
Abstract
Telomerase reverse transcriptase (TERT) is a self-antigen that is expressed constitutively in many tumours, and is, therefore, an important target for anticancer immunotherapy. In the past 10 years, trials of immunotherapy with TERT-based vaccines have demonstrated only modest benefits. In this Perspectives, I discuss the possible immunological reasons for this limited antitumour efficacy, and propose that advances in our understanding of the genetics and biology of the involvement of TERT in cancer provides the basis for renewed interest in TERT- based immunotherapy. Telomerase and TERT are expressed in cancer cells at every stage of tumour evolution, from the cancer stem cell to circulating tumour cells and tumour metastases. Many cancer types also harbour cells with mutations in the TERT promoter region, which increase transcriptional activation of this gene. These new findings should spur new interest in the development of TERT-based immunotherapies that are redesigned in line with established immunological considerations and working principles, and are tailored to patients stratified on the basis of TERT-promoter mutations and other underlying tumour characteristics. Thus, despite the disappointment of previous clinical trials, TERT offers the potential for personalized immunotherapy, perhaps in combination with immune-checkpoint inhibition.
Similar content being viewed by others
Main
Telomerase reverse transcriptase (TERT) is a component of the ribonucleoprotein telomerase, a unique cellular enzyme: via reverse transcription of its own RNA template (TERC), telomerase synthesizes the tandem 5′-TTAGGG-3′ exonucleotide repeats of telomeric DNA, which prevents chromosome attrition resulting from incomplete semiconservative DNA replication at chromosomal ends1. Thus, the discovery of telomerase and telomerase-mediated extension of telomeric DNA solved the end-replication problem — that is, the mechanism by which telomeric DNA is maintained2. In addition, this discovery paved the way to elucidate the 'end-protection problem' and the mechanisms by which telomere elongation prevents the severe consequences of a cellular response to exposed DNA ends3, such as end-to-end joining, DNA recombination, or DNA repair, which would lead to unstable chromosomes and, ultimately, aneuploidy4. As telomeres shorten progressively with successive cell divisions in the absence of TERT expression (which is restricted to certain cell types, predominantly stem or germ cells), telomere length is considered to mirror the replicative history of a cell lineage5.
Telomerase imparts cells with the capacity to continuously replicate, which is often referred to as 'cell immortality' (Ref. 6). When TERT is overexpressed together with simian virus 40 large T antigen and RAS oncoproteins in human cells, the cells become 'immortal', are malignantly transformed, and can form tumours in vivo7. Moreover, using the canonical telomeric repeat amplification protocol (TRAP) assay, telomerase activity is detected in >85% of human tumours of various histological types8, but not in normal tissues. Modest levels of telomerase activity are detected in tissues with high self-renewal capacity, such as the bone marrow, testes, gastrointestinal crypt epithelium, and hair follicles9,10,11. In a minority of tumours of mesenchymal origin (soft-tissue sarcoma and osteosarcoma), as well as pancreatic neuroendrocrine neoplasms and gliomas12, telomere length is maintained by one or more alternative, TERT-independent mechanisms. These mechanisms are referred to as 'alternative lengthening of telomeres' (ALT), and involve copying telomeric template DNA via homologous recombination13. Nevertheless, TERT is an important immunological target for anticancer therapy because of its widespread overexpression in human cancers, throughout the entire trajectory of the disease (see 'Recentring TERT in tumour evolution' section of this Perspectives).
TERT immunology in cancer
Human TERT is a self-antigen that consists of 1,132 amino acids14. Soon after its amino acid sequence was deduced more than 15 years ago, several laboratories probed the antigenicity and immunogenicity of TERT15,16 — antigenicity refers to the property of being recognized by the adaptive immune system, whereas immunogenicity refers to the capacity to induce an adaptive immune response. As TERT is an intracellular protein that is not expressed on the external cell surface, it can only be recognized by T cells as short peptides comprising 8–16 amino acids, which are processed inside the cell before being exported to, and presented at, the cell surface in the context of major histocompatibility complex (MHC) molecules. The initial experiments into TERT immunology focused on TERT-peptide binding to MHC class I (MHC I) molecules, which are expressed by almost all cell types and, when bound to a target antigen, can induce the activity of CD8+ cytotoxic T lymphocytes (CTLs) expressing a complementary T-cell receptor (TCR). Thus, the initial questions were essentially whether endogenous TERT could be processed and presented in the context of MHC I to become the target of CD8+ T lymphocytes, and thereby activate cytotoxic T-cell responses. The first two immunogenic TERT peptides discovered (p540 and p865) were identified based on a predicted high-affinity interaction with human histocompatibility antigen (HLA)-A*02 molecules, a MHC I serotype group. By probing the surface of cancer cells using CD8+ T cells induced with these TERT peptides in vitro, investigators determined that the peptides are expressed on histologically distinct HLA-A*02+/+, TERT-expressing cancer cell lines15, and primary cancer cells16; antibodies targeting MHC I blocked T-cell killing of the cancer cells, implying that the reaction was mediated by the MHC I–peptide complexes. TCR-mimic antibodies directed at the same TERT peptides were also found to bind HLA-A*02+/TERT+ cell lines17. Other groups, however, were unable to detect the HLA-A*02-restricted, dominant TERT peptide (p540) on the surface of cancer cells18,19,20. This discrepancy could be attributed to a number of factors, including methodological variation between different laboratories (for instance, different groups have reported disparate results with the same anti-HLA-A*02–TERT-peptide complex antibody17,19,21); a lack of stringent specificity-control with regard to the CD8+ T cells used to probe model cancer cells (cold target inhibition, a competitive assay that ensures the specificity of cell recognition, was used in only one study15); and the use of different cell lines, each with presumably different immunopeptidomes generated in the endoplasmic reticulum. In line with the latter interpretation, p540 was found to be preferentially destroyed during antigen processing, as it contains a proteasome cleavage site18. Thus, p540 might be expressed at only the low end of natural antigen-presentation levels (∼10 copies per cell)22,23. Over the years, further evidence showed that TERT is processed endogenously, and that TERT peptides are presented by cancer cells that express HLA-A*03, HLA-A*24, and HLA-B*7 MHC I molecules24,25,26,27,28.
Anticancer immunity can also be efficiently mediated by CD4+ T cells that recognize antigens in the context of MHC class II (MHC II) molecules29. Predicting MHC II-restricted peptide antigens is more complex than predicting MHC I peptides, as 3,658 MHC II alleles are identified in the Immuno Polymorphism Database (IPD) and international Immunogenetics (IMGT)/HLA database: https://www.ebi.ac.uk/ipd/imgt/hla/). Indeed, few bona fide MHC II-binding TERT peptides have been identified and characterized; although some of those that have been identified are both promiscuous (bind to multiple MHC II alleles), and are produced endogenously in cancer cells30,31,32. Thus, cancer cells can present TERT peptides that can be recognized by either CD4+ or CD8+ T cells.
In vitro, TERT protein has been demonstrated to be readily immunogenic for peripheral blood T lymphocytes harvested from healthy individuals and patients with cancer, suggesting that TERT-reactive T-cell precursors exist in the blood and are not deleted in the thymus. This finding is important because immunization does not result in the de novo creation of antigen-specific T cells, but rather the selective expansion of reactive clones that pre-exist in the T-cell repertoire15,16,24,26,28,33. The in vivo immunogenicity of TERT has also been interrogated in preclinical models using a variety of approaches, including synthetic TERT peptides15,24,28, dendritic cells (DCs) transfected with TERT mRNA34,35,36, and DCs transduced with TERT-adenovirus37, TERT-encoding lentivirus vectors38, and plasmid DNA encoding TERT39. In selected instances, the induction of T-cell responses was associated with inhibition of tumour growth34,38,39,40,41.
Antigenicity and immunogenicity are merely two aspects of a larger immunological equation. Any T-cell response in vivo also depends on the size of the available T-cell repertoire for a given protein. Indeed, the identification of TERT peptides with high-affinity for MHC molecules that are immunogenic for peripheral blood T cells in vitro provides no clue as to the actual size and breadth of the anti-TERT T-cell repertoire that exists in vivo. Using flow cytometry, investigators have detected CD8+ T cells with specificity for TERT in the blood of variable proportions of patients with chronic myeloid leukaemia (∼80%), breast cancer (∼75%), lung cancer (∼40%), colorectal cancer (∼20%), and hepatocellular carcinoma (∼10%)42,43,44,45,46. In one study in patients with solid tumours, limiting-dilution analyses resulted in estimated TERT-specific T-cell-precursor frequencies as high as 1:298–1:540 (Ref. 47), suggesting the presence of spontaneous immune responses to TERT in some patients. Moreover, evidence indicates that spontaneous CD4+-T-cell responses against promiscuous TERT peptides occur in 38% of patients with lung cancer32. The available T-cell repertoire for TERT in patients with cancer seems to be enriched over that of healthy individuals32,43,47; however, limited information — if any — exists on the functional status of these T cells, when in the course of cancer development their populations expand, and whether they could re-expand following TERT vaccination.
Immune tolerance is a major determinant of an individual's unique T-cell repertoire. During ontogeny, immune tolerance shapes the T-cell repertoire via elimination of T-cell precursors that express TCRs with high-affinity for MHC–peptide complexes (signal 1), while T-cell precursors with low–moderate-affinity TCRs are spared. Tumour growth can also promote peripheral immune tolerance if antigen-presenting cells activate T cells in the absence of co-stimulatory molecules (signal 2). In addition, certain T-cell specificities can be lost over time owing to T-cell senescence and exhaustion48, or as a result of remodelling of cancer-cell immunogenicity by immune editing49. The roles that these factors have in determining the immunological responses to TERT in patients with cancer remain largely unknown.
Autoimmunity and TERT vaccines
At the outset of the decade-long quest for a successful therapeutic TERT-based vaccine against cancer, a recurrent concern raised was the potential for this approach to result in collateral damage to host tissues. For example, T lymphocytes and B lymphocytes are known to express telomerase during clonal expansion50; therefore, will these cells be attacked by TERT-specific T cells during such immunotherapy? The dynamics between clonal expansion of lymphocytes, telomerase activity, and TERT expression has been analysed, and telomerase expression in activated murine CD4+ T cells was found to be induced in an antigen-specific and CD28–B7-mediated co-stimulation-dependent manner51. This finding highlights the possibility that CTLs specific for TERT could potentially engage in 'fratricidal' killing of T cells, with systemic consequences. In humans, however, T-cell activation through ligation of the CD3 subunit of the TCR results in TERT phosphorylation and relocation from the cytoplasm to the nucleus, without a net increase in TERT-protein levels52. Thus, following priming, the total amount of TERT protein in human T cells remains constant. On the other hand, human memory T cells have shorter telomeres compared with their naive counterparts, implying decreased telomerase activity53. Furthermore, terminally differentiated T cells, such as pre-senescent CD27−/CD28− T cells, do not express telomerase48. Taken together, these observations suggest an age-dependent decrease in telomerase, and hence TERT, expression in human T lymphocytes, with a diminished risk of autoimmunity.
