
Abstract
Since the identification of the first SCN5A mutation associated with long QT syndrome in 1995, several mutations in this gene for the α subunit of the cardiac sodium channel have been identified in a heterogeneous subset of cardiac rhythm syndromes, including Brugada syndrome, progressive cardiac conduction defect, sick sinus node syndrome, atrial fibrillation and dilated cardiomyopathy. Robust clinical evidence has been accompanied by bench studies performed in different models spanning from in vitro expression systems to transgenic mice. Together, these studies have helped establish genotype–phenotype correlations and have shaped our understanding of the role of the cardiac sodium channel in health and in disease. Remarkably, these advances in understanding have impacted on clinical management by allowing us to start developing gene-specific risk stratification schemes and mutation-specific management strategies. In this Review, we summarize the current understanding of the molecular mechanism of SCN5A-associated inherited arrhythmias, focusing on the most recent development of mutation-specific management in SCN5A-associated long QT syndrome type 3. We also briefly discuss arrhythmia-causing mutations in the genes encoding the β subunit of the cardiac sodium channel and in those encoding proteins in the associated macromolecular complex.
Key Points
-
The α subunit of the cardiac sodium channel is encoded by the SCN5A gene; most mutations that affect the sodium current in the heart are found in this gene
-
Mutations in the SCN5A gene are associated with long QT syndrome, Brugada syndrome, progressive cardiac conduction defect, sick sinus node syndrome, atrial fibrillation, dilated cardiomyopathy and overlapping syndromes
-
Mutations in genes encoding the β subunit of the cardiac sodium channel and in those encoding proteins in the associated macromolecular complex can also cause arrhythmias
-
The biophysical characterization of SCN5A mutations allows us to explain, at least in part, the clinical phenotypes observed in patients
-
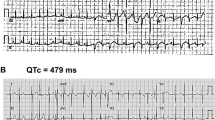
A gene-specific approach is required for risk stratification and treatment of patients with long QT syndrome type 3
-
In vitro studies of SCN5A mutations have identified sets of biophysical properties that might be used to predict patients' responses to therapy
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout








Similar content being viewed by others

References
Wang, Q. et al. SCN5A mutations associated with an inherited cardiac arrhythmia, long QT syndrome. Cell 80, 805–811 (1995).
Inherited Arrhythmias Database. [online January 2000] http://www.fsm.it/cardmoc/ (accessed 25 February 2009).
Chen, Q. et al. Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nat. 392, 293–296 (1998).
Schott, J. J. et al. Cardiac conduction defects associate with mutations in SCN5A. Nature Genet. 23, 20–21 (1999).
Benson, D. W. et al. Congenital sick sinus syndrome caused by recessive mutations in the cardiac sodium channel gene (SCN5A). J. Clin. Invest. 112, 1019–1028 (2003).
Makiyama, T. et al. A novel SCN5A gain-of-function mutation M1875T associated with familial atrial fibrillation. J. Am. Coll. Cardiol. 52, 1326–1334 (2008).
Bezzina, C. R. et al. Compound heterozygosity for mutations (W156X and R225W) in SCN5A associated with severe cardiac conduction disturbances and degenerative changes in the conduction system. Circ. Res. 92, 159–168 (2003).
Napolitano, C., Rivolta, I. & Priori, S. G. Cardiac sodium channel diseases. Clin. Chem. Lab. Med. 41, 439–444 (2003).
George, A. L. Jr. Inherited disorders of voltage-gated sodium channels. J. Clin. Invest. 115, 1990–1999 (2005).
Clancy, C. E. & Kass, R. S. Inherited and acquired vulnerability to ventricular arrhythmias: cardiac Na+ and K+ channels. Physiol. Rev. 85, 33–47 (2005).
Kass, R. S. & Moss, A. J. Mutation-specific pharmacology of the long QT syndrome. Handb. Exp. Pharmacol. 171, 287–304 (2006).
Napolitano, C., Bloise, R. & Priori, S. G. Gene-specific therapy for inherited arrhythmogenic diseases. Pharmacol. Ther. 110, 1–13 (2006).
Balser, J. R. The cardiac sodium channel: gating function and molecular pharmacology. J. Mol. Cell Cardiol. 33, 599–613 (2001).
Watanabe, H. et al. Sodium channel beta1 subunit mutations associated with Brugada syndrome and cardiac conduction disease in humans. J. Clin. Invest. 118, 2260–2268 (2008).
Medeiros-Domingo, A. et al. SCN4B-encoded sodium channel beta4 subunit in congenital long-QT syndrome. Circulation 116, 134–142 (2007).
Abriel, H. & Kass, R. S. Regulation of the voltage-gated cardiac sodium channel Nav1.5 by interacting proteins. Trends Cardiovasc. Med. 15, 35–40 (2005).
