
Key Points
-
Single-strand breaks (SSBs) are the most common lesions arising in cells, and chromosomal single-strand break repair (SSBR) is a rapid and efficient process.
-
In addition to the rapid 'global' SSBR processes that remove SSBs throughout the genome and throughout interphase, there might be S-phase specific processes that operate at replication forks in conjunction with homologous recombination.
-
Two of the proteins that repair damaged DNA termini during global SSBR (tyrosyl-DNA phosphodiesterase 1 and aprataxin) are mutated in the hereditary genetic diseases spinocerebellar ataxia with axonal neuropathy 1 (SCAN1) and ataxia oculomotor apraxia 1 (AOA1), implicating unrepaired SSBs in progressive neurological dysfunction.
-
Whereas post-mitotic cells seem to be dependent on global SSBR for genetic integrity, proliferating cells can additionally use replication-coupled SSBR. This might explain why SCAN1 and AOA1 are not associated with elevated genetic instability and cancer.
Abstract
Hereditary defects in the repair of DNA damage are implicated in a variety of diseases, many of which are typified by neurological dysfunction and/or increased genetic instability and cancer. Of the different types of DNA damage that arise in cells, single-strand breaks (SSBs) are the most common, arising at a frequency of tens of thousands per cell per day from direct attack by intracellular metabolites and from spontaneous DNA decay. Here, the molecular mechanisms and organization of the DNA-repair pathways that remove SSBs are reviewed and the connection between defects in these pathways and hereditary neurodegenerative disease are discussed.
Similar content being viewed by others


Main
Single-strand breaks (SSBs) are discontinuities in one strand of the DNA double helix and are usually accompanied by loss of a single nucleotide and by damaged 5′- and/or 3′-termini at the site of the break. If not repaired rapidly or appropriately, chromosomal SSBs pose a serious threat to genetic stability and cell survival. Consequently, cells have evolved rapid and efficient mechanisms for their repair. The likely importance of SSB repair (SSBR) is highlighted by the observation that two of the proteins involved in this process are mutated in hereditary neurodegenerative disease.
Here, I describe the source and structure of endogenous cellular SSBs, and outline the ways in which these lesions can have an impact on cell fate. I also present our current working models for SSBR and review the association between defects in these processes and human disease.
Source and structure of endogenous SSBs
One of the most common sources of SSBs is oxidative attack by endogenous reactive oxygen species (ROS). In the case of free radicals from hydrogen peroxide (H2O2), which is a physiologically relevant source of ROS, SSBs occur three orders of magnitude more frequently than double-strand breaks (DSBs)1. SSBs can occur directly by disintegration of the oxidized sugar or indirectly during the DNA base-excision repair (BER) of oxidized bases, abasic sites, or bases that are damaged or altered in other ways2,3,4. SSBs can also arise as a result of erroneous or abortive activity of cellular enzymes such as DNA topoisomerase 1 (TOP1). TOP1 creates a 'cleavage complex' intermediate containing a DNA nick in order to relax DNA during transcription and DNA replication5. These intermediates are normally transient and are rapidly resealed by TOP1. However, collision with RNA or DNA polymerases, or close proximity to other types of DNA lesion, can convert cleavage complexes into TOP1-linked SSBs (TOP1–SSBs) or TOP1-linked DSBs (TOP1–DSBs), in which TOP1 is covalently linked to the 3′-terminus of the DNA strand break6. This type of break has become the focus of much attention in recent years, because defects in its repair are associated with the human genetic disease spinocerebellar ataxia with axonal neuropathy 1 (SCAN1)7.
SSBs and cell fate
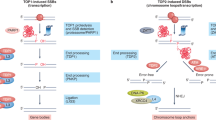
Chromosomal SSBs can have an impact on cell fate in a number of ways if they are not repaired rapidly or appropriately (Fig. 1). The most likely consequence of unrepaired SSBs in proliferating cells is the blockage or collapse of DNA replication forks during the S phase of the cell cycle, possibly leading to the formation of DSBs8,9. Even though cells possess a remarkable ability to accurately repair this type of DSB using homologous recombination (HR) (see below), acute increases in cellular SSB levels might saturate this pathway, leading to genetic instability and/or cell death. In non-proliferating cells, such as post-mitotic neurons, cell death induced by SSBs might involve stalling of RNA polymerases during transcription; this is because SSBs can block RNA polymerase progression in vitro, particularly if they possess damaged termini10,11,12,13. Alternatively, under some physiological situations, high levels of single-strand breakage might induce cell death through excessive activation of the SSB sensor protein poly(ADP-ribose) polymerase 1 (PARP1)14,15. Under these conditions, prolonged activation of PARP1 leads to depletion of cellular NAD+ and ATP and/or release of apoptosis-inducing factor (AIF) from mitochondria. This type of cell death is relevant to a number of pathological conditions involving oxidative stress, including diabetes, arthritis, and post-ischaemic brain or heart damage resulting from stroke or heart attack, respectively.

Single-strand breaks (SSBs) can arise in a variety of ways, including directly from disintegration of deoxyribose, indirectly as normal intermediates of base-excision repair (BER), or as abortive intermediates of topoisomerase 1 (TOP1) activity. If they are not repaired rapidly or appropriately, SSBs can collapse replication forks, block transcription or promote excessive activation of the SSB sensor protein poly(ADP-ribose) polymerase 1 (PARP1). Red circles denote damaged DNA termini. pADPr, poly(ADP-ribose).
Mechanisms of chromosomal SSBR
Although endogenous SSBs arise from a variety of different sources, there is extensive overlap between the enzymes used to remove these breaks. Consequently, the different mechanisms of repairing SSBs from different sources (for example, those arising as normal intermediates of BER versus those arising directly from sugar disintegration) are considered sub-pathways of SSBR. Most SSBs are repaired by a rapid global SSBR process that can be divided into four basic steps: SSB detection, DNA end processing, DNA gap filling and DNA ligation (Fig. 2).

