumably by dissociating ribosomes from mRNP complexes13,14,15,16,30.
Once RNA is extracted from the isolated mRNP complexes, there are several methods that may be used to identify the global set of mRNA targets. As far as we have been able to assess using RT-PCR, the mRNAs recovered using RIP-Chip are intact and of full length. Originally, spotted arrays with a few hundred genes were used to develop these procedures13. But alternative detection technologies as well as newer generations of arrays have been introduced that have also been applied to RIP-Chip. For example, a new technology that may be used to detect mRNA targets involves massively parallel sequencing such as single molecule platform sequencing (SMPS)31. These high-throughput sequencing methods are currently more expensive, but they offer a more complete and unbiased assessment of the global population of RNAs associated with a RNP complex. Another more recent technology is 'tiling arrays', which consist of oligonucleotides spanning the sequences of entire chromosomes32. Tiling arrays, when combined with RIP-Chip, can detect coding as well as small noncoding RNA species present within RNP complexes. Moreover, tiling arrays can allow the detection of discrete splice variants that associate with RBPs, and in combination with partial digestion or fragmentation of the mRNA, may allow precise mapping of RBP binding sites within the RNAs to which they are directly bound (S.A. Tenenbaum, P. Kapranov, T.R. Gingeras and J.D.K.,unpublished observations). Additionally, RIP-Chip, together with microarrays designed to detect microRNAs, can be used to identify the microRNAs that copurify with RNP complexes (P.J. Lager, J.M. Thomson, U. Ohler, S.M. Hammond and J.D.K., unpublished data)33.
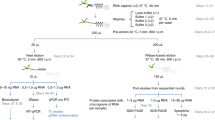
The protocols outlined here concern the preparation of cell extracts and the recovery of messenger ribonucleoprotein (mRNP) complexes to identify subsets of mRNAs and other noncoding RNAs en masse that are potentially coregulated. To prepare high-quality materials that contain intact pre-mRNPs or mature mRNPs, good biochemical practices are essential. This includes keeping materials on ice and adding inhibitors of ribonucleases (RNases) and proteases at all steps except where noted. For this protocol, preparing the mRNA lysate and coating protein A/G beads with antibody may be performed in advance of the immunoprecipitation reaction and RNA precipitation, or all steps can be performed sequentially as a single procedure.
The RIP-Chip protocol was also developed to identify RNA components associated with mRNP complexes under changing cellular conditions while minimizing some of the concerns such as sequence bias and high backgrounds known to be a problem with other protocols mentioned above. When designing a RIP-Chip experiment there are several issues to be considered for customizing optimal performance. First, prior knowledge of the biochemical properties of the mRNP complex, including specificity for individual targets and behavior of the complex, can be valuable in optimizing the protocol. For example, 'is the RBP of interest abundant?', 'does it have known sequence preferences?' and 'in which cellular compartment is it localized?' It can be useful to have prior knowledge of the approximate affinity of the RBP for its RNA substrates, although affinities in vivo are rarely known. To date, RIP-Chip has proved quite robust with dozens of RBPs, but the protocol may not be optimal for detecting weak associations or indirect interactions, as little prior knowledge is available in this regard. Another potential concern with all protocols like RIP-Chip is that the endpoint detection method can be expensive, and it may therefore be astute to incorporate a series of pilot experiments using single RNA detection methods (e.g., RT-PCR) while testing multiple control RNAs before investing in a genome-wide experiment13,14.