Napolitano, C. et al. Genetic testing in the long QT syndrome: development and validation of an efficient approach to genotyping in clinical practice. JAMA 294, 2975–2980 (2005).
Splawski, I. et al. Spectrum of mutations in long-QT syndrome genes. KVLQT1, HERG, SCN5A, KCNE1, and KCNE2. Circulation 102, 1178–1185 (2000).
Priori, S. G. et al. Association of long QT syndrome loci and cardiac events among patients treated with beta-blockers. JAMA 292, 1341–1344 (2004).
Bennett, P. B., Yazawa, K., Makita, N. & George, A. L. Jr . Molecular mechanism for an inherited cardiac arrhythmia. Nature 376, 683–685 (1995).
Wang, D. W., Yazawa, K., George, A. L. Jr & Bennett, P. B. Characterization of human cardiac Na+ channel mutations in the congenital long QT syndrome. Proc. Natl Acad. Sci. USA 93, 13200–13205 (1996).
Clancy, C. E., Tateyama, M., Liu, H., Wehrens, X. H. & Kass, R. S. Non-equilibrium gating in cardiac Na+ channels: an original mechanism of arrhythmia. Circulation 107, 2233–2237 (2003).
Nuyens, D. et al. Abrupt rate accelerations or premature beats cause life-threatening arrhythmias in mice with long-QT3 syndrome. Nat. Med. 7, 1021–1027 (2001).
Fabritz, L. et al. Effect of pacing and mexiletine on dispersion of repolarisation and arrhythmias in DeltaKPQ SCN5A (long QT3) mice. Cardiovasc. Res. 57, 1085–1093 (2003).
Tian, X. L. et al. Mechanisms by which SCN5A mutation N1325S causes cardiac arrhythmias and sudden death in vivo. Cardiovasc. Res. 61, 256–267 (2004).
Brugada, P. & Brugada, J. Right bundle branch block, persistent ST segment elevation and sudden cardiac death: a distinct clinical and electrocardiographic syndrome. A multicenter report. J. Am. Coll. Cardiol. 20, 1391–1396 (1992).
Bai, R. et al. Yield of genetic screening in inherited cardiac channelopathies: how to prioritize access to genetic testing. Circ. Arrhythmia Electrophysiol. 2, 6–15 (2009).
Antzelevitch, C. et al. Brugada syndrome: report of the second consensus conference: endorsed by the Heart Rhythm Society and the European Heart Rhythm Association. Circulation 111, 659–670 (2005).
Deschenes, I. et al. Electrophysiological characterization of SCN5A mutations causing long QT (E1784K) and Brugada (R1512W and R1432G) syndromes. Cardiovasc. Res. 46, 55–65 (2000).
Valdivia, C. R. et al. A novel SCN5A arrhythmia mutation, M1766L, with expression defect rescued by mexiletine. Cardiovasc. Res. 55, 279–289 (2002).
Valdivia, C. R. et al. A trafficking defective, Brugada syndrome-causing SCN5A mutation rescued by drugs. Cardiovasc. Res. 62, 53–62 (2004).
Pfahnl, A. E. et al. A sodium channel pore mutation causing Brugada syndrome. Heart Rhythm 4, 46–53 (2007).
Herfst, L. J., Rook, M. B. & Jongsma, H. J. Trafficking and functional expression of cardiac Na+ channels. J. Mol. Cell Cardiol. 36, 185–193 (2004).
Bezzina, C. R. & Tan, H. L. Pharmacological rescue of mutant ion channels. Cardiovasc. Res. 55, 229–232 (2002).
Rivolta, I. et al. Inherited Brugada and long QT-3 syndrome mutations of a single residue of the cardiac sodium channel confer distinct channel and clinical phenotypes. J. Biol. Chem. 276, 30623–30630 (2001).
Baroudi, G., Carbonneau, E., Pouliot, V. & Chahine, M. SCN5A mutation (T1620M) causing Brugada syndrome exhibits different phenotypes when expressed in Xenopus oocytes and mammalian cells. FEBS Lett. 467, 12–16 (2000).
Wang, D. W., Makita, N., Kitabatake, A., Balser, J. R. & George, A. L. Jr. Enhanced Na+ channel intermediate inactivation in Brugada syndrome. Circ. Res. 87, E37–E43 (2000).
Grant, A. O. Electrophysiological basis and genetics of Brugada syndrome. J. Cardiovasc. Electrophysiol. 16 (Suppl. 1), S3–S7 (2005).
Yan, G. X. & Antzelevitch, C. Cellular basis for the Brugada syndrome and other mechanisms of arrhythmogenesis associated with ST-segment elevation. Circulation 100, 1660–1666 (1999).
Antzelevitch, C. Heterogeneity and cardiac arrhythmias: an overview. Heart Rhythm 4, 964–972 (2007).