Single strand breaks (SSBs) can arise indirectly during base-excision repair (BER) by enzymatic incision at an apurinic–apyrimidinic (AP) site by AP endonuclease I (APE1) or by the lyase activity of a bifunctional DNA glycosylase. AP sites arise by spontaneous or induced (for example, at oxidized deoxyribose) base loss or by enzymatic excision of damaged (for example, alkylated, oxidized or deaminated) bases. SSBs can also arise directly by reactive oxygen species (ROS)-induced disintegration of oxidized deoxyribose (sugar damage), and during abortive topoisomerase 1 (TOP1) activity involving collision with RNA polymerase (RNAP) or close proximity to other types of DNA lesion, which creates a TOP1-linked SSB (TOP1–SSB). Direct breaks are detected by poly(ADP-ribose) polymerase 1 (PARP1) binding and activation, which promotes rapid access by, and accumulation of, downstream repair factors (see text for details). Poly(ADP-ribose) glycohydrolase (PARG) restores PARP1 to its pre-activated state in preparation for subsequent rounds of SSB detection. Note that SSBs arising during BER or induced by TOP1 might not require PARP1 for detection because these breaks are created in a scheduled fashion by cellular enzymes (APE1 in BER) or might be 'detected' by collision with RNAP (TOP1–SSBs). Once a SSB has been detected, it undergoes end processing. Damaged termini (red circles) that are present at indirect, BER-induced SSBs are repaired by APE1, DNA polymerase (Pol) β, polynucleotide kinase 3′-phosphatase (PNKP) and aprataxin (APTX). Direct, sugar-damage induced SSBs are repaired by APE1, PNKP and APTX. TOP1–SSBs are repaired by tyrosyl-DNA phosphodiesterase 1 (TDP1), which removes TOP1 from the 3′-termini at such breaks, resulting in a 3′-phosphate terminus, which is subsequently repaired by PNKP. PNKP also repairs the 5′-hydroxy termini present at TOP1 breaks. The end-processing mechanism is followed by gap filling. At most SSBs, Pol β inserts the missing nucleotide, this is termed short-patch repair. Under some circumstances (for example, an oxidized deoxyribose phosphate that cannot be repaired by Pol β) gap filling might be extended for ∼2–12 nucleotides (nt) by Pol β, Pol δ and/or Pol ε (Pol δ/ε) during long-patch repair. Note that long-patch repair involves the removal of the damaged 5′-terminus as a flap of two or more displaced nucleotides by flap endonuclease 1 (FEN1), in a reaction stimulated by PARP1 and proliferating cell nuclear antigen (PCNA). Also note that TOP1–SSBs are DNA nicks and therefore might not require a gap-filling step. DNA ligation is the final step in SSB repair. Short-patch repair sites are mainly ligated by DNA ligase 3 (LIG3), and long-patch repair sites are mainly ligated by DNA ligase 1 (LIG1). α,β, 3′-α,β unsaturated aldehyde; Ade, aldehyde; AMP, 5′-AMP; dRP, 5′-deoxyribose phosphate; OH, 5′-hydroxyl; P, 3′-phosphate; PG, 3′-phosphoglycolate.
SSB detection. SBs arising directly from disintegration of oxidized deoxyribose are primarily detected by PARP1, although contributions from other members of the PARP superfamily are possible16,17,18. PARP1 rapidly binds to and is activated by DNA strand breaks, and subsequently modifies itself and other target proteins with branched chains of poly(ADP-ribose) (pADPr) of up to several hundred ADP-ribose units in length. The binding and activity of PARP1 at DNA breaks is transient, because poly(ADP-ribosylated) PARP1 rapidly dissociates from DNA and pADPr is rapidly degraded by poly(ADP-ribose) glycohydrolase (PARG), thereby restoring PARP1 and other poly(ADP-ribosylated) proteins to their de-ribosylated state in preparation for subsequent rounds of SSB detection and signalling19. PARP1 is also activated at SSBs that arise indirectly during BER20,21,22, but it is less clear why such breaks require 'detecting' as they arise as part of a co-ordinated repair process in which DNA intermediates might be passed from one enzyme to another in a molecular relay23,24,25. Perhaps some SSB intermediates become uncoupled from this relay and so require PARP1 to re-engage the repair machinery. Alternatively, perhaps PARP1 fulfils a different role during BER, downstream of SSB detection (see below). It is similarly unclear whether PARP1 is involved in detecting abortive TOP1–SSBs.
A role for PARP activity in accelerating chromosomal SSBR is suggested both by inhibitor studies20,21,26,27,28,29 and by experiments using cells that are selectively depleted or deleted of PARP1 (Refs 30,31,32,33). PARG is also required for rapid rates of SSBR, suggesting that optimal SSBR rates require that levels of pADPr are dynamic and tightly regulated30,34. PARP1 might accelerate SSBR by promoting the focal accumulation or stability of SSBR protein complexes at chromosomal SSBs through interaction with dedicated pADPr-binding motifs35,36,37,38,39,40. Arguably the most important of these is X-ray repair cross-complementing protein 1 (XRCC1), which functions as a molecular scaffold that interacts with, stabilizes, and stimulates multiple enzymatic components of the SSBR process41. Note that the impact of PARP1 on focal accumulation of SSBR proteins at chromosomal SSBs does not necessarily reflect their initial recruitment, which mechanistically might be different and might require recognition of their cognate DNA substrates42. PARP1 might also accelerate chromosomal SSBR by regulating chromatin structure: histone proteins are targets for poly(ADP-ribosylation), and pADPr synthesis can disrupt nucleosomes and can regulate higher-order chromatin compaction43,44,45,46,47. Other possible roles include promoting DNA gap filling during long-patch repair (see below)48,49,50, generating ATP for the final step of DNA ligation51,52, and inhibiting illegitimate recombination events and/or unwanted nucleolytic activity22,53. Finally, because PARP1 is also a transcriptional regulator, it remains possible that it accelerates SSBR rates indirectly, by affecting the level of one or more SSBR proteins50.
DNA end processing. The 3′- and/or 5′-termini of most, if not all, SSBs are 'damaged' and must be restored to conventional 3′-hydroxyl (3′-OH) and 5′-phosphate moieties for gap filling and DNA ligation to occur. This is the most enzymatically diverse step of SSBR, as indicated by the large number of enzymes that are available for this process (Fig. 2). This, in turn, reflects the variety of damaged termini that can arise (Fig. 3). End processing is a crucial stage of SSBR because DNA breaks with abnormal termini are particularly cytotoxic. For example, it is the removal of 5′-deoxyribose phosphate (dRP) residues by DNA polymerase (Pol) β that accounts for its importance in maintaining cellular resistance to DNA base damage, rather than its role as a DNA polymerase during DNA gap filling (see below)54. In addition, over-expression of DNA glycosylase enzymes that initiate BER can actually increase the sensitivity of cells to DNA base damage, and their deletion can decrease this sensitivity, most probably because of changes in steady-state levels of SSB intermediates55,56,57. The cytotoxicity of damaged termini might in part reflect a greater ability to block progression of RNA polymerases, and/or difficulties encountered by cells in dealing with blocked termini at collapsed replication forks.

For each type of single-strand break (SSB) the mechanism of damage is summarized, with the causative agent(s) in parentheses. Ade, aldehyde: AP, apurinic–apyrimidinic; APE1, AP endonuclease I; APTX, aprataxin; BER, base-excision repair; dRP, 5′-deoxyribose phosphate; FEN1, flap endonuclease 1; LPR, long-patch repair; P, phosphate; PNKP, polynucleotide kinase 3′-phosphatase; Pol β, DNA polymerase β; ROS, reactive oxygen species; TDP1, tyrosyl-DNA phosphodiesterase 1; TOP1, topoisomerase 1.