Materials
Reagents
-
1 M dithiothreitol (DTT; Fermentas, cat. no. RO861)
-
Ethylenediamine tetraacetic acid, EDTA (EM Science, cat. no. 4005)
-
Glycogen (Roche, cat. no. 10901393001)
-
1 M HEPES (pH 7.0; Sigma, cat. no. H3375)
-
Igepal Nonidet P-40 (NP40; Sigma, cat. no. I-30201)
-
1 M KCl (Mallinckrodt, cat. no. 8648)
-
1 M MgCl2 (EMD Bioscience, cat. no. MX0045-1)
-
NT2 buffer (see REAGENT SETUP)
-
Polysome lysis buffer (see REAGENT SETUP)
-
Protease inhibitor cocktail tablets (Roche, cat. no. 11697498001)
-
Protein A, immobilized on Sepharose (Sigma, cat. no. P3391)
-
Proteinase K (Roche, cat. no. 1964364)
-
RNase Out RNase inhibitor, 100 units/ml (Invitrogen, cat. no. 10777-019)
-
1 M NaCl (Mallinckrodt, cat. no. 7581)
-
Sodium dodecyl sulfate (SDS; EMD Bioscience, cat. no. 7910)
-
1 M Tris-HCl (pH 7.4; JT Baker, cat. no. 4103-01)
-
Trizol (Invitrogen, cat. no. 15596-026)
-
Urea (Mallinckrodt, cat. no. 8648)
-
Vanadyl ribonucleoside complexes (VRC; New England Biolabs, cat. no. S1402S)
Reagent Setup
-
Solutions Prepare all of the above solutions and buffers in RNase-DNase–free H2O.
-
Polysome lysis buffer
-
100 mM KCl
-
5 mM MgCl2
-
10 mM HEPES (pH 7.0)
-
0.5% NP40
-
1 mM DTT
-
100 units ml−1 RNase Out
-
400 μM VRC
-
Protease inhibitor cocktail
-
To make 5 ml of polysome lysis buffer, add 50 μl of 1 M HEPES (pH 7.0), 500 μl of 1 M KCl, 25 μl of 1 M MgCl2 and 25 μl of NP40 to 4.7 ml of RNase-DNase–free H2O. Add 50 μl of 1 M DTT, 12.5 μl of 100 U/ml RNase Out, 200 μl of Protease inhibitor cocktail (dissolve tablets according to the manufacturer's instructions) and 10 μl of 200 mM VRCs at the time of use.
-
NT2 buffer
-
50 mM Tris-HCl (pH 7.4)
-
150 mM NaCl
-
1 mM MgCl2
-
0.05% NP40
-
To make 50 ml of NT2 buffer, add 2.5 ml of 1 M Tris (pH 7.4), 7.5 ml of 1 M NaCl, 50 μl of 1 M MgCl2 and 25 μl NP40 to 40 ml of RNase-DNase–free H2O.
Procedure
Preparation of mRNP lysate
Timing ∼1 h
-
1
Collect enough tissue culture cells to generate 2–5 mg of total protein per RIP. Typically, this is comparable to 5–20 × 106 mammalian cells. Pellet by centrifugation (∼1,000g) for 10 min at 4 °C, washing several times with 10 ml of ice cold phosphate-buffered saline (PBS) in a conical tube. Alternatively, whole tissue may be ground using a mechanical homogenizer. Additionally, individual cells derived by micro-dissection may be used. The total amount of protein used per RIP must be optimized based upon the abundance of the RNA-binding protein being investigated as well as the planned method of RNA detection.
-
2
Resuspend final cell pellet with an approximately equal volume of polysome lysis buffer supplemented with RNase inhibitors and protease inhibitors (see REAGENT SETUP). Clumps of cells should be broken up by pipetting up and down several times. Allow mRNP lysate to incubate on ice for 5 min and store at −100 °C. Lysate may be stored for several months at −100 °C. The lysis of certain cell types can be enhanced by pumping the lysate through a small gauge syringe needle.
Critical Step
Immediate freezing of the lysate is essential to complete the lysis process as well as preventing adventitious binding. Additional freeze-thaw cycles should be avoided to prevent protein and RNA degradation.
Antibody coating of protein A/G beads
Timing ∼15 min
-
3
At 4 °C, pre-swell protein-A Sepharose beads in NT2 supplemented with 5% BSA to a final ratio of 1:5 for at least 1 h before use. Protein G or A/G Sepharose beads may be substituted depending upon the isotype of the antibody being used.
Pause point
Pre-swollen beads may be stored for several months at 4 °C when supplemented with 0.02% sodium azide.
-
4
In a 1.5 ml microcentrifuge tube, add 250–500 μl of protein A–BSA slurry. After a pulse centrifugation this should yield a pelleted bead volume of ∼50 μl at the bottom of the microcentrifuge tube.
