
Abstract
A detailed protocol is described allowing the generation of essentially pure populations of glutamatergic neurons from mouse embryonic stem (ES) cells. It is based on the culture of ES cells that are kept undifferentiated by repeated splitting and subsequently amplified as non-adherent cell aggregates. Treatment with retinoic acid causes these ES cells to essentially become neural progenitors with the characteristics of Pax6-positive radial glial cells. As they do in vivo, these progenitors differentiate in glutamatergic pyramidal neurons that form functional synaptic contacts and can be kept in culture for long periods of time. This protocol does not require the use of ES lines expressing resistance or fluorescent markers and can thus be applied in principle to any wild-type or mutant ES line of interest. At least 2 weeks are required from starting ES cell culture until plating progenitors and differentiating neurons establish synaptic transmission within about 10 days.
Similar content being viewed by others


Introduction
A significant problem in neurobiology is the lack of relevant culture systems allowing the generation of defined populations of cells reflecting the property of post-mitotic, process-bearing neurons of the central nervous system (CNS). The unique success of PC12 cells over three decades illustrates how useful cell culture systems can be to study questions related to the biochemistry of neuronal differentiation in the peripheral nervous system (PNS)1. This cell line was originally derived from a tumor of the adrenal medulla and because PC12 cells divide in culture, unlimited quantities of homogeneous material became available for biochemical studies1. Moreover, the addition of nerve growth factor causes them to exit the cell cycle and to elongate processes, thus allowing key aspects of neuronal differentiation to be studied under defined conditions1. As PC12 cells induced to differentiate with nerve growth factor resemble sympathetic neurons of the PNS, but not of neurons of the CNS, there is a need for new cell culture systems to be developed. In theory, mouse ES cells represent an attractive option as they rapidly divide and can generate any cell type, including of course CNS neurons. Also, and unlike PC12 cells, their genome can be readily and specifically altered by well-established techniques, thus allowing the impact of mutations involved in neuronal differentiation and function to be studied under defined conditions. Although a number of previous studies (see in particular refs. 3, 4, 5, 6, 7, 8, 9, 10, 11 and references therein) indicate that neurons and glial cells can be generated in vitro from ES cells, the spontaneous tendency of ES cells to start differentiation programs even in the presence of LIF has been a major obstacle in the development of robust ES cell-based systems for biochemical studies. Indeed, homogeneity is a key aspect of useful in vitro cellular systems and most published differentiation protocols result in the generation of heterogeneous neuronal populations.
We previously reported that the problem of heterogeneity can be solved by selecting ES cells for their ability to rapidly divide, thus eliminating cells that have begun to differentiate12. When homogeneous ES cells are treated with retinoic acid (RA), they all become progenitor cells with the characteristics of radial glial cells. Similar to their behavior in vivo13, these progenitors go on to differentiate into glutamatergic neurons that are functionally connected12. Their potential has also been tested in transplantation studies, revealing the developmental restriction expected for this type of neural progenitor. In particular, when transplanted in the prospective spinal cord of chick embryos, the progenitors generated by this protocol give rise to motoneurons and specific subtypes of interneurons in the anterior spinal cord, but not to dorsal phenotypes or sensory neurons in dorsal root ganglia14. That the fate of the progenitors is restricted was indicated by control experiments using ES cells not treated with RA in a similar setting, and these experiments revealed that as expected, mouse ES cells have a much broader developmental potential in the chick embryo, and can for example become sensory neurons expressing appropriate markers when leaving the site of transplantation. When transplanted in the prospective dorsal telencephalon, these progenitors develop the characteristics of glutamatergic neurons (Nikoletopoulou and Barde, unpublished results). Since its publication, this protocol has been used extensively and successfully with several standard mouse ES lines with indistinguishable results, as well as with a variety of genetically modified ES cells and ES cell lines isolated from the blastocysts of wild-type mice and of mice carrying specific mutations (see examples in Table 1). The detailed protocol presented here leads to cultures that are over 90% homogeneous. It also fulfills three important criteria that have been proposed for using ES cells as a model for lineage-specific differentiation: efficiency and reproducibility, recapitulation of a developmental program and appropriate functional properties of the differentiated cells11. At least 2 weeks are required from thawing ES cells until neural progenitors can be plated (see the flow chart in Fig. 1), and we describe here conditions allowing long-term culture of ES cell-differentiated neurons that can be used to study a number of processes of interest to neurobiologists, either with wild-type or with genetically modified or virally infected cells (see Fig. 2).

The four main steps and the corresponding cell types (ES cells, CAs, progenitor cells, neurons) are shown with a minimal time frame for each step. Maturation of neurons can be followed for up to 8 weeks. The neurons exhibit synaptic activity that can be recorded by standard electrophysiological techniques and they begin to develop spines after about 3 weeks in culture.

Genetically modified ES cells as well as ES cell lines isolated from transgenic mouse lines have been successfully used in our differentiation protocol. Obviously, engineered ES cell lines giving rise to interesting neuronal phenotypes in vitro can be used to establish transgenic mouse lines. Neural progenitors can be frozen after dissociation of CAs for later use. In addition, they can be transfected by lentivirus infection with high efficiency to express different cDNAs during neuronal maturation. The homogeneity of the neuronal culture allows gene and protein expression profiling. Neurons can also be plated in high-throughput formats, for example, 384-well plates.
As all cells in the aggregates express similar antigenic markers14, we now avoid the terminology “embryoid body” as it refers to a mixture of cells containing progenitors of ecto-, meso- and endodermal lineages9.