The common types of damaged termini and the principal SSBR enzymes involved in their repair are summarized above (Figs 2,3), but several aspects merit further attention. 3′-phosphate and 3′-phosphoglycolate termini are noteworthy because together they constitute the majority of direct SSBs induced by ROS, and because 3′-phosphate is a major substrate for polynucleotide kinase 3′-phosphatase (PNKP)58,59,60 and 3′-phosphoglycolate for apurinic–apyrimidinic (AP) endonuclease I (APE1; also known as APEX1)61,62,63,64,65. By contrast, SSBs arising indirectly during BER typically harbour 5′-dRP termini created by cleavage of abasic sites by APE1. As discussed above, 5′-dRP termini are substrates for the AP lyase activity of Pol β54,66. However, if Pol β cannot remove the 5′-dRP moiety (for example, if the dRP is oxidized2,67) then the 5′-terminus can be displaced during long-patch gap filling and removed by flap endonuclease 1 (FEN1) as a single-strand flap. It is worth noting that SSBs that arise during BER might alternatively possess a 3′-phosphate or 3′-α,β-unsaturated aldehyde terminus, rather than a 5′-dRP terminus, if they are created by cleavage of an abasic site through DNA glycosylase AP lyase activity rather than through APE1 activity. These termini are substrates for PNKP and APE1, respectively63,64,68.
Two other types of damaged terminus that warrant additional attention are 3′- and 5′-termini linked covalently to TOP1 and AMP, respectively. TOP1–SSBs arise from abortive TOP1 activity and are processed by tyrosyl-DNA phosphodiesterase 1 (TDP1)69,70. 5′-AMP–SSBs, at which AMP is covalently linked to 5′-phosphate through a pyrophosphate bond, arise from abortive DNA-ligase activity at existing SSBs and are processed by aprataxin (APTX)71,72. Although the occurrence of 5′-AMP termini in vivo remains to be demonstrated, it is intriguing that both TDP1 and APTX seem to process SSBs that arise as a result of failed or abortive activity of endogenous enzymatic activities, particularly because both TDP1 and APTX are mutated in hereditary neurodegenerative diseases (see below).
Finally, it is important to note that XRCC1 has a particularly important role during DNA end processing. XRCC1 directly interacts with PNKP58,73, APTX74,75,76,77, and Pol β39,78, and indirectly, through DNA ligase IIIα, with TDP1 (Refs 7,79). The repair of 5′-hydroxyl and/or 3′-phosphate termini is rate limiting for SSBR in XRCC1-mutant Chinese hamster ovary (CHO) cells and cell extracts58,80. In the case of PNKP, it is clear that the interaction with XRCC1 stimulates both DNA kinase and DNA phosphatase activity in vitro, possibly by increasing damage discrimination by PNKP and by displacing the enzyme from its reaction product58,73,81. The interaction of XRCC1 with PNKP, APTX and Pol β also promotes the accumulation of these enzymes at oxidative chromosomal breaks induced by H2O2 and ultraviolet A (UVA) laser in vivo37,73,82.
DNA gap filling. After damaged 3′-termini at SSBs have been restored to their conventional hydroxyl configuration, gap filling can occur (Fig. 2). This often involves insertion of the single nucleotide that is missing (short-patch repair), but at some SSBs gap filling might continue for two or more nucleotides, with FEN1 removing the displaced 5′-residue either one at a time or as a single-stranded flap (long-patch repair)47 (Fig. 1). Early studies using permeabilized cells suggest that multiple DNA polymerases, including Pol β, are involved in the repair of SSBs, with the choice of DNA polymerase influenced by the source and type of SSB. In vitro studies using DNA molecules with defined types of break similarly implicate Pol β in gap filling, both at direct SSBs arising from oxidative damage and at indirect SSBs during BER, and show that Pol δ and/or Pol ε (Pol δ/ε) can also conduct this role47. Direct measurements of chromosomal repair rates in mouse embryonic fibroblasts (MEFs) confirm that Pol β is required for rapid repair of methyl methanesulphonate (MMS)-induced SSBs during BER83,84. By contrast, a requirement for Pol β for rapid SSBR rates at oxidative SSBs has not been observed83. This might reflect the availability of an alternative short-patch DNA polymerases such as Pol λ or Pol ι, which possess dRP lyase activity, for repair of oxidative SSBs, or alternatively the use of long-patch DNA polymerases85,86,87,88. A number of accessory proteins might also be important for gap filling during SSBR. For example, PARP1 and FEN1 stimulate long-patch gap filling by Pol β in vitro49,89, and the RFC–PCNA complex — made up of the replication factor C (RFC) clamp loader and the proliferating cell nuclear antigen (PCNA) clamp — might promote long-patch gap filling by Pol δ/ε90,91,92. In addition, XRCC1 interacts with PCNA93,94 and Pol β39,78 and is important for the accumulation of RFC, PCNA and Pol β at direct SSBs induced by UVA laser damage in cells37,90.
DNA ligation. The final step of SSBR is DNA ligation (Fig. 2). Three human DNA ligase genes have been identified (LIG1, LIG3, and LIG4), which encode five different polypeptides (see Ref. 95 for a recent review). LIG3 encodes three polypeptides, denoted DNA ligase IIIα (LIG3α), DNA ligase IIIβ (LIG3β), and a mitochondrial isoform (mtLIG3). Of these, LIG1, LIG3α, and mtLIG3 are implicated in SSBR. mtLIG3 interacts with Pol γ and is required for integrity of the mitochondrial genome96,97, whereas LIG1 and LIG3α seem to be the enzymes of choice during nuclear long-patch and short-patch SSBR, respectively47. LIG3α is stable and active as a recombinant protein in vitro, but the nuclear enzyme requires constitutive interaction with XRCC1 for stability and for accumulation at sites of oxidative chromosome damage98,99. By contrast, LIG1 requires interaction with PCNA for accumulation at such sites98, although one might also expect a dependency on XRCC1 because XRCC1 is required for PCNA accumulation at sites of oxidative damage37. It is important to note that our understanding of the requirement for specific DNA ligases during SSBR is derived largely from in vitro assays, and that the level of functional overlap between LIG1 and LIG3α during SSBR in living cells remains to be determined.
All of the proteins encoded by LIG3 possess an amino-terminal zinc finger (ZNF) homologous to the two that are present in PARP1 (Ref. 100). The LIG3 ZNF is largely dispensable for binding and ligation of simple nicked DNA substrates in vitro, although a weak effect (∼2-fold) on these activities is observed under some experimental conditions39,95,101,102,103. The dispensability of the ZNF for simple-nick ligation most probably reflects the presence of an efficient nick-binding activity within the conserved catalytic core of the enzyme. However, the ZNF has a major impact on binding and ligation of DNA breaks that are located near to unusual secondary structure or to other intermediates of DNA repair, suggesting that one function of the ZNF is to broaden the range of substrates targeted by LIG3α95,101,102. The ZNF might thus be important for repair of chromosomal breaks in repetitive regions of the nuclear and/or mitochondrial genome, or at multiply-damaged sites or clustered lesions. Intriguingly, the ZNF is required for efficient DSB end joining even in the absence of proximal secondary structure, suggesting that this domain might have a major role in alternative non-homologous end joining, in which LIG3 is believed to participate104,105.
