
Key Points
-
The present genetic diversity of HIV is the result of multiple cross-species transfers to humans from African non-human primates.
-
HIV-1 has its origin in chimpanzees and is the result of three separate transfers to humans; by contrast, HIV-2 is most closely related to strains in the sooty mangabey and is the result of at least four transfers.
-
The most recent common ancestor of HIV-1 group M (the virus that causes the vast majority of infections globally) is estimated to have existed in the 1930s, although more 'fossil' viruses are needed to confirm this.
-
HIV-1 subtypes arise as the consequence of founder events and incomplete sampling.
-
Over the course of an infection, positive natural selection is the dominant mode of HIV evolution within a single patient.
-
Over the epidemic as a whole, stochastic processes are more important for the evolution of the virus between patients. There is little evidence that fitness differences among strains determine their population structure and distribution.
-
Genetic recombination is a fundamental mechanism for the evolution of HIV.
-
The development of drug treatments has greatly extended life expectancy and quality of life for those who are HIV positive but the development of drug resistance has been a major setback.
-
Recombination can rapidly bring together resistance mutations for multiple drugs during combination therapy.
-
The development of a vaccine for HIV has been a frustratingly slow process with the few candidate vaccines that have made it to phase III trials demonstrating no efficacy.
-
The high rate of evolution means that the pattern of diversity of HIV is changing at an alarming rate.Vaccines are therefore aiming at a constantly moving target.
Abstract
Understanding the evolution of the human immunodeficiency virus (HIV) is crucial for reconstructing its origin, deciphering its interaction with the immune system and developing effective control strategies. Although it is clear that HIV-1 and HIV-2 originated in African primates, dating their transmission to humans is problematic, especially because of frequent recombination. Our ability to predict the spread of drug-resistance and immune-escape mutations depends on understanding how HIV evolution differs within and among hosts and on the role played by positive selection. For this purpose, extensive sampling of HIV genetic diversity is required, and is essential for informing the design of HIV vaccines.
Similar content being viewed by others


Main
AIDS is arguably the most serious infectious disease to have affected humankind. Not only are an estimated 42 million people carrying the virus at present1, but its case fatality rate is close to 100%, making it an infection of devastating ferocity. In 2002 alone, 5 million people became infected with the causative agent — the human immunodeficiency virus (HIV) — and, of these, 70% live in sub-Saharan Africa. Although a succession of antiviral agents has made HIV/AIDS a more manageable disease in some industrialized nations, and several vaccines are about to enter Phase III clinical trials, HIV will doubtless continue to impose a terrible burden of morbidity and mortality (Box 1).
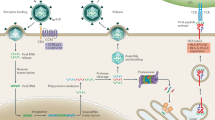
Although the development of anti-HIV drugs and vaccines has often been a frustrating exercise, and many aspects of HIV pathogenesis remain unclear, research into the origins and evolution of the virus has proven more fruitful. Ironically, one of the main reasons that successful treatment of HIV infection is so difficult — its rapid rate of evolutionary change — has allowed us to reconstruct the evolutionary history of HIV with great precision. More importantly, establishing the ground rules that underpin the evolution of HIV will lead to better vaccines and antiviral agents. Here, we review the current evidence documenting the origin of HIV, discuss how the virus evolves in relation to the human immune system and argue that evolutionary ideas are essential for the successful control of the virus. Basic aspects of the biology of HIV are illustrated in Fig. 1.

Although the human immunodeficiency virus (HIV) is able to infect a variety of cell types, AIDS results from the depletion of CD4+ T-HELPER LYMPHOCYTE CELLS, a key component of the human immune system. The env (envelope) gene encodes the proteins of the outer envelope of the virus, the gag (group-specific antigen) gene encode the components of the inner capsid protein, whereas the pol (polymerase) gene codes for the enzymes (such as REVERSE TRANSCRIPTASE) that are used in viral replication.
The origins of HIV
The key to understanding the origin of HIV was the discovery that closely related viruses — the simian immunodeficiency viruses (SIVs) — were present in a wide variety of African primates2. Collectively, HIV and SIV comprise the primate lentiviruses, and SIVs have been isolated in more than 20 African primate species. Importantly, in no case (other than laboratory-associated infections of Asian macaque monkeys) has it been shown that the SIVs cause disease in their hosts, although only a few studies of their natural history in wild populations have been undertaken.
Molecular phylogenies of HIV and SIV. The evolutionary history of HIV-1 and HIV-2 has been reconstructed in great detail by inferring phylogenetic trees of the primate lentiviruses. It was soon discovered that the two human viruses are related to different SIVs and therefore have different evolutionary origins (Fig. 2). Specifically, HIV-1 is most closely related to SIVcpz, which is found in some sub-species of chimpanzee (Pan troglodytes troglodytes and Pan troglodytes schweinfurthii) that inhabit parts of equatorial Western and Central Africa, respectively3,4. SIVcpz from P. t. troglodytes is of most interest because it shares the closest relationship with the abundant HIV-1 M group. The geographical range of P. t. troglodytes also encompasses the region in Africa that has the greatest genetic diversity of HIV-1, containing groups M, N and O; such a distribution is expected if this was where HIV-1 first emerged. By contrast, HIV-2 is most closely related to SIVsm5, which is found at high prevalence in sooty mangabey monkeys (Cercocebus atys). As with HIV-1, sooty mangabeys are most frequent in the regions of West Africa where HIV-2 is likely to have emerged.

Because both the human immunodeficiency virus type 1 (HIV-1) and HIV-2 lineages (red branches) fall within the simian immunodeficiency viruses (SIVs) that are isolated from other primates, they represent independent cross-species transmission events. The tree and other evidence also indicate that HIV-1 groups M, N and O represent separate transfers from chimpanzees (SIVcpz), again because there is a mixing of the HIV-1 and SIV lineages. Similarly, HIV-2 seems to have been transferred from sooty mangabey monkeys (SIVsm) on many occasions, although this is best documented in other phylogenetic analyses2. The tree was reconstructed using a MAXIMUM LIKELIHOOD METHOD on an alignment of 34 published nucleotide sequences of the viral polymerase (pol) gene (Fig. 1), excluding third codon positions (full details from the authors on request). Other abbreviations for viruses and their primate hosts are as follows: SIVcol, black and white colobus; SIVdrl, drill; SIVgsn, greater spot-nosed monkey; SIVlhoest, L'Hoest monkey; SIVmac, macaque; SIVmnd, mandrill; SIVmon, Campbell's mona monkey; SIVrcm, red-capped monkey; SIVsab, Sabaeus monkey; SIVsun, sun-tailed monkey; SIVsyk, Sykes' monkey; SIVtan, tantalus monkey; SIVver, vervet monkey. For clarity, only some subtypes of HIV-1 and HIV-2 are shown. All gene sequences were taken from GenBank (see online links box).