Materials
Reagents
-
Critical
All reagents and materials used must be sterile.
-
Water (Aqua ad inject; Braun), used for all cell culture work
-
Dulbecco's PBS (Gibco, cat. no. 14190)
-
DMEM (Gibco, cat. no. 21969035)
-
FCS (Gibco, personal reservation of a whole batch, EU-approved origin)
Critical
Thaw at 4 °C overnight and heat-inactivate at 55 °C for 30 min, store inactivated aliquots at −20 °C and thaw directly before use only at room temperature.
-
Leukemia inhibitory factor (LIF; Chemicon, cat. no. ESG1107), 107 U ml−1 LIF, 1:100 in DMEM.
Critical
Store aliquots at −20 °C (even though Chemicon recommends storage at 4 °C).
-
L-Glutamine (Gibco, cat. no. 25030024), 100 × (i.e., 200 mM), use 1:100, store aliquots at −20 °C
-
Non-essential amino acids (Gibco, cat. no. 11140035), use 1:100, store at 4 °C
-
β-Mercaptoethanol (Sigma, cat. no. M-7522)
Caution
β-Mercaptoethanol is toxic by inhalation, ingestion and skin contact. Personal protection: gloves, safety glasses and good ventilation.
-
Gelatine (Sigma, cat. no. G-1890)
-
Trypsin-EDTA (1 × ; Gibco, cat. no. 25300-054) for splitting ES cells; store at 4 °C after thawing at 4 °C; use within 2 weeks.
Critical
Do not leave at room temperature.
-
RA (Sigma, cat. no. R-2625)
-
Boric acid (Riedel de Haen, cat. no. 31146)
-
Poly-DL-ornithine (PORN) hydrobromide 3000-15000 (Sigma, cat. no. P-8638)
-
Laminin (Roche, cat. no. 11234217001), store aliquots at −20 °C
Critical
Thaw aliquots only once immediately before use.
-
Trypsin 0.05% (w/v) in 0.05% (w/v) EDTA/PBS (Sigma, cat. no. T-8802, 100 mg TPCK-treated to inhibit chymotrypsin) for dissociation of cell aggregates (CAs)
Critical
Prepare directly from powder before use.
-
Insulin (Sigma, cat. no. I-6634, 100 mg), stock solution 5 mg ml−1 in water (acidified with one drop of 25% HCl/10 ml to dissolve it), store at −20 °C
-
Transferrin (Sigma, cat. no. T-1147, human apo-transferrin, 100 mg), stock solution 2 mg ml−1 in water, store at −20 °C
-
Progesterone (Sigma, cat. no. P-8783, 5 g), stock solution 2 mM in EtOH, store at −20 °C, working solution 20 μM dilution of stock solution in water, store at −20 °C
-
Putrescine (Sigma, cat. no. P-5780), stock solution 100 μM in water, store at −20 °C
-
Sodium selenite (Sigma, cat. no. S-5261, 25 g), stock solution 300 μM in water, store at 4 °C
-
F-12 (Ham) nutrient mixture + L-glutamine (Gibco, cat. no. 21765029)
-
BSA (Sigma, cat. no. A-9418, Powder Fraction V 100 g), aliquots 10 mg ml−1, store at −20 °C
-
Ingredients for complete medium, aqueous solutions: L-alanine (Sigma, cat. no. A-7627) 2 mg ml−1, biotin (Sigma, cat. no. B-4501) 0.1 mg ml−1, L-carnitine (Sigma, cat. no. C-0283) 2 mg ml−1, ethanolamine (Sigma, cat. no. E-9508) 1 mg ml−1, D-galactose (Sigma, cat. no. G-0625) 15 mg ml−1, L-proline (Sigma, cat. no. P-0380) 7.76 mg ml−1, putrescine (Sigma, cat. no. P-7505) 16.1 mg ml−1, Na-pyruvate (Sigma, cat. no. P-5280) 25 mg ml−1, Na-selenite (Sigma, cat. no. S-1382) 0.016 mg ml−1, vitamin B12 (Sigma, cat. no. V-2876) 0.34 mg ml−1, zinc sulfate (Sigma, cat. no. Z-4750) 0.194 mg ml−1, catalase (Sigma, cat. no. C-40) 16 mg ml−1, glutathione (Sigma, cat. no. G-6013) 1 mg ml−1, superoxide dismutase (Sigma, cat. no. S-2515) 2.5 mg ml−1; ethanolic solutions: linoleic acid (Sigma, cat. no. L-1376) 100 mg ml−1, linolenic acid (Sigma, cat. no. L-2376) 100 mg ml−1, progesterone (Sigma, cat. no. P-8783) 0.63 mg ml−1, all-trans retinol (Sigma, cat. no. R-7632) 10 mg ml−1, retinylacetate (Sigma, cat. no. R-7882) 10 mg ml−1, tocopherol (Sigma, cat. no. T-3251) 100 mg ml−1, tocopherolacetate (Sigma, cat. no. T-3001) 100 mg ml−1.
Critical
Freeze stock solutions of aqueous solutions only once. Ethanol solutions are stable for about 6 months and can be frozen several times.
-
DMSO (Sigma, cat. no. D-2650)
Equipment
-
Tissue-culture plates for ES cell culture (recommended): 60 mm (Nunc, cat. no. 150288) and 100 mm (Corning, cat. no. 430167)
-
Bacteriological Petri dishes (Greiner, Bio-one 94/16 with vents, cat. no. 633102).