Human B lymphocytes in germinal centres of lymph organs have markedly longer telomeres — presumably resulting from higher levels of telomerase and TERT expression — than naive and memory B cells54,55. Thus, one would predict that TERT-specific CTLs could eliminate B cells during the germinal-centre reaction; however, human CTLs with specificity for a low-affinity TERT peptide do not lyse autologous CD40-activated B lymphocytes in vitro24. Additionally, CTLs that target high-affinity TERT peptides do not kill bone-marrow-derived HLA-matched CD34+ haematopoietic stem cells (HSCs)15, despite evidence that most bone marrow HSCs express telomerase56. Nevertheless, caution is needed when using TERT-directed immunotherapy, as indicated by the results of a preclinical study in mice, in which three cycles of adoptive T-cell therapy with mouse TERT-specific CD8+ T cells caused transient, self-resolving B-cell lymphopenia57. This effect might be attributable to either differences in the regulation of TERT expression between mouse and human tissues, or the aggressive therapeutic regimen used in the mouse model (transfer of 5 × 106 TERT-specific CD8+ T cells, high-dose IL-2, and immunization of the mice with an adenovirus-vector encoding the TERT antigen recognized by the transferred T cells). In a prior report, the same group demonstrated that mice actively immunized with TERT had no B-cell abnormalities39, suggesting that adoptive T-cell therapy could be associated with a greater risk of adverse events.
Collectively, the concerns that activated T cells, B lymphocytes, and HSCs would be targeted by TERT-specific T cells, albeit valid in principle, seem to be mitigated by the transient nature and low level of TERT expression in these cell types, and the fact that the induction of telomerase activity is not associated with a parallel increase in levels of TERT protein (at least in T cells). Congruently, preclinical studies revealed no abnormalities in the spleen and lymph nodes of HLA-A*02-transgenic mice vaccinated with a low-affinity TERT analogue peptide40.
Therapeutic TERT-vaccine trials
Following the discoveries discussed in the previous sections, therapeutic TERT-based vaccination was rapidly pursued in patients with different types of cancer. Indeed, a total of 23 clinical studies with published data have investigated this anticancer strategy: 18 phase I/I–II and four phase II studies, and one phase III trial (Table 1). Various vaccination approaches were used in these studies.
Overall, synthetic peptides tailored to induce either CD4+ or CD8+ T-cell responses via their affinity for MHC II and MHC I molecules, respectively, have been the prevalent immunogen used: this approach was used in 13 of the phase I/I–II, three of the four phase II studies, and in the sole phase III trial. Many studies (13 out of 23) have been conducted in HLA-A*02+ patients, as this is the most-frequent MHC I allele in white individuals (∼45% of whom express this HLA serotype)58. Only five studies involved MHC II-restricted peptides. In six studies, cells (dendritic cells or B lymphocytes) transfected with RNA or DNA, or cultured with apoptotic tumour cells, were used to vaccinate patients. Concomitant chemotherapy was used in only three phase I/I–II studies, but in three of the four phase II studies as well as in the sole phase III trial.
In the phase I/I–II studies, a TERT-specific T-cell response in ≥50% of the evaluable patients vaccinated was reported in 16 of the 18 studies (Table 1). Objective clinical responses were reported in four phase I/I–II studies with distinct designs, in a variable percentage (8–71%) of the evaluable vaccinees59,60,61,62,63. No objective responses were reported in the other phase I/I–II studies in which clinical outcomes were assessed (n = 6)64,65,66,67,68,69. Disease stabilization in >50% of the evaluable patients vaccinated was reported in two studies (rates of 67% and 83%)59,69, whereas lower rates of disease stabilization (16–48%) have been observed in seven studies (Table 1). Cumulatively, the findings of the phase I/I–II studies showed that, although therapeutic TERT-based vaccination can induce specific T-cell responses in many of those vaccinated, the effect on tumour size was minimal: temporary disease stabilization was generally the best clinical result (Table 1).
Three of the four phase II clinical trials have been conducted in patients with a single type of cancer and vaccination was combined with chemotherapy (Table 1). Synthetic peptide immunogens were used in all but one of the phase II studies. Development of a TERT-specific T-cell response in >50% vaccinees (range 55–80%) was reported in three out the four studies61,70,71. Objective responses were reported in only one phase II study, comprising a complete response in a patient with breast cancer and liver metastases and a partial response in a patient with metastatic hepatocellular carcinoma71. Temporary disease stabilization was reported in three of the four studies, with rates that ranged from 33% to 57% in those vaccinated (Table 1)70,71,72.
In the sole phase III trial73, investigators evaluated the efficacy of TERT-peptide vaccination plus chemotherapy in patients with pancreatic cancer. In this three-arm randomized study that involved 1,062 patients, the median survival durations were not significantly different between the sequential or concurrent chemoimmunotherapy groups compared with the chemotherapy-only group, with an overall lack of benefit in terms of both median time to progression (4.5 months and 6.6 months, respectively, versus 6.4 months) and overall survival (6.9 months and 8.4 months, respectively, versus 7.9 months)73. Overall, objective responses were observed in 63 (18%) of 358 patients in the chemotherapy-alone group, 31 (9%) of 350 patients in the sequential chemoimmunotherapy group, and 55 (16%) of 354 patients in the concurrent chemoimmunotherapy group (Table 1).
In the few studies (six) with evaluable data, the immune response to TERT vaccination was generally found to correlate with clinical benefit, in terms of overall survival (Table 2). In fact, responders typically had overall survival durations that were significantly prolonged and approached or exceeded double that observed for nonresponders. Future studies will need to establish whether clinical benefit in immunological responders also correlates with the induction of T cells that infiltrate the tumour, as observed in a patient included in a phase I study59.
Collectively, the data from these clinical trials performed to date indicate that therapeutic TERT-based vaccination has limited anticancer efficacy: the various immunogens reportedly induce T-cell responses to TERT in patients with cancer, but this effect is typically insufficient to control tumour growth or disease progression. In general, however, these clinical studies confirmed that the risk of adverse events following vaccination targeting TERT is minimal or nonexistent. For instance, no lymphopenia was observed in patients with prostate cancer who were vaccinated using a combination of high-affinity and low-affinity TERT peptides74, nor in patients with myeloma who received infusion of TERT-reactive CD8+ T-cell expanded ex vivo and boosted by TERT-peptide vaccination75.
Interrogation of the ClinicalTrial.gov (https://clinicaltrials.gov/) and WHO International Clinical Trial Registry Platform (http://www.who.int/ictrp/en/) databases reveals that an additional 16 new clinical trials of TERT vaccines are in progress, including phase I, phase II, and phase III trials. Most of these studies are, however, based on the same working principles as those used in the completed studies reviewed herein. Thus, a reasonable expectation is that similar trends will be found.
Lessons learned
Why isn't therapeutic vaccination against TERT more effective? Can lessons be learned from the results of the work conducted during the past decade? Is it possible to improve efficacy through better vaccine design, thus making therapeutic TERT vaccination a worthwhile option for patients with cancer? Multiple reasons might explain why the initial experience with therapeutic TERT vaccination has been largely disappointing; in the following paragraphs, I outline the immunological considerations that I believe received too little attention in previous approaches to TERT vaccination, but could potentially lead to marked improvements in future trials.
First, is TERT-peptide presentation altered in cancer cells? One might expect that patient-to-patient and cell-to-cell variability in the presentation of TERT peptides exists. The immunopeptidome, the multitude of peptides generated intracellularly and sorted in specialized organelles (for example, the endoplasmic reticulum), is not a mirror of the proteome or the transcriptome, and therefore, its content cannot be predicted. Indeed, the composition of the immunopeptidome is subject to, among other factors, plastic remodelling based on the cellular metabolic activity76. In addition, epigenetic changes might also affect the immunopeptidome. For instance, treatment of human tumour cells with the HDAC inhibitor trichostatin A has been shown to result in a threefold decrease in the levels of high-affinity TERT peptide–MHC I complexes displayed at the surface, and reduced cell killing by CD8+ T cells21.
Second, is the available T-cell repertoire skewed in response to TERT vaccination? T cells with the highest affinity for self-peptides are removed owing to the thymic immune-tolerance mechanisms. High-affinity anti-TERT T cells might preferentially interact with TERT peptides with high affinity for the MHC molecule (that is, as more-stable binders), resulting in their depletion from the T-cell repertoire, such that they can no longer contribute to the vaccine-induced T-cell responses. Evidence indicates that pruning of self-antigen-specific T-cell lineages, rather than outright deletion, occurs in humans77, although no information exists on the size of the preimmune, high-affinity T-cell repertoire in cancer-free individuals. Nevertheless, a way around this obstacle would be to select low-affinity peptides, artificially increase their MHC-binding affinity, and then empirically identify the peptides with improved immunogenicity24. One such TERT peptide, pY572, is an analogue of a peptide with low affinity for HLA-A*02 (p572); pY572 induces TERT-specific CTL in humans, shares a crossreactive T-cell repertoire with the parental peptide, results in the lysis of tumour cells in vitro24, and has been included in two vaccine formulations66,74.
Third, were previous vaccination approaches optimal to maximize the induction of T-cell responses? In many published studies, TERT peptides were used straightforwardly, without consideration of CD4+ T-cell help. The necessity to include peptides that activate both CD4+ T-helper cells and CD8+ CTLs in the same immunogen in order to achieve effective vaccination was first shown with a lipopeptide vaccine against the hepatitis B virus78. The induction of CD8+ T cells in a 'helpless mode' has subsequently been found to yield CD8+ T-cell responses that are poorly maintained, with low numbers of precursor T-cell that expand poorly after antigen restimulation79,80. Likewise, the induction of antitumour CD4+ T-cell responses using a self-peptide alone, as used in many of the TERT-vaccine trials reported to date, would inevitably be ineffective. As recently reviewed elsewhere29, cooperation between two CD4+ T cells, which is based on associative recognition of antigen (the 'help for helpers' paradigm), is a basic immunological principle that should be an essential consideration in the design of therapeutic anticancer vaccines.