Molecular phylogenies also show that there have been many cross-species transmissions to humans, because there is a mixing of HIV and SIV lineages (Fig. 2). Although the vagaries of sampling make it difficult to determine exactly how many cross-species transfers have occurred, for HIV-2 this number might be at least four2, whereas three jumps from chimpanzees to humans are thought to explain the current diversity of HIV-1, such that groups M, N and O each have an independent origin3,6. However, inter-specific recombination, which might be common among the primate lentiviruses, greatly complicates this analysis7. For example, HIV-1 group N seems to be a recombinant between a SIVcpz strain and a virus related to the ancestor of group M (Ref. 3), but this event occurred before the establishment of group M and N in humans. SIVcpz is itself a composite of viruses, the descendants of which are now found in red-capped mangabeys (Cercocebus torquatus, SIVrcm) and greater spot-nosed monkeys (Cercocebus nictitans, SIVgsn)8.
Dating the evolution of primate lentiviruses. To fully understand the evolutionary history of HIV, it is also necessary to determine its timescale. At face value, the wide host-species range of the SIVs, as well as their low VIRULENCE and strong association with specific species9, argues for an ancient evolutionary history, perhaps representing virus–host co-divergence over several million years. Indeed, in some cases the phylogeny of the SIVs matches that of their primate hosts, as expected under a co-divergence model10. However, there are an increasing number of instances in which host and virus phylogenies are mismatched, implying more recent cross-species transfer in the SIVs8,11,12. If cross-species transfer is in fact the main mode of evolution, then any resemblance between host and virus trees might be because related primate species are often found in adjacent geographical ranges, or because host switching is most likely to occur between closely related host species13.
The timescale of evolution inferred from viral MOLECULAR CLOCKS also seems incompatible with long-term co-divergence. If co-divergence were true, then the divergence times of SIVs should broadly match those of their hosts, going back millions of years. However, all molecular clock estimates of primate lentivirus evolution are orders of magnitude more recent than this14, and the rates of mutation and replication are similar among these viruses15,16. Therefore, if molecular clocks are accurate, then the evolutionary timescale for each epidemic of HIV-1 and HIV-2 is measured only in decades. Several methods are available to measure substitution rates, and therefore divergence times, in RNA viruses, although the most reliable estimates come from analysing the temporal distribution of nodes on trees (Box 2). Application of these (and similar) methods have led to suggestions that the M group of HIV-1 originated in the 1930s, with a range of ∼10 years on either side17,18,19 (but see Box 2). A broadly similar evolutionary timescale has been proposed for HIV-2 (Ref. 20).
Although most estimates for the time of origin of HIV-1 are consistent, all can be subject to the same systematic bias. Once again, recombination might contribute to this error. Recombination has complex effects on the estimation of divergence times, by increasing apparent variation in rates among nucleotide sites and reducing genetic distances between sequences21,22. In these circumstances, perhaps the only reliable indicators of the timescale of HIV evolution are archival viral samples. The earliest HIV-1 M group sequence that is available was sampled in the Democratic Republic of Congo in 1959 (Ref. 23). That this sequence falls some distance from the root of the M group tree is strong evidence that the diversification of these viruses occurred before this time (Box 2). Accurately dating HIV evolution will require the analysis of more such 'fossil' viruses.
Emergence of the human diseases. By what mechanisms did HIV jump to humans? Although several theories have been proposed, there is little evidence to suggest that anything other than entirely 'natural' processes are responsible for its emergence2. In particular, given the frequency with which primate bushmeat that is sold in African markets is infected with SIV24, it is easy to envisage how individuals that are involved in the slaughter of animals or the preparation of food could become infected. Indeed, it is likely that SIV jumped into humans many times before the transmission pathways leading to the current AIDS epidemics were established, and that these incipient infections occurred in isolated rural communities and soon burnt out because of a lack of susceptible hosts.
It is also important to explain why HIV-1 forms subtypes, such that virus sequences tend to fall into distinct clusters with approximately equal genetic distances between them (10–30%, depending on the genes compared). These clusters are most likely to be produced by a combination of FOUNDER EFFECTS and incomplete sampling. In particular, intensive viral collection from West and Central Africa has now uncovered strains that fall between the previously described subtypes25. This indicates that these parts of Africa were the source of the strains that ignited successful epidemics in other localities, in Africa and beyond, and that the subtype structure of the HIV-1 tree to a large extent reflects sampling bias26. For example, most HIV-1 strains that were isolated in North America and Europe fall into subtype B, and their relative similarity reflects their recent common origin from a founder in, or from, Africa. Under the evolutionary timescale proposed for group M, the individual subtypes would have diversified in the last 40 years or so17,27, although there is debate as to whether subtype B originated in the 1960s or the 1970s28.
The phylogenesis of HIV-1 is therefore a dynamic process, such that subtypes will disappear and new epidemics arise (Fig. 3). More importantly, the current recommendations of what constitutes a subtype will become meaningless as phylogenetic complexity increases through the continued spread of the epidemic, with recombination among currently recognized types and more widespread sampling. The mean genetic divergence of subtype A, B, C and D genomes isolated in 1999 (9.8%, 10.0%, 7.9% and 8.5%, respectively) each equal that of the whole group M epidemic in 1985 (8.8%). The genetic divergence of the entire group M has increased to 14.9% in the same period (data from the HIV Database; see online links box). Although some of this increase can be ascribed to increased sampling, the substitution rate that this implies (∼0.002 substitutions per site, per year) lies within the range calculated previously for the viral env (envelope) and gag (group-specific antigen) genes (0.0028 and 0.0019, respectively)17.

The current global genetic diversity of HIV-1 group M is the result of several historical events in which geographically isolated epidemics (lower coloured triangles) were founded from strains that were present in a source population (large base triangle), most likely in the west of Central Africa25. Within each of these epidemics, frequent mixing of strains results in a complex recombinant structure (arrows within triangles). Subsequently, owing to global travel, the geographical ranges of these epidemics have increasingly overlapped, resulting in inter-subtype circulating recombinant forms (arrow between triangles), the number and mosaic complexity of which has steadily been increasing.
Evolutionary processes
Within-host evolution. One of the earliest and most striking observations made about HIV is the extensive genetic variation that the virus has within individual hosts, particularly in the hypervariable regions of the env gene29; this variation makes HIV one of the fastest evolving of all organisms. Such rapid evolution is the result of an explosive combination of factors. First, the virus experiences a high rate of mutation, with reverse transcriptase making ∼0.2 errors per genome during each replication cycle30, and further errors occurring during transcription from DNA by RNA Pol II polymerase. Second, HIV has remarkable replicatory dynamics: it has a viral generation time of ∼2.5 days and produces ∼1010–1012 new VIRIONS each day31. Finally, frequent recombination and natural selection further elevate its rate of evolutionary change.