Critical
We strongly recommend Greiner bacteriological Petri dishes as all other bacteriological dishes we tested did not work (e.g., Falcon, Corning). CAs had a strong tendency to adhere to all other dishes. Pay attention to the catalog number; different Greiner bacteriological Petri dishes were also tested and CAs adhered to them.
-
Tissue-culture plates for neuronal culture: 60 mm (Nunc, cat. no. 150288), 24-well (Nunc, cat. no. 142475) and 12-well (Nunc, cat. no. 150628), glass coverslips (Assistent ∅14 mm, cat. no. 1001)
-
40 μm nylon cell strainer (BD Falcon, cat. no. 352340)
-
Cryotubes (Nunc, cat. no. 377267)
-
Stericup 0.22 μm (Millipore, cat. nos. SCGP4 05RE 500 μl, SCGP4 02RE 250 μl)
Reagent setup
-
Gelatine solution 0.2% in water (Braun, see above), sterilize directly by autoclaving.
-
ES cell culture medium DMEM supplemented with 15% FCS, 103 U ml−1 LIF (1:100), 2 mM L-glutamine, 1 × non-essential amino acids, 5 μl per 500 ml β-mercaptoethanol.
Critical
All media should be stored for less than 1 week, or even better used within 2–3 days; pH color indicator is important: the N2 medium in particular can turn pink just after 2 days, and if this occurs, discard.
-
CA cell culture medium DMEM supplemented with 10% FCS, 2 mM L-glutamine, 1 × non-essential amino acids and 5 μl per 500 ml β-mercaptoethanol.
-
RA Dissolve 5 mM in DMSO; store at −20 °C.
Critical
Prepare in a dark hood owing to light sensitivity. Thaw aliquots in the dark directly before use.
-
PORN Prepare stocks 0.5 mg ml−1 in borate buffer (150 mM, pH 8.3 with NaOH); store at 4 °C.
Critical
Do not store stock solution at 4 °C for longer than 1 week and use 1:5 freshly diluted in water.
-
N2 medium 125 ml DMEM, 1.25 ml L-glutamine (2 mM), 125 ml F-12, 1.25 ml insulin (25 μg ml−1), 6.25 ml transferrin (50 μg ml−1), 0.25 ml progesterone (20 nM), 0.25 ml putrescine (100 nM), 25 μl sodium selenite (30 nM) and 1.25 ml BSA (50 μg ml−1) (2.5 ml penicillin/streptomycin (1% vol/vol) can also be added).
Critical
The pH of the medium is critical (7.0–7.8 preferred, should never be higher than 8.0 at room temperature); discard when color is pink.
-
Complete medium Dissolve 100 μl per 100 ml L-alanine (2 μg ml−1), 100 μl per 100 ml biotin (0.1 μg ml−1), 100 μl per 100 ml L-carnitine (2 μg ml−1), 100 μl per 100 ml ethanolamine (1 μg ml−1), 100 μl per 100 ml D-galactose (15 μg ml−1), 100 μl per 100 ml L-proline (7.76 μg ml−1), 100 μl per 100 ml putrescine (16.1 μg ml−1), 100 μl per 100 ml Na-pyruvate (25 μg ml−1), 100 μl per 100 ml Na-selenite (0.016 μg ml−1), 100 μl per 100 ml vitamin B12 (0.34 μg ml−1), 100 μl per 100 ml zinc sulfate (0.194 μg ml−1), 16 μl per 100 ml catalase (2.56 μg ml−1), 100 μl per 100 ml glutathione (1 μg ml−1), 100 μl per 100 ml superoxide dismutase (2.5 μg ml−1), 1 μl per 100 ml linoleic acid (1 μg ml−1), 1 μl per 100 ml linolenic acid (1 μg ml−1), 1 μl per 100 ml progesterone (6.3 ng ml−1), 1 μl per 100 ml all-trans retinol (100 ng ml−1), 1 μl per 100 ml retinylacetate (100 ng ml−1), 1 μl per 100 ml tocopherol (1 μg ml−1) and 1 μl per 100 ml tocopherolacetate (1 μg ml−1) in 30 ml DMEM and add 1 g BSA, 2 mg transferrin, 1.6 mg insulin and 2 mM L-glutamine (penicillin/streptomycin 1% (vol/vol) can also be added, but see below); fill up to 400 ml DMEM. No F12 nutrient mixture is used.
Critical
It is critical to dissolve insulin as indicated above as it needs the acidification brought about by BSA in the 30 ml solution.
Critical
Complete medium can be stored for a few weeks if L-glutamine is not added; add L-glutamine directly before use.
-
Preparation of glass coverslips Rinse coverslips for 48 h in 65% nitric acid (Fluka, cat. no. 84382) while shaking in a beaker, wash carefully under running tap water, wash with 100% ethanol, air-dry coverslips separately on Whatman filter paper and sterilize with UV. Prepared coverslips can be either stored in the beaker in ethanol or after sterilizing separately in multiwell plates or the like.
Critical
Coverslips should not stick together when drying and the glass surface must remain completely clean.
Caution
Concentrated nitric acid is highly aggressive and its vapors are potentially harmful. It should be collected in glass and not in metal vessels and disposed off as toxic chemical.
-
Coating of plates and coverslips for neuronal differentiation Cover the surface with sufficient PORN solution overnight at 37 °C in the incubator (e.g., 3 ml on 60 mm, 0.5 ml per well on 24-well dishes). Wash three times with water (Braun, see above). We recommend to do the washings with water and not with PBS owing to crystallization. 60 mm dishes: add laminin directly to PBS in the dish, 20 μl per 4 ml PBS, and shake well to distribute evenly. 12-well, 24-well plates: prepare a solution with 100 μl laminin in 12 ml PBS, add 0.5 ml per well and incubate for at least 2 h at 37 °C in incubator; use the plates within 1 week when left at 37 °C in incubator, but preferably freshly coated. Aspirate laminin before use. Plates can be used immediately without being washed.
