
Abstract
The ability to isolate oligodendroglial precursor cells (OPCs) provides a powerful means to characterize their differentiation, properties and potential for myelin repair. Although much knowledge is available for isolation of OPCs from the rat central nervous system, preparation and maintenance of mouse OPCs has been until recently a challenge owing to difficulties in obtaining a sufficient quantity of purified OPCs. Here, we describe protocols to prepare highly enriched rat OPCs and nearly homogenous mouse OPCs. The mouse method generates predominantly OPCs from cortical neural progenitor cells as clonal aggregates called “oligospheres” by taking advantage of molecular genetic tools. Isolated OPCs can be further differentiated into oligodendrocytes. Collectively, we describe simple and efficient methods for the preparation and in vitro maintenance of enriched OPCs from rats and mice. Isolation and culture of a large, homogenous population of rodent OPCs should significantly facilitate studies on OPC lineage progression and their utility in myelin repair after injury.
Similar content being viewed by others
Introduction
Oligodendrocytes, the myelinating glia of the central nervous system (CNS), play a crucial role in facilitating the rapid conduction of neuronal action potentials1 and supporting axonal survival2. Oligodendrocytes are generated from OPCs, which proliferate and migrate throughout the CNS during late embryonic development, and later differentiate into mature myelinating oligodendrocytes3,4. Several distinct stages during oligodendrocyte maturation have been identified for oligodendroglial lineage cells in vitro3,5. These include proliferating OPCs, characterized by expression of progenitor cell markers such as the platelet-derived growth factor alpha receptor (PDGFαR) with a bipolar or tripoIar morphology, intermediate immature oligodendrocytes, expressing markers recognized by the O4 antibody with a multipolar morphology, and finally mature myelinating oligodendrocytes, expressing myelin-specific proteins such as myelin basic protein. When transplanted into the brain of hypomyelinated hosts, oligodendrocyte precursors can migrate a significant distance, and give rise to a large number of mature oligodendrocytes6,7, as well as myelinate axons8. Simple methods for the isolation and purification of workable quantities of OPCs not only aid in efforts to better understand oligodendrocyte development, function and axon–oligodendroglial interactions but also provide an indispensable tool for myelin repair research. Several methods for isolation of rat OPCs from the CNS have been described, such as immunopanning5,9,10, fluorescence-activated cell sorting (FACS) by exploiting cell surface-specific antigens5,11, differential gradient centrifugation7,8,12 or a shaking method based on differential adherent properties of glia13,14, which permits the separation of rat OPCs from the astroglial cells in the mixed glial culture by shearing forces.
In contrast to rat OPCs, mouse OPCs have proven more difficult to isolate. Mouse OPCs do not share all of the cell surface antigens with their rat counterparts such as A2B5 (ref. 15), impeding approaches such as immunopanning and cell sorting (FACS) as described for rat OPC isolation. In addition, mouse OPCs tend to differentiate in in vitro mixed glial cultures. They are also relatively difficult to separate from astrocytes by shaking methods. Most importantly, as many transgenic and knockout studies have been carried out in mice, it becomes increasingly crucial to develop a simple procedure for the isolation and purification of ample amounts of OPCs from mouse CNS tissues. The fact that multipotent neural progenitor cells can give rise to oligodendroglial lineage-restricted precursors16,17 suggests a new avenue for the generation of OPCs from neural progenitor cells. Several studies described methods to generate self-renewing OPCs from neural progenitor/stem cells in different species such as dog and rodents18,19,20,21. As PDGFαR expression identifies OPCs in the CNS4,22, we availed ourselves of PDGFαR-GFP knock-in mice23, where a nuclear-localized green fluorescent protein (GFP) was knocked into the PDGFαR locus, to discern OPCs. Thus, by tracking GFP expression, we were able to monitor the formation of, and isolate, mouse PDGFαR+ OPCs from cortical progenitor cells under different culture conditions. Thus, here we describe technically simple procedures to prepare a large, highly enriched population of OPCs from rats or mice. These methods allow isolation of rat OPCs using a selective detachment procedure13 with modifications24, and the generation of large numbers of mouse OPCs through formation of “oligospheres” from neurospheres using embryonic multipotent cortical progenitor cells. The OPCs isolated by the procedures described below can be induced to differentiate into immature oligodendroblasts and then into mature oligodendrocytes. These methods will facilitate the in vitro use of OPCs to address such issues as effects of various molecules on OPC differentiation and axon–oligodendroglia interactions.
Materials
Reagents
-
C57B6/J (The Jackson laboratory) and PDGFαR-GFP knock-in mice23
Caution
Experiments involving live animals must conform to national and institutional regulations.
-
Sprague–Dawley rats (Harlan Industries Inc.)
Caution
Experiments involving live animals must conform to national and institutional regulations.