Two key evolutionary questions arise when considering within-host genetic variation; how much of this diversity is shaped by IMMUNE SELECTION, and what is the relationship between genetic diversity and clinical outcome? Although it is possible that stochastic forces (GENETIC DRIFT) are involved in intra-host evolution, especially when the viral EFFECTIVE POPULATION SIZE is small (perhaps owing to population subdivision32), and when advantageous mutations are at low frequency, there is strong evidence that natural (positive) selection is the driving force of intra-host evolution. So, HIV successively FIXES mutations that allow it to evade immune responses, especially in the env gene. Host immune-selection pressure could be generated by neutralizing antibodies33 — for example, in the guise of an evolving GLYCAN SHIELD34, by T-helper cells35 or by CYTOTOXIC T LYMPHOCYTES (CTLs)36,37,38,39. CTL escape has been particularly well characterized, especially in SIV models40,41, where it can have a large impact on disease outcome42. Positive selection is also easily detectable in computational analyses of HIV gene sequences, in which the per site rate of NONSYNONYMOUS SUBSTITUTION (dN) exceeds that of SYNONYMOUS SUBSTITUTION (dS)35,43,44, and in the structure of intra-host phylogenies (see below). The remarkable strength of immune selection was revealed in a recent analysis of evolutionary dynamics in 50 HIV-1 patients45. Most fixed env amino-acid changes in these patients confer a selective advantage, with an average of one adaptive fixation event every ∼2.5 months. Under these criteria, HIV shows stronger positive selection than any other organism studied so far.
AIDS pathogenesis. Less clear is the role, if any, played by viral evolution in the development of AIDS in HIV-infected individuals. One early hypothesis was that there was a direct link between genetic (or, more specifically, antigenic) variation and pathogenesis, such that the immune system was unable to suppress each replicating variant and the patient succumbed to AIDS46. However, studies on larger numbers of patients have painted a far more complex picture of the relationship between genetic diversity and disease status. Although there is some evidence that rates of nonsynonymous substitution vary according to disease status, the direction of the correlation and its underlying causes are more uncertain47,48,49. This could, in part, be because analyses of intra-host sequence variation have failed to distinguish between total genetic variation and selectively advantageous changes, the latter being key to understanding the interaction between host and virus. Moreover, recombination again complicates the analysis, as it might produce false-positive evidence for natural selection in measures of dN/dS50,51. How to tease apart the respective roles of selection and recombination in shaping genetic diversity is a key area for future research in evolutionary genetics.
It is also important to determine whether the evolution of virulent viruses within each host tips the balance towards AIDS. The first indication that this might be the case was that AIDS patients more frequently harboured viruses that formed SYNCYTIA in vitro (SI strains) than those who were asymptomatic52. More recently, the development of SI strains has been associated with a broadening of co-receptor usage. Although HIV preferentially infects CD4+ cells, it also utilizes CHEMOKINE co-receptors, most notably CCR5 and CXCR4. Viral strains that infect macrophages use the CCR5 receptor (known as R5 strains), dominate during the early years of HIV infection and do not induce syncytia in vitro (NSI strains)53. A broadening of co-receptor usage — so that strains also using the CXCR4 receptor emerge (known as X4 strains) — generally occurs later in infection48. These T-cell tropic SI strains are associated with a higher rate of CD4+ cell decline and therefore a more rapid progression to AIDS54. However, many HIV-infected patients progress to AIDS without the appearance of X4 viruses. Moreover, HIV-1 subtype C, now the most common subtype in sub-Saharan Africa, is mainly composed of R5 virus strains55, but causes AIDS with the same frequency as other subtypes. Consequently, although viruses of higher virulence often appear during intra-host viral evolution, this alone is not responsible for the development of AIDS.
Taken together, most studies of intra-host HIV evolution indicate that the extent and structure of viral genetic diversity is more a marker of the arms race between host and virus than the cause of AIDS itself. In particular, a strong host immune response generates a strong selective reply from the virus, as measured in dN/dS. Therefore, the greatest positive selection in HIV is generally seen in patients with longer asymptomatic phases35,45.
Evolution within and among hosts. The persistent nature of HIV infection means that evolution occurs both within and among hosts. Strikingly, intra-host and inter-host evolution seem to be very different processes, with positive selection dominating in the former but not the latter. This is evident in the structure of phylogenetic trees (Fig. 4). Intra-host HIV phylogenies have a strong temporal structure, reflecting the successive fixation of advantageous mutations and the extinction of unfavourable lineages. By contrast, those trees that track viral evolution among hosts show little evidence for continual positive selection. Rather, they depict the (neutral) spatial and temporal diffusion of the virus, with viral lineages co-existing for extended time periods. Indeed, there is little evidence that fitness differences determine subtype structure and distribution. For example, experimental studies have revealed that subtype C viruses consistently have lower in vitro fitness than those assigned to subtype B (Ref. 56). Although caution should be shown when extrapolating from the laboratory to nature, this indicates that the high prevalence of subtype C in sub-Saharan Africa is the result of its chance entry into populations with high rates of partner exchange. However, it is unclear whether the success of HIV-1 group M, relative to groups N and O, is the result of some intrinsic property of the virus that enhances transmissibility, or because the founding virus from group M was fortunate enough to find itself in populations in which the epidemiological conditions were ideal for transmission.

The tree was constructed using the NEIGHBOUR-JOINING METHOD on envelope gene-sequence data that was taken from nine HIV-infected patients48 (a total of 1,195 sequences, 822 base pairs in length), with those viruses sampled from each patient depicted by a different colour. In each case, intra-host HIV evolution is characterized by continual immune-driven selection, such that there is a successive selective replacement of strains through time, with relatively little genetic diversity at any time point. By contrast, there is little evidence for positive selection at the population level (bold lines connecting patients), so that multiple lineages are able to coexist at any time point. A major BOTTLENECK is also likely to occur when the virus is transmitted to new hosts.
Why is natural selection a less potent force among hosts than within them? The first factor is the bottleneck that accompanies inter-host transmission, which greatly reduces genetic diversity. Evidence for a strong bottleneck at transmission is the homogeneity of the virus during primary infection57,58, although this could depend on the mode of transmission59. The second important factor concerns the behavioural aspects of HIV transmission. HIV is predominantly a sexually transmitted disease, and so the extensive variation in rates of partner exchange will, in combination with the transmission bottleneck, generate strong genetic drift. As a result, strains with advantageous mutations could, by chance, find themselves in individuals with low rates of partner exchange and so will not be transmitted far in the population. Of more debate is whether a bottleneck has a selective component, so that strains that are better adapted to new hosts (such as R5 strains) competitively establish themselves in primary infection60, or whether it is entirely neutral61 and thereby only magnifies the effects of genetic drift.
Finally, some advantageous mutations, such as those conferring CTL escape, might not appear until relatively late in infection62. If these late-escape mutants do not arise until after most individuals have transmitted the virus, natural selection will be less effective at the population level. As a consequence, HIV strains might not readily adapt to the HLA HAPLOTYPE distributions of their local populations63, because some CTL-escape mutants have little opportunity for further transmission. The data presented to support the adaptation of HIV to HLA haplotypes at the population level only considered within-host evolution, albeit in a large number of patients, and did not measure the effect of transmission. Indeed, the fact that repeated individual adaptation was observed in these patients indicates that the HIV population as a whole was not adapted to the host HLA distribution. Moreover, although certain CTL-escape mutants can be transmitted through the population64, it is possible that CTL-escape mutations that are passed to individuals with the 'wrong' HLA background will sometimes be deleterious and removed by purifying selection. In summary, inter-host HIV evolution is not merely intra-host evolution played out over a longer timescale, and the evolutionary process that occurs within hosts will not select for viruses with enhanced transmissibility.