Organization and regulation of SSBR
As indicated in the text above and in Fig. 2, each of the four basic steps of SSBR involves multiple protein–protein interactions. Arguably the most important of these are the interactions between XRCC1 and enzymatic components of the repair process. XRCC1 accelerates the overall process of SSBR, which it does by stabilizing (for example, LIG3α)99,106 and/or stimulating (for example, PNKP)58,81 its protein partners. XRCC1 also facilitates the accumulation of its protein partners at sites of chromosomal DNA damage, although it remains to be determined whether or not this reflects the initial recruitment of these proteins to DNA breaks, which mechanistically might occur by a different process.
It is not obvious why higher eukaryotes use XRCC1 to accelerate global rates of SSBR — lower eukaryotes such as yeast do not need this protein. One possibility is that this factor has arisen in response to dramatic increases in genome size. Because the frequency of SSBs arising per cell per day is intrinsically related to DNA content, it is possible that higher eukaryotes require faster global rates of SSBR to maintain their steady-state level of SSBs below a certain threshold. The organism with the smallest genome that contains XRCC1 is the social amoeba Dictyostelium, the genome of which is three times larger (34 Mb) than that of budding yeast, which lacks a recognizable XRCC1.
It remains to be established whether SSBR proteins are organized into one or more constitutive protein complexes or whether SSBR is organized into a sequential series of rapid but transient protein–protein interactions at SSBs. Both are most probably true. For example, the interactions between XRCC1 and PNKP73, APTX74,76, and aprataxin and PNK-like factor (APLF)107,108 are largely constitutive because they are mediated by forkhead-associated (FHA) domain-mediated interactions with threonine and/or serine residues that are phosphorylated by the constitutively active protein kinase, CK2. Similarly, levels of LIG3α are reduced ∼5-fold in cells lacking XRCC1 or in those that express a form of XRCC1 that cannot bind LIG3a, suggesting that at least 80% of cellular LIG3α is constitutively bound to XRCC1 (Ref. 99). Moreover, the level of these complexes increases little, if at all, in response to acute increases in SSB levels. By contrast, the interaction between XRCC1 and PARP1 clearly increases after DNA damage, consistent with the preferential interaction of XRCC1 with auto-modified PARP1 (Refs 40,48,109). This interaction thus enables the accumulation of XRCC1, and, consequently, those SSBR enzymes that interact with this scaffold protein, at chromosomal breaks.
Although SSBR is a housekeeping process, cellular SSBR capacity might still be regulated. For example, some cell types moderately increase the levels of SSBR proteins (for example, XRCC1 and APE1) in response to genotoxins, oxidative stress, or ischemia injury in the brain, probably by changes in gene expression110,111,112. SSBR enzymes might also be regulated post-translationally113 or even proteolytically as indicated by reduced steady-state levels of LIG3α in cells that lack XRCC1 (Ref. 99). This might reflect a general cellular mechanism for removing excess (that is, unbound) amounts of specific subunits of a multi-protein complex, thereby maintaining the stoichiometry of those subunits. This might be particularly important for proteins that could engage in spurious or non-specific DNA metabolic activity if not chaperoned by a protein partner. Alternatively, the degradation of SSBR proteins by the proteosome might reflect the removal of those SSBR proteins that are not actively engaged in chromosomal SSBR114.
SSBR and the cell cycle
The SSBR processes described above most probably operate throughout the genome and throughout the cell cycle to rapidly detect and remove the majority of chromosomal SSBs, and are therefore denoted 'global SSBR'. However, it is possible that cellular SSBR capacity and mechanism is regulated to some extent depending on cell-cycle status. For example, it has been suggested that XRCC1 expression is regulated by FOXM1 and E2F1 — transcription factors that regulate a variety of genes required for DNA replication and proliferation110,115. XRCC1 has also been reported to be downregulated in terminally differentiated muscle cells, although how applicable this observation is to other non-proliferating cells remains to be determined116. There is also mounting evidence that S-phase cells possess one or more additional SSBR processes for repairing SSBs, possibly once they are encountered by the DNA replication machinery93,117,118,119,120 (Fig. 4). The molecular detail of this hypothetical process (which we have previously denoted replication-coupled SSBR, RC-SSBR)41,121 is unclear, but is probably similar to the long-patch subpathway of global SSBR described in Fig. 2 because many of the enzymes involved in the long-patch repair process (for example, Pol δ/ε, PCNA, LIG1 and FEN1) are core components of the replication machinery. RC-SSBR is likely to operate in conjunction with HR because SSBs that are encountered by a replication fork can be converted into DSBs8,9 (Fig. 4b). Consequently, RC-SSBR and HR might function together as an effective back-up mechanism that ensures that SSBs that escape global repair do not irrevocably block DNA replication. RC-SSBR might also involve some global SSBR factors, such as XRCC1 and possibly components of the global SSBR end-processing machinery. However, a key feature of this process is likely to be the availability of structure-specific nucleases that can remove a variety of damaged termini (Fig. 4). As proposed previously122,123 and discussed below, this feature of RC-SSBR and HR might in part account for the lack of genetic instability and cancer in human genetic diseases such as ataxia-oculomotor apraxia 1 (AOA1) and SCAN1 (see below) in which global SSBR proteins are mutated. Thus, whereas global SSBR might be the primary determinant of genetic integrity and survival in non-cycling cells in response to SSBs, RC-SSBR (in conjunction with HR) might be the major determinant of these end-points in proliferating cells.

a | A replication fork (Rep) approaches a single-strand break (SSB) with damaged termini (red lines denote nascent strands, red ovals denote damaged termini). Note that in this example the SSB is in the leading-strand template, but similar outcomes are applicable to a SSB in the lagging-strand template. b | RC-SSBR and template switching. The replication fork collapses by replication 'run-off' creating a one-ended double-strand break (DSB; green box) in one sister chromatid and leaving a residual SSB in the other (blue box). Note that the SSB shown here possesses a single-strand gap but alternatively might possess a nick or a single-strand flap, depending on the proximity of the terminus of the adjacent nascent strand (in the example depicted this is the 5′-terminus of the nearest Okazaki fragment). The 5′-terminus of the DSB is processed and resected by CtIP together with MRN in preparation for template switching. End processing of the SSB terminus is conducted by global SSBR factors (TDP1, PNKP, APTX and DNA polymerase (Pol) β) or alternatively by structure-specific nucleases such as ERCC1–XPF at 3′-termini (as shown here), FEN1 at 5′-flapped termini, or the MRN complex at 5′- and possibly 3′-termini. DNA gap filling during RC-SSBR involves PCNA, the replicative polymerases Pol δ and/or Pol ε (Pol δ/ε) and associated factors, and DNA ligation by LIG1. Following RC-SSBR, the replication fork is reformed by RAD51-mediated template switching. Note that, following RC-SSBR and homologous recombination (HR), proliferating cells might be more able to tolerate loss of certain global SSBR end-processing factors (for example, TDP1 or APTX). Also note PARP1 and XRCC1 might fulfil an important but undefined role during RC-SSBR. c | The SSB is converted into a two-ended DSB by re-initiation of DNA synthesis downstream of the SSB or by approach of a replication fork from the other direction. Note that in this scenario, RC-SSBR is not required because the two-sided DSB can be repaired by HR (as shown) or by non-homologous end joining DSB repair (DSBR) pathways. APTX, aprataxin; CtIP, CTBP-interacting protein; ERCC1, excision repair cross-complementing rodent repair deficiency, complementation group 1; FEN1, flap endonuclease 1; LIG1, DNA ligase 1; MRN, Mre11–Rad50–Nbs1; PARP1, poly(ADP-ribose) polymerase 1; PCNA, proliferating cell nuclear antigen; PNKP, polynucleotide kinase 3′-phosphatase; TDP1, tyrosyl-DNA phosphodiesterase 1; XPF, xeroderma pigmentosum, complementation group F (also known as ERCC4); XRCC1, X-ray repair cross-complementing protein 1.