Fourth, was vaccination performed with nonpersisting vaccine in adjuvant formulations? Vaccination using peptides in adjuvants (incomplete Freund's adjuvant or Montanide® ISA adjuvants) that create antigen depots can result in sequestration of tumour-specific T cells at the injection site, thus diminishing tumour-infiltration and favouring apoptosis of the T cells, as shown in preclinical models81. Specifically Montanide® ISA adjuvants were used in six TERT-vaccine trials19,66,71,75,82,83, with variable results with respect to the immune responses observed post-vaccination.
Fifth, is the class of responding T cells optimal for tumour-cell targeting? Both CD8+ and CD4+ T cells can induce antitumour responses. The effectiveness of the antitumour immune response might not only be dependent on the number of T cells activated; the quality of T cells induced by therapeutic vaccination is also relevant. Studies in mice have shown that while T cells at different stages of differentiation can mediate antitumour activity, central memory (TCM)84 and memory T cells with stem-cell-like properties (TSCM)85, confer superior protection, at least with respect to CD8+ T cells. Few of the TERT-vaccine trials performed to date have used vaccines optimized to generate these classes of T cells, for example, through low-dose immunization in the context of associative recognition of antigen (that is, CD4+ T-cell help to CD8+ or CD4+ T cells).
Sixth, are TERT-specific T cells induced by therapeutic vaccination 'anergized' systemically or locally in the tumour microenvironment? Even if all the prerequisites for optimal induction of protective antitumour T-cell responses are satisfied, a vaccine might not necessarily induce TERT-specific T cells if they are rendered anergic, or nonfunctional, in vivo. Early studies of anticancer vaccines revealed that circulating melanoma-antigen-specific T cells induced by peptide vaccination are quiescent cells, with low expression of genes associated with T-cell activation, proliferation, and effector function86. Whether this is also the case for the TERT-specific T cells generated in vaccinees in previous TERT vaccine trials is unknown, but this aspect of the immune response should be considered in future studies.
Finally, the timing of therapeutic vaccination needs to be gauged relative to the course of the disease. Vaccination of patients with very advanced-stage disease might limit both the immune response generated and antitumour effectiveness of any response. This consideration would favour the idea that immune intervention should take place early in the course of the disease, more as a 'pre-emptive' strike than as a curative approach in patients with advanced-stage disease. Preferably, patients should be vaccinated at the disease stage at which the tumour burden is much lower and more localized, and tumour immunosuppressive mechanisms might be less established. In the advanced-stage-disease setting, one could consider the adoptive transfer of TERT-specific T cells, followed by TERT vaccination75, with the expectation that passively administered T cells would initiate a process of tumour destruction that would promote the development and improve the effectiveness of subsequently active anti-TERT immunity upon vaccination.
The tumour microenvironment has a major role in determining the success of therapeutic vaccination, although the functional aspects of the tumour microenvironment in the anti-TERT immune response are, by and large, unexplored. In one report, investigators demonstrated prevalent tumour-specific regulatory T (Treg) cells and Treg-cell-dependent inhibition of memory responses to TERT antigens ex vivo in patients with colon carcinoma, but no information on tumour-infiltrating T cells was provided87. Theoretically, however, the tumour microenvironment would not be expected to have unique characteristics (for example, accumulation of cells able to inhibit T-cell function or upregulation of negative regulators on T cells) that would affect TERT vaccination. The tumour microenvironment is often enriched in T-cells that express the immune-checkpoint proteins cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4), programmed cell-death protein 1 (PD-1) or its ligand programmed cell death 1 ligand 1 (PD-L1), and/or other inhibitory molecules88,89,90; regulatory/suppressor CD4+ and CD8+ T cells (Treg cells)91,92; myeloid cells with both proinflammatory and immunosuppressive characteristics (such as tumour-associated macrophages and myeloid-derived suppressor cells)93,94; and B cells with tumour-promoting regulatory functions95,96. For example, we are now beginning to appreciate the negative effects of the endoplasmic-reticulum-stress response in the tumour microenvironment97. As a result of this response, bone-marrow-derived macrophages and DCs acquire proinflammatory/suppressive characteristics, secrete arginase 1, and have a reduced capacity for antigen presentation98. Together, these environmental changes negatively affect the activation and expansion of naive T cells98. Thus, controlling the tumour microenvironment, by either targeting these cell types directly or interfering with the pathways that result in their dysregulation, at the time of vaccination is likely to be not only important, but also necessary. The role of the tumour microenvironment in restraining antitumour immunity is exemplified by the results with single-agent99,100,101,102,103,104,105,106 or dual-agent therapy107,108 with immune-checkpoint inhibitors. The clinical responses observed with these treatments indicate that releasing the break on naturally acquired immune responses to tumour antigens is enough to result in deep and durable tumour responses in some patients, demonstrating the capacity for tumour antigens, which could theoretically include TERT, to evoke effective immunity.
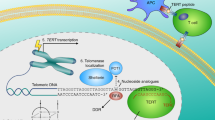
Immunosuppressive cell types in the tumour microenvironment can be manipulated using various strategies. Immune-checkpoint inhibitors can restore the activity of exhausted T cells and regulate Treg-cell activity — for example, via CTLA-4, glucocorticoid-induced TNF-receptor family related protein (GITR), or OX40 (Refs 109,110). Vaccination-induced Treg cells might counter-suppress the anticancer T-cell response generated111,112, but can potentially be opposed in a variety of ways: administration of a single low-dose of cyclophosphamide before anticancer vaccination with non-TERT vaccines; and treatment with thalidomide can reduce the frequency of Treg cells113. Myeloid cells with proinflammatory immunosuppressive characteristics might be controlled with nitro-aspirin; doxorubicin; all-trans retinoic acid (ATRA); inhibitors of cyclooxygenase 2 (COX-2), arginase 1, or phosphodiesterase type 5 (PDE5); or selective inhibitors of the endoplasmic-reticulum-stress response97,114,115. Finally, tumour-promoting B cells within the tumour microenvironment could be targeted with anti-CD20 antibodies (such as rituximab) or B-cell-kinase inhibitors, such as the Bruton tyrosine kinase inhibitor ibrutinib. Combining such therapies with TERT-based vaccination might improve therapeutic responses and, thus, patient outcomes, and this approach will be interesting to explore in future studies (Fig. 1).

In the laboratory: vaccine formulation and optimization based on established immunological principles to maximize the expansion of CD8+ and CD4+ T cells from the pool of TERT-specific precursors in the available T-cell repertoire, leveraging vaccine design and delivery features — collectively, the vaccine platform. In the clinic: patient selection for vaccination on the basis of genetic alterations in the TERT-promoter region, and further prioritized based on tumour tissue of origin and the related rate of stem-cell division, telomerase-expressing circulating tumour cells, and the stage of disease. Ideally, TERT vaccination should be used as a 'pre-emptive strike', rather than a curative approach. In the patients: the aim of immunization is to induce a wide spectrum anti-TERT T-cell response, involving both CD8+ and CD4+ T cells, in secondary lymphoid organs. Consideration should be given to the generation of long-term memory responses, preferentially of the central memory or stem-like memory type, by modulating antigen dose or administering an agonistic antibody to OX40 or 4-1BB, co-stimulatory molecules expressed by T cells. In the tumour microenvironment: the best objective clinical responses are expected when TERT immunization is associated with control of the tumour microenvironment. Thus, efforts should be made to modify the immunosuppressive tumour microenvironment and its pleiotropic effects, which are known to derail autochthonous and vaccine-induced antitumour T-cell responses. Several approaches can be considered, used concomitantly or sequentially with the vaccine. APC, antigen-presenting cell; ATRA, all-trans retinoic acid; COX-2, cyclooxygenase 2; CTLA-4, cytotoxic T lymphocyte-associated antigen 4; ER, endoplasmic reticulum; GITR, glucocorticoid-induced TNFR-family-related gene; LAG3, lymphocyte-activation gene 3; MHC, major histocompatibility complex; miRNA, microRNA; NO-aspirin, nitro-aspirin; PD-1, programmed cell-death protein 1; PD-L1, programmed cell death 1 ligand 1; TERT, telomerase reverse transcriptase; Treg, regulatory T.
These considerations are not unique to TERT vaccination, and apply to vaccines against other tumour antigens expressed in a high proportion of human cancers, such as MUC1, mutated p53, mutated KRAS, and NY-ESO1 (also known as cancer/testis antigen 1), which are observed in >60%, 5–48%, 9–30%, and 20–40% of all tumours, respectively116,117,118,119. Some of these antigens have undergone intense clinical experimentation with regard to vaccine development in the past 10–15 years. For these antigens, a reassessment of vaccine and trial design along the lines discussed in this Perspectives would be timely, in order to capitalize on knowledge and expertise accrued to date.
Taken together, these considerations indicate that, if cancer immunotherapy targeting TERT is justified, optimization of immunogenicity and the quality of T-cell responses (Box 1) should be the focus of new research efforts. In addition, the importance of assessing and quantifying the existence of T-cells precursors specific for TERT before immunotherapy should be a key consideration. A new generation of TERT-DNA vaccines delivered via electroporation has already been tested successfully in nonhuman primates, and induced a broad spectrum of anti-TERT responses120. Nevertheless, renewed interest in other forms of TERT immunotherapy is now warranted on the basis of two important developments: firstly, TERT upregulation has been associated with every stage of tumour evolution; secondly, whole-genome sequencing (WGS) of cancers has revealed mutations and rearrangements that affect TERT transcription. The former finding makes TERT an ideal target antigen on tumour cells based not only on expression level, percentage of positive cells, and number of patients with antigen-positive cancers, but also owing to antigen expression on cancer stem cells (CSCs)121. The latter observations might permit selection of patients for TERT-based therapy on the basis of genomic abnormalities that increase TERT expression. In addition, the developments in our understanding and therapeutic targeting of tumour immunosuppressive mechanisms made over the past 5 years could potentially be leveraged to exploit the associations of TERT with cancer and thus advance TERT-based immunotherapy.
Recentring TERT in tumour evolution
To date, TERT immunotherapy has been based on the early discovery that bulk tumours are generally telomerase positive. Importantly, more-recent findings indicate that TERT is expressed at every stage of the cancer process, from the incipient CSCs and/or tumour-initiating cells to advanced metastatic cancer cells, and has essential roles at each stage of tumorigenesis.