Recombination and HIV diversity. Genetic recombination is an integral part of the HIV lifecycle, occurring when reverse transcriptase switches between alternative genomic templates during replication. As already mentioned, the recombination rate of HIV is one of the highest of all organisms, with an estimated three recombination events occurring per genome per replication cycle65, thereby exceeding the mutation rate per replication. The discovery that most infected cells harbour two or more different proviruses66, and the evidence for dual infection67,68, set the stage for recombination to have a central role in generating HIV diversity. Indeed, recombination has now been detected at all phylogenetic levels: among primate lentiviruses7,8, among HIV-1 groups69, among subtypes70 and within subtypes71. Prevalent inter-subtype recombinants are denoted 'circulating recombinant forms' (CRFs). There are 15 currently recognized CRFs that show a broad range of complexity and are widely distributed. In some geographical regions, CRFs account for at least 25% of all HIV infections72. Probably because it is more difficult to detect, the role of intra-subtype recombination has traditionally been downplayed. However, recent population-genetic studies indicate that recombination is also a pervasive force within subtypes71,73.
As hinted at earlier, recombination has important implications for understanding the HIV epidemic. In particular, many evolutionary inferences about HIV are made after the reconstruction of phylogenies, which can be greatly affected by recombination. Therefore, analyses of phylogenetic relationships, the timing of events, demographic processes or natural selection, are all potentially affected by recombination21,73,74. For example, although the data set used for dating the origin of HIV-1 M group to the 1930s was 'cleaned' for known recombinants before analysis17, the presence of recombination still seems evident, raising concerns about the accuracy of this estimate75. Because recombination is so frequent, it cannot be factored out by simply identifying recombinants and excluding those from the analysis. Indeed, today's subtypes might comprise old and successful recombinant lineages that trace back to a shared ancestral population (Fig. 3). HIV should therefore be studied with methods that are robust to the occurrence of recombination, or that explicitly take recombination into account. The development of such methods will doubtless prove difficult, but is necessary to make reliable inferences on many aspects of HIV-1 evolution. A simple way to start might be through the use of network approaches for phylogenetic inference, in which individual sequences are allowed to have many ancestors, and which provide a good alternative to traditional trees76,77.
Within individual hosts, recombination interacts with selection and drift to produce complex population dynamics, and perhaps provides an efficient mechanism for the virus to escape from the accumulation of deleterious mutations or to jump between ADAPTIVE PEAKS. Specifically, recombination might accelerate progression to AIDS78 and provide an effective mechanism (coupled with mutation) to evade drug therapy, vaccine treatment79 or immune pressure80,81. For example, vaccines without STERILIZING activity (such as CTL-inducing vaccines) or drug therapies with inconsistent application might offer the chance for SUPERINFECTION, in turn allowing recombination to produce viruses that carry many vaccine or drug escape mutations. Clearly, more studies are needed to quantify in vivo the role of recombination in generating important HIV diversity.
Consequences of HIV evolution
The evolution of drug resistance. The emergence of drug resistance in HIV has perhaps been the single largest setback in the treatment of AIDS. Once touted as a cure for HIV infection82, there is now less optimism that highly active antiretroviral therapy (HAART), involving combinations of drugs that act against different aspects of the viral life cycle, can rid the body of virus altogether. Mathematical models that predicted the eradication of virus from a patient within 2–3 years83 failed to adequately take into account two key aspects of HIV biology: viral reservoirs and evolution. No doubt, the development of HAART has greatly extended life expectancy and quality of life for those suffering from AIDS who can access these expensive drugs. The failure of HAART as a cure has led to many important lessons concerning our understanding of HIV biology84.
The discovery of latent reservoirs of HIV-1 in patients on HAART85 was one of the key components in understanding the ability of the virus to persist long after the initiation of therapy86. These reservoirs can serve to replenish the main pool of replicating virus and are now known from a variety of cell types, including CD4+ T lymphocytes, follicular dendritic cells87 and macrophages88, and are housed in various tissues throughout the body89. However, key questions remain regarding when and how these reservoirs become stocked with virus, whether or not replication is ongoing in or near the reservoirs, and whether the reservoirs are restocked with virus from subsequent replication. Although drug therapy is efficient at controlling viral levels in the plasma, the viruses in reservoirs are protected from interaction with drugs and can be extremely long-lived. It is therefore crucial to gain a clearer picture of the role and function of reservoirs in HIV infection.
Although the importance of viral evolution in immune escape is well understood, the evolutionary potential of HIV was severely underestimated by those proposing HAART therapy as a cure for AIDS. This occurred for two reasons. First, whereas the evolution of drug resistance in single drug therapy (such as AZT) had been documented, conventional wisdom was that the new triple-drug combination therapy would prove too difficult to evolve a solution to, because mutations were believed to occur singly and to accumulate in an ordered fashion90. However, this ignored recombination, which allows the virus to accumulate and exchange drug-resistant mutations in a nonlinear fashion, leading to rapid evolution of drug-resistant mutants, even between different reservoirs81 (Fig. 5). Second, early studies from patients on HAART concluded that the virus was not undergoing evolution in those with 'undetectable' levels of viral load85,91. In fact, the data from these studies clearly indicated that the virus was evolving; sequences from different time points were separated by measurable branch lengths, indicating that mutational changes had accumulated. Evidently, it was only a matter of time before the virus hit the mutations that would confer drug resistance92,93. Thankfully, through computational and mathematical approaches, we are now gaining a better understanding of the evolutionary dynamics of HIV-1 and its reaction to drug therapy94.

Hypothetical example showing how recombination will be an important mechanism to generate drug resistance in HIV. In this figure, two different HIV strains that are resistant to drug A (in red) and drug B (in blue) recombine to produce a new strain that is resistant to both drugs.
There is now a growing database of HIV mutations that confer drug resistance (see HIV drug resistance database in online links box) and their implications for drug resistance are, of course, enormous. More worryingly, there is evidence that some drug-resistant mutants show a greater infectivity, and in some cases a higher replication rate, compared with viruses without drug-resistant mutations95. Drug-resistant mutations are now being seen in drug-naive patients96, perhaps indicative of adaptation of the virus to a changed environment in parts of the world where drug treatment is widespread. However, it is unclear how many of these cases are simply the result of contact with a drug-treated individual, as in most cases drug-resistance mutations have an overall fitness cost in the absence of drugs97.