Critical
Even distribution of laminin is essential; laminin should be added to each well with shaking up to the 12-well plate format; 24-well plates and plates with multiple wells can be coated with a stock solution of laminin. For 96-well plates, we prefer to prepare a solution of laminin in N2 medium added to the wells just before adding the cell suspension. Laminin-coated dishes should never dry out, as this rapidly denatures laminin. The cell suspension should be added as rapidly as possible after removing the solution.
Procedure
Culture of ES cells
-
1
Prepare gelatine-coated plates by covering them with a 0.2% gelatine solution for at least 10 min in the incubator.
-
2
Culture ES cells on feeder cells (inactivated mouse embryonic fibroblasts (MEFs)) on tissue-culture dishes (60 mm) in ES cell culture medium at 37 °C and 7% CO2. Prepare MEFs and generate and inactivate feeder cells following standard procedures. In general, we used inactivation of MEFs by mitomycin. All media are prewarmed at 37 °C.
-
3
If feeder cells are inactivated with mitomycin, thoroughly wash them with medium and let them sit at least 1 h in the incubator in ES medium before adding ES cells.
-
4
Thaw a vial of about 3 × 106 ES cells quickly by resuspending the cells in 10 ml ES medium and centrifuging for 5 min at 180g at room temperature. Resuspend the cell pellet in ES medium again and plate the cells on a 60 mm cell culture dish with feeders.
-
5
Change ES medium the next day. (see Fig. 3)
Figure 3: Morphology of ES cells at different stages of the procedure. 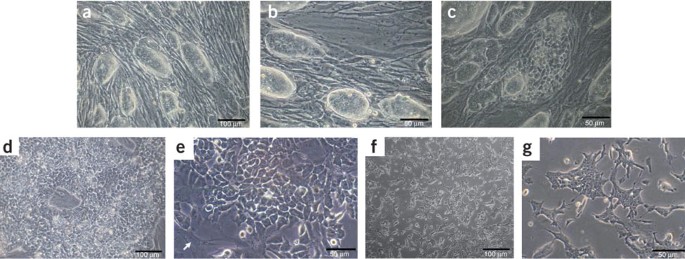
(a–c) ES cells on feeder cells. ES cells form oval, shiny colonies with clear boundaries (a,b). They should not exceed the size illustrated here for a culture 2 days after splitting. The ES cells should be split again at this point at a ratio of about 1:5. Good, pluripotent ES cell cultures reach the same density and morphology again after 2 days. Spontaneous differentiation of ES cell colonies on feeders. c illustrates a differentiated ES cell colony. The round and shiny shape disappeared and distinct cells can be seen. A good ES cell culture should not show such colonies. In addition, most other ES cell colonies on this picture do not show clear boundaries, are of very different sizes, some are flat and not shiny. Scale bars, 50 and 100 μm. (d–g) ES cells cultured without feeder cells. The morphology of ES cells cultured without feeder cells is very important and representative examples are shown. d and e show different magnification of ES cells that have been split twice without feeders. The pictures illustrate the results 2 days after the second splitting. At this stage, ES cells have to be split again without feeders or CA formation can be started when the ES cells have reached this appearance without feeder cells. The arrow in e at the lower left corner points to a remaining fibroblast. f and g show ES cells 1 day after the third splitting without feeder cells; thus cells are not yet as confluent as in d, but fibroblasts can hardly be detected at the third passage without feeders. In contrast to the oval, shiny appearance of ES cell colonies on feeders, ES cells without feeders flatten out and single cells with a uniform morphology, including nuclei and nucleoli, should be clearly distinguishable. Scale bars, 50 and 100 μm.
Critical Step
Reproducible and good-quality ES cell differentiation strongly depends on the quality of ES cells used, as well as on the way they are cultured. The density of ES cells is one of the most crucial factors. The cells should be split at ratios between 1:3 and 1:6 every 2 days. FCS batches have to be checked and selected for their quality for successful differentiation similar to the quality test for germline transmission. During the last years, we have used several different batches of FCS that all led to comparable results. Cells must be passaged every 48 h on feeders and the culture medium changed daily. The temperature of the incubator should be kept constant at 37 °C, and especially not higher as this dramatically affects the success of the procedure. Another CO2 content might be possible, but we did not explore thoroughly, for example, 10% was successful as well. Splitting ratios and frequencies are crucial and obviously depend on the growth rate of the particular passage and the morphology of the cells. Colonies of ES cells should form oval, shiny structures with clear boundaries (see examples in Fig. 3a,b). We observed that flat-looking colonies (see Fig. 3c) cannot be used. Also, they should never be cultured at too low a density. Fast proliferation of ES cells is essential; the cells should grow at a rate such that they need to be subcultured at a ratio of about 1:5 every 2 days. If high-quality ES cell culture is performed, successful differentiations also with ES cells of high passage number will occur.
-
6
After 2 days split cells: quickly wash twice with PBS, cover the plate with a thin film of 1 × trypsin-EDTA solution and incubate for 3 min in the incubator.
-
7
Resuspend ES cells in 10 ml fresh ES medium by thoroughly pipetting up and down several times and centrifuge for 5 min at 180g at room temperature.
-
8
Resuspend cell pellet in fresh ES medium again by pipetting up and down several times. Good dissociation of the cells is important, the goal being to obtain single cell culture or aggregates of 2–3 cells. Larger cell clumps should not remain.