-
Dulbecco's modified Eagle's media(DMEM; Invitrogen/Gibco 11960) without L-glutamine and sodium pyruvate
-
100 × N2 supplement (Invitrogen 17502048)
-
DMEM/F12 (Invitrogen/Gibco 11330-032)
-
Fetal bovine serum (FBS; Hyclone SH300700)
-
L-Glutamine (Sigma G8540)
-
Sodium pyruvate (Sigma P2256)
-
Bovine serum albumin (BSA; Sigma A9647)
-
Apo-transferrin (Sigma T2252)
-
Insulin (Sigma I6634)
-
Sodium selenite (Sigma S5261)
-
D-Biotin (Sigma B4501)
-
Hydrocortisone (Sigma H0888)
-
Human PDGF-AA (Peprotech 100-13A)
-
Basic FGF (Peprotech 100-18B)
-
Human recombinant epidermal growth factor (EGF; Peprotech, 100-15)
-
Ciliary neurotrophin factor (CNTF; Peprotech, 450-50)
-
N-acetyl-L-cysteine (NAC; Sigma A-8199)
-
Triiodothyronine (Sigma, T-2752)
-
Poly-D,L-ornithine (Sigma P0421)
-
Poly-D-lysine (Sigma P0899)
-
Circle cover glass (12 mm diameter) (Carolina Biological; 63-3009)
-
Penicillin/streptomycin (Invitrogen 15140)
-
Hanks balanced salt solution (HBSS; Invitrogen 14025)
-
DNase I (Sigma D5025)
-
Trypsin: L-1-tosylamido-2-phenylethylchloromethyl ketone (TPCK)-treated trypsin (Sigma T1426)
-
Trypan blue (0.04% (w/v), Sigma T8154)
-
Phenylmethylsulfonyl fluoride (Sigma P7626-5G)
-
Dulbecco's phosphate-buffered saline (DPBS) without Mg and Ca (Invitrogen 14190-144)
Equipment
-
Humidified tissue culture incubator (37 °C, 5% CO2)
-
Laminar flow hood
-
Dissecting microscope (MZ6; Leica)
-
Water bath at 37 °C
-
Microdissecting instruments (sterilized): small dissecting scissors; Dumont forceps—straight and angled; curved microdissecting scissors; spatula
-
Tabletop centrifuge
-
Hemocytometer
-
Orbital shaker (Barnstead Digital Orbital Shaker, cat. no. SHKE2000)
-
Oven for orbital shaker (Bellco Benchtop incubator)
-
15 ml plastic conical tubes (Falcon 352097)
-
50 ml plastic conical tubes (Falcon 352070)
-
Glass pipettes (Fisher 13-678-27F)
-
T75 cm2 tissue culture flask with plug-seal (Fisher 13-680-59)
-
6- or 24-well tissue culture plates (Nunc)
-
Sterile medium filters (0.22 μm)
-
10 cm Petri dish (Fisher 08-757-13)
-
70 μm cell strainer (Falcon 352350)
-
20 or 50 μm sterile screening fabric nylon mesh (Sefar America; 03-20/14 or 03-50/31)
Reagent setup
-
DMEM20S DMEM, 4 mM L-glutamine, 1 mM sodium pyruvate, 20% FBS, 50 U ml−1 penicillin and 50 μg ml−1 streptomycin. Shelf life: 2 weeks at 4 °C.
-
Basal chemically defined medium DMEM, 4 mM L-glutamine, 1 mM sodium pyruvate, 0.1% BSA, 50 μg ml−1 Apo-transferrin, 5 μg ml−1 insulin, 30 nM sodium selenite, 10 nM D-biotin and 10 nM hydrocortisone. Shelf life: 2 weeks at 4 °C.
-
OPC medium Basal chemically defined medium (BDM) containing 10 ng ml−1 PDGF-AA and 10 ng ml−1 bFGF. Shelf life: 2 weeks at 4 °C
Critical
Growth factors should be prepared as 1,000 × stocks and stored as aliquots at −80 °C. They should be added to BDM just before OPC plating or medium change.
-
NAC stock solution (1,000 ×) Dissolve 50 mg NAC (Sigma A8199) in 10 ml DMEM and adjust the pH to 7 with 1 N HCl. Make 40 μl aliquots; store at −20 °C.
-
Oligodendrocyte differentiation medium BDM containing 15 nM triiodothyronine, 10 ng ml−1 CNTF and 1 × NAC. Shelf life: 2 weeks at 4 °C.
Note: CNTF and NAC can enhance oligodendrocyte survival25.
-
Neural culture medium DMEM/F12 supplemented with 25 μg ml−1 insulin, 100 μg ml−1 Apo-transferrin, 20 nM progesterone, 60 μM putrescine and 30 nM sodium selenite. Shelf life: 2 weeks at 4 °C.
-
Neurosphere growth medium Neural culture medium (NCM) supplemented with 20 ng ml−1 bFGF and 20 ng ml−1 EGF. Shelf life: 1 week at 4 °C.
Critical
The biological activity of growth factors in serum-free media (SFM) decreases with time. For optimal results, add growth factors to SFM on the day of preparation. Generally, we store SFM with growth factors at 4 °C up to 7 days for the passage of neurospheres.
-
B104 growth medium (For the growth of B104 neuroblastoma cells) DMEM/F12 supplemented with 10% FBS.
-
N2 medium DMEM/F12 supplemented with 1 × N2.