TERT is expressed in CSCs. Studies in mice and humans point to the fact that telomerase and TERT are important in enabling the self-renewal of stem and progenitor cells. Mice deficient in the telomerase RNA component (TERC) demonstrate defective stem-cell function, which can be rescued by reintroducing functional telomerase and by abrogating p53 activity122. Furthermore, in a subset of patients with dyskeratosis congenita, mutations in telomerase (TERT or TERC) that inactivate its enzymatic activity lead to bone-marrow failure123. These observations suggest that telomerase expression underlies the self-renewal capacity of stem cells.
Abundant evidence indicates that telomerase is associated with cancer, but which cells account for telomerase positivity in a bulk tumour mass is not entirely clear (a tumour with a volume of 1 cm3 might comprise a mixture of 109 heterogeneous cells), and whether these TERT-positive cells derive from a population of cells with tumour-initiating properties is unknown. CSCs are hypothesized to possess the exclusive ability to self-renew and propagate the tumour124. Paradoxically, CSCs have been shown to have short telomeres, compared with those in bulk tumour cells125. This finding suggests that altered telomerase levels in CSCs that formed early in tumour evolution maintain telomeres at low levels sufficient to oppose quiescence and/or senescence, or the DNA-damage response that can result in apoptosis. Of note, TERT overexpression has been demonstrated in CD133+ CSCs derived directly from CD133+ primary astrocytic glioblastoma126; however, these cells were found to give rise to CD133− progenitor cells with low telomerase levels, which were nontumorigenic and had progressive telomere shortening during repeated rounds of cell division. TERT overexpression has also been identified in androgen-receptor-negative and androgen-insensitive CD44+ prostate cancer cells derived from surgical specimens (early localized tumours)127,128; the authors hypothesized that these cells represent tumour-initiating cells that underlie tumour growth and resistance to androgen-deprivation therapy127,128. Interestingly, probing of induced pluripotent stem cells (iPS) generated from human prostate cancer cells with TCR-mimic antibodies demonstrates that these cells process and present the dominant p540 or p865 TERT peptides (M. Zanetti, unpublished data). The implications are profound. The demonstration that prostate-cancer-derived iPS cells process and present TERT peptides implies that CSCs, similarly to differentiated cancer cells, are susceptible to immune attack. This finding is important, as CSCs and progenitor cells are widely believed to be resistant to conventional therapies. Thus, the development of immunotherapies that effectively eradicate these cell types, through targeting of TERT, would represent a major step towards preventing cancer relapse.
TERT in CTCs. Circulating tumour cells (CTCs) can be detected in the blood of patients with various cancers, and are thought to be shed from the primary tumour into the general circulation via the lymphatics, or through ruptures in the walls of capillaries and small blood vessels. Increasing attention is being placed on the use of CTCs for diagnostic and prognostic purposes, and these cells harbour information for genomic interrogation. Telomerase expression has been demonstrated in CTCs in the blood of patients with prostate, ovarian, breast, and metastatic bladder cancer, or non-small-cell lung cancer129,130,131,132. No report exists that demonstrates TERT-antigen presentation by CTCs, although one could reasonably assume that CTCs process and present TERT peptides, and would, consequently, be recognized by CD8+ T cells once they seed a distal tissue. If this assumption proves to be correct, TERT vaccination might promote removal of CTCs, thus preventing colonization of other tissues — in turn, reducing the likelihood of relapse and improving survival. Moreover, discriminating CTCs on the basis of high and low TERT expression or TERT-protein levels might be important in efforts to stratify patients with cancer for immunotherapy (Fig. 1). Hence, whether TERT is expressed in CTCs should be a key question for further studies.
TERT is required for epithelial-to-mesenchymal transition. Epithelial-to-mesenchymal transition (EMT), a process whereby differentiated neoplastic cells undergo transcriptional phenotypic inversion, occurs in premetastatic cancer cells, and is also associated with the acquisition of stem-cell-like characteristics133. Cells that have undergone EMT display not only enhanced motility, but also a CSC phenotype (self-renewal) that manifests through a process of cell dedifferentiation134. Thus, a plausible prediction is that factors that promote EMT also promote the transcriptional activation of TERT, and vice versa. Indeed, findings indicate that TERT overexpression promotes, and its inhibition suppresses, EMT135; EMT mediated by transforming growth factor-β1 and β-catenin is abolished by small interfering RNA silencing of TERT expression and, therefore, EMT seems to require TERT activation. Clearly, this requirement renders TERT an important target in efforts to control EMT immunologically.
Genetic aberration of TERT
The transcriptional regulation of TERT expression is complex, involving multiple factors and a broad range of mechanisms, and has been extensively reviewed elsewhere136. The human TERT promoter lacks both TATA and CAAT boxes that are involved in the regulation of many genes, but is highly GC-rich. The promoter is inactive in normal and 'pre-immortal' cells, but is activated by derepression in 'immortal' cells, such as stem and progenitor cells, and a subset of tumour cells. The TERT promoter contains binding sites for transcription factors putatively involved in its regulation, including the oncoproteins MYC, Sp1, the human papillomavirus 16E6 protein, and steroid-hormone receptors (oestrogen and androgen receptors). On the other hand, numerous negative regulators of TERT transcription have been identified, including p53, E2F1/4/5, hypomethylated retinoblastoma-associated protein, and Wilms tumour protein136.
In the past 3 years, WGS studies have revealed a disease-segregating, highly-penetrant, causal germ-line mutation in the noncoding promoter region of TERT in a family with hereditary melanoma, as well as somatic TERT-promoter mutations in >70% of melanoma cell lines and tissues from patients with metastatic melanoma137,138,139. These unexpected findings led to the speculation that TERT-promoter mutations might be responsible for increased TERT expression and telomerase activity in many cancers. Indeed, the TERT-promoter mutations generate a sequence with affinity for ETS/TCF family transcription factors140, with data indicating that the mutated regions are bound predominantly by the transcription factor GA-binding protein (GABP)141. Prevalent TERT-promoter mutations have been reported in other cancer types, including glioblastoma (∼80%), bladder cancer (>60%), hepatocellular carcinoma (∼50%), grade II–III gliomas (10–44%), and thyroid carcinoma (11–21%)142,143,144,145,146,147. In fact, the results of WGS analyses have established that TERT-promoter mutations are the most-prevalent mutations in noncoding regions of cancer genomes148,149. The effects of promoter mutations on TERT transcription have been investigated using TERT–luciferase-gene reporter constructs, revealing that the mutations result in a twofold-to-fivefold increase in TERT transcriptional activity138,145. Similarly, TERT transcription is increased in hepatocellular carcinoma cells harbouring promoter mutations at this locus, compared with those from the normal liver, or cirrhotic lesions144. Notably, this study also revealed that TERT-promoter mutations are among the earliest genetic alterations associated with neoplastic transformation144, highlighting the potential importance of such mutations in tumorigenesis. This finding also indicates that TERT-promoter aberrations might be clonal mutations — that is, mutations that are common to almost all cancer cells within a tumour. Of note, earlier in 2016, neoantigens arising from clonal mutations were shown to be the key targets of immunotherapy150; thus, whether clonal expression of TERT antigens can be exploited for immunotherapy should be established in future studies.
In patients with cancer, the most-frequent, mutually exclusive TERT-promoter mutations are −124C>T and −146C>T, but other promoter sites can harbour somatic mutations (CC>TT tandem mutations at −124/−125 and −135/−139 positions). The exact mechanism(s) by which TERT-promoter mutations are generated is not known; however, these novel findings underscore the possible role of these mutations in adaptive mechanisms that drive tumorigenesis. In fact, in the setting of telomerase-deficiency and telomere dysfunction, conditional re-expression of the Tert gene exacerbates tumour growth and metastasis in a mouse model of Tp53−/−/Pten−/− prostate cancer151, and facilitates the progression of T-cell lymphoma that arises spontaneously in Atm−/− mice152. Interestingly, ALT was noted in the context of telomerase suppression in the Atm−/− mouse model152, and this finding might highlight a potential resistance mechanism to TERT-based immunotherapy. Of relevance, in humans the TERT promoter is a common integration site for several viruses, including hepatitis B virus153,154, hepatitis C virus155, and human papillomavirus156 — which can lead to activation of the TERT gene in cis.
A causal nexus between TERT-promoter mutations and tumorigenesis has been investigated. In human bladder cancer cell lines the presence of promoter mutations was found to correlate with higher levels of TERT mRNA157; in turn, patients with TERT-promoter mutations had worse disease-specific survival than those without such mutations in two independent cohorts of patients with bladder cancer157. The results of additional studies showed that patients with TERT-promoter mutations have a more-aggressive disease course and shorter survival, compared with those without these mutations142,146,158. The mechanism by which TERT-promoter mutations promote tumorigenesis has been further clarified by demonstrating that they prevent silencing of the TERT gene, thus increasing its transcriptional levels and suppressing telomere shortening in vivo159.
In addition to TERT-promoter mutations, recurrent genomic rearrangements in a chromosomal region proximal to TERT have been reported160. These rearrangements are present in some patients with high-risk neuroblastoma, induce strong transcriptional upregulation of TERT, and correlate with a poor clinical outcome160. Moreover, associations between TERT-promoter mutations and genetic polymorphisms at the TERT promoter have also been reported. For example, some patients with glioblastoma who are homozygous for the rs2853669 C-allele, which is situated in a putative ETS2 binding site in the TERT promoter close to the C228T and C250T mutation hotspots, also have tumours with TERT-promoter mutations161. These patients had markedly shorter overall survival durations than those with the wild-type allele (11 months versus 20 months, P = 0.002, and 12 months versus 20 months, P = 0.04, for the C228T and C250T mutations, respectively)161. This relationship, therefore, warrants further investigation.
Collectively, genomic investigations indicate that in some patients with cancer, TERT-promoter mutations yield greater TERT expression and possibly higher levels of TERT protein. From an immunological viewpoint, these mutations are relevant to the composition of the 'HLA ligandome' — that is, the number and range of MHC–peptide complexes presented at the cell surface. Any correlation that exists between mRNA transcription and the HLA ligandome in cancer tissues might only be weak162,163, and a single human cell can display ∼120,000 MHC I molecules on its surface22; nevertheless, a target cell need only express 1–3 specific MHC–peptide complexes to be functionally recognized by the corresponding CD8+ T cells164. High-resolution mass-spectroscopy-based approaches will be needed to address this issue, in the context of cells with TERT-promoter mutations.