Pause point
ES cells can be frozen by resuspending the cells after trypsinization and centrifugation in ES medium + 10% DMSO. Our best results have been achieved using a NalgeneCryo 1 °C Freezing Container (cat. no. 5100-0001).
-
9
Culture ES cells on feeders on 60 mm cell culture dishes for subsequent passages by splitting them about 1:5 every second day and changing the medium every other day.
Critical Step
STEP Thorough dissociation after trypsinization is especially important.
-
10
Culture ES cells on feeder cells (following Steps 5–8) for at least two passages after thawing. Subsequently, culture ES cells without feeder cells on gelatine-coated plates for at least two, preferably three, passages according to Steps 5–8.
-
11
Split ES cells 1:5 for the first passage without feeder cells (e.g., take half of one 60 mm cell culture dish with ES cells on feeders and transfer to a 100 mm cell culture dish without feeders) and subsequently at ratios of about 1:3–1:5.
Critical Step
The first passage without feeders is a key step and the success of the differentiation procedure strongly depends on the density of the ES cells in this passage. The ES cells should occupy at least one-third of the plate 1 day after first splitting without feeders. This is why we recommend to split them at ratios between 1:3 and 1:5. The morphology of the ES cells without feeder cells is crucial and ES cells should grow in a homogeneous monolayer with ES cells of uniform size (see Fig. 3d–g). Cells should neither be clumpy nor should they become large and flat, which is a typical sign of the beginning of differentiation. A high proliferative status of the ES cells is essential for successful differentiation in our protocol and is achieved when a splitting rate of about 1:3–1:5 every 2 days is reached. However, splitting ratios for highly proliferative ES cells (on feeder layer cells as well as without feeders) can vary depending on the ES cell line and have to be optimized for each new ES cell line generated. In our experience, the splitting ratios given in this protocol apply to the vast majority of the ES cell lines we used. If uncertainties arise, the next step of CA formation can be performed in parallel with different splitting ratios of the ES cells. At least 2–3 passages of ES cells without feeder cells are successful; differentiation is more likely to occur at higher numbers of passages without feeder cells resulting in lower quality of the procedure; only do five passages of ES cells without feeders at the most for better quality.

Formation of CAs
-
12
Trypsinize ES cells as in Steps 6 and 7, resuspend in CA medium and count the cells. Reliable counting of cell numbers can be achieved with the CASY Cell Counter&Analyser System (Schärfe System), but also in a counting chamber like a hemocytometer. When using hemocytometers, attention should be paid to only count live cells and not debris, as only then the correct ES cell number can be used for CA formation.
-
13
Plate 4 × 106 cells onto bacteriological Greiner Petri dishes in 15 ml CA medium. (See Fig. 4)
Figure 4: Morphology of cells following plating. 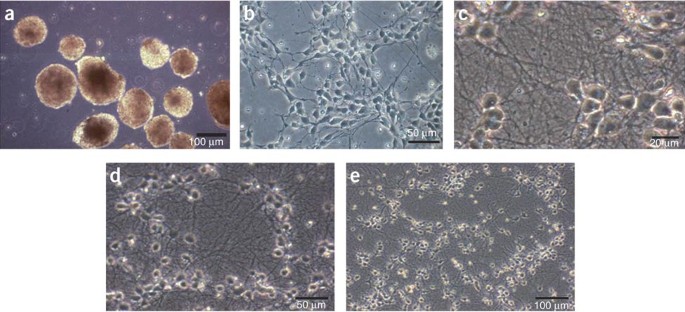
(a) Cellular aggregates. CAs shortly before dissociation and plating of neural progenitors. Although the shape of different CAs is quite homogeneous in this illustration, more irregular shapes are frequently observed. We found that these also lead to successful differentiations. Also aggregation of several CAs to tube-like structures has no impact on differentiation. However, CAs must be in suspension and must not stick to the culture plates. Scale bar, 100 μm. (b) Progenitor cells 20 h after plating dissociated CAs. Neural progenitors 20 h after dissociating CAs and plating of the cells are shown. Cells are still spindle-shaped, although this particular morphology is more obvious 2 h after plating (not shown). Neurites start to rapidly grow out and to form a network over the next few days. In this illustration, the density corresponds to about 1/3 of the plate covered with cells, which is appropriate for successful long-term neuronal cultures. Scale bar, 50 μm. (c–e) Neurons 11 days after plating. A dense neuritic network has formed. Neuronal cell bodies are shiny and should not cluster, but be fairly uniformly distributed as illustrated. Neurites should be branched and curved but never straight, which is a sign that they do not adhere well to the substrate or too many non-neuronal cells are underneath. Neurites that do not adhere well to the substrate tend to form bundles with other neurites, a feature that is not compatible with long-term cultures. Scale bars, 20, 50 and 100 μm, respectively.