SSBR and hereditary genetic disease
Ataxia-oculomotor apraxia 1. AOA1 is an autosomal recessive spinocerebellar ataxia syndrome that resembles Friedreich ataxia and ataxia-telangiectasia (AT) neurologically, but which lacks non-neurological features such as immunodeficiency and telangiectasia124. Initial studies also suggested that AOA1 lacks the overt cellular and chromosomal sensitivity to ionizing radiation that accompanies AT125. Although subsequent studies have revealed considerable clinical variation within AOA1, characteristic features of AOA1 seem to be variable onset (1–16 years), cerebellar atrophy and ataxia (uncoordinated movement and gait), late axonal peripheral neuropathy, and oculomotor apraxia (limited eye movement on command). In addition, a number of other features appear in AOA1, including cognitive impairment, hypercholesterolaemia, hypoalbuminaemia and involuntary movements (choreoathetosis and dystonia). In a large study covering approximately half of the Portuguese population, AOA1 accounted for ∼21% (22 patients in 11 families) of 107 individuals with recessive spinocerebellar ataxia, a frequency second only to Friedreich ataxia (38%)126.
Five of the families included in the study described above were subsequently linked to a locus designated as AOA1 (Ref. 127), along with five Japanese patients with early-onset cerebellar ataxia with hypoalbuminaemia (EOCA-HA). A recent study of 14 French, Italian and Algerian patients with AOA1 from nine families supports the likelihood that, although cerebellar atrophy, ataxia, and sensorimotor axonal neuropathy are common to most patients with AOA1, the presence and severity of other features is more variable (∼85% in the case of oculomotor apraxia)128. Variation in clinical impact and/or severity of AOA1 is further suggested by reported clinical overlap with other neurological diseases and conditions, such as multiple system atrophy (MSA) and ataxia with coenzyme Q10 (coQ10) deficiency129,130,131. Intriguingly, Baba et al. reported that two patients with clinical features resembling the cerebellar subtype of MSA (MSA-C) harboured novel heterozygous mutations in APTX (Fig. 5; mutations K153E and S242N). AOA1 accounts for 5–10% of all autosomal recessive cerebellar ataxias and has a variable age of onset (typically 1–18 years), with a mean of ∼5 years127,128. Unlike AT cells, AOA1 cells are only mildly sensitive, if at all, to ionizing radiation and other genotoxins, and exhibit normal cell-cycle checkpoint control and chromosome stability following ionizing radiation74. Consistent with these observations, elevated cancer incidence has not been reported in patients with AOA1.

Known domains are indicated, along with the location of established direct interactions with X-ray repair cross-complementing protein 1 (XRCC1) and XRCC4. The red box denotes the position of the catalytic histidine triad (HIT). The black box denotes the additional 14 amino acids that are present at the amino-terminal in the longest (356 aa) alternative splice variant. Asterisks denote mutations that are associated with particularly late-onset and/or mild clinical presentation. Parentheses denote the countries in which each mutation has been reported. Δ denotes deletion, followed by its location. fs, denotes frameshift: the nucleotide mutations associated with the frameshifts are; V230fs, 689insT; N280fs, 840delT; A292fs, splice-site mutation 875-1G>A (IVS7-1 G>A). Citations for mutations are omitted for brevity. DSBR, double-strand break repair; FHA, forkhead associated; ZNF, zinc finger; NLS, nuclear localization signal; SSBR, single-strand break repair.
In late 2001, the gene that is mutated in AOA1 was identified and designated APTX132,133. The protein product of APTX, aprataxin, was initially characterized as a polypeptide of 342 residues. However, the longest form of this polypeptide might include an extra 14 amino acids at the amino terminus, owing to alternative splicing at the 5′ end of the gene134. The prevalence and function of the extra sequence is unclear, but as it is located immediately upstream of a putative mitochondrial targeting sequence (K.W.C., unpublished observations) it might influence the sub-cellular localization of APTX.
APTX contains a central histidine triad (HIT) domain and is a member of the HIT-domain superfamily of nucleotide hydrolases and/or transferases. The amino terminus of APTX exhibits homology to PNKP133, and like PNKP encodes a divergent FHA domain122. Two splice variants of APTX were originally reported, one of which encoded a protein lacking the FHA domain. The FHA domains of both APTX and PNKP facilitate constitutive interactions with CK2-phosphorylated XRCC1 (Refs 73,74,75,76,77, 122, 135). It is noteworthy that a third member of this divergent FHA-domain family has been identified (APLF, also known as PALF and Xip1) and shown to bind CK2-phosphorylated XRCC1 (Refs 107, 108, 136). In fact, all three FHA-domain proteins are also sequestered into the DSB repair (DSBR) machinery through FHA domain-mediated interaction with CK2-phosphorylated XRCC4 (Refs 74, 108, 137). It is thus highly likely that APTX, PNKP, and APLF have roles both in SSBR and DSBR. APTX also associates with PARP1 and tumour suppressor protein p53, and with the nucleolar proteins nucleolin, nucleophosmin and upstream-binding factor 1 (UBF1)75,138. The association and partial co-localization of APTX with nucleolar proteins is also mediated by the FHA domain, although whether these associations are direct or indirect remains to be determined. Nevertheless, the association of APTX with nucleolar proteins might indicate that SSBR and/or DSBR is particularly important at sites of high transcriptional activity, perhaps to prevent SSBs from blocking gene expression.
Most of the mutations identified in AOA1 so far are located within the HIT domain or just upstream of the C-terminal ZNF motif, consistent with a crucial requirement for the HIT domain for normal neurological function (Fig. 5). Many of these mutations greatly reduce the stability and/or cellular level of APTX and might therefore be functional null alleles75,139,140. Some mutations seem to be associated with later disease onset and/or milder clinical features133,141,142,143, which in some cases seem to have less effect on APTX stability and/or activity139.