TERT immunotherapy in rebound
Upregulation of TERT in cancer cells at all stages of differentiation is well established. Cells that accumulate initial cancer-promoting mutations are 'immortalized' by the activation of telomerase, and tumour-initiating cells and progenitor cells require telomerase for self-renewal126,127,128. Telomerase reactivation is not only necessary for tumour-cell resistance to apoptosis, but also for the initiation of local invasion and cancer progression. Mobilization and extravasation require TERT, and its overexpression promotes EMT134,165. Thus, TERT is a potential immunological target throughout the evolution and progression of cancer. TERT overexpression promotes the canonical functions of telomerase — telomere elongation and prevention of the DNA-damage response — that are critical for the survival and proliferation of cancer cells. Via its noncanonical functions, for example, the activation of β-catenin, TERT also confers resistance to antigrowth signals, and can affect all nine recognized hallmarks of cancer165. Thus, I feel that we should continue in our efforts to pursue TERT as a target for anticancer therapy, exploiting our improved understanding of both TERT and cancer immunology.
The discovery that mutations in the TERT promoter are frequently associated with certain types of cancer, and are, overall, the most-prevalent mutations in noncoding regions of cancer genomes, only strengthens the idea that TERT is a critically important therapeutic target for anticancer therapy. TERT-promoter mutations prevent silencing of the TERT gene, increasing its transcription; although TERT-protein expression might not be directly proportional to the level of heightened transcription157, even a twofold increase in TERT levels would generate more peptides, potentially making tumour cells more susceptible to T-cell killing. Experiments will need to interrogate, on a quantitative basis, TERT-peptide processing and presentation in the context of TERT-promoter mutations.
Subsequently, a reasonable proposal would be to use WGS or promoter-targeted sequencing to guide the selection of patients on the basis of TERT-promoter mutations, on the assumption that the tumour cells present in these patients have a constitutively higher content of the target antigen and more-aggressive growth characteristics. A way to test this hypothesis would be to vaccinate patients with bladder cancer or hepatocellular carcinoma (where the frequency of TERT-promoter mutations is on average one in two), and see if, at comparable levels of induction of anti-TERT T-cell responses, patients with TERT-promoter mutations have a more-favourable clinical outcome, or at least a greater degree of improvement (as such patients might already have a poorer prognosis), than vaccinees without such mutations.
At a time of burgeoning interest in personalized immunotherapy166,167, a practical approach based on the systematic identification of TERT-promoter mutations, on a cancer-type and individual-patient basis, followed by therapeutic TERT vaccination, could represent a convenient and effective alternative option (Box 2). By taking into account the frequency of TERT-promoter mutations in different cancer types142,143,144,149, variations in the propensity of cancer formation in different tissues (which are related to the rates of stem-cell division in the particular tissues)168, genomic rearrangements proximal to TERT160, and high numbers of telomerase-expressing cancer progenitor cells127, one could potentially identify cancer types in which TERT immunotherapy would have the highest likelihood of clinical success. On the basis of these criteria, TERT-based immunotherapies should be tested for efficacy, on a prioritized basis, in sporadic melanoma, hepatocellular carcinoma, bladder carcinoma, glioblastoma, neuroblastoma, and prostate adenocarcinoma. Ideally, one would want to probe for TERT expression and presentation on an individual-patient basis. TERT-protein abundance in tumour samples can be assessed using commercially available antibodies, although the performance of these antibodies in biomarker assays should be validated by reference laboratories. TERT presentation by MHC molecules could be assessed with anti-MHC–TERT-peptide-complex antibodies, but this would require generating antibodies for TERT peptides bound to each main HLA serotype169. CTCs should be systematically probed for telomerase expression — an approach that will require the development of new technology. Furthermore, prospective studies should use massively parallel sequencing of T-cell receptor CDR3 Vβ amplicons to analyse spectratypes in vaccinees, on an individual basis170.
The efficacy of therapeutic vaccination will also depend on controlling negative immune regulation at the time of vaccination. A combined approach with immune-checkpoint inhibitors, such as antibodies targeting CTLA-4, PD-1, or PD-L1171,172,173, could result in expansion of TERT-specific T-cell precursor populations, and/or reactivation of anergic or exhausted autochthonous T-cells in the tumour microenvironment. By 'releasing the brakes and simultaneously stepping on the accelerator', this combination-therapy approach could unleash the full potential of TERT vaccination, particularly in view of the fact that chemotherapy-induced tumour regression is synergized by a pre-existing TERT-reactive T cells174, and that tumour regression after therapeutic PD-1 blockade requires pre-existing CD8+ T-cells that are negatively regulated by PD-1–PD-L1 interactions175. Checkpoint-inhibitor monotherapy is effective in selected cancer types and only a fraction (17–40%) of patients have objective responses100,101,105,176, and response has been shown to depend on the tumour mutational burden (>100 mutations per tumour)176,177,178. A higher objective response rate (∼60%) has been observed in patients with melanoma treated with two checkpoint inhibitors — the anti-PD-1 antibody nivolumab and the anti-CTLA-4 antibody ipilimumab — in combination, but drug-related adverse events of grade 3 or 4 also occurred at a high frequency (in ∼55% of patients)107. In this context, determining if immune-checkpoint inhibition used in combination with TERT vaccination generates similar or better results would be of interest, not only in cancers in which non-synonymous mutations are prevalent179, but also in cancers with low mutation rates. In patients with high levels of TERT and TERT-antigen presentation, an alternative approach might be to use adoptive T-cell approaches, such as chimeric antigen receptor (CAR) T-cell therapy that enables effective and selective targeting of antigens expressed or presented prominently on the surface of tumour cells — with or without immune-checkpoint inhibition180,181. Limited data is available, however, on this approach in patients with solid tumours, suitable T-cell therapies will need to be developed, and such a strategy is costly and logistically challenging.
Conclusions
Despite the limited success of past clinical trials, TERT-based immunotherapy might offer opportunities for personalized intervention if vaccine design and immunization modalities are optimized based on the immunological and more general considerations discussed herein; patients selection is prioritized on the basis of TERT-promoter mutations and genomic rearrangements proximal to TERT (molecular profiling); and perhaps in combination with immune-checkpoint inhibitors. I believe that this approach could elevate cancer immunotherapy to new standards of success, possibly beyond the limits of immune-checkpoint inhibition alone. The type of precision/personalized intervention proposed herein should be pursued as an alternative to contemporary approaches to personalized immunotherapy that are based on neoantigen peptides identified by whole-exome sequencing; although initial proof of concept in humans has already been provided for the latter approach182,183, one can predict that it will have more limited large-scale clinical applicability, with higher costs and considerable organizational challenges.
References
Blackburn, E. H. Telomerases. Annu. Rev. Biochem. 61, 113–129 (1992).
Bryan, T. M. & Cech, T. R. Telomerase and the maintenance of chromosome ends. Curr. Opin. Cell Biol. 11, 318–324 (1999).
de Lange, T. How telomeres solve the end-protection problem. Science 326, 948–952 (2009).
Davoli, T. & de Lange, T. Telomere-driven tetraploidization occurs in human cells undergoing crisis and promotes transformation of mouse cells. Cancer Cell 21, 765–776 (2012).
Hayflick, L. The limited in vitro lifetime of human diploid cell strains. Exp. Cell Res. 37, 614–636 (1965).
Bodnar, A. G. et al. Extension of life-span by introduction of telomerase into normal human cells. Science 279, 349–352 (1998).
Hahn, W. C. et al. Creation of human tumour cells with defined genetic elements. Nature 400, 464–468 (1999).
Kim, N. W. et al. Specific association of human telomerase activity with immortal cells and cancer. Science 266, 2011–2015 (1994).
Yui, J., Chiu, C. P. & Lansdorp, P. M. Telomerase activity in candidate stem cells from fetal liver and adult bone marrow. Blood 91, 3255–3262 (1998).
Bachor, C., Bachor, O. A. & Boukamp, P. Telomerase is active in normal gastrointestinal mucosa and not up-regulated in precancerous lesions. J. Cancer Res. Clin. Oncol. 125, 453–460 (1999).
Ramirez, R. D., Wright, W. E., Shay, J. W. & Taylor, R. S. Telomerase activity concentrates in the mitotically active segments of human hair follicles. J. Invest. Dermatol. 108, 113–117 (1997).
Heaphy, C. M. et al. Prevalence of the alternative lengthening of telomeres telomere maintenance mechanism in human cancer subtypes. Am. J. Pathol. 179, 1608–1615 (2011).
Cesare, A. J. & Reddel, R. R. Alternative lengthening of telomeres: models, mechanisms and implications. Nat. Rev. Genet. 11, 319–330 (2010).
Nakamura, T. M. et al. Telomerase catalytic subunit homologs from fission yeast and human. Science 277, 955–959 (1997).
Minev, B. et al. Cytotoxic T cell immunity against telomerase reverse transcriptase in humans. Proc. Natl Acad. Sci. USA 97, 4796–4801 (2000).
Vonderheide, R. H., Hahn, W. C., Schultze, J. L. & Nadler, L. M. The telomerase catalytic subunit is a widely expressed tumor-associated antigen recognized by cytotoxic T lymphocytes. Immunity 10, 673–679 (1999).
Lev, A. et al. Isolation and characterization of human recombinant antibodies endowed with the antigen-specific, major histocompatibility complex-restricted specificity of T cells directed toward the widely expressed tumor T-cell epitopes of the telomerase catalytic subunit. Cancer Res. 62, 3184–3194 (2002).
Ayyoub, M. et al. Lack of tumor recognition by hTERT peptide 540-548-specific CD8+ T cells from melanoma patients reveals inefficient antigen processing. Eur. J. Immunol. 31, 2642–2651 (2001).
Parkhurst, M. R. et al. Immunization of patients with the hTERT:540-548 peptide induces peptide-reactive T lymphocytes that do not recognize tumors endogenously expressing telomerase. Clin. Cancer Res. 10, 4688–4698 (2004).
Purbhoo, M. A. et al. The HLA A*0201-restricted hTERT(540-548) peptide is not detected on tumor cells by a CTL clone or a high-affinity T-cell receptor. Mol. Cancer Ther. 6, 2081–2091 (2007).
Pellicciotta, I. et al. Presentation of telomerase reverse transcriptase, a self-tumor antigen, is down-regulated by histone deacetylase inhibition. Cancer Res. 68, 8085–8093 (2008).