Critical Step
The number of ES cells used in the CA formation step is essential for homogeneous neuronal differentiation and can be different depending on the ES cell line. A total of 4 × 106 cells represent the number that was found to be successful with most ES cell lines and to result in good quality, homogeneous neuronal cultures. By contrast, aggregates formed with 1 × 106 ES cells did not lead to the generation of neurons. This was also observed with the very same ES cells generating pure neuronal cultures when used at 4 × 106, instead of 1 × 106 cells. The brand of tissue culture plates to grow the CAs is also crucial (see EQUIPMENT). We found that 15 ml is the optimal volume to grow the CAs in 10 cm dishes; using less medium also leads to lower differentiation quality. About 2 × 107 cells should be harvested when dissociating the CAs as proliferation during CA formation is an important factor. Although the formation of non-adherent CAs is an essential part of the procedure, it is still unclear why it is so crucial to the success of the protocol. Cell–cell contact as opposed to contact of ES cells with the substrate may be one of the critical elements. As there is substantial cell division during this aggregation phase, especially before the addition of RA, it is possible that this step also helps selecting for cells with similar characteristics. We note that the spontaneous formation of cell clumps by aggregation leads to very different shapes of CAs (Fig. 4a) ranging from small spheres to elongated, tube-like structures. We found these various shapes to be of no predictive value with regard to the success of the procedure. Also the size and aggregation of multiple CAs do not show any correlation with the outcome of the neuronal differentiation.
-
14
Change CA medium after 2 days of aggregation by transferring the entire CA suspension into a 50 ml Falcon tube. Transfer carefully using a pipette so as not to disintegrate the CAs. Let CAs settle by leaving them for about 3–5 min under the hood. Remove the supernatant and resuspend the CAs gently with 15 ml CA medium again by pipetting up and down twice. Pipettes with narrow openings should not be used.
-
15
Change CA medium again after 4 days and add RA 1:1,000 from 5 mM stock solution (final concentration 5 μM) to the CA medium for resuspending CAs. RA is light-sensitive and thus should not be left under light for long periods of time.
-
16
Change CA medium after 6 days of aggregation as in Step 14, but resuspend again in CA medium with RA.

Dissociation of CAs and neuronal differentiation
-
17
Coat appropriate plates with PORN and laminin, 60 mm per 12-well/24–well, etc. (see REAGENT SETUP). These should be ready for day 8, when CAs are dissociated after the start of CA formation and cells are plated on PORN/laminin-coated plates to allow differentiation of neuronal precursors (see Step 23). For immunofluorescence analysis, place pre-treated coverslips (see REAGENT SETUP) at the bottom of the dish before coating.
-
18
Dissociate CAs on day 8 of CA formation. Wash CAs twice with PBS, proceeding according to Step 14 when changing CA medium.
-
19
Prepare fresh 0.05% trypsin solution in 0.05% EDTA/PBS with trypsin powder directly before use (during second PBS wash of CAs).
-
20
Trypsinize CAs by incubating them in 0.5 ml of 0.05% trypsin solution for 3 min at 37 °C in the water bath. During incubation, gently shake the Falcon tube a few times. Disintegration of the CAs can be readily observed.
-
21
Resuspend dissociated CAs in 10 ml CA medium to inactivate trypsin (by the serum in CA medium). Dissociate CAs by pipetting up and down with a 10 ml plastic pipette until no CA clumps can be seen any more. Pipettes with openings corresponding to those of 5 ml plastic pipettes can also be used, as well as fire-polished Pasteur pipettes. Centrifuge for 5 min at 180g at room temperature. Remove the supernatant as completely as possible.
Critical Step
CAs must readily disintegrate, and if they do not, the neuronal differentiation is unlikely to be successful. Complete dissociation is essential at this step. The use of a defined concentration of trypsin is necessary at this step as using prepared trypsin will not disintegrate CAs properly. Also, it is important to remove the supernatant completely after centrifugation of the dissociated CAs to eliminate even traces of serum that influence neuronal differentiation when plating the neuronal precursors.
Pause point
Dissociated CAs can be frozen by resuspending cells in CA medium + 10% DMSO. When thawing the cells, resuspend them quickly in N2 medium and centrifuge for 5 min at 180g at room temperature. Quick removal of DMSO is essential to recover high numbers of neuronal precursors. Up to 50% of cells may nonetheless be lost after thawing; thus, optimal cell numbers should be tested by dilution series.
-
22
Resuspend cell pellet in N2 medium thoroughly. Filter the cell suspension through a 40 μm nylon cell strainer by applying drop by drop; avoid applying pressure.
-
23
Count cells and plate them at about 2 × 105 cells per cm2 on PORN/laminin-coated plates, for example 4.5 × 106 cells on a 60 mm dish in sufficient amount of N2 medium, for example 3 ml medium, and 0.45 × 106 cells per well of 24-well plate in 0.5 ml. We again use the CASY Cell Counter&Analyser System (Schärfe System) to determine cell numbers.
Critical Step
Plating density and homogeneous distribution of neuronal progenitors is crucial. Different densities should be tried to optimize the differentiation, which is especially important for long-term culture. If the progenitors are plated too diluted, they will not survive for very long, but if they are plated at too high a density a stable network of neurites will not develop. As an approximation, about 30% of the plate should be covered with cells 24 h after plating (see Fig. 4b).
-
24
Change N2 medium 2 h after plating.
-
25
Change N2 medium again after 1 day (approximately 24 h).
-
26
Change medium to complete medium after 2 days (approximately 48 h).
Critical Step
Use of complete medium is recommended, even if it has a highly similar composition to B27 supplement. However, in our experience, the percentage of non-neuronal cells in a culture 7 days after plating of dissociated CAs is higher in B27 than in complete medium. As our Pax6-positive progenitors also express GLAST as a marker of radial glial cells14, B27 suppresses the other potential of radial glial cells to differentiate into astrocytes. Do not add complete medium earlier than after 36 h, preferably after 48 h.
Long-term culture of differentiated neurons
-
27
Cell densities for long-term culture should be optimized by dilution series (Fig. 4c–e). Change the complete medium again on days 4, 8 and 12. Never change the medium entirely again after this time, but only add some complete medium at later stages, for example once or twice 2 ml. Some medium can be removed if the plate or the well is full. Medium should never become yellow.