On the basis of sequence comparisons and substrate specificity, APTX seems to be a discrete branch of the HIT-domain superfamily144. Indeed, although APTX can hydrolyse substrates typical of either the fragile histidine triad (Fhit) or the histidine triad nucleotide-binding protein (Hint) branch of HIT-domain proteins, releasing AMP from diadenosine tetraphosphate or AMP-lysine, respectively, its catalytic activity is low (Kcat < 0.03 s−1)139,144. APTX has also been reported to process 3′-phosphate and 3′-phosphoglycolate termini, raising the possibility that it is an end-processing factor. However, the activity of APTX on such substrates is also low (Kcat ∼0.0003–0.003 s-1). A more likely physiological substrate for APTX are 5′-AMP termini71,72. DNA strand breaks in which the 5′-terminus is linked to AMP are normal intermediates of DNA ligation, but if they arise before 3′-DNA end processing has occurred ligation is inhibited. APTX can remove AMP from the 5′-terminus of DNA breaks at such 'abortive' DNA-ligation events, and is thus a DNA deadenylase that can 'proofread' the DNA ligase reaction (see Ref. 145 for a recent review). In addition to the HIT domain, the C-terminal ZNF is important for APTX activity on 5′-AMP, most probably to increase the affinity and/or specificity of APTX for 5′-AMP substrates72. The ability to process 5′-AMP termini is an elegant activity, although it remains to be determined whether this type of terminus arises in vivo.
Spinocerebellar ataxia with axonal neuropathy 1 (SCAN1). Takashima et al. identified an autosomal recessive ataxia that they denoted SCAN1 (Ref. 146). Like AOA1, patients with SCAN1 lack chromosomal instability and cancer predisposition but exhibit cerebellar atrophy and peripheral neuropathy. Mild hypercholesterolaemia and hypoalbuminaemia are similarly present. SCAN1 has a later age of onset (∼15 years) than AOA1 and does not involve cognitive decline, on the basis of the limited number of patients available to date. SCAN1 is associated with a mutation in TDP1, an end-processing factor that repairs TOP1–SSBs. The level of TOP1 strand breaks is induced exogenously by treatment with camptothecin, which increases the half-life of TOP1 cleavage complexes, and endogenously by 'trapping' of cleavage complexes by other DNA lesions147,148,149,150,151,152. The second observation might explain why TOP1-dependent strand breaks are also induced by H2O2 and ionizing radiation153,154,155,156. It is worth noting, however, that although TOP1-linked termini are likely to be the primary physiological substrate of TDP1, this protein might also process other types of 3′-termini, and possibly 5′-termini, particularly at DSBs157,158,159,160.
To date, SCAN1 is restricted to nine patients from a single Saudi Arabian family, three of which have been examined in detail146. Affected individuals possess a common homozygous mutation (H493R) within the second HKD motif of the TDP1 active site (Fig. 6). The ability of TDP1 to remove TOP1 peptide and create 3′-phosphate termini is reduced ∼25-fold in experiments using recombinant mutated TDP1 and ∼100-fold in experiments using SCAN1-cell extracts7,161. The greater defect in SCAN1 extracts is likely to reflect the 2–3-fold reduction in TDP1 protein levels in these cells, owing presumably to instability of the mutant protein. Intriguingly, the mutant TDP1 protein can partially process TOP1-linked termini, removing TOP1 while remaining trapped covalently on the 3′-terminus161,162. The extent to which mutant TDP1 converts TOP1-breaks into TDP1-breaks is concentration dependent, and the relative proportion of unprocessed (and therefore TOP1-associated) versus partially processed (and therefore TDP1-associated) termini that accumulate in SCAN1 cells is unclear. Consequently, the relative contribution of these termini to the neurological defects in SCAN1 is unknown. It is worth noting, however, that in Tdp1−/− mice, which can accumulate TOP1-linked but not TDP1-linked termini, there is an age-dependent decrease in cerebellar size, consistent with an effect of TOP1-linked termini on neurological function163.

Known domains are indicated, along with the location of a direct interaction with ligase 3 (LIG3). The red boxes denote the position of the two HKD catalytic active sites. The H493R mutation in SCAN1 is indicated in red. NTD, N-terminal domain; PDD, phosphodiesterase domain.
SSBs and/or DSBs: causes of SCAN1 and AOA1?
Both SCAN1 lymphoblastoid cells and Tdp1−/− primary mouse post-mitotic neurons display significantly reduced global rates of chromosomal SSBR7,156,163. This is true not only following exposure to camptothecin and ionizing radiation, but also following treatment with H2O2. Because H2O2 is a physiologically relevant oxidizing agent, these observations provide compelling support for the idea that SCAN1 neurons might possess higher steady-state levels of this lesion. By contrast, neither non-replicating SCAN1 lymphoblastoid cells nor Tdp1−/− primary mouse post-mitotic neurons display a measurable defect in DSBR following treatment with camptothecin, ionizing radiation, or H2O27,156,163. This might indicate that the frequency of TDP1 DSB substrates is too rare to detect by existing cell-based assays, or that the role of TDP1 is highly redundant during DSBR in non-cycling cells. The situation is more complex in AOA1, however. APTX constitutively interacts with XRCC1 and XRCC4, and AOA1 cells exhibit mild to moderate hypersensitivity to genotoxins, consistent with a role for APTX in both SSBR and DSBR74,75. However, AOA1 cells lack a measurable defect in DSBR, and the evidence for a defect in SSBR is conflicting75,82,164. Thus, although it is likely that APTX is involved in the repair of both SSBs and DSBs, the relationship between this disease and either or both of these lesions remains to be established.
SSBs and cancer
One of the most striking features of SCAN1 and AOA1 is the absence of increased genetic instability and cancer. At first glance this seems surprising, given that unrepaired SSBs can result in potentially clastogenic DSBs during DNA replication. As we have proposed previously7,122,165 and discussed above, this might in part reflect that proliferating cells possess alternative end-processing factors (for example, the structure-specific nucleases ERCC1–XPF, FEN1 and the MRN (Mre11–Rad50–Nbs1) complex) that efficiently and accurately remove DNA strand breaks during DNA replication (Fig. 4). Consistent with this idea, lymphoblastoid cells from patients with SCAN1 have a 3-fold increased frequency of 'spontaneous' sister-chromatid exchange, a hallmark of HR7. In short, it is likely that the roles of TDP1 and APTX are redundant in proliferating cells, at physiological levels of SSBs at least. Genetic instability in SCAN1 and AOA1 might be further limited by committing to apoptosis those few cells in which a strand break avoids repair and/or is involved in a genetic rearrangement. These arguments do not exclude an effect of SSBR on cancer frequency in other circumstances, however. Mutations in proteins with a role in RC-SSBR, or with a more extensive involvement in global SSBR such that the steady-state levels of SSBs is elevated above that which HR can tolerate, might have an impact on genetic instability. Although many such mutations are likely to be embryonic lethal because they are incompatible with rapid cell proliferation during embryonic development, hypomorphic or somatic mutations in such genes could affect cancer incidence. For example, the possible correlation between cancer incidence and a number of XRCC1 polymorphisms is the focus of intense epidemiological investigation, and mutations in Pol β have been identified in ∼30% of human tumours, some of which can affect polymerase fidelity and/or activity during SSBR166.