Viral genetic diversity and vaccine design. The development of an HIV vaccine has been frustratingly slow. Most attention has been directed towards the viral envelope region and comprises a variety of approaches, including recombinant proteins, synthetic peptides, recombinant viral vectors, recombinant bacterial vectors, recombinant particles, and whole-killed and live-attenuated HIV. The latter two have not progressed to clinical trials owing to an unfavourable benefit/risk ratio, which is further supported by experimental evidence from humans and simian models98,99.
Not least of the hurdles in the development of an effective HIV vaccine is the clinical trial process to show efficacy against the virus. Phase I and II vaccine trials are performed on small numbers of volunteers at relatively low risk for HIV-1 infection and attempt to establish a record of safety and provide valuable immunogenicity data. Phase III trials, on the other hand, are large-scale natural experiments on populations with high incidence and high risk for HIV-1 infection. Although more than 60 Phase I and II trials have taken place for more than 30 candidate vaccines, only two, developed by VaxGen (see online links box), have progressed to Phase III trials. The results from the first US-based Phase III trial showed that the vaccine had no efficacy for the target population experiencing subtype B infection. However, there was evidence of some efficacy in certain minority subpopulations, although the sample size in these categories was limited100. A similar (no efficacy) result was recently announced from their Phase III trial in Thailand.
The other intriguing result from the US trial was that the sampling greatly increased the estimates of HIV genetic diversity across that country100. As such, these data make it clear that, despite attempts to characterize genetic diversity in the USA for HIV vaccine development101, our database of HIV variation is still woefully inadequate, even for understanding diversity in a single subtype. We have relied on the published HIV database to serve as the basis for population-genetic and diversity studies, even though these data have largely been collected for medical studies. Recently, it has been suggested that reconstructions of ancestral HIV sequences or simple consensus sequences could be used to develop vaccine strains101,102. The intent of these techniques is to obtain a strain that is, as best as possible, similar to all the strains of a given subtype and therefore will provide maximum possible coverage. Although these approaches are based on evolutionary reasoning, the long development and testing cycle of a new vaccine will mean that the genetic diversity of HIV might have changed considerably in the intervening time. It is therefore crucial, for the development of successful vaccines, to better understand the impact of drug resistance in HIV, and to accurately document the overall evolution of this virus and, especially, to continually monitor genetic diversity of HIV on a global perspective. The use of carefully designed sampling strategies, concomitant with our improving understanding and modelling of the molecular evolution and epidemiology of this virus, will allow us to begin to predict how this diversity will change.
Conclusions
As this review has shown, great progress has been made in our understanding of the origins and evolution of HIV. However, it is clear that there are a number of unanswered questions and areas for which future research will be highly beneficial. Perhaps the issue of greatest importance is to fully determine the extent of the difference between viral evolution within and among hosts. With this knowledge, we will be better able to predict the long-term spread of drug resistance and CTL-escape mutations, as well as the likely impact of vaccination.

References
UNAIDS. AIDS Epidemic Update 2002, <http://www.unaids.org/worldaidsday/2002/press/Epiupdate.HtmlReport> (2002).
Hahn, B. H., Shaw, G. M., de Cock, K. M. & Sharp, P. M. AIDS as a zoonosis: scientific and public health implications. Science 287, 607–614 (2000). Important review of the origin of HIV-1 and HIV-2. Clearly shows that AIDS is a zoonotic disease with its origins in the primate lentiviruses.
Gao, F. et al. Origin of HIV-1 in the chimpanzee Pan troglodytes troglodytes. Nature 397, 436–441 (1999).
Santiago, M. L. et al. SIVcpz in wild chimpanzees. Science 295, 465–465 (2002).
Gao, F. et al. Human infection by genetically diverse SIVsm-related HIV-2 in West Africa. Nature 358, 495–499 (1992).
Sharp, P. M. et al. The origins of acquired immune deficiency syndrome viruses: where and when. Phil. Trans. Roy. Lond. B 356, 867–876 (2001).
Salemi, M. et al. Mosaic genomes of the six major primate lentiviruses lineages revealed by phylogenetic analysis. J. Virol. 77, 7202–7213 (2003).
Bailes, E. et al. Hybrid origin of SIV in chimpanzees. Science 300, 1713 (2003). Recent sequence analysis paper showing that chimpanzee SIVcpz has a hybrid origin, with two monkey SIVs acting as 'parental' strains.
Allan, J. S. et al. Species-specific diversity among simian immunodeficiency viruses from African green monkeys. J. Virol. 65, 2816–2828 (1991).
Beer, B. E. et al. Simian immunodefiency virus (SIV) from sun-tailed monkeys (Ceropithecus solatus): evidence for host-dependent evolution of SIV within the C. lhoesti superspecies. J. Virol. 73, 7734–7744 (1999).
Bibollet-Ruche, F. et al. Simian immunodeficiency virus infection in a patas monkey (Erythrocebus patas): evidence for cross-species transmission from African green monkeys (Cercopithecus aethiops sabaeus) in the wild. J. Virol. 77, 773–781 (1996).
Courgnaud, V. et al. Partial molecular characterization of two simian immunodeficiency virus (SIV) from African colobids: SIVwrc from western red colobus (Piliocolobus badius) and SIVolc from olive colobus (Procolobus verus). J. Virol. 77, 744–748 (2003).
Charleston, M. A. & Robertson, D. L. Preferential host switching by primate lentiviruses can account for phylogenetic similarity with the primate phylogeny. Syst. Biol. 51, 528–535 (2002).
Sharp, P. M. et al. Origins and evolution of AIDS viruses: estimating the timescale. Biochem. Soc. Trans. 28, 275–282 (2000).
Müller-Trutwin, M. C. et al. The evolutionary rate of nonpathogenic simian immunodeficiency virus (SIVagm) is in agreement with a rapid and continuous replication in vivo. Virology 223, 89–102 (1996).
Rey-Cuillé, M. A. et al. Simian immunodeficiency virus replicates to high levels in sooty mangabeys without inducing disease. J. Virol. 72, 3872–3886 (1998).
Korber, B. et al. Timing the ancestor of the HIV-1 pandemic strains. Science 288, 1789–1796 (2000). High-profile paper that provides a timescale for the origin and evolution of HIV-1.
Salemi, M. et al. Dating the radiation of HIV-1 group M in 1930s using a new method to uncover clock-like molecular evolution. FASEB J. 15, 276–278 (2000).
Yusim, K. et al. Using human immunodeficiency virus type 1 sequences to infer historical features of the acquired immune deficiency syndrome epidemic and human immunodeficiency virus evolution. Phil. Trans. R. Soc. Lond. B 356, 855–866 (2001).
Lemey, P. et al. Tracing the origin and history of the HIV-2 epidemic. Proc. Natl Acad. Sci. USA 100, 6588–6592 (2003).
Schierup, M. H. & Hein, J. Recombination and the molecular clock. Mol. Biol. Evol. 17, 1578–1579 (2000).
Worobey, M. A novel approach to detecting and measuring recombination: new insights into evolution in viruses, bacteria, and mitochondria. Mol. Biol. Evol. 18, 1425–1434 (2001).
Zhu, T. F. et al. An African HIV-1 sequence from 1959 and implications for the origin of the epidemic. Nature 391, 594–597 (1998).