RNA, protein and immunocytochemistry
-
28
RNA extractions and protein lysates can be prepared at desired time points by conventional methods. Immunocytochemistry can be performed on glass coverslips according to standard technology. Fix cells with 4% paraformaldehyde for 10 min.
Troubleshooting
Troubleshooting advice can be found in Table 2.
Timing
The protocol comprises four distinct steps: (i) culture of ES cells, on feeder cells and without feeders; (ii) formation of CAs; (iii) dissociation of CAs and plating of dissociated cells on PORN/laminin-coated dishes; and (iv) neuronal differentiation and culture of neurons. Steps 1–3 take 2–3 weeks, followed by Step 4 of differentiating the neuronal precursors into mature neurons that depends on the individual need for the degree of maturity of neurons.
Anticipated results
The protocol results in homogeneous cultures of glutamatergic neurons. Good-quality cultures consist of about 90–95% glutamatergic neurons characterized by VGLUT expression in addition to β-tubulinIII, MAP2 and Tau12. Cells with neuronal morphology appear within less than 2 days after dissociation of CAs. Seven days after dissociation, about 90–95% of the cells are VGLUT-positive neurons12, including less than 1% of the cells positive for Isl-1, GABA, TH and ChAT. At this time point, less than 1% of GFAP-positive cells can be identified and hardly any oligodendrocytes can be seen. Neurons start to express synaptophysin 5 days after plating. GluR1 starts to be expressed slightly earlier, whereas expression of Trk receptors starts to be clearly expressed only at 5–7 days with a strong signal at 11 days. In contrast, expression of the p75 neurotrophin receptor decreases during differentiation12. PSD-95 can be detected only at later stages, starting at 11 days of differentiation with clear signals after 21 days. The expression of the amyloid precursor proteins is already clearly detectable on progenitors and subsequently keeps increasing during the course of neuronal differentiation12. Electrical activity has been recorded by whole-cell patch-clamp recordings starting at about 12 days, and patching was found to be difficult before that time12. In 3-week-old cultures, about 5% of GABAergic neurons can be detected by vGAT expression. Neuronal precursors are identified as radial glial cells by expression of Pax6, RC2, BLBP, nestin, vimentin and a spindle-shaped morphology. Expression of Pax6 rapidly decreases when neurons differentiate12. Quantitative PCR using RNA extracted from different lines indicates that neurons differentiate synchronously (see Fig. 5 for a time course of Pax6 and TrkB mRNA expression). Also note that similar mRNA expression levels are detected with different ES cell lines. These results not only demonstrate that the protocol can be applied for ES cell lines of different origins with similar results, but also that the cells differentiate in synchrony.

Three different ES cell lines are shown. J1 and G30 are explained in Table 1. E3#8 are E3 ES cells with a GFP cDNA inserted in one allele of the Mapt locus and a human amyloid precursor protein cDNA in the second allele. A 400 ng portion of total RNA in duplicates was used for RT-PCR (Stratagene, cat. no. 600085) according to the manufacturer's instructions. Following real-time PCR for Pax6 and TrkB including 18S, Ct values of triplicates were averaged and normalized according to Pfaffl16. A relative expression was arbitrarily set as 1 for the first reliable detection of the genes (CAs for Pax6, 4 days after plating dissociated CAs for TrkB). (a) Pax6 is highly expressed in the progenitor cells at the time of CA dissociation. It rapidly decreases (the 2- and 6-h time points are not illustrated here). Already 1 day after plating of neural progenitors, expression of Pax6 is hardly detectable in any of the lines, illustrating the fact that differentiation occurs synchronously. (b) TrkB mRNA expression is robust 4 days after dissociating the CAs and increases with the maturation of neurons. All lines show the same levels and time course of TrkB expression reflecting synchronous differentiation.
As infection of ES cells, especially by mycoplasms, impairs the quality of the culture, we never use penicillin/streptomycin in the ES medium. This facilitates the early detection of contamination, which we recommend to perform on a routine basis, especially for mycoplasms. In particular, each ES cell line should be tested for the presence of mycoplasms (e.g., PCR kits from Stratagene). In addition, the karyotype of ES lines should also be checked on a routine basis. Both mycoplasm infection and karyotype influence the quality of differentiation.
Downscaling of CA formation in 24-well plates is also possible and was achieved by calculating and keeping the ratio of cell numbers to surface area of plate and medium volume. This can be especially useful when different treatments are compared at the CA stage. In addition, freezing-dissociated CA progenitors facilitate the establishment of neuronal cultures at a desired time (see Fig. 2).
Importantly, we have also observed that neural progenitors and neurons can be efficiently infected with lentiviruses (Fig. 2) to acutely express cDNAs (or siRNAs) at various stages of neuronal maturation. Lentiviruses with GFP expressed under the control of the PGK (phosphoglycerate kinase) promoter revealed a high infection rate and expression level in neurons. Cells can be infected from 1 day onwards (after CA dissociation) and subsequently at different time points with more than 90% of the neurons expressing the gene of interest after about 5 days. Expression remained stable for several weeks.
With time in culture, neurons mature and develop synaptic activity that can be readily monitored by electrical recordings. Spines begin to form at around 3 weeks in culture as detected following transfection with actin-GFP cDNAs or phalloidin staining.