Coronel, R. et al. Right ventricular fibrosis and conduction delay in a patient with clinical signs of Brugada syndrome: a combined electrophysiological, genetic, histopathologic, and computational study. Circulation 112, 2769–2777 (2005).
Remme, C. A. et al. Overlap syndrome of cardiac sodium channel disease in mice carrying the equivalent mutation of human SCN5A-1795insD. Circulation 114, 2584–2594 (2006).
Stokoe, K. S. et al. Effects of flecainide and quinidine on arrhythmogenic properties of Scn5a± murine hearts modelling the Brugada syndrome. J. Physiol. 581, 255–275 (2007).
Wang, D. W., Viswanathan, P. C., Balser, J. R., George, A. L. Jr & Benson, D. W. Clinical, genetic, and biophysical characterization of SCN5A mutations associated with atrioventricular conduction block. Circulation 105, 341–346 (2002).
Tan, H. L. et al. A sodium-channel mutation causes isolated cardiac conduction disease. Nature 409, 1043–1047 (2001).
Kyndt, F. et al. Novel SCN5A mutation leading either to isolated cardiac conduction defect or Brugada syndrome in a large French family. Circulation 104, 3081–3086 (2001).
Shirai, N. et al. A mutant cardiac sodium channel with multiple biophysical defects associated with overlapping clinical features of Brugada syndrome and cardiac conduction disease. Cardiovasc. Res. 53, 348–354 (2002).
Papadatos, G. A. et al. Slowed conduction and ventricular tachycardia after targeted disruption of the cardiac sodium channel gene SCN5A. Proc. Natl Acad. Sci. USA 99, 6210–6215 (2002).
Royer, A. et al. Mouse model of SCN5A-linked hereditary Lenegre's disease: age-related conduction slowing and myocardial fibrosis. Circulation 111, 1738–1746 (2005).
Smits, J. P. et al. A mutation in the human cardiac sodium channel (E161K) contributes to sick sinus syndrome, conduction disease and Brugada syndrome in two families. J. Mol. Cell Cardiol. 38, 969–981 (2005).
Lei, M. Zhang, H., Grace, A. A. & Huang, C. L. SCN5A and sinoatrial node pacemaker function. Cardiovasc. Res. 74, 356–365 (2007).
Olson, T. M. & Keating, M. T. Mapping a cardiomyopathy locus to chromosome 3p22-p25. J. Clin. Invest. 97, 528–532 (1996).
Ge, J. et al. Molecular and clinical characterization of a novel SCN5A mutation associated with atrioventricular block and dilated cardiomyopathy. Circ. Arrhythmia Electrophysiol. 1, 83–92 (2008).
Shi, R. et al. The cardiac sodium channel mutation delQKP 1507–1509 is associated with the expanding phenotypic spectrum of LQT3, conduction disorder, dilated cardiomyopathy, and high incidence of youth sudden death. Europace 10, 1329–1335 (2008).
Letsas, K. P. et al. Prevalence of paroxysmal atrial fibrillation in Brugada syndrome: a case series and a review of the literature. J. Cardiovasc. Med. (Hagerstown) 8, 803–806 (2007).
Bezzina, C. et al. A single Na+ channel mutation causing both long-QT and Brugada syndromes. Circ. Res. 85, 1206–1213 (1999).
Grant, A. O. et al. Long QT syndrome, Brugada syndrome, and conduction system disease are linked to a single sodium channel mutation. J. Clin. Invest. 110, 1201–1209 (2002).
Priori, S. G. et al. The elusive link between LQT3 and Brugada syndrome: the role of flecainide challenge. Circulation 102, 945–947 (2000).
Wei, J. et al. Congenital long-QT syndrome caused by a novel mutation in a conserved acidic domain of the cardiac Na+ channel. Circulation 99, 3165–3171 (1999).
Priori, S. G. et al. Natural history of Brugada syndrome: insights for risk stratification and management. Circulation 105, 1342–1347 (2002).
Makita, N. et al. The E1784K mutation in SCN5A is associated with mixed clinical phenotype of type 3 long QT syndrome. J. Clin. Invest. 118, 2219–2229 (2008).
Remme, C. A. & Wilde, A. A. SCN5A overlap syndromes: no end to disease complexity? Europace 10, 1253–1255 (2008).
Priori, S. G. Inherited arrhythmogenic diseases: the complexity beyond monogenic disorders. Circ. Res. 94, 140–145 (2004).
Priori, S. G. et al. Clinical and genetic heterogeneity of right bundle branch block and ST-segment elevation syndrome: a prospective evaluation of 52 families. Circulation 102, 2509–2515 (2000).