Peeters, M. et al. Risk to human health from a plethora of Simian immunodeficiency viruses in primate bushmeat. Emerg. Infect. Dis. 8, 451–457 (2002).
Vidal, N. et al. Unprecedented degree of HIV-1 group M genetic diversity in the Democratic Republic of Congo suggests that the HIV-1 pandemic originated in Central Africa. J. Virol. 74, 10498–10507 (2000).
Rambaut, A., Robertson, D. L., Pybus, O. G., Peeters, M. & Holmes, E. C. Phylogeny and the origin of HIV-1. Nature 410, 1047–1048 (2001). Shows how the subtype structure of the tree for the HIV-1 M group is produced by a combination of incomplete sampling and local founder effects.
Robbins, K. E. et al. US human immunodeficiency virus type 1 epidemic: date of origin, population history, and characterization of early strains. J. Virol. 77, 6359–6366 (2003).
Smith, U. R., Kuiken, C. & Korber, B. T. Recent evolutionary history of human immunodeficiency type 1 subtype B–response. J. Mol. Evol. 56, 643–644 (2003).
Holmes, E. C., Zhang, L. Q., Simmonds, P., Ludlam, C. A. & Leigh Brown, A. J. Convergent and divergent sequence evolution in the surface envelope glycoprotein of human immunodeficiency virus type 1 within a single infected patient. Proc. Natl Acad. Sci. USA 89, 4835–4839 (1992).
Preston, B. D., Poiesz, B. J. & Loeb, L. A. Fidelity of HIV-1 reverse transcriptase. Science 242, 1169–1171 (1988).
Perelson, A. S., Neumann, A. U., Markowitz, M., Leonard, J. M. & Ho, D. D. HIV-1 dynamics in vivo: virion clearance rate, infected cell life-span, and viral generation time. Science 271, 1582–1586 (1996). Presents an analysis that shows the remarkable replicatory dynamics of HIV.
Frost, S. D. W., Dumaurier, M. J., Wain-Hobson, S. & Leigh Brown, A. J. Genetic drift and within-host metapopulation dynamics of HIV-1 infection. Proc. Natl Acad. Sci. USA 98, 6975–6980 (2001).
Richman, D. D., Wrin, T., Little, S. J. & Petropoulos, C. J. Rapid evolution of the neutralizing antibody response to HIV type 1 infection. Proc. Natl Acad. Sci. USA 100, 4144–4149 (2003).
Wei, X. et al. Antibody neutralization and escape by HIV-1. Nature 422, 307–312 (2003).
Ross, H. A. & Rodrigo, A. G. Immune-mediated positive selection drives human immunodeficiency virus type 1 molecular variation and predicts disease duration. J. Virol. 76, 11715–11720 (2002).
Kelleher, A. D. et al. Clustered mutations in HIV-1 gag are consistently required for escape from HLA-B27 restricted CTL responses. J. Exp. Med. 193, 375–385 (2001).
Ogg, G. S. et al. Quantitation of HIV-1 specific cytotoxic T lymphocytes and plasma load of viral RNA. Science 279, 2103–2106 (1998).
Phillips, R. E. et al. Human immunodeficiency virus genetic variation that can escape cytotoxic T cell recognition. Nature 354, 453–459 (1991).
Yusim, K. et al. Clustering patterns of cytotoxic T-lymphocyte epitopes in human immunodeficiency virus type 1 (HIV-1) proteins reveal imprints of immune evasion on HIV-1 global variation. J. Virol. 76, 8757–8768 (2002).
Allen, T. M. et al. Tat-specific cytotoxic T lymphocytes select for SIV escape variants during resolution of primary viraemia. Nature 407, 386–390 (2000).
O'Connor, D. H. et al. Acute phase cytotoxic T lymphocyte escape is a hallmark of simian immunodeficiency virus infection. Nature Med. 8, 493–499 (2002).
Barouch, D. H. et al. Eventual AIDS vaccine failure in a rhesus monkey by viral escape from cytotoxic T lymphocytes. Nature 415, 335–339 (2002).
Bonhoeffer, S., Holmes, E. C. & Nowak, M. A. Causes of HIV diversity. Nature 376, 125 (1995).
Zanotto, P. M. D. A., Kallas, E. G., de Souza, R. F. & Holmes, E. C. Genealogical evidence for positive selection in the nef gene of HIV-1. Genetics 153, 1077–1089 (1999).
Williamson, S. Adaptation in the env gene of HIV-1 and evolutionary theories of disease progression. Mol. Biol. Evol. 20, 1318–1325 (2003). Important paper that presents an analysis of the rate of adaptive evolution in a large number of HIV patients and how this relates to disease progression.
Nowak, M. A. et al. Antigenic diversity threshold and the development of AIDS. Science 254, 963–969 (1991).
Markham, R. B. et al. Patterns of HIV-1 evolution in individuals with differing rates of CD4 T cell decline. Proc. Natl Acad. Sci. USA 95, 12568–12573 (1998).
Shankarappa, R. et al. Consistent viral evolutionary changes associated with the progression of human immunodeficiency virus type 1 infection. J. Virol. 73, 10489–10502 (1999). An extensive analysis of the dynamics of intra-host HIV evolution in a multi-patient study.
Wolinksy, S. M. et al. Adaptive evolution of human immunodeficiency virus-type 1 during the natural course of infection. Science 272, 537–542 (1996).
Anisimova, M., Nielsen, R. & Yang, Z. Effect of recombination on the accuracy of the likelihood ratio method for detecting positive selection at amino acid sites. Genetics 164 1229–1236 (2003).
Shriner, D., Nickle, D. C., Jensen, M. A. & Mullins, J. I. Potential impact of recombination on sitewise approaches for detecting positive natural selection. Genet. Res. 81, 115–121 (2003).
Cheng-Mayer, C., Seto, D., Tateno, M. & Levy, J. A. Biological features of HIV-1 that correlate with virulence in the host. Science 240, 80–82 (1988).
Bjorndal, A. et al. Coreceptor usage of primary human immunodeficiency virus type 1 isolates varies according to biological phenotype. J. Virol. 71, 7478–7487 (1997).
Zhang, L. Q. et al. Chemokine coreceptor usage by diverse primary isolates of human immunodeficiency virus type 1. J. Virol. 72, 9301–9312 (1998).
Peeters, M. et al. Evidence for differences in MT2 cell tropism according to genetic subtypes of HIV-1: syncytium-inducing variants seem rare among subtype C HIV-1 viruses. J. Acq. Imm. Def. Syn. Human Retro. 20, 115–121 (1999).
Ball, S. C. et al. Comparing the ex vivo fitness of CCR5-tropic human immunodeficiency virus type 1 isolates of subtypes B and C. J. Virol. 77, 1021–1038 (2003).
Delwart, E. et al. Homogeneous quasispecies in 16 out of 17 individuals during very early HIV-1 primary infection. AIDS 16, 189–195 (2002).