-
B104 neuroblastoma conditioned medium (B104 CM) Culture B104 neuroblastoma cells in B104 growth medium until confluent. Wash with 1 × Puck's BSS and feed with N2 medium. After 4 days, collect the medium, add phenylmethylsulfonyl fluoride to a final concentration of 1 μg ml−1 and mix quickly by swirling. Centrifuge at 2,000g for 30 min in a swinging bucket centrifuge at 4 °C. Filter the supernatant with a 0.22 μm pore size filter system and retain the filtered supernatant. This is B104 CM. Shelf life: up to 6 months at −80 °C.
Critical
The B104 CM should be aliquoted and stored at −80 °C for later use. To minimize variation between cultures, B104-CM from each batch should be tested before use. Freeze–thaw cycles should be minimized with no more than two freeze–thaw cycles.
-
Oligosphere medium 7 parts of NCM:3 parts of B104CM (7:3 = vol:vol): this oligosphere media can be stored at 4 °C for 2 weeks.
-
10 × Puck's BSS Add NaCl 80 g, KCl 4 g, Na2HPO4 ·7H2O 0.9 g, KH2PO4 0.4 g and glucose 10 g in 1,000 ml triple-distilled water and filter-sterilize (0.22 μm) and store at 4 °C. The working solution is 1 × Puck's BSS. Make this by diluting stock solution 1:10 with sterile triple-distilled water in the tissue culture hood for sterility.
-
Trypsin stock solution 0.25% (w/v) trypsin in HBSS and stored in aliquots at −20 °C.
-
DNase I stock 0.20 mg ml−1 in HBSS (20 ×) and stored in aliquots at −20 °C. Final concentration should be 10 μg ml−1 after dilution in HBSS.
Equipment setup
-
Poly- D -lysine-coated flasks Dilute a 100 × stock of poly-D-lysine (10 mg ml−1 in 0.5% BSA in DPBS, stored as aliquots at −20 °C) with 1 × DPBS and filter-sterilize (0.22 μm). Coat culture flasks and plates with the 1 × poly-D-lysine coating solution (∼7 ml per flask) for 1–2 h in 37 °C incubator or overnight at room temperature (RT; 22 °C). Remove coating solution, wash three times with sterile ddH2O and dry completely in a tissue culture hood, and store with caps screwed tightly at RT. The coated flask can be stored at RT for at least 4 weeks.
-
Poly- D,L -ornithine-coated plates Dilute a 100 × stock of poly-D,L-ornithine (5 mg ml−1 in PBS, stored in aliquots at −80 °C) with 1 × DPBS and filter-sterilize (0.22 μm). Add sufficient quantity of 1 × coating solution to cover the surface of culture plates and incubate for 1–2 h at 37 °C or overnight at RT. Remove solution, wash three times with ddH2O and air-dry in a tissue culture hood. After complete drying, coated plates can be stored at RT for at least 4 weeks.
Procedure
Dissection and plating of cerebral cortices
-
1
Follow option A for neonatal rats and option B for mouse embryos. Note that neonatal mouse pups, for example at P0–2, can be used as described for neonatal rat pups. However, the efficiency to form oligospheres is low when using the neonatal brain as compared to the embryonic brain.
-
A
Dissection, plating and culture of neonatal rat cortices
-
i
In a laminar flow hood, pour ice-cold HBSS into two 10 cm Petri dishes placed in a tray filled with ice.
-
ii
Decapitate a P1–2 rat pup with large scissors and place the head in cold HBSS.
Critical Step
Anesthetize the pups first by burying them in ice for 1–5 min.
-
iii
Submerge one head in a Petri dish containing clean ice-cold 70% ethanol briefly, rinse off the ethanol by submerging the head in ice-cold HBSS and then transfer it into another Petri dish containing ice-cold HBSS. Repeat for the remaining heads in the litter (approximately 10–15 pups can be processed at a time). Change to a clean Petri dish containing fresh ice-cold HBSS if the buffer becomes bloody.
Caution
Keep 70% ethanol away from flame to prevent fire hazard.
-
iv
Hold the nose portion of one head with Dumont forceps in a clean Petri dish. Use a curved microdissecting scissors to gently cut skin along the midline advancing toward the nose, and then cut the skull.
Caution
The tip of the scissors should point away from the brain to avoid damaging the brain. The skull at this age is very soft.
-
v
Make two lateral cuts at the base of the skull by inserting the scissors where the spinal cord was severed at the foramen magnum. Fold back the two sides of the skull with forceps, scoop out brain and cut off cerebellum with a spatula.
-
vi
Divide the cerebrum along the midline into two cerebral hemispheres and subsequently cut off olfactory bulbs, basal ganglia below the cerebral cortex and the hippocampus with Dumont forceps (Fig. 1). Place the isolated cerebral cortex in a clean Petri dish containing HBSS on ice.