Mohler, P. J. et al. Nav1.5 E1053K mutation causing Brugada syndrome blocks binding to ankyrin-G and expression of Nav1.5 on the surface of cardiomyocytes. Proc. Natl Acad. Sci. USA 101, 17533–17538 (2004).
Vatta, M. et al. Mutant caveolin-3 induces persistent late sodium current and is associated with long-QT syndrome. Circulation 114, 2104–2112 (2006).
Ueda, K. et al. Syntrophin mutation associated with long QT syndrome through activation of the nNOS-SCN5A macromolecular complex. Proc. Natl Acad. Sci. USA 105, 9355–9360 (2008).
London, B. et al. Mutation in glycerol-3-phosphate dehydrogenase 1 like gene (GPD1-L) decreases cardiac Na+ current and causes inherited arrhythmias. Circulation 116, 2260–2268 (2007).
Zipes, D. P. et al. ACC/AHA/ESC 2006 Guidelines for Management of Patients With Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death: a report of the American College of Cardiology/American Heart Association Task Force and the European Society of Cardiology Committee for Practice Guidelines (writing committee to develop Guidelines for Management of Patients With Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death): developed in collaboration with the European Heart Rhythm Association and the Heart Rhythm Society. Circulation 114, e385–e484 (2006).
Schwartz, P. J. et al. Long QT syndrome patients with mutations of the SCN5A and HERG genes have differential responses to Na+ channel blockade and to increases in heart rate. Implications for gene-specific therapy. Circulation 92, 3381–3386 (1995).
Chang, C. C. et al. A novel SCN5A mutation manifests as a malignant form of long QT syndrome with perinatal onset of tachycardia/bradycardia. Cardiovasc. Res. 64, 268–278 (2004).
Kambouris, N. G. et al. Phenotypic characterization of a novel long-QT syndrome mutation (R1623Q) in the cardiac sodium channel. Circulation 97, 640–644 (1998).
Benhorin, J. et al. Effects of flecainide in patients with new SCN5A mutation: mutation-specific therapy for long-QT syndrome? Circulation 101, 1698–1706 (2000).
Abriel, H., Wehrens, X. H., Benhorin, J., Kerem, B. & Kass, R. S. Molecular pharmacology of the sodium channel mutation D1790G linked to the long-QT syndrome. Circulation 102, 921–925 (2000).
Ruan, Y., Liu, N., Bloise, R., Napolitano, C. & Priori, S. G. Gating properties of SCN5A mutations and the response to mexiletine in long-QT syndrome type 3 patients. Circulation 116, 1137–1144 (2007).
Priori, S. G. et al. Genetics of Long QT, Brugada and other channelopathies. In Cardiac Electrophysiology (eds Zipes, D. P. et al.) 462–470 (Elsevier, Philadelphia, 2003).
Makiyama, T. et al. High risk for bradyarrhythmic complications in patients with Brugada syndrome caused by SCN5A gene mutations. J. Am. Coll. Cardiol. 46, 2100–2106 (2005).
Makita, N. et al. Congenital atrial standstill associated with coinheritance of a novel SCN5A mutation and connexin 40 polymorphisms. Heart Rhythm 2, 1128–1134 (2005).
Takehara, N. et al. A cardiac sodium channel mutation identified in Brugada syndrome associated with atrial standstill. J. Intern. Med. 255, 137–142 (2004).
Zhang, Y. et al. Correlations between clinical and physiological consequences of the novel mutation R878C in a highly conserved pore residue in the cardiac Na+ channel. Acta Physiol. (Oxf.) 194, 311–323 (2008).
Tan, B. H. et al. A novel C-terminal truncation SCN5A mutation from a patient with sick sinus syndrome, conduction disorder and ventricular tachycardia. Cardiovasc. Res. 76, 409–417 (2007).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
About this article
Cite this article
Ruan, Y., Liu, N. & Priori, S. Sodium channel mutations and arrhythmias. Nat Rev Cardiol 6, 337–348 (2009). https://doi.org/10.1038/nrcardio.2009.44
Issue Date:
DOI: https://doi.org/10.1038/nrcardio.2009.44
This article is cited by
-
Identification of SCN7A as the key gene associated with tumor mutation burden in gastric cancer
BMC Gastroenterology (2022)
-
FAT10 protects against ischemia-induced ventricular arrhythmia by decreasing Nedd4-2/Nav1.5 complex formation
Cell Death & Disease (2021)
-
Disease-associated HCN4 V759I variant is not sufficient to impair cardiac pacemaking
Pflügers Archiv - European Journal of Physiology (2020)
-
Comparing human iPSC-cardiomyocytes versus HEK293T cells unveils disease-causing effects of Brugada mutation A735V of NaV1.5 sodium channels
Scientific Reports (2019)
-
Drug-Induced Brugada Syndrome in a Psychiatric Patient: a Case Report
SN Comprehensive Clinical Medicine (2019)