Zhu, T. F. et al. Genotypic and phenotypic characterization of HIV-1 in patients with primary infection. Science 261, 1179–1181 (1993).
Dickover, R. E., Garratty, E. M., Plaeger, S. & Bryson, Y. J. Perinatal transmission of major, minor, and multiple maternal human immunodeficiency virus type 1 variants in utero and intrapartum. J. Virol. 75, 2194–2203 (2001).
Wolinksy, S. M. et al. Selective transmission of human immunodeficiency virus type-1 variants from mothers to infants. Science 255, 1134–1137 (1992).
Verhofstede, C. et al. Diversity of the human immunodeficiency virus type 1 (HIV-1) env sequence after vertical transmission in mother-child pairs infected with HIV-1 subtype A. J. Virol. 77, 3050–3057 (2003).
Goulder, P. J. R. et al. Late escape from an immunodominant cytotoxic T-lymphocyte response associated with progression to AIDS. Nature Med. 3, 212–217 (1997).
Moore, C. B. et al. Evidence for HIV-1 adaptation to HLA-restricted immune responses at the population level. Science 296, 1439–1443 (2002). Extensive study that shows how HIV-1 adapts to HLA haplotypes at the intra-host level. However, the paper does not consider whether intra-host 'escape' mutants are transmitted among hosts.
Goulder, P. J. R. et al. Evolution and transmission of stable CTL escape mutations in HIV infection. Nature 412, 334–337 (2001).
Zhuang, J. et al. Human immunodeficiency virus type 1 recombination: rate, fidelity, and putative hot spots. J. Virol. 76, 11273–11282 (2002). Recent paper that shows the extremely high rate of recombination in HIV.
Jung, A. et al. Multiply infected spleen cells in HIV patients. Nature 418, 144 (2002).
Jost, S. et al. A patient with HIV-1 superinfection. N. Engl. J. Med. 347, 731–736 (2002).
Koelsch, K. K. et al. Clade B HIV-1 superinfection with wild-type virus after primary infection with drug-resistant clade B virus. AIDS 17, F11–6 (2003).
Takehisa, J. et al. Human immunodeficiency virus type 1 intergroup (M/O) recombination in cameroon. J. Virol. 73, 6810–6820 (1999).
Robertson, D. L., Sharp, P. M., McCutchan, F. E. & Hahn, B. H. Recombination in HIV-1. Nature 374, 124–126 (1995).
Crandall, K. A. & Templeton, A. R. in The Evolution of HIV (ed. Crandall, K. A.) 153–176 (The Johns Hopkins University Press, Baltimore, 1999).
Pandrea, I. et al. Analysis of partial pol and env sequences indicates a high prevalence of HIV type 1 recombinant strains circulating in Gabon. AIDS Res. Hum. Retroviruses 18, 1103–1116 (2002).
McVean, G., Awadalla, P. & Fearnhead, P. A coalescent-based method for detecting and estimating recombination from gene sequences. Genetics 160, 1231–1241 (2002).
Posada, D. & Crandall, K. A. The effect of recombination on the accuracy of phylogeny estimation. J. Mol. Evol. 54, 396–402 (2002).
Schierup, M. H. & Forsberg, R. in Proceedings of the conference: Origin of HIV and emerging persistent viruses (Accademia Nazionale dei Lincei, Rome, 2003).
Posada, D. & Crandall, K. A. Intraspecific gene genealogies: trees grafting into networks. Trends Ecol. Evol. 16, 37–45 (2001).
Wain-Hobson, S., Renoux-Elbe, C., Vartanian, J. P. & Meyerhans, A. Network analysis of human and simian immunodeficiency virus sequence sets reveals massive recombination resulting in shorter pathways. J. Gen. Virol. 84, 885–895 (2003).
Liu, S. L. et al. Selection for human immunodeficiency virus type 1 recombinants in a patient with rapid progression to AIDS. J. Virol. 76, 10674–10684 (2002).
Nájera, R., Delgado, E., Pérez-Alvarez, L. & Thomson, M. M. Genetic recombination and its role in the development of the HIV-1 pandemic. AIDS 16, S3–16 (2002).
Kellam, P. & Larder, B. A. Retroviral recombination can lead to linkage of reverse transcriptase mutations that confer increased zidovudine resistance. J. Virol. 69, 669–674 (1995).
Morris, A. et al. Mosaic structure of the human immunodeficiency virus type 1 genome infecting lymphoid cells and the brain: evidence for frequent in vivo recombination events in the evolution of regional populations. J. Virol. 73, 8720–8731 (1999).
Wain-Hobson, S. Down or out in blood and lymph? Nature 387, 123–124 (1997).
Perelson, A. S. et al. Decay characteristics of HIV-1-infected compartments during combination therapy. Nature 387, 188–191 (1997).
Cohen, O. J. & Fauci, A. S. Transmission of multidrug-resistant human immunodeficiency virus — the wake-up call. N. Engl. J. Med. 339, 341–343 (1998).
Finzi, D. et al. Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science 278, 1295–1300 (1997).
Blankson, J. N., Persaud, D. & Siliciano, R. F. The challenge of viral reservoirs in HIV-1 infection. Ann. Rev. Med. 53, 557–593 (2002).
Smith, B. A. et al. Persistence of infectious HIV on follicular dendritic cells. J. Immunol. 166, 690–696 (2001).
Igarashi, T. et al. Macrophage are the principal reservoir and sustain high virus loads in rhesus macaques after the depletion of CD4+ T cells by a highly pathogenic simian immunodeficiency virus/HIV type 1 chimera (SHIV): implications for HIV-1 infections of humans. Proc. Natl Acad. Sci. USA 98, 658–663 (2001).
Clements, J. et al. The central nervous system as a reservoir for simian immunodeficiency virus (SIV): steady-state levels of SIV DNA in brain from acute through asymptomatic infection. J. Infect. Dis. 186, 905–913 (2002).
Molla, A. et al. Ordered accumulation of mutations in HIV protease confer resistance to ritonavir. Nature Med. 2, 760–766 (1996).
Wong, J. K. et al. Recovery of replication-competent HIV despite prolonged suppression of plasma viremia. Science 278, 1291–1295 (1997).
Chun, T. -W., Davey, R. T., Engel, D., Lane, H. C. & Fauci, A. S. AIDS: re-emergence of HIV after stopping therapy. Nature 401, 874–875 (1999). Important paper that explores the development of drug resistance in HIV.
Martinez-Picado, J. et al. Antiretroviral resistance during successful therapy of HIV type 1 infection. Proc. Natl Acad. Sci. USA 97, 10948–10953 (2000).
Ribeiro, R. M. & Bonhoeffer, S. Production of resistant HIV mutants during antiretroviral therapy. Proc. Natl Acad. Sci. USA 97, 7681–7686 (2000).
Simon, V. et al. Infectivity and replication capacity of drug-resistant human immunodeficiency virus type 1 variants isolated during primary infection. J. Virol. 77, 7736–7745 (2003).