Figure 1: Dissection of the neonatal rodent cortex. 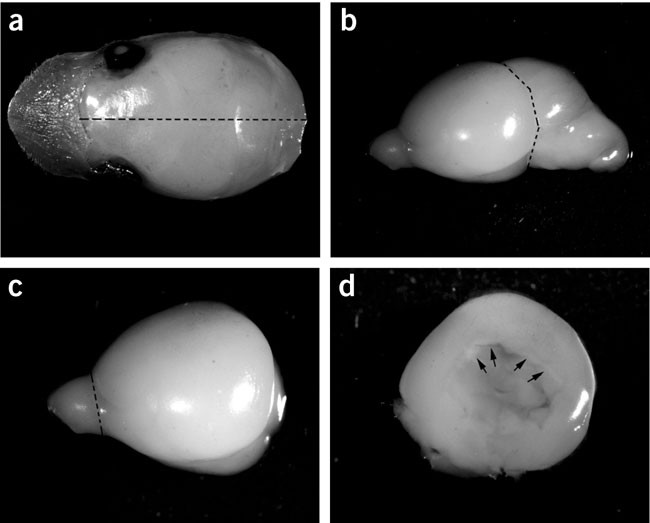
(a) Hold the nose portion of the head with sterile forceps, gently remove skin, skull and take out the brain with a sterile spatula. Dotted line shows midline cut through the brain. (b) Half of the brain cut along the midline showing the cerebral hemisphere, the cerebellum and the olfactory bulb. Dotted line indicates the cut for the removal of cerebellum. (c) Dorsal view of one cerebral hemisphere with dotted line indicating the cut for the removal of the olfactory bulb. (d) The cerebral cortex after the removal of basal ganglia and the arrows indicates the place to dissect out the hippocampus.
-
vii
Repeat Steps iv–vi for the remaining heads.
-
viii
Take one cortex. Remove the meninges with forceps with fine tips under a dissection microscope. Repeat with the remaining cortices.
-
ix
Place all meninges-free cortices into one clean Petri dish on ice.
-
x
Dice the cortical tissues with a sterilized razor blade into ∼1 mm3 chunks.
-
xi
Add 13.6 ml HBSS, 0.8 ml DNase I stock solution (0.2 mg ml−1) and 0.6 ml trypsin stock solution (0.25%) into the Petri dish.
-
xii
Incubate for 15 min in the tissue culture incubator at 37 °C.
-
xiii
Pipette the contents of the Petri dish into a 50 ml sterile centrifuge tube, wash away the residual tissues in the Petri dish with 5 ml DMEM20S and transfer into the 50 ml tube to stop trypsinization.
-
xiv
Collect cells/tissues by centrifugation in a swinging bucket at 100g (∼800 r.p.m.) for 5 min.
-
xv
Carefully aspirate the supernatant with a Pasteur pipette. Discard the supernatant.
-
xvi
Add 20 ml DMEM20S to the 50 ml tube.
-
xvii
Triturate and dissociate the pellet with a 10 ml glass pipette until nearly homogenous.
-
xviii
Let the tissue settle for ∼10 min on ice.
-
xix
Pass the tissue suspension (avoid the pellet) through a 70 μm nylon cell strainer placed on a 50 ml conical tube and collect the flow-through.
-
xx
Add 20 ml DMEM20S to the settled tissue and triturate again.
-
xxi
Pass the suspension through a 70 μm nylon cell strainer and collect in a 50 ml tube.
-
xxii
Combine the filtered cell suspensions in a T75 poly-D-lysine-coated flask and make up the volume to 100 ml with DMEM20S (approximately 10 ml DMEM20S per animal).
-
xxiii
Plate 10 ml cell suspension per coated flask (approximately 10 million cells per flask).
-
xxiv
Spread cell suspension over the entire flask surface and date the flask. Incubate the flask in a tissue culture incubator with 5% CO2 at 37 °C.
-
xxv
Feed every 2–3 days with complete medium change of 10 ml DMEM20S for 10 days.
Timing 3.5 h to 10 days
-
i
-
B
Dissection, plating and culture of mouse embryonic cortices
-
i
In the laminar flow hood, remove the embryos,for example at embryonic day (E)14.5–17.5, from timed pregnant mice and place them in a clean Petri dish containing cold HBSS on ice.
-
ii
Decapitate the embryos, remove the skin of the telencephalic bulb and skull gently from the head by holding the neck region with a forceps.
-
iii
Squeeze off the telencephalic bulb by 45° angled Dumont forceps. Remove the medial portion and retain the lateral part of the cerebral cortex. Place the cortex in a clean Petri dish containing HBSS on ice and remove the meninges with the forceps.
-
iv
Repeat Steps iii and iv for the remaining heads if oligospheres are needed on a bulk scale.
-
v
Cut each cortex into 2–3 pieces and transfer them to ice-cold neurosphere growth medium (0.5 ml per brain) with 20 ng ml−1 EGF and 20 ng ml−1 bFGF.
-
vi
Dissociate the cortices by mechanical trituration with a fire-polished glass Pasteur pipette (about 35 strokes) until the cell suspension has no or very few small clumps.
-
vii
Leave suspension on ice for 2 min. Pass the suspension through a 50 μm nylon pouch placed on 15 ml conical tube to obtain single cell suspension.
-
viii
Count the cells with a hemocytometer and add 5 × 104 cells per ml cell suspension to each well of a six-well plate (4 ml per well of neurosphere growth medium). Incubate the plate in a tissue culture incubator with 5% CO2 at 37 °C.
-
ix
Feed every 2 days by replacing half of the medium with fresh neurosphere growth medium for ∼4 days.
Timing 2 h to 4 days
-
i
-
A

OPC isolation and culture
-
2
Follow option A for rat OPCs and option B for mouse OPCs.