-
5
Add antibody to bead slurry and incubate for 2–18 hours, tumbling end over end at 4 °C. The volume of antibody added to the beads is dependent upon antibody titer, but this amount should be more than enough to pull down all available protein being investigated.
Pause point
This mixture may be stored for several weeks at 4 °C when supplemented with 0.02% sodium azide.
Critical Step
In parallel, a control antibody must be used to assess background RNA levels. Typically, this is an isotype-matched antibody or whole normal sera from a matched species. The amount of control antibody should be equal to the amount of immunoprecipitating antibody being used.
-
6
Immediately before use, wash antibody-coated beads with 1 ml of ice-cold NT2 buffer 4–5 times. To wash, spin down beads by pulsing in an ultracentrifuge at 4 °C, remove liquid with hand pipettor or aspirator and resuspend in ice-cold NT2 buffer by flicking the tube several times with a finger. This washing removes unbound antibody as well as contaminants such as RNases, which may be present in the antibody mixture, and is one of the reasons we pre-bind the antibody to beads.
-
7
After the final wash, resuspend beads in 850 μl of ice-cold NT2 buffer. Add 200 units of an RNase inhibitor (5 μl RNase Out), 2 μl (to final concentration of 400 μM) Vanadyl ribonucleoside complexes, 10 μl of 100 mM DTT and EDTA to 20 mM.
Immunoprecipitation reaction and RNA precipitation
Timing ∼6 h, including binding incubation
-
8
Thaw mRNP lysate on ice and centrifuge at 15,000g for 15 min to clear lysate of large particles. Transfer cleared supernatant to microfuge tube and store on ice. Additionally, pre-clearing of lysate with beads may be used to reduce background, if necessary. This may, however, reduce signal.
-
9
Add 100 μl of cleared lysate to antibody mixture prepared in Step 7.
Critical Step
This dilution of lysate is important to reduce adventitious binding.
-
10
Immediately flick tube several times with a finger to mix, and centrifuge briefly at 8,000–10,000g to pellet beads. Remove 100 μl of supernatant to represent total cellular mRNA.
-
11
Incubate for 4 h at 4 °C tumbling end over end. Alternatively, incubate 2 h at room temperature (18–25 °C) and times as short as 15 min have worked well in some cases.
-
12
Pellet beads and save supernatant for later analysis if desired. Supernatant may be stored at –20 °C for several months.
-
13
Wash beads 4–5 times with 1 ml of ice-cold NT2 buffer by pulsing in an ultracentrifuge and removing supernatant with a hand pipettor or an aspirator.
Critical Step
Thorough washing is critical for reducing background. NT2 buffer may be supplemented with urea, sodium deoxycholate or SDS to increase stringency depending upon the RNA-binding protein being investigated. All tubes should be kept on ice as much as possible while working quickly during the washing process to reduce degradation.
-
14
Resuspend the beads in 100 μl of NT2 buffer. NT2 buffer can also be supplemented with 30 μg of proteinase K to release the RNP components. Incubate mixture for 30 min at 55 °C, flicking the tube occasionally using a finger.
-
15
Release the RNP components and isolate the RNA from the immunoprecipitated pellet by adding either Trizol reagent (Invitrogen) or phenol-chloroform-isoamyl alcohol directly to the beads. Precipitate RNA and resuspend in a volume appropriate for subsequent detection method. Addition of glycogen (20 μg) as a carrier to the precipitation reaction aids in making the RNA pellet more readily visible and aids in recovery of RNA.
-
16
After release of the RNP components, one can isolate the proteins associated with the complex and submit them for standard mass spectroscopy or other proteomics identification procedures. Such information can be very useful in the functional analysis subsequent to the identification of RNA subsets as described above.
Troubleshooting
If after RNA isolation and analysis there is adequate signal from total RNA but poor or no signal from RNA isolated through RIP-Chip, there are several steps that may be taken to ascertain possible causes. After confirming that there is adequate expression of the protein of interest within the cell, it is necessary to confirm that this protein was recovered via RIP-Chip. To do this, RIP-Chip is performed exactly as described above but stopped before Step 14 (before the addition of proteinase K). The washed beads are resuspended and boiled in Laemmli buffer to recover associated proteins. Western blot analysis is performed using this material to determine if the RBP of interest has been effectively recovered through RIP-Chip.