Engelhard, V. H. The contributions of mass spectrometry to understanding of immune recognition by T lymphocytes. Int. J. Mass Spectrom. 259, 32–39 (2007).
Bossi, G. et al. Examining the presentation of tumor-associated antigens on peptide-pulsed T2 cells. Oncoimmunology 2, e26840 (2013).
Hernandez, J. et al. Identification of a human telomerase reverse transcriptase peptide of low affinity for HLA-A2.1 that induces CTL and mediates lysis of tumor cells. Proc. Natl Acad. Sci. USA 99, 12275–12280 (2002).
Scardino, A. et al. HER-2/neu and hTERT cryptic epitopes as novel targets for broad spectrum tumor immunotherapy. J. Immunol. 168, 5900–5906 (2002).
Vonderheide, R. H. et al. Characterization of HLA-A3-restricted cytotoxic T lymphocytes reactive against the widely expressed tumor antigen telomerase. Clin. Cancer Res. 7, 3343–3348 (2001).
Arai, J. et al. Identification of human telomerase reverse transcriptase-derived peptides that induce HLA-A24-restricted antileukemia cytotoxic T lymphocytes. Blood 97, 2903–2907 (2001).
Cortez-Gonzalez, X. et al. Immunogenic HLA-B7 restricted peptides of hTRT. Int. Immunol. 18, 1707–1718 (2006).
Zanetti, M. Tapping CD4 T cells for cancer immunotherapy: the choice of personalized genomics. J. Immunol. 194, 2049–2056 (2015).
Schroers, R. et al. Human telomerase reverse transcriptase-specific T-helper responses induced by promiscuous major histocompatibility complex class II-restricted epitopes. Clin. Cancer Res. 9, 4743–4755 (2003).
Schroers, R., Huang, X. F., Hammer, J., Zhang, J. & Chen, S. Y. Identification of HLA DR7-restricted epitopes from human telomerase reverse transcriptase recognized by CD4+ T-helper cells. Cancer Res. 62, 2600–2605 (2002).
Godet, Y. et al. Analysis of spontaneous tumor-specific CD4 T-cell immunity in lung cancer using promiscuous HLA-DR telomerase-derived epitopes: potential synergistic effect with chemotherapy response. Clin. Cancer Res. 18, 2943–2953 (2012).
Dupont, J., Latouche, J. B., Ma, C. & Sadelain, M. Artificial antigen-presenting cells transduced with telomerase efficiently expand epitope-specific, human leukocyte antigen-restricted cytotoxic T cells. Cancer Res. 65, 5417–5427 (2005).
Nair, S. K. et al. Induction of cytotoxic T cell responses and tumor immunity against unrelated tumors using telomerase reverse transcriptase RNA transfected dendritic cells. Nat. Med. 6, 1011–1017 (2000).
Saeboe-Larssen, S., Fossberg, E. & Gaudernack, G. mRNA-based electrotransfection of human dendritic cells and induction of cytotoxic T lymphocyte responses against the telomerase catalytic subunit (hTERT). J. Immunol. Methods 259, 191–203 (2002).
Su, Z. et al. Enhanced induction of telomerase-specific CD4+ T cells using dendritic cells transfected with RNA encoding a chimeric gene product. Cancer Res. 62, 5041–5048 (2002).
Frolkis, M. et al. Dendritic cells reconstituted with human telomerase gene induce potent cytotoxic T-cell response against different types of tumors. Cancer Gene Ther. 10, 239–249 (2003).
Adotevi, O. et al. Targeting human telomerase reverse transcriptase with recombinant lentivector is highly effective to stimulate antitumor CD8 T-cell immunity in vivo. Blood 115, 3025–3032 (2010).
Mennuni, C. et al. Preventive vaccination with telomerase controls tumor growth in genetically engineered and carcinogen-induced mouse models of cancer. Cancer Res. 68, 9865–9874 (2008).
Gross, D. A. et al. High vaccination efficiency of low-affinity epitopes in antitumor immunotherapy. J. Clin. Invest. 113, 425–433 (2004).
Dosset, M. et al. Universal cancer peptide-based therapeutic vaccine breaks tolerance against telomerase and eradicates established tumor. Clin. Cancer Res. 18, 6284–6295 (2012).
Gannage, M. et al. Ex vivo characterization of multiepitopic tumor-specific CD8 T cells in patients with chronic myeloid leukemia: implications for vaccine development and adoptive cellular immunotherapy. J. Immunol. 174, 8210–8218 (2005).
Titu, L. V. et al. Cytotoxic T-cell immunity against telomerase reverse transcriptase in colorectal cancer patients. Oncol. Rep. 12, 871–876 (2004).
Amarnath, S. M. et al. In vitro quantification of the cytotoxic T lymphocyte response against human telomerase reverse transcriptase in breast cancer. Int. J. Oncol. 25, 211–217 (2004).
Mizukoshi, E. et al. Cytotoxic T cell responses to human telomerase reverse transcriptase in patients with hepatocellular carcinoma. Hepatology 43, 1284–1294 (2006).
Karanikas, V. et al. Naturally occurring tumor-specific CD8+ T-cell precursors in individuals with and without cancer. Immunol. Cell Biol. 88, 575–585 (2010).
Filaci, G. et al. Frequency of telomerase-specific CD8+ T lymphocytes in cancer patients. Blood 107, 1505–1512 (2006).
Akbar, A. N. & Henson, S. M. Are senescence and exhaustion intertwined or unrelated processes that compromise immunity? Nat. Rev. Immunol. 11, 289–295 (2011).
Schreiber, R. D., Old, L. J. & Smyth, M. J. Cancer immunoediting: integrating immunity's roles in cancer suppression and promotion. Science 331, 1565–1570 (2011).
Hodes, R. J., Hathcock, K. S. & Weng, N. P. Telomeres in T and B cells. Nat. Rev. Immunol. 2, 699–706 (2002).
Hathcock, K. S., Weng, N. P., Merica, R., Jenkins, M. K. & Hodes, R. Cutting edge: antigen-dependent regulation of telomerase activity in murine T cells. J. Immunol. 160, 5702–5706 (1998).
Liu, K., Hodes, R. J. & Weng, N. Cutting edge: telomerase activation in human T lymphocytes does not require increase in telomerase reverse transcriptase (hTERT) protein but is associated with hTERT phosphorylation and nuclear translocation. J. Immunol. 166, 4826–4830 (2001).
Weng, N. P., Levine, B. L., June, C. H. & Hodes, R. J. Human naive and memory T lymphocytes differ in telomeric length and replicative potential. Proc. Natl Acad. Sci. USA 92, 11091–11094 (1995).
Weng, N. P., Granger, L. & Hodes, R. J. Telomere lengthening and telomerase activation during human B cell differentiation. Proc. Natl Acad. Sci. USA 94, 10827–10832 (1997).
Liu, K. et al. Constitutive and regulated expression of telomerase reverse transcriptase (hTERT) in human lymphocytes. Proc. Natl Acad. Sci. USA 96, 5147–5152 (1999).
Morrison, S. J., Prowse, K. R., Ho, P. & Weissman, I. L. Telomerase activity in hematopoietic cells is associated with self-renewal potential. Immunity 5, 207–216 (1996).
Ugel, S. et al. Autoimmune B-cell lymphopenia after successful adoptive therapy with telomerase-specific T lymphocytes. Blood 115, 1374–1384 (2010).
Sette, A. & Sidney, J. Nine major HLA class I supertypes account for the vast preponderance of HLA-A and -B polymorphism. Immunogenetics 50, 201–212 (1999).
Vonderheide, R. H. et al. Vaccination of cancer patients against telomerase induces functional antitumor CD8+ T lymphocytes. Clin. Cancer Res. 10, 828–839 (2004).
Brunsvig, P. F. et al. Telomerase peptide vaccination: a phase I/II study in patients with non-small cell lung cancer. Cancer Immunol. Immunother. 55, 1553–1564 (2006).
Brunsvig, P. F. et al. Telomerase peptide vaccination in NSCLC: a phase II trial in stage III patients vaccinated after chemoradiotherapy and an 8-year update on a phase I/II trial. Clin. Cancer Res. 17, 6847–6857 (2011).
Kyte, J. A. et al. Telomerase peptide vaccination combined with temozolomide: a clinical trial in stage IV melanoma patients. Clin. Cancer Res. 17, 4568–4580 (2011).
Vik-Mo, E. O. et al. Therapeutic vaccination against autologous cancer stem cells with mRNA-transfected dendritic cells in patients with glioblastoma. Cancer Immunol. Immunother. 62, 1499–1509 (2013).
Su, Z. et al. Immunological and clinical responses in metastatic renal cancer patients vaccinated with tumor RNA-transfected dendritic cells. Cancer Res. 63, 2127–2133 (2003).
Bernhardt, S. L. et al. Telomerase peptide vaccination of patients with non-resectable pancreatic cancer: a dose escalating phase I/II study. Br. J. Cancer 95, 1474–1482 (2006).
Bolonaki, I. et al. Vaccination of patients with advanced non-small-cell lung cancer with an optimized cryptic human telomerase reverse transcriptase peptide. J. Clin. Oncol. 25, 2727–2734 (2007).
Berntsen, A. et al. Therapeutic dendritic cell vaccination of patients with metastatic renal cell carcinoma: a clinical phase 1/2 trial. J. Immunother. 31, 771–780 (2008).
Schlapbach, C., Yerly, D., Daubner, B., Yawalkar, N. & Hunger, R. E. Telomerase-specific GV1001 peptide vaccination fails to induce objective tumor response in patients with cutaneous T cell lymphoma. J. Dermatol. Sci. 62, 75–83 (2011).
Staff, C., Mozaffari, F., Frodin, J. E., Mellstedt, H. & Liljefors, M. Telomerase (GV1001) vaccination together with gemcitabine in advanced pancreatic cancer patients. Int. J. Oncol. 45, 1293–1303 (2014).
Ellebaek, E. et al. Metastatic melanoma patients treated with dendritic cell vaccination, interleukin-2 and metronomic cyclophosphamide: results from a phase II trial. Cancer Immunol. Immunother. 61, 1791–1804 (2012).
Kotsakis, A. et al. Clinical outcome of patients with various advanced cancer types vaccinated with an optimized cryptic human telomerase reverse transcriptase (TERT) peptide: results of an expanded phase II study. Ann. Oncol. 23, 442–449 (2012).
Greten, T. F. et al. A phase II open label trial evaluating safety and efficacy of a telomerase peptide vaccination in patients with advanced hepatocellular carcinoma. BMC Cancer 10, 209 (2010).