-
A
Isolation and culture of rat OPCs
-
i
At ∼10 days after plating, mixed glial cultures will be confluent, and phase-dark, process-bearing OPCs appear on top of phase-gray bed layers of astrocytes. The culture is ready to be shaken to obtain OPCs.
-
ii
Remove culture flasks from the incubator.
-
iii
Screw tightly the plug-seal caps. Note that the viability of OPCs is unaffected by the closed environment of the shaking procedure.
-
iv
Secure the flasks to a Styrofoam board on a horizontal orbital shaker (autoclave tape holds better than regular paper tape).
-
v
Pre-shake the flasks on the shaker for 1 h at 200 r.p.m. at 37 °C to remove microglial cells.
-
vi
Discard the medium from the flasks by aspiration.
-
vii
Add 10 ml DMEM20S to each flask.
-
viii
Tighten the plug-seal caps and secure the flasks to the orbital shaker. Shake the flasks at 200 r.p.m. overnight at 37 °C (∼18–20 h).
Critical Step
A completely closed environment with presumably a low O2 level allows OPCs to detach easily from the astrocyte layer in mixed glia culture.
-
ix
Remove flasks from the shaker and sterilize the surface of flasks by spraying with 70% ethanol to prevent contamination.
-
x
Collect the cell suspension from each flask by a pipette and transfer to an untreated Petri dish (1 flask per Petri dish). Add 10 ml DMEM20S to each flask if they will be shaken again, and return them to the tissue incubator. The culture medium should be completely changed every 2–3 days to allow more OPCs to grow on the astrocyte layer. Mixed glia can be shaken a second time if one continues to culture them for an additional week after the first shake using the same procedure. In our experience, there is no functional difference in OPCs obtained from the first shake and the second shake, although the yield of OPCs from the second shake is lower. Discard flasks that have been shaken twice.
Critical Step
The Petri dishes used here must be untreated. They should not be treated for tissue culture because OPCs tend to attach to treated Petri dishes, resulting in very low yield of OPCs. We use Petri dishes from Fisher (cat. no. 08-757-13).
-
xi
Incubate Petri dishes for 30–60 min in tissue culture incubator at 37 °C for differential adhesion of contaminating microglia and astrocytes. Note that microglia and astrocytes attach to the Petri dish more efficiently than OPCs.
-
xii
Gently swirl the Petri dish and collect cell suspension into a 50 ml tube.
-
xiii
Flame a surgical forceps (hemostats) and attach to it a sterile screening pouch (20 μm pore size), and place over a clean 50 ml conical tube.
-
xiv
Pass the cell suspension slowly through the sieves into the 50 ml tube.
-
xv
Centrifuge the cell suspension for 10 min at 100g (∼800 r.p.m.) in a swinging bucket.
-
xvi
Carefully remove the supernatant by pipette without dislodging the pellet. Suspend and dissociate the pellet in a small amount of the remaining medium (∼0.5 ml).
Critical Step
The pellet is very loose, so exercise great care when removing the supernatant. Also, OPCs should be dissociated thoroughly to single cells. Otherwise, they tend to grow in clumps. Avoid generating air bubbles when dissociating.
-
xvii
Count living cells using the Trypan blue exclusion assay with a hemocytometer. Living cells do not take up the dye and are colorless under brightfield.
-
xviii
Dilute the OPC suspension with the OPC medium to the desired concentration and plate onto poly-D,L-ornithine-coated plates to achieve a density of 1 × 104 per cm2.
-
xix
Incubate the plates in a tissue culture incubator at 37 °C.
-
xx
Feed cells every other day with half of medium change with BDM containing 20 ng ml−1 PDGF-AA and 20 ng ml−1 bFGF for 7–10 days. Cells are primarily A2B5+, O4+ but O1− and MBP− oligodendrocyte precursors after 7 days in vitro24 (Fig. 2).
Figure 2: Highly enriched primary rat OPC cultures. 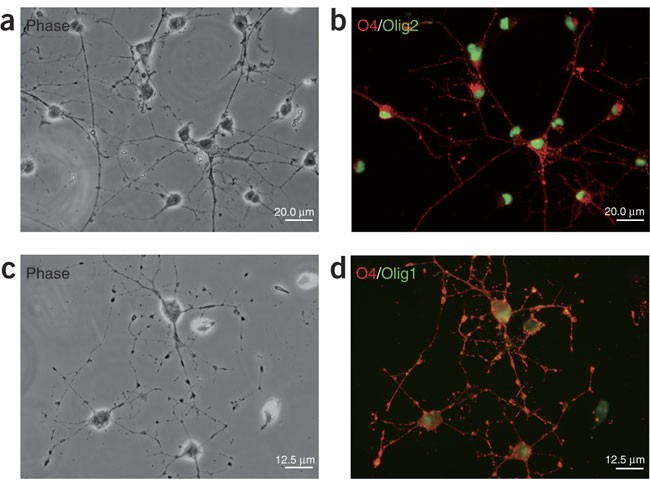
(a,c) Phase contrast micrographs of living OPCs derived from neonatal rat cortices. OPCs are of typical bipolar or tripolar morphology. (b,d) OPCs are immunopositive for O4 (red) and the oligodendrocyte lineage-specific transcription factors (green: in panel b, Olig2; in panel d, Olig1).