If the protein is not detected, one possible explanation is inaccessibility of the epitope to which the immunoprecipitating antibody is directed. Therefore, one may use an epitope-tagged RBP for mRNP isolation13,14. Alternatively, antibodies recognizing different epitopes may be used for RIP-Chip. In general, this has not been an issue because most RBPs are present on the surfaces of RNPs and polyclonal antibodies (e.g., many human autoantibodies against RBPs) are presented to the immune systems as RNP complexes3,9,30. Even under conditions in which the RBP or RNP-associated protein is detected in the cell extract, RIP-Chip may need to be further optimized for the specific mRNP being investigated. As noted above, more stringent washing may be achieved by adding optimally determined concentrations of urea, SDS, deoxycholate or other detergents and chaotropic agents to the wash buffer to reduce the background. If an RNA component of the mRNP is known, one can reverse transcribe the RNA and perform PCR to quickly optimize the RIP wash conditions14. In this manner, a large number of conditions may be quickly screened to reduce background nonspecific binding. Alternatively, quantitative real-time PCR may be used to discern PCR cycle differences that are not obtainable when using nonquantitative PCR. We have also found that multiprobe RNase protection assays offer a rapid way to optimize RIP13,14. As mentioned above, correct choices of variations of these methods are best served with knowledge of at least certain components of the mRNP. For example, binding of members of the RNA-recognition family (RRM) and Pumilio family of RNA binding proteins are not affected by the presence of urea, whereas other RBPs may be affected adversely. Further caution should be taken to make sure that the antibody or ligand tag are not affected by harsh washing conditions.
Timing
Steps 1-2: 1 h
Steps 3-7: 15 min
Steps 8-15: 6 h
Anticipated Results
Depending on the method used to detect RNA targets, the exact results and strategy of global analysis may vary. When dealing with large data sets of putative mRNA targets first identifying the biological function(s) of the RBP with respect to a single or small set of mRNA species can be advantageous. Certain functions may become more readily obvious when looking at the entire list of targets than when focusing on a single gene. This can be accomplished by running the entire list of mRNA targets through gene ontology (GO) programs, such as Panther (http://www.pantherdb.org) or Webgestalt (http://genereg.ornl.gov/webgestalt/)34, which search annotated GO categories and provide statistical analysis indicating enriched GO categories. In this type of analysis it is important to compare the target mRNAs of the RBP to mRNAs expressed in that cell type rather than to a general global list of mRNAs for that organism. Examples of this type of analysis have been demonstrated in yeast27, Drosophila28 and mammalian cells13,34,35.
Furthermore, it is more probable to identify sequence or structural motifs common to an entire set of targets when using large lists of targets than when dealing with only a few targets. These motifs, however, frequently contain significant variability and are often poorly characterized, thus making their identification difficult with small data sets. This type of analysis has been done effectively with Hu proteins13,36, the Fragile X Mental Retardation protein35 and Pumilio proteins27,28.
One of the limitations of the RIP-Chip procedure is that it is often difficult to distinguish 'direct' from 'indirect' RNA binders. Unfortunately, unbiased methods to make this distinction are still less than satisfactory. In general, however, the field has enjoyed abundant success in identifying mRNAs within isolated RNP complexes that can be validated as bone fide targets using a variety of methods including: (i) the use of alternative reagents, (ii) multiple cell types, (iii) using wide latitudes of reagent concentration, (iv) using alternative detection methods (e.g., microarrays, QRT-PCR, northern blotting), (v) genetic validation and (vi) noting predictable structural and functional relationships among the mRNA subsets. Therefore as noted above, it is always appropriate to optimize the conditions for preparing lysates and the antibody ligand or epitope tag that function best with the RNP or RBP of interest. As demonstrated and reviewed in several articles, the methods described by Tenenbaum et al., when properly optimized have performed very well for many investigators with minimal alteration5,6,13,14,15,16,19,27,28,29,35,36.
Change history
31 August 2006
In the version of the article originally published, in the last sentence of the ANTICIPATED RESULTS section, the callout should be to reference 19 instead of 18. The error has been corrected in the HTML and PDF versions of the article.