Pitisuttithum, P. et al. Safety and immunogenicity of combinations of recombinant subtype E and B human immunodeficiency virus type 1 envelope glycoprotein 120 vaccines in healthy Thai adults. J. Infect. Dis. 188, 219–227 (2003).
Back, N. K. et al. Reduced replication of 3TC-resistant HIV-1 variants in primary cells due to a processivity defect of the reverse transcriptase enzyme. EMBO J. 15, 4040–4049 (1996).
Baba, T. W. et al. Human neutralizing monoclonal antibodies of the IgG1 subtype protect against mucosal simian-human immunodeficiency virus infection. Nature Med. 6, 200–206 (2000).
Greenough, T. C., Sullivan, J. L. & Desrosiers, R. C. Declining CD4 T-cell counts in a person infected with nef-deleted HIV-1. N. Engl. J. Med. 340, 236–237 (1999).
Berman, P. Preliminary results of VAX004: The first large scale field trial of an HIV vaccine. in Keystone Conference: HIV Vaccine (Banf, CA, 2003).
Gaschen, B. et al. Diversity considerations in HIV-1 vaccine selection. Science 296, 2354–2360 (2003).
Nickle, D. C. et al. Consensus and Ancestral State HIV Vaccines. Science 299, 1515–1518 (2003).
Barré-Sinoussi, F. et al. Isolation of a T-lymphotropic retrovirus from a patient at risk for acquired immune deficiency syndrome (AIDS). Science 220, 868–871 (1983). Ground-breaking paper that reports the isolation of HIV for the first time.
Walker, P. R., Worobey, M., Rambaut, A., Holmes, E. C. & Pybus, O. G. Sexual transmission of HIV in Africa. Nature 422, 679 (2003).
Drummond, A. J., Pybus, O. G. & Rambaut, A. Inference of viral evolutionary rates from molecular sequences. Adv. Parasit. 54, 331–358 (2003). Review that describes the methods for estimating rates of evolution from virus sequences that have been sampled over time.
Acknowledgements
We thank O. Pybus and D. Robertson for some useful discussion, and A. Rodrigo and two anonymous referees for constructive comments on the draft of this manuscript. Support in the form of a research fellowship from the Royal Society (to A.R.), from the 'Ramón y Cajal' programme of the Spanish government (to D.P.), and from a National Institutes of Health grant (to K.A.C and D.P.) is gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
Andrew Rambaut, David Posada and Keith A. Crandall have undertaken consultancy work for VaxGen, which involved genetic analysis of HIV sequences after the completion of the Phase III trials. Edward C. Holmes has no competing interests.
Related links
Related links
DATABASES
CDC database of infectious diseases
GenBank
LocusLink
FURTHER INFORMATION
Software and links for estimating rates of viruses
Software and links for estimating recombination
Glossary
- T-HELPER LYMPHOCYTE CELL
-
A type of lymphocyte that alerts the immune system to produce other cells as a response against a specific infection.
- REVERSE TRANSCRIPTASE
-
An enzyme that is used by retroviruses and retrotransposons to synthesize DNA from an RNA template.
- MAXIMUM LIKELIHOOD METHOD
-
A method that selects the tree that has the highest probability of explaining the sequence data, under a specific model of substitution (changes in the nucleotide or amino-acid sequence).
- VIRULENCE
-
The ability to cause disease by breaking down the protective mechanisms of the host.
- MOLECULAR CLOCK
-
The principle that any gene or protein has a near-constant rate of evolution in all branches of a clade, which means that the amount of sequence divergence between two sequences will be proportional to the amount of time elapsed since their shared ancestor existed.
- FOUNDER EFFECTS
-
A situation in which a new population is founded by a small number of incoming individuals. Similar to a bottleneck, the founder effect severely reduces genetic diversity, increasing the effect of random drift.
- VIRION
-
A mature virus particle, consisting of the genome enclosed in a protein shell and constituting the infectious form of the virus.
- IMMUNE SELECTION
-
The process of natural selection of those virus variants that can successfully evade or manipulate the immune system.
- GENETIC DRIFT
-
Changes in the frequency of a genetic variant in a population owing to chance alone.
- EFFECTIVE POPULATION SIZE
-
(Ne). Formulated by Wright in 1931, Ne reflects the size of an idealized population that would experience drift in the same way as the actual (census) population. Ne can be lower than census population size owing to various factors, including a history of population bottlenecks and reduced recombination.
- FIXATION
-
The increase in the frequency of a genetic variant in a population to 100%.
- GLYCAN SHIELD
-
A continuously evolving pattern of glycosolation of the HIV envelope protein that hinders antibody binding but does not reduce viral fitness.
- CYTOTOXIC T LYMPHOCYTES
-
A type of lymphocyte that can destroy virus-infected cells.
- NONSYNONYMOUS SUBSTITUTION
-
A change in nucleotide sequence that alters the encoded amino acid.
- SYNONYMOUS SUBSTITUTION
-
A nucleotide change in a DNA sequence that does not result in a change in the encoded amino acid.
- SYNCYTIUM
-
A multinucleate cell in which the nuclei are not separated by cell membranes.
- CHEMOKINES
-
Small molecules that have a central role in inflammatory responses and trigger migration and activation of phagocytic cells and lymphocytes.
- NEIGHBOUR-JOINING METHOD
-
An algorithm that uses genetic distances to construct a phylogeny by the sequential addition of taxa.
- BOTTLENECK
-
A severe reduction in population size that causes the loss of genetic variation. The role of random drift is increased, whereas the power of selection is reduced, by bottlenecks.
- HLA HAPLOTYPE
-
The particular pattern of alleles at the human major histocompatibility complex (MHC) loci, which defines which antigens are recognized by T cells.
- ADAPTIVE PEAKS
-
If all morphological variation or all possible phenotypes are considered as a landscape, some will have particularly high fitness (peaks).
- STERILIZING IMMUNITY
-
An immune response whereby the infection is entirely prevented or rapidly cleared, leaving no detectable trace.
- SUPERINFECTION
-
The infection by a second strain of virus in a patient that is already infected.
Rights and permissions
About this article
Cite this article
Rambaut, A., Posada, D., Crandall, K. et al. The causes and consequences of HIV evolution. Nat Rev Genet 5, 52–61 (2004). https://doi.org/10.1038/nrg1246
Issue Date:
DOI: https://doi.org/10.1038/nrg1246
This article is cited by
-
HIV infection
Nature Reviews Disease Primers (2023)
-
A large population sample of African HIV genomes from the 1980s reveals a reduction in subtype D over time associated with propensity for CXCR4 tropism
Retrovirology (2022)
-
Immunoescape of HIV-1 in Env-EL9 CD8 + T cell response restricted by HLA-B*14:02 in a Non progressor who lost twenty-seven years of HIV-1 control
Retrovirology (2022)
-
Fc receptors and the diversity of antibody responses to HIV infection and vaccination
Genes & Immunity (2022)
-
Epidemic spreading under mutually independent intra- and inter-host pathogen evolution
Nature Communications (2022)