SSBs and neurodegeneration
The existence of dedicated pathways for dealing with unrepaired SSBs during S phase might explain why SCAN1 and AOA1 are not associated with measurably increased genetic instability and cancer. However, this cannot explain why the impact of these diseases on post-mitotic cells is largely restricted to the nervous system. One possibility is that neurons might be more dependent on TDP1 and APTX for DNA end processing than are other post-mitotic cells, owing to a more limited availability of alternative end-processing factors. Another factor we have previously postulated is the high level of oxidative stress encountered by the nervous system, which consumes ∼20% of inhaled oxygen and possesses low levels of antioxidant enzymes122,165,167. In addition, there is a high transcriptional demand in post-mitotic neurons, which might further increase the dependency of these cells on SSBR. Finally, the limited regenerative capacity of neurons, compared with other non-cycling cell types that are more readily replaced by precursors, might render this tissue particularly sensitive to cell dysfunction or loss. Further analyses of mouse models of these diseases will hopefully shed light on these crucial issues.
Conclusions and future questions
SSBs are the most common lesions arising in cells, and chromosomal SSBR is a rapid and efficient process. Most breaks are removed within minutes by a global pathway that is accelerated by the SSB sensor protein PARP1 and the molecular scaffold protein XRCC1. Two of the proteins that are implicated in DNA end-processing during global SSBR, TDP1 and APTX, are mutated in the hereditary genetic diseases SCAN1 and AOA1, respectively, implicating unrepaired SSBs in progressive neurological dysfunction. Whereas post-mitotic cells seem to be dependent on global SSBR for genetic integrity, proliferating cells might possess an additional SSBR pathway that operates at replication forks in conjunction with HR. This might allow proliferating cells to tolerate SSBs that escape global SSBR, at physiological levels of damage at least, perhaps explaining why SCAN1 and AOA1 are not associated with elevated genetic instability and cancer. Many questions remain, however. For example, do unrepaired DSBs contribute to these diseases, or are they strictly SSBR-defective syndromes? What is the nature of the damaged DNA termini that lead to neuronal dysfunction, and can we detect their accumulation in vivo? How do unrepaired SSBs lead to neuronal dysfunction, and why are these diseases selectively associated with defects in specific regions of the nervous system? Last, but not least, are defects in SSBR associated with other neurological disorders in which oxidative stress is an etiological factor (for example, Parkinson disease and Alzheimer disease), and do SSBs contribute to neurological decline during normal human ageing?
References
Bradley, M. O. & Kohn, K. W. X-ray induced DNA double strand break production and repair in mammalian cells as measured by neutral filter elution. Nucleic Acids Res. 7, 793–804 (1979).
Demple, B. & DeMott, M. S. Dynamics and diversions in base excision DNA repair of oxidized abasic lesions. Oncogene 21, 8926–8934 (2002).
Hegde, M. L., Hazra, T. K. & Mitra, S. Early steps in the DNA base excision/single-strand interruption repair pathway in mammalian cells. Cell Res. 18, 27–47 (2008).
Pogozelski, W. K. & Tullius, T. D. Oxidative strand scission of nucleic acids: routes initiated by hydrogen abstraction from the sugar moiety. Chem. Rev. 98, 1089–1108 (1998).
Wang, J. C. Cellular roles of DNA topoisomerases: a molecular perspective. Nature Rev. Mol. Cell Biol. 3, 430–440 (2002).
Pommier, Y. et al. Repair of and checkpoint response to topoisomerase I-mediated DNA damage. Mutat. Res. 532, 173–203 (2003).
El-Khamisy, S. F. et al. Defective DNA single-strand break repair in spinocerebellar ataxia with axonal neuropathy-1. Nature 434, 108–113 (2005). This paper provides the first direct connection between defects in SSBR and neurological disease.
Kouzminova, E. A. & Kuzminov, A. Fragmentation of replicating chromosomes triggered by uracil in DNA. J. Mol. Biol. 355, 20–33 (2006).
Kuzminov, A. Single-strand interruptions in replicating chromosomes cause double-strand breaks. Proc. Natl Acad. Sci. USA 98, 8241–8246 (2001). These authors provide supporting evidence for the concept that unrepaired SSBs can lead to DSBs during DNA replication.
Bendixen, C., Thomsen, B., Alsner, J. & Westergaard, O. Camptothecin-stabilized topoisomerase I-DNA adducts cause premature termination of transcription. Biochemistry 29, 5613–5619 (1990).
Zhou, W. & Doetsch, P. W. Effects of abasic sites and DNA single-strand breaks on prokaryotic RNA polymerases. Proc. Natl Acad. Sci. USA 90, 6601–6605 (1993). This paper, together with references 12–13, highlights the seminal concept that SSBs can block transcription.
Zhou, W. & Doetsch, P. W. Transcription bypass or blockage at single-strand breaks on the DNA template strand: effect of different 3′ and 5′ flanking groups on the T7 RNA polymerase elongation complex. Biochemistry 33, 14926–14934 (1994).
Kathe, S. D., Shen, G. P. & Wallace, S. S. Single-stranded breaks in DNA but not oxidative DNA base damages block transcriptional elongation by RNA polymerase II in HeLa cell nuclear extracts. J. Biol. Chem. 279, 18511–18520 (2004).
Heeres, J. T. & Hergenrother, P. J. Poly(ADP-ribose) makes a date with death. Curr. Opin. Chem. Biol. 11, 644–653 (2007).
Moroni, F. Poly(ADP-ribose) polymerase 1 (PARP-1) and postischemic brain damage. Curr. Opin. Pharmacol. 8, 96–103 (2008).
D'Amours, D., Desnoyers, S., D'Silva, I. & Poirier, G. G. Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem. J. 342, 249–268 (1999).
Ame, J. C., Spenlehauer, C. & de Murcia, G. The PARP superfamily. Bioessays 26, 882–893 (2004).
Kim, M. Y., Zhang, T. & Kraus, W. L. Poly(ADP-ribosyl)ation by PARP-1: 'PAR-laying' NAD+ into a nuclear signal. Genes Dev. 19, 1951–1967 (2005).
Davidovic, L., Vodenicharov, M., Affar, E. B. & Poirier, G. G. Importance of poly(ADP-ribose) glycohydrolase in the control of poly(ADP-ribose) metabolism. Exp. Cell Res. 268, 7–13 (2001).
Morgan, W. F. & Cleaver, J. E. Effect of 3-aminobenzamide on the rate of ligation during repair of alkylated DNA in human fibroblasts. Cancer Res. 43, 3104–3107 (1983).
Durkacz, B. W., Omidiji, O., Gray, D. A. & Shall, S. (ADP-ribose)n participates in DNA excision repair. Nature 283, 593–596 (1980).
Parsons, J. L., Dianova, II, Allinson, S. L. & Dianov, G. L. Poly(ADP-ribose) polymerase-1 protects excessive DNA strand breaks from deterioration during repair in human cell extracts. Febs J. 272, 2012–2021 (2005).
Mol, C. D., Izumi, T., Mitra, S. & Tainer, J. A. DNA-bound structures and mutants reveal abasic DNA binding by APE1 DNA repair and coordination. Nature 403, 451–456 (2000).
Rice, P. A. Holding damaged DNA together. Nature Struct. Biol. 6, 805–806 (1999). References 23 and 24 describe a conceptual framework for the organization of BER.
Wilson, S. H. & Kunkel, T. A. Passing the baton in base excision repair. Nature Struct. Biol. 7, 176–178 (2000).