References
Ideker, T. et al. Integrated genomic and proteomic analyses of a systematically perturbed metabolic network. Science 292, 929–934 (2001).
Griffin, T.J. et al. Complementary profiling of gene expression at the transcriptome and proteome levels in Saccharomyces cerevisiae. Mol. Cell. Proteomics 1, 323–333 (2002).
Keene, J.D. Ribonucleoprotein infrastructure regulating the flow of genetic information between the genome and the proteome. Proc. Natl. Acad. Sci. USA 98, 7018–7024 (2001).
Keene, J.D. & Tenenbaum, S.A. Eukaryotic mRNPs may represent posttranscriptional operons. Mol. Cell 9, 1161–1167 (2002).
Hieronymus, H. & Silver, P.A. A systems view of mRNP biology. Genes Dev. 18, 2845–2860 (2004).
Keene, J.D. & Lager, P.J. Posttranscriptional operons and regulons coordinating gene expression. Chr. Res. 13, 327–337 (2005).
Moore, M.J. From birth to death: the complex lives of eukaryotic mRNAs. Science 309, 1514–1518 (2005).
Cilley, C.D. & Williamson, J.R. PACE Analysis of RNA-peptide interactions. In Methods in Molecular Biology (S.R. Haynes, ed.) 129–142 (Humana Press, Totowa, New Jersey, 1999)
Gao, F., Carson, C., Levine, T.D. & Keene, J.D. Selection of a subset of mRNAs from 3′UTR combinatorial libraries using neuronal RNA-binding protein, Hel-N1. Proc. Natl. Acad. Sci. USA 91, 11207–11211 (1994).
SenGupta, D.J. et al. A three-hybrid system to detect RNA-protein interactions in vivo. Proc. Natl. Acad. Sci. USA 93, 8496–8501 (1996).
Bernstein, D.S., Buter, N., Stumpf, C. & Wickens, M. Analyzing mRNA-protein complexes using a yeast three-hybrid system. Methods 26, 123–141 (2002).
Wang, Z.F. et al. The protein that binds the 3′ end of histone mRNA: a novel RNA-binding protein required for histone pre-mRNA processing. Genes Dev. 10, 3028–3040 (1996).
Tenenbaum, S.A., Carson, C.C., Lager, P.J. & Keene, J.D. Identifying mRNA subsets in mRNP complexes using cDNA arrays. Proc. Natl. Acad. Sci. USA 97, 14085–14090 (2000).
Tenenbaum, S.A., Lager, P.J., Carson, C.C. & Keene, J.D. Ribonomics: identifying mRNA subsets in mRNP complexes using antibodies to RNA-binding proteins and genomic arrays. Methods 26, 191–198 (2002).
Tenenbaum, S.A., Carson, C.C., Atasoy, U. & Keene, J.D. Genome-wide regulatory analysis combining en masse nuclear run-ons (emRUNs) and ribonomic profiling. Gene 317, 79–87 (2003).
Eystathioy, T. et al. A phosphorylated cytoplasmic autoantigen, GW182, associates with a unique population of human mRNAs within novel cytoplasmic speckles. Mol. Biol. Cell 13, 1338–1351 (2002).
Kaneko, S. & Manley, J.L. The mammalian RNA polymerase II C-terminal domain interacts with RNA to suppress transcription-coupled 3′ end formation. Mol. Cell 20, 91–103 (2005).
Mili, S. & Steitz, J.A. Evidence for reassociation of RNA-binding proteins after cell lysis: implications for the interpretation of immunoprecipitation analyses. RNA 10, 1692–1694 (2004).
Penalva, L.O.F. Burdick, M.D., Lin, S.M., Sutterluety, H. & Keene, J.D. RNA-binding proteins to assess gene expression states of co-cultivated cells in response to tumor cells. Mol. Can. 3, 24–35 (2004).
Penalva, O.F., Tenenbaum, S.A. & Keene, J.D. Gene expression analysis of messenger RNP complexes. Meth. Mol. Biol. 257, 125–134 (2004).
Roy, P.J., Stuart, J.M., Lund, J. & Kim, S.K. Chromosomal clustering of muscle-expressed genes in Caenorhabditis elegans. Nature 418, 975–979 (2002).