Timing 24 h to 7 days
-
i
-
B
Generation of mouse OPCs from neurospheres
-
i
At around the 4th day after the formation of neurospheres (sphere size around 200–300 μm in diameter), the culture is ready for oligosphere induction. Note that generally neurosphere formation takes about 3–5 days for the embryonic cortical tissue and for the neonatal tissue it takes about 10 days.
-
ii
Gradually change the EGF/bFGF-containing neurosphere growth medium to B104 CM-containing oligosphere medium by replacing one-fourth of the former medium with the latter medium every other day for 2 weeks. Note that during the transition period (weeks 1–2), the number and size of spheres do not change significantly. However, the number of GFP fluorescent cells should increase significantly in the spheres. After 2 weeks, at least 95% cells in the spheres should show intense GFP fluorescence. As the cells are PDGFαR-GFP+, signifying oligodendrocyte precursors, the spheres are now referred to as oligospheres. By comparing the oligosphere medium (NCM plus B104CM) with NCM alone, NCM plus 20 ng ml−1 bFGF, 20 or 40 ng ml−1 PDGF-AA, we observed that the B104CM-containing oligosphere medium is most effective in induction of oligosphere formation from neurospheres.
-
iii
The oligospheres should now be dissociated. They can be either mechanically dissociated by a fire-polished pipette as described above followed by passage through a 50 μm nylon mesh to obtain single cells or dissociated by enzymatic treatment with trypsin. Enzymatic dissociation: incubate oligospheres with 0.5 ml of 0.05% trypsin at 37 °C for 5 min; halt with 4 ml oligosphere medium; spin down the cells at 120g (∼1,000 r.p.m.) for 5 min at RT and pass cells through a 50 μm nylon mesh to obtain cell suspension.
-
iv
Culture the cell suspension in the oligosphere medium on an uncoated plate at a density of ∼3 × 104 cells per ml. Oligospheres should form again after 5–7 days. Alternatively, the cell suspension can be plated on the poly-ornithine-coated plates for OPC proliferation in the OPC medium or differentiation in the oligodendrocyte differentiation medium.
Caution
While passaging oligospheres, some oligospheres tend to attach to the bottom of the plate and assume typical early oligodendrocyte morphology. Generally, we take only the free-floating oligospheres for passaging. Avoid excessive formation of air bubbles while doing mechanical dissociation of neurospheres or oligospheres, as they will reduce cell viability.
Timing ∼14 days
-
i
-
A

Troubleshooting
Culturing rat OPCs: a low yield of rat OPCs is often a result of (i) unknowingly using Petri dishes treated for tissue culture when removing contaminating microglia and astrocytes by differential adhesion; (ii) accidentally removing some OPCs as the OPC pellet is very loose. Do not completely remove all the supernatant. Always leave some medium above the loose pellet.
Culturing of neurospheres: a low yield of neurospheres is often a result of harsh dissociation of cortices by generating many air bubbles. Use a fire-polished Pasteur pipette with gentle trituration. Another possible reason is that cell suspension is plated at a very high cell density, as the dead and dying cells will prevent the generation of neurospheres. Always count the live cell number before plating.
Oligosphere generation: if cells in spheres are not completely PDGFαR+ or GFP+, continue to culture with B104CM-containing oligosphere medium. It is also possible that the quality of B104CM is not sufficient to induce oligosphere formation. Replace with a new or fresh batch of B104CM. Alternatively, FACS sorting of GFP+ cells from cell suspension after passaging the spheres can facilitate the isolation of the homogenous population of PDGFαR-GFP-positive OPCs. If few oligospheres are generated, the cells from oligosphere passaging can be plated at higher density (e.g., 1 × 105 cells per ml). Higher density plating will yield more oligospheres.
Timing
(1)Dissection and plating of cerebral cortices
(A)Dissection and plating of neonatal rat cortices: 3.5 h to 10 days
Steps i–ix, dissection of rat neonatal cortices: 2.5 h
Steps x–xxiii, plating of rat neonatal cortices: 1 h
Steps xxiv and xxv, culture of rat mixed glia : ∼ 10 days
(B)Dissection and plating of mouse embryonic cortices: 2 h to 4 days
Steps i–v, dissection of mouse embryonic cortices: 1.5 h
Steps vi–vii, plating of mouse embryonic cortices: 0.5 h
Steps viii–ix, culture of mouse neurospheres: ∼ 4 days
(2)OPC isolation and culture
(A)Isolation and culture of rat OPCs: 24 h to 7 days
Steps i–vii, pre-shake of mixed glial culture: 1.5 h
Steps viii–xvii, isolating and plating of OPCs: ∼22.5 h
Steps xviii–xx, culture of rat OPCs: ∼7 days
(B)Preparation of mouse OPCs from neurospheres: ∼14 days
Steps i and ii, mouse oligosphere generation from neurospheres: ∼14 days
Steps iii and iv, mouse OPC plating and culture: 1 h to days
Anticipated results
For rat OPC generation, after 7 days in vitro, cells are primarily A2B5+, O4+ but O1− and MBP− oligodendrocyte precursors (Fig. 2). Contaminating microglia and astrocytes are routinely less than 2−3% each24.