Outlook
The method described in this protocol creates new opportunities to dissect molecular mechanisms acting during normal CNS development, as well as in neurodegenerative processes. For example, ES cells can be isolated from well-established transgenic models or engineered to introduce candidate wild-type cDNAs or disease-relevant mutations. One key aspect of our system is the homogeneity of the cultures, thus allowing meaningful gene and protein expression profiling and comparison between wild-type and mutant cells (Fig. 2). Candidate drugs can also be tested on CNS neurons in a high-throughput format.
The protocol could be modified to try and replace RA as a trigger of neuronal differentiation by more physiological stimuli, including for example those modulating the Wnt pathway. Recent transplantation studies with progenitors generated by RA treatment indicate that they are not fated to generate exclusively caudal cells and can effectively and meaningfully populate the brain when transplanted in either the prospective ventral or dorsal chick telencephalon (Nikoletopoulou and Barde, manuscript in preparation). Note that our procedure avoids the use of exogenous growth factors following CA dissociation, and while fibroblast growth factor and epidermal growth factor have been successfully used to grow ES cell-derived progenitors with the characteristics of radial glial cells10, these progenitors generate heterogeneous cell populations10. Trying to achieve homogeneous differentiation of neuronal types other than glutamatergic neurons, such as spinal cord motoneurons or dopaminergic neurons, is also an important objective for the future. This would appear feasible as there are clear indications in the literature that this can be achieved either by using instructive factors such as sonic hedgehog and/or ES cells engineered with transcription factors7,8.
In the future, it will also be important to see the degree to which human ES cells could also be used to generate defined and homogeneous populations of human neurons. This will most likely require substantial modifications of the procedure described here as the conditions necessary to amplify human ES cells that remain pluripotent seem to be quite different from those established for the mouse11,15.
References
Greene, L.A. & Tischler, A.S. Establishment of a noradrenergic clonal line of rat adrenal pheochromocytoma cells which respond to nerve growth factor. Proc. Natl. Acad. Sci. USA 73, 2424–2428 (1976).
Bain, G., Kitchens, D., Yao, M., Huettner, J.E. & Gottlieb, D.I. Embryonic stem cells express neuronal properties in vitro . Dev. Biol. 168, 342–357 (1995).
Li, M., Pevny, L., Lovell-Badge, R. & Smith, A. Generation of purified neural precursors from embryonic stem cells by lineage selection. Curr. Biol. 8, 971–974 (1998).
Okabe, S., Forsberg-Nilsson, K., Spiro, A.C., Segal, M. & McKay, R.D. Development of neuronal precursor cells and functional postmitotic neurons from embryonic stem cells in vitro . Mech. Dev. 59, 89–102 (1996).
Rathjen, J. & Rathjen, P.D. Mouse ES cells: experimental exploitation of pluripotent differentiation potential. Curr. Opin. Genet. Dev. 11, 587–594 (2001).
Smith, A.G. Embryo-derived stem cells: of mice and men. Annu. Rev. Cell Dev. Biol. 17, 435–462 (2001).
Wichterle, H., Lieberam, I., Porter, J.A. & Jessell, T.M. Directed differentiation of embryonic stem cells into motor neurons. Cell 110, 385–397 (2002).
Kim, J.Y. et al. Dopaminergic neuronal differentiation from rat embryonic neural precursors by Nurr1 overexpression. J. Neurochem. 85, 1443–1454 (2003).
Wobus, A.M. & Boheler, K.R. Embryonic stem cells: prospects for developmental biology and cell therapy. Physiol. Rev. 85, 635–678 (2005).
Conti, L. et al. Niche-independent symmetrical self-renewal of a mammalian tissue stem cell. PLoS Biol. 3, e283 (2005).
Keller, G. Embryonic stem cell differentiation: emergence of a new era in biology and medicine. Genes Dev. 19, 1129–1155 (2005).
Bibel, M. et al. Differentiation of mouse embryonic stem cells into a defined neuronal lineage. Nat. Neurosci. 7, 1003–1009 (2004).
Malatesta, P. et al. Neuronal or glial progeny: regional differences in radial glia fate. Neuron 37, 751–764 (2003).
Plachta, N., Bibel, M., Tucker, K.L. & Barde, Y.A. Developmental potential of defined neural progenitors derived from mouse embryonic stem cells. Development 131, 5449–5456 (2004).
Pera, M.F. & Trounson, A.O. Human embryonic stem cells: prospects for development. Development 131, 5515–5525 (2004).
Pfaffl, M. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29, 2002–2007 (2001).
Tucker, K.L., Meyer, M. & Barde, Y.A. Neurotrophins are required for nerve growth during development. Nat. Neurosci. 4, 29–37 (2001).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
About this article
Cite this article
Bibel, M., Richter, J., Lacroix, E. et al. Generation of a defined and uniform population of CNS progenitors and neurons from mouse embryonic stem cells. Nat Protoc 2, 1034–1043 (2007). https://doi.org/10.1038/nprot.2007.147
Published:
Issue Date:
DOI: https://doi.org/10.1038/nprot.2007.147
This article is cited by
-
Dissecting the roles of MBD2 isoforms and domains in regulating NuRD complex function during cellular differentiation
Nature Communications (2023)
-
Transcriptional repression by a secondary DNA binding surface of DNA topoisomerase I safeguards against hypertranscription
Nature Communications (2023)
-
MLL3/MLL4 methyltransferase activities control early embryonic development and embryonic stem cell differentiation in a lineage-selective manner
Nature Genetics (2023)
-
ZFP462 safeguards neural lineage specification by targeting G9A/GLP-mediated heterochromatin to silence enhancers
Nature Cell Biology (2023)
-
Tissue engineering of the gastrointestinal tract: the historic path to translation
Journal of Biological Engineering (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