Niranjanakumari, S., Lasda, E., Brazas, R. & Garcia-Blanco, M.A. Reversible cross-linking combined with immunoprecipitation to study RNA-protein interactions in vivo. Methods 26, 182–190 (2002).
Ule, J., Jensen, K., Mele, A. & Darnell, R.B. CLIP: a method for identifying protein-RNA interaction sites in living cells. Methods 37, 376–386 (2005).
Zang, Z., Edenberg, H.J. & Davis, R.L. Isolation of mRNA from specific tissues of Drosophilia by mRNA tagging. Nuc. Acids Res. 33, e148 (2005).
Kunitomo, H., Uesugi, H., Kohara, Y. & Iino, Y. Identification of ciliated sensory neuron-expressed genes in Caenorhabditis elegans using targeted pull-down of poly(A) tails. Genome Biol. 6, R17 (2005).
Ule, J. et al. CLIP identifies Nova-regulated RNA networks in the brain. Science 302, 1212–1215 (2003).
Gerber, A.P., Herschlag, D. & Brown, P.O. Extensive association of functionally and cytotopically related mRNAs with Puf family RNA-binding proteins in yeast. PLoS Biol. 2, E79 (2004).
Gerber, A.P., Luschnig, S., Kransow, M.A., Brown, P.O. & Herschlag, D. Genome-wide identification of mRNAs associated with the translational regulator PUMILIO in Drosophila melanogaster. Proc. Natl. Acad. Sci. USA 103, 4487–4492 (2006).
Penalva, P.O. & Keene, J.D. Biotinylated tags for recovery and characterization of ribonucleoprotein complexes. Biotechniques 37, 604–610 (2004).
Antic, D., Lu, N. & Keene, J.D. ELAV tumor antigen, Hel-N1, increases translation of neurofilament M mRNA and induces formation of neurites in human teratocarcinoma cells. Genes Dev. 13, 449–461 (1999).
Zhu, J., Shendure, J., Mitra, R.D. & Church, G.M. Single molecule profiling of alternative pre-mRNA splicing. Science 301, 836–838 (2003).
Kapranov, P. et al. Large-scale transcriptional activity in chromosomes 21 and 22. Science 296, 916–919 (2002).
Thomson, J.M., Parker, J., Perou, C.M. & Hammond, S.M. A custom microarray platform for analysis of microRNA gene expression. Nat. Methods. 1, 47–53 (2004).
Zhang, B., Kirov, S. & Snoody, J. WebGestalt: and integrated system for exploring gene sets in various biological contexts. Nucleic Acids Res. 33, W741–748 (2005).
Brown, V. et al. Microarray identification of Fragile X-Mental Retardation protein (FMRP)-associated brain mRNAs and altered mRNA translational profiles in fragile X syndrome. Cell 107, 477–487 (2001).
Lopez de Silanes, I., Zhan, M., Lal, A., Yang, X. & Gorospe, M. Identification of a target RNA motif for RNA-binding protein HuR. Proc. Natl. Acad. Sci. USA 101, 2987–2992 (2004).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
J.D.K. holds stock in Ribonomics, Inc., a company that owns patents for the RIP-Chip technology.
Rights and permissions
About this article
Cite this article
Keene, J., Komisarow, J. & Friedersdorf, M. RIP-Chip: the isolation and identification of mRNAs, microRNAs and protein components of ribonucleoprotein complexes from cell extracts. Nat Protoc 1, 302–307 (2006). https://doi.org/10.1038/nprot.2006.47
Published:
Issue Date:
DOI: https://doi.org/10.1038/nprot.2006.47
This article is cited by
-
RIP-PEN-seq identifies a class of kink-turn RNAs as splicing regulators
Nature Biotechnology (2024)
-
NAP-seq reveals multiple classes of structured noncoding RNAs with regulatory functions
Nature Communications (2024)
-
3D genomics and its applications in precision medicine
Cellular & Molecular Biology Letters (2023)
-
LINE-1 regulates cortical development by acting as long non-coding RNAs
Nature Communications (2023)
-
On the utility of microfluidic systems to study protein interactions: advantages, challenges, and applications
European Biophysics Journal (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