For mouse OPC generation, the embryonic cortex such as at E14.5 yields the highest number of oligospheres (Fig. 3). However, the neonatal cortex (P0–2) also can yield large number of oligospheres. The difference is that the neonatal cortical brain tissue requires a longer time (10 days) to form neurospheres when compared to embryonic cortical brain tissue (3–5 days).

Stages of oligosphere formation from neurospheres generated from cortical neural progenitor cells of mouse E14.5 embryos. (a) Neurosphere formation at stage 1; (b) early phase of oligosphere formation at stage 2; (c) initial appearance of oligospheres at stage 3 and (d) oligosphere formation at stage 4, at which essentially all the cells are positive for PDGFαR/GFP, indicating that they consist of OPCs.
During oligosphere formation, we are usually able to observe four stages (Fig. 3a–d). During neurosphere formation, we detect few GFP+ cells in the neurosphere (Fig. 3a) (stage 1). After gradual replacement of the neurosphere growth medium containing B104CM, we can observe a dramatic increase in GFP+ cells in spheres during the first week (stage 2) and the second week (stage 3) of culture. In the third week of culture, essentially all cells in the spheres become GFP+/PDGFαR+ as oligospheres (stage 4).
Oligospheres were also plated on poly-L-ornithine-coated coverslips in the presence of the OPC medium. Cells from oligospheres are able to migrate away to form individual OPCs with bipolar and tripolar morphologies (Fig. 4a–f). These GFP+ OPCs can be confirmed with OPC markers such as PDGFαR and Olig2 expression by immunohistochemistry (Fig. 4g–j). With this method, nearly homogenous oligodendrocyte precursors (>98%) can be prepared from mouse embryonic cortical tissues. No neuronal cells indicated by expression of Tuj1 were detected in the isolated OPC population, although approximately 2% of cells represent other neuroglial cells or early progenitors as evidenced by expression of nestin and GFAP (not shown). In addition, by mechanical or enzymatic dissociation, the oligosphere can be cultured and passaged in the OPC medium without differentiation. They can be further induced to form differentiated oligodendrocytes (e.g., O4+) when cultured in the oligodendrocyte differentiation medium (Fig. 4k).

Oligospheres plated on the poly-ornithine-coated coverslip exhibit cells migrating away from oligospheres. Phase-contrast (a) and fluorescence image (b) of a representative oligosphere, demonstrating that essentially all cells are GFP-positive. (c–f) Dissociated cells from oligospheres display typical bipolar or tripolar morphology of early OPCs (c), and are GFP+/PDGFαR+ as shown by intense nuclear GFP expression (d). Higher magnification images of the OPCs with bipolar and tripolar morphology (arrows) are shown in e and f. (g–j) GFP-expressing OPCs (g) generated from oligospheres are immunostained for OPC markers Olig2 (h) and PDGFαR (i). (j) Overlay of the expression of GFP, Olig2 and PDGFαR in OPCs. (k) OPCs (PDGFαR-GFP+, arrows) were induced to become differentiated O4+ oligodendrocytes (red) when cultured in the oligodendrocyte differentiation medium for 48 h. Scale bar in a–d in 100 μm.
Multiple approaches have been developed for OPC purification; each has its own merits. Compared to other OPC isolation methods such as costly immunopanning5,9,10 and technically demanding differential gradient centrifugation7,8,12, the method for OPC preparation through oligosphere generation from both mouse and rat neural progenitor cells is simple and straightforward20,21,26. It can be easily followed by a researcher without prior OPC isolation experience. In addition, bulk and economical OPC generation can be achieved without a requirement for specific cell surface antibodies and equipment.
References
Miller, G. Neuroscience. The dark side of glia. Science 308, 778–781 (2005).
Lappe-Siefke, C. et al. Disruption of Cnp1 uncouples oligodendroglial functions in axonal support and myelination. Nat. Genet. 33, 366–374 (2003).
Pfeiffer, S.E., Warrington, A.E. & Bansal, R. The oligodendrocyte and its many cellular processes. Trends Cell Biol. 3, 191–197 (1993).
Miller, R. & Reynolds, R. in Myelin Biology and Disorders (ed. Lazzarini, R.A.) 289–310 (Elsevier Academic Press, New York, 2004).
Gard, A.L., Williams, W.C., II & Burrell, M.R. Oligodendroblasts distinguished from O-2A glial progenitors by surface phenotype (O4+GalC−) and response to cytokines using signal transducer LIFR beta. Dev. Biol. 167, 596–608 (1995).
Warrington, A.E., Barbarese, E. & Pfeiffer, S.E. Differential myelinogenic capacity of specific developmental stages of the oligodendrocyte lineage upon transplantation into hypomyelinating hosts. J. Neurosci. Res. 34, 1–13 (1993).
Vitry, S., Avellana-Adalid, V., Lachapelle, F. & Evercooren, A.B. Migration and multipotentiality of PSA-NCAM+ neural precursors transplanted in the developing brain. Mol. Cell. Neurosci. 17, 983–1000 (2001).
Duncan, I.D., Paino, C., Archer, D.R. & Wood, P.M. Functional capacities of transplanted cell-sorted adult oligodendrocytes. Dev. Neurosci. 14, 114–122 (1992).
Gard, A.L. & Pfeiffer, S.E. Glial cell mitogens bFGF and PDGF differentially regulate development of O4+GalC− oligodendrocyte progenitors. Dev. Biol. 159, 618–630 (1993).