Middleton, G. et al. Gemcitabine and capecitabine with or without telomerase peptide vaccine GV1001 in patients with locally advanced or metastatic pancreatic cancer (TeloVac): an open-label, randomised, phase 3 trial. Lancet Oncol. 15, 829–840 (2014).
Cortez-Gonzalez, X. & Zanetti, M. Telomerase immunity from bench to bedside: round one. J. Transl. Med. 5, 12 (2007).
Rapoport, A. P. et al. Combination immunotherapy using adoptive T-cell transfer and tumor antigen vaccination on the basis of hTERT and survivin after ASCT for myeloma. Blood 117, 788–797 (2011).
Caron, E. et al. The MHC I immunopeptidome conveys to the cell surface an integrative view of cellular regulation. Mol. Syst. Biol. 7, 533 (2011).
Yu, W. et al. Clonal deletion prunes but does not eliminate self-specific αβ CD8+ T lymphocytes. Immunity 42, 929–941 (2015).
Vitiello, A. et al. Development of a lipopeptide-based therapeutic vaccine to treat chronic HBV infection. I. Induction of a primary cytotoxic T lymphocyte response in humans. J. Clin. Invest. 95, 341–349 (1995).
Shedlock, D. J. & Shen, H. Requirement for CD4 T cell help in generating functional CD8 T cell memory. Science 300, 337–339 (2003).
Langlade-Demoyen, P. et al. Role of T cell help and endoplasmic reticulum targeting in protective CTL response against influenza virus. Eur. J. Immunol. 33, 720–728 (2003).
Hailemichael, Y. et al. Persistent antigen at vaccination sites induces tumor-specific CD8+ T cell sequestration, dysfunction and deletion. Nat. Med. 19, 465–472 (2013).
Mavroudis, D. et al. A phase I study of the optimized cryptic peptide TERT572y in patients with advanced malignancies. Oncology 70, 306–314 (2006).
Fenoglio, D. et al. A multi-peptide, dual-adjuvant telomerase vaccine (GX301) is highly immunogenic in patients with prostate and renal cancer. Cancer Immunol. Immunother. 62, 1041–1052 (2013).
Klebanoff, C. A. et al. Central memory self/tumor-reactive CD8+ T cells confer superior antitumor immunity compared with effector memory T cells. Proc. Natl Acad. Sci. USA 102, 9571–9576 (2005).
Gattinoni, L. et al. A human memory T cell subset with stem cell-like properties. Nat. Med. 17, 1290–1297 (2011).
Monsurro, V. V. et al. Quiescent phenotype of tumor-specific CD8+ T cells following immunization. Blood 104, 1970–1978 (2004).
Bonertz, A. et al. Antigen-specific TREGS control T cell responses against a limited repertoire of tumor antigens in patients with colorectal carcinoma. J. Clin. Invest. 119, 3311–3321 (2009).
Chambers, C. A., Kuhns, M. S., Egen, J. G. & Allison, J. P. CTLA-4-mediated inhibition in regulation of T cell responses: mechanisms and manipulation in tumor immunotherapy. Annu. Rev. Immunol. 19, 565–594 (2001).
Keir, M. E., Butte, M. J., Freeman, G. J. & Sharpe, A. H. PD-1 and its ligands in tolerance and immunity. Annu. Rev. Immunol. 26, 677–704 (2008).
Sharma, P. & Allison, J. P. The future of immune checkpoint therapy. Science 348, 56–61 (2015).
Wang, R. F. CD8+ regulatory T cells, their suppressive mechanisms, and regulation in cancer. Hum. Immunol. 69, 811–814 (2008).
Mougiakakos, D., Choudhury, A., Lladser, A., Kiessling, R. & Johansson, C. C. Regulatory T cells in cancer. Adv. Cancer Res. 107, 57–117 (2010).
Mahadevan, N. R. & Zanetti, M. Tumor stress inside out: cell-extrinsic effects of the unfolded protein response in tumor cells modulate the immunological landscape of the tumor microenvironment. J. Immunol. 187, 4403–4409 (2011).
Gajewski, T. F., Schreiber, H. & Fu, Y. X. Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 14, 1014–1022 (2013).
Affara, N. I. et al. B cells regulate macrophage phenotype and response to chemotherapy in squamous carcinomas. Cancer Cell 25, 809–821 (2014).
Shalapour, S. et al. Immunosuppressive plasma cells impede T-cell-dependent immunogenic chemotherapy. Nature 521, 94–98 (2015).
Rodvold, J. J., Mahadevan, N. R. & Zanetti, M. Immune modulation by ER stress and inflammation in the tumor microenvironment. Cancer Lett. http://dx.doi.org/10.1016/j.canlet.2015.09.009 (2015).
Mahadevan, N. R. et al. Cell-extrinsic effects of tumor ER stress imprint myeloid dendritic cells and impair CD8+ T cell priming. PLoS ONE 7, e51845 (2012).
Hodi, F. S. et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 363, 711–723 (2010).
Robert, C. et al. Nivolumab in previously untreated melanoma without BRAF mutation. N. Engl. J. Med. 372, 320–330 (2015).
Schadendorf, D. et al. Pooled analysis of long-term survival data from phase II and phase III trials of ipilimumab in unresectable or metastatic melanoma. J. Clin. Oncol. 33, 1889–1894 (2015).
Topalian, S. L. et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 366, 2443–2454 (2012).
Topalian, S. L. et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J. Clin. Oncol. 32, 1020–1030 (2014).
Hamid, O. et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N. Engl. J. Med. 369, 134–144 (2013).
Gettinger, S. N. et al. Overall survival and long-term safety of nivolumab (anti-programmed death 1 antibody, BMS-936558, ONO-4538) in patients with previously treated advanced non-small-cell lung cancer. J. Clin. Oncol. 33, 2004–2012 (2015).
Powles, T. et al. MPDL3280A (anti-PD-L1) treatment leads to clinical activity in metastatic bladder cancer. Nature 515, 558–562 (2014).
Postow, M. A. et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N. Engl. J. Med. 372, 2006–2017 (2015).
Antonia, S. et al. Safety and antitumour activity of durvalumab plus tremelimumab in non-small cell lung cancer: a multicentre, phase 1b study. Lancet Oncol. 17, 299–308 (2016).
Wing, K. et al. CTLA-4 control over Foxp3+ regulatory T cell function. Science 322, 271–275 (2008).
Nishikawa, H. & Sakaguchi, S. Regulatory T cells in cancer immunotherapy. Curr. Opin. Immunol. 27, 1–7 (2014).
Klebanoff, C. A., Gattinoni, L. & Restifo, N. P. CD8+ T-cell memory in tumor immunology and immunotherapy. Immunol. Rev. 211, 214–224 (2006).
Filaci, G. et al. CD8+ CD28− T regulatory lymphocytes inhibiting T cell proliferative and cytotoxic functions infiltrate human cancers. J. Immunol. 179, 4323–4334 (2007).
Giannopoulos, K. et al. The high frequency of T regulatory cells in patients with B-cell chronic lymphocytic leukemia is diminished through treatment with thalidomide. Leukemia 22, 222–224 (2008).
Wesolowski, R., Markowitz, J. & Carson, W. E. 3rd. Myeloid derived suppressor cells — a new therapeutic target in the treatment of cancer. J. Immunother. Cancer 1, 10 (2013).
Marvel, D. & Gabrilovich, D. I. Myeloid-derived suppressor cells in the tumor microenvironment: expect the unexpected. J. Clin. Invest. 125, 3356–3364 (2015).
Finn, O. J. et al. MUC-1 epithelial tumor mucin-based immunity and cancer vaccines. Immunol. Rev. 145, 61–89 (1995).
Cox, A. D., Fesik, S. W., Kimmelman, A. C., Luo, J. & Der, C. J. Drugging the undruggable RAS: mission possible? Nat. Rev. Drug Discov. 13, 828–851 (2014).
Olivier, M., Hollstein, M. & Hainaut, P. TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb. Perspect. Biol. 2, a001008 (2010).
Gnjatic, S. et al. NY-ESO-1: review of an immunogenic tumor antigen. Adv. Cancer Res. 95, 1–30 (2006).
Yan, J. et al. Highly optimized DNA vaccine targeting human telomerase reverse transcriptase stimulates potent antitumor immunity. Cancer Immunol. Res. 1, 179–189 (2013).
Cheever, M. A. et al. The prioritization of cancer antigens: a national cancer institute pilot project for the acceleration of translational research. Clin. Cancer Res. 15, 5323–5337 (2009).
Flores, I. & Blasco, M. A. A p53-dependent response limits epidermal stem cell functionality and organismal size in mice with short telomeres. PLoS ONE 4, e4934 (2009).
Goldman, F. D. et al. Characterization of primitive hematopoietic cells from patients with dyskeratosis congenita. Blood 111, 4523–4531 (2008).
Clarke, M. F. et al. Cancer stem cells — perspectives on current status and future directions: AACR workshop on cancer stem cells. Cancer Res. 66, 9339–9344 (2006).
Shay, J. W. & Wright, W. E. Role of telomeres and telomerase in cancer. Semin. Cancer Biol. 21, 349–353 (2011).
Beier, F. et al. Identification of CD133−/telomeraselow progenitor cells in glioblastoma-derived cancer stem cell lines. Cell. Mol. Neurobiol. 31, 337–343 (2011).
Xu, T., He, K., Wang, L. & Goldkorn, A. Prostate tumor cells with cancer progenitor properties have high telomerase activity and are rapidly killed by telomerase interference. Prostate 71, 1390–1400 (2011).
Finones, R. R. et al. Early human prostate adenocarcinomas harbor androgen-independent cancer cells. PLoS ONE 8, e74438 (2013).
Fizazi, K. et al. High detection rate of circulating tumor cells in blood of patients with prostate cancer using telomerase activity. Ann. Oncol. 18, 518–521 (2007).
Gauthier, L. R. et al. Detection of circulating carcinoma cells by telomerase activity. Br. J. Cancer 84, 631–635 (2001).
Soria, J. C. et al. The molecular detection of circulating tumor cells in bladder cancer using telomerase activity. J. Urol. 167, 352–356 (2002).
Sapi, E., Okpokwasili, N. I. & Rutherford, T. Detection of telomerase-positive circulating epithelial cells in ovarian cancer patients. Cancer Detect. Prev. 26, 158–167 (2002).
Mani, S. A. et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 133, 704–715 (2008).