Barres, B.A. & Raff, M.C. Proliferation of oligodendrocyte precursor cells depends on electrical activity in axons. Nature 361, 258–260 (1993).
Behar, T., McMorris, F.A., Novotny, E.A., Barker, J.L. & Dubois-Dalcq, M. Growth and differentiation properties of O-2A progenitors purified from rat cerebral hemispheres. J. Neurosci. Res. 21, 168–180 (1988).
Goldman, J.E., Geier, S.S. & Hirano, M. Differentiation of astrocytes and oligodendrocytes from germinal matrix cells in primary culture. J. Neurosci. 6, 52–60 (1986).
McCarthy, K.D. & de Vellis, J. Preparation of separate astroglial and oligodendroglial cell cultures from rat cerebral tissue. J. Cell Biol. 85, 890–902 (1980).
Szuchet, S. & Yim, S.H. Characterization of a subset of oligodendrocytes separated on the basis of selective adherence properties. J. Neurosci. Res. 11, 131–144 (1984).
Fanarraga, M.L., Sommer, I. & Griffiths, I.R. O-2A progenitors of the mouse optic nerve exhibit a developmental pattern of antigen expression different from the rat. Glia 15, 95–104 (1995).
Lu, Q.R. et al. Sonic hedgehog-regulated oligodendrocyte lineage genes encoding bHLH proteins in the mammalian central nervous system. Neuron 25, 317–329 (2000).
Johe, K.K., Hazel, T.G., Muller, T., Dugich-Djordjevic, M.M. & McKay, R.D. Single factors direct the differentiation of stem cells from the fetal and adult central nervous system. Genes Dev. 10, 3129–3140 (1996).
Avellana-Adalid, V., Nait-Oumesmar, B., Lachapelle, F. & Baron-Van Evercooren, A. Expansion of rat oligodendrocyte progenitors into proliferative “oligospheres” that retain differentiation potential. J. Neurosci. Res. 45, 558–570 (1996).
Zhang, S.C., Lipsitz, D. & Duncan, I.D. Self-renewing canine oligodendroglial progenitor expanded as oligospheres. J. Neurosci. Res. 54, 181–190 (1998).
Zhang, S.C., Lundberg, C., Lipsitz, D., O'Connor, L.T. & Duncan, I.D. Generation of oligodendroglial progenitors from neural stem cells. J. Neurocytol. 27, 475–489 (1998).
Vitry, S., Avellana-Adalid, V., Hardy, R., Lachapelle, F. & Baron-Van Evercooren, A. Mouse oligospheres: from pre-progenitors to functional oligodendrocytes. J. Neurosci. Res. 58, 735–751 (1999).
Woodruff, R.H., Tekki-Kessaris, N., Stiles, C.D., Rowitch, D.H. & Richardson, W.D. Oligodendrocyte development in the spinal cord and telencephalon: common themes and new perspectives. Int. J. Dev. Neurosci. 19, 379–385 (2001).
Soriano, P. The PDGF alpha receptor is required for neural crest cell development and for normal patterning of the somites. Development 124, 2691–2700 (1997).
Li, J. et al. Novel role of vitamin k in preventing oxidative injury to developing oligodendrocytes and neurons. J. Neurosci. 23, 5816–5826 (2003).
Mayer, M. & Noble, M. N-acetyl-L-cysteine is a pluripotent protector against cell death and enhancer of trophic factor-mediated cell survival in vitro . Proc. Natl. Acad. Sci. USA 91, 7496–7500 (1994).
Zhang, S.C., Ge, B. & Duncan, I.D. Adult brain retains the potential to generate oligodendroglial progenitors with extensive myelination capacity. Proc. Natl. Acad. Sci. USA 96, 4089–4094 (1999).
Acknowledgements
This study was funded by grants from National Multiple Sclerosis Society, March of Dimes Birth Defect Foundation and National Institutes of Health (R01 NS050389) to Q.R.L., and United Cerebral Palsy Foundation (to J.L.). Q.R.L. is a Harry Weaver Neuroscience Scholar and a Basil O'Conner Scholar.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
About this article
Cite this article
Chen, Y., Balasubramaniyan, V., Peng, J. et al. Isolation and culture of rat and mouse oligodendrocyte precursor cells. Nat Protoc 2, 1044–1051 (2007). https://doi.org/10.1038/nprot.2007.149
Published:
Issue Date:
DOI: https://doi.org/10.1038/nprot.2007.149
This article is cited by
-
The aminosteroid U73122 promotes oligodendrocytes generation and myelin formation
Acta Pharmacologica Sinica (2024)
-
Pyruvate Dehydrogenase-Dependent Metabolic Programming Affects the Oligodendrocyte Maturation and Remyelination
Molecular Neurobiology (2024)
-
Systemic and intrinsic functions of ATRX in glial cell fate and CNS myelination in male mice
Nature Communications (2023)
-
Culture Protocol and Transcriptomic Analysis of Murine SVZ NPCs and OPCs
Stem Cell Reviews and Reports (2023)
-
Pearls and Pitfalls of Isolating Rat OPCs for In Vitro Culture with Different Methods
Cellular and Molecular Neurobiology (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