Scheel, C. & Weinberg, R. A. Cancer stem cells and epithelial-mesenchymal transition: concepts and molecular links. Semin. Cancer Biol. 22, 396–403 (2012).
Liu, Z. et al. Telomerase reverse transcriptase promotes epithelial–mesenchymal transition and stem cell-like traits in cancer cells. Oncogene 32, 4203–4213 (2013).
Daniel, M., Peek, G. W. & Tollefsbol, T. O. Regulation of the human catalytic subunit of telomerase (hTERT). Gene 498, 135–146 (2012).
Horn, S. et al. TERT promoter mutations in familial and sporadic melanoma. Science 339, 959–961 (2013).
Huang, F. W. et al. Highly recurrent TERT promoter mutations in human melanoma. Science 339, 957–959 (2013).
Heidenreich, B. et al. Telomerase reverse transcriptase promoter mutations in primary cutaneous melanoma. Nat. Commun. 5, 3401 (2014).
Patton, E. E. & Harrington, L. Cancer: trouble upstream. Nature 495, 320–321 (2013).
Bell, R. J. et al. Cancer. The transcription factor GABP selectively binds and activates the mutant TERT promoter in cancer. Science 348, 1036–1039 (2015).
Killela, P. J. et al. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc. Natl Acad. Sci. USA 110, 6021–6026 (2013).
Liu, X. et al. Highly prevalent TERT promoter mutations in bladder cancer and glioblastoma. Cell Cycle 12, 1637–1638 (2013).
Nault, J. C. et al. High frequency of telomerase reverse-transcriptase promoter somatic mutations in hepatocellular carcinoma and preneoplastic lesions. Nat. Commun. 4, 2218 (2013).
Huang, D. S. et al. Recurrent TERT promoter mutations identified in a large-scale study of multiple tumour types are associated with increased TERT expression and telomerase activation. Eur. J. Cancer 51, 969–976 (2015).
Eckel-Passow, J. E. et al. Glioma groups based on 1p/19q, IDH, and TERT promoter mutations in tumors. N. Engl. J. Med. 372, 2499–2508 (2015).
Vinagre, J. et al. Frequency of TERT promoter mutations in human cancers. Nat. Commun. 4, 2185 (2013).
Melton, C., Reuter, J. A., Spacek, D. V. & Snyder, M. Recurrent somatic mutations in regulatory regions of human cancer genomes. Nat. Genet. 47, 710–716 (2015).
Weinhold, N., Jacobsen, A., Schultz, N., Sander, C. & Lee, W. Genome-wide analysis of noncoding regulatory mutations in cancer. Nat. Genet. 46, 1160–1165 (2014).
McGranahan, N. et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 351, 1463–1469 (2016).
Ding, Z. et al. Telomerase reactivation following telomere dysfunction yields murine prostate tumors with bone metastases. Cell 148, 896–907 (2012).
Hu, J. et al. Antitelomerase therapy provokes ALT and mitochondrial adaptive mechanisms in cancer. Cell 148, 651–663 (2012).
Horikawa, I. & Barrett, J. C. cis-activation of the human telomerase gene (hTERT) by the hepatitis B virus genome. J. Natl Cancer Inst. 93, 1171–1173 (2001).
Paterlini-Brechot, P. et al. Hepatitis B virus-related insertional mutagenesis occurs frequently in human liver cancers and recurrently targets human telomerase gene. Oncogene 22, 3911–3916 (2003).
Zhu, Z. et al. Hepatitis C virus core protein enhances telomerase activity in Huh7 cells. J. Med. Virol. 82, 239–248 (2010).
Ferber, M. J. et al. Integrations of the hepatitis B virus (HBV) and human papillomavirus (HPV) into the human telomerase reverse transcriptase (hTERT) gene in liver and cervical cancers. Oncogene 22, 3813–3820 (2003).
Borah, S. et al. Cancer. TERT promoter mutations and telomerase reactivation in urothelial cancer. Science 347, 1006–1010 (2015).
Rachakonda, P. S. et al. TERT promoter mutations in bladder cancer affect patient survival and disease recurrence through modification by a common polymorphism. Proc. Natl Acad. Sci. USA 110, 17426–17431 (2013).
Chiba, K. et al. Cancer-associated TERT promoter mutations abrogate telomerase silencing. eLife 4, e07918 (2015).
Peifer, M. et al. Telomerase activation by genomic rearrangements in high-risk neuroblastoma. Nature 526, 700–704 (2015).
Mosrati, M. A. et al. TERT promoter mutations and polymorphisms as prognostic factors in primary glioblastoma. Oncotarget 6, 16663–16673 (2015).
Milner, E., Barnea, E., Beer, I. & Admon, A. The turnover kinetics of major histocompatibility complex peptides of human cancer cells. Mol. Cell Proteom. 5, 357–365 (2006).
Weinzierl, A. O. et al. Distorted relation between mRNA copy number and corresponding major histocompatibility complex ligand density on the cell surface. Mol. Cell Proteom. 6, 102–113 (2007).
Sykulev, Y., Joo, M., Vturina, I., Tsomides, T. J. & Eisen, H. N. Evidence that a single peptide–MHC complex on a target cell can elicit a cytolytic T cell response. Immunity 4, 565–571 (1996).
Low, K. C. & Tergaonkar, V. Telomerase: central regulator of all of the hallmarks of cancer. Trends Biochem. Sci. 38, 426–434 (2013).
Schumacher, T. N. & Schreiber, R. D. Neoantigens in cancer immunotherapy. Science 348, 69–74 (2015).
Rosenberg, S. A. & Restifo, N. P. Adoptive cell transfer as personalized immunotherapy for human cancer. Science 348, 62–68 (2015).
Tomasetti, C. & Vogelstein, B. Cancer etiology. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science 347, 78–81 (2015).
Greenbaum, J. et al. Functional classification of class II human leukocyte antigen (HLA) molecules reveals seven different supertypes and a surprising degree of repertoire sharing across supertypes. Immunogenetics 63, 325–335 (2011).
Freeman, J. D., Warren, R. L., Webb, J. R., Nelson, B. H. & Holt, R. A. Profiling the T-cell receptor beta-chain repertoire by massively parallel sequencing. Genome Res. 19, 1817–1824 (2009).
Topalian, S. L., Drake, C. G. & Pardoll, D. M. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell 27, 450–461 (2015).
Nguyen, L. T. & Ohashi, P. S. Clinical blockade of PD1 and LAG3 — potential mechanisms of action. Nat. Rev. Immunol. 15, 45–56 (2015).
Mahoney, K. M., Rennert, P. D. & Freeman, G. J. Combination cancer immunotherapy and new immunomodulatory targets. Nat. Rev. Drug Discov. 14, 561–584 (2015).
Godet, Y., Dosset, M., Borg, C. & Adotevi, O. Is preexisting antitumor CD4 T cell response indispensable for the chemotherapy induced immune regression of cancer? Oncoimmunology 1, 1617–1619 (2012).
Tumeh, P. C. et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 515, 568–571 (2014).
Van Allen, E. M. et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 350, 207–211 (2015).
Snyder, A. et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N. Engl. J. Med. 371, 2189–2199 (2014).
Rizvi, N. A. et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 348, 124–128 (2015).
Alexandrov, L. B. et al. Signatures of mutational processes in human cancer. Nature 500, 415–421 (2013).
Jackson, H. J., Rafiq, S. & Brentjens, R. J. Driving CAR T-cells forward. Nat. Rev. Clin. Oncol. http://dx.doi.org/10.1038/nrclinonc.2016.36 (2016).
Khalil, D. N., Smith, E. L., Brentjens, R. J. & Wolchok, J. D. The future of cancer treatment: immunomodulation, CARs and combination immunotherapy. Nat. Rev. Clin. Oncol. 13, 273–290 (2016).
Tran, E. et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science 344, 641–645 (2014).
Carreno, B. M. et al. Cancer immunotherapy. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science 348, 803–808 (2015).
Su, Z. et al. Telomerase mRNA-transfected dendritic cells stimulate antigen-specific CD8+ and CD4+ T cell responses in patients with metastatic prostate cancer. J. Immunol. 174, 3798–3807 (2005).
Millard, F. E., Gerloni, M., Darrah, D., Monsurro, V. & Zanetti, M. Phase I study of transgenic B lymphocyte immunization (TLI) against telomerase in androgen-independent prostate cancer (PC) [abstract]. J. Clin. Onc. 22 (Suppl.) 2519 (2004).
Kitawaki, T. et al. Cross-priming of CD8+ T cells in vivo by dendritic cells pulsed with autologous apoptotic leukemic cells in immunotherapy for elderly patients with acute myeloid leukemia. Exp. Hematol. 39, 424–433.e2 (2011).
Hunger, R. E. et al. Vaccination of patients with cutaneous melanoma with telomerase-specific peptides. Cancer Immunol. Immunother. 60, 1553–1564 (2011).
Acknowledgements
I wish to thank Dr X. Cortez-Gonzalez for patiently assembling and reviewing the clinical-trial information presented, and my many colleagues at the University of California, San Diego, and the J. Craig Venter Institute, La Jolla, California, USA, who provided comments and helpful suggestions during the preparation of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
M.Z. is a named inventor for the Issued US patent #7388071 (Telomerase as cancer antigen), and serves as Medical Advisor to Invectys, a Paris-based biotechnology company that is developing a TERT vaccine.
Rights and permissions
About this article

Cite this article
Zanetti, M. A second chance for telomerase reverse transcriptase in anticancer immunotherapy. Nat Rev Clin Oncol 14, 115–128 (2017). https://doi.org/10.1038/nrclinonc.2016.67
Published:
Issue Date:
DOI: https://doi.org/10.1038/nrclinonc.2016.67
This article is cited by
-
The regulations of telomerase reverse transcriptase (TERT) in cancer
Cell Death & Disease (2024)
-
Combining old and new concepts in targeting telomerase for cancer therapy: transient, immediate, complete and combinatory attack (TICCA)
Cancer Cell International (2023)
-
Distinct mutational pattern of T-cell large granular lymphocyte leukemia combined with pure red cell aplasia: low mutational burden of STAT3
Scientific Reports (2023)
-
NIPU: a randomised, open-label, phase II study evaluating nivolumab and ipilimumab combined with UV1 vaccination as second line treatment in patients with malignant mesothelioma
Journal of Translational Medicine (2021)
-
Targeting neoantigens for cancer immunotherapy
Biomarker Research (2021)