Durkacz, B. W., Shall, S. & Irwin, J. The effect of inhibition of (ADP-ribose)n biosynthesis on DNA repair assayed by the nucleoid technique. Eur. J. Biochem. 121, 65–69 (1981).
James, M. R. & Lehmann, A. R. Role of poly(adenosine diphosphate ribose) in deoxyribonucleic acid repair in human fibroblasts. Biochemistry 21, 4007–4013 (1982).
Lehmann, A. R. & Broughton, B. C. Poly(ADP-ribosylation) reduces the steady-state level of breaks in DNA following treatment of human cells with alkylating agents. Carcinogenesis 5, 117–119 (1984).
Schraufstatter, I. U. et al. Hydrogen peroxide-induced injury of cells and its prevention by inhibitors of poly(ADP-ribose) polymerase. Proc. Natl Acad. Sci. USA 83, 4908–4912 (1986).
Fisher, A., Hochegger, H., Takeda, S. & Caldecott, K. W. Poly (ADP-ribose) polymerase-1 accelerates single-strand break repair in concert with poly (ADP-ribose) glycohydrolase. Mol. Cell Biol. 27, 5597–5605 (2007).
Le Page, F., Schreiber, V., Dherin, C., De Murcia, G. & Boiteux, S. Poly(ADP-ribose) polymerase-1 (PARP-1) is required in murine cell lines for base excision repair of oxidative DNA damage in the absence of DNA polymerase beta. J. Biol. Chem. 278, 18471–18477 (2003).
Trucco, C., Oliver, F. J., de Murcia, G. & Menissier-de Murcia, J. DNA repair defect in poly(ADP-ribose) polymerase-deficient cell lines. Nucleic Acids Res. 26, 2644–2649 (1998).
Ding, R., Pommier, Y., Kang, V. H. & Smulson, M. Depletion of poly(ADP-ribose) polymerase by antisense RNA expression results in a delay in DNA strand break rejoining. J. Biol. Chem. 267, 12804–12812 (1992).
Gao, H. et al. Altered poly(ADP-ribose) metabolism impairs cellular responses to genotoxic stress in a hypomorphic mutant of poly(ADP-ribose) glycohydrolase. Exp. Cell Res. 313, 984–996 (2007).
Pleschke, J. M., Kleczkowska, H. E., Strohm, M. & Althaus, F. R. Poly(ADP-ribose) binds to specific domains in DNA damage checkpoint proteins. J. Biol. Chem. 275, 40974–40980 (2000).
El-Khamisy, S. F., Masutani, M., Suzuki, H. & Caldecott, K. W. A requirement for PARP-1 for the assembly or stability of XRCC1 nuclear foci at sites of oxidative DNA damage. Nucleic Acids Res. 31, 5526–5533 (2003).
Lan, L. et al. In situ analysis of repair processes for oxidative DNA damage in mammalian cells. Proc. Natl Acad. Sci. USA 101, 13738–13743 (2004).
Okano, S., Lan, L., Caldecott, K. W., Mori, T. & Yasui, A. Spatial and temporal cellular responses to single-strand breaks in human cells. Mol. Cell Biol. 23, 3974–3981 (2003).
Caldecott, K. W., Aoufouchi, S., Johnson, P. & Shall, S. XRCC1 polypeptide interacts with DNA polymerase beta and possibly poly (ADP-ribose) polymerase, and DNA ligase III is a novel molecular 'nick-sensor' in vitro. Nucleic Acids Res. 24, 4387–4394 (1996).
Masson, M. et al. XRCC1 is specifically associated with poly(ADP-ribose) polymerase and negatively regulates its activity following DNA damage. Mol. Cell Biol. 18, 3563–3571 (1998).
Caldecott, K. W. XRCC1 and DNA strand break repair. DNA Repair (Amst.) 2, 955–969 (2003).
Dianov, G. L. & Parsons, J. L. Co-ordination of DNA single strand break repair. DNA Repair (Amst.) 6, 454–460 (2007).
Poirier, G. G., deMurcia, G., Jongstra-Bilen, J., Niedergang, C. & Mandel, P. Poly(ADP-ribosy)lation of polynucleosomes causes relaxation of chromatin structure. Proc. Natl Acad. Sci. USA 79, 3423–3427 (1982).
Tulin, A. & Spradling, A. Chromatin loosening by poly(ADP)-ribose polymerase (PARP) at Drosophila puff loci. Science 299, 560–562 (2003).
Tulin, A., Stewart, D. & Spradling, A. C. The Drosophila heterochromatic gene encoding poly(ADP-ribose) polymerase (PARP) is required to modulate chromatin structure during development. Genes Dev. 16, 2108–2119 (2002).
Mathis, G. & Althaus, F. R. Release of core DNA from nucleosomal core particles following (ADP-ribose)n-modification in vitro. Biochem. Biophys. Res. Commun. 143, 1049–1054 (1987).
Caldecott, K. W. Mammalian single-strand break repair: Mechanisms and links with chromatin. DNA Repair (Amst.) 6, 443–453 (2006).
Dantzer, F. et al. Base excision repair is impaired in mammalian cells lacking Poly(ADP- ribose) polymerase-1. Biochemistry 39, 7559–7569 (2000).
Prasad, R. et al. DNA polymerase beta-mediated long patch base excision repair. Poly(ADP-ribose) polymerase-1 stimulates strand displacement DNA synthesis. J. Biol. Chem. 276, 32411–32414 (2001).
Sanderson, R. J. & Lindahl, T. Down-regulation of DNA repair synthesis at DNA single-strand interruptions in poly(ADP-ribose) polymerase-1 deficient murine cell extracts. DNA Repair (Amst.) 1, 547–558 (2002).
Oei, S. L. & Ziegler, M. ATP for the DNA ligation step in base excision repair is generated from poly(ADP-ribose). J. Biol. Chem. 275, 23234–23239 (2000).
Petermann, E., Ziegler, M. & Oei, S. L. ATP-dependent selection between single nucleotide and long patch base excision repair. DNA Repair (Amst.) 2, 1101–1114 (2003).
Lindahl, T., Satoh, M. S., Poirier, G. G. & Klungland, A. Post-translational modification of poly(ADP-ribose) polymerase induced by DNA strand breaks. Trends Biochem. Sci. 20, 405–411 (1995).
Sobol, R. W. et al. The lyase activity of the DNA repair protein beta-polymerase protects from DNA-damage-induced cytotoxicity. Nature 405, 807–810 (2000). This is an important paper that demonstrated the significance of Pol b in repairing damaged SSB termini.
Roth, R. B. & Samson, L. D. 3-Methyladenine DNA glycosylase-deficient Aag null mice display unexpected bone marrow alkylation resistance. Cancer Res. 62, 656–660 (2002).
Trivedi, R. N. et al. Human methyl purine DNA glycosylase and DNA polymerase beta expression collectively predict sensitivity to temozolomide. Mol. Pharmacol. 13 May 2008 (doi:10.1124/mol.108.045112).
Sobol, R. W. et al. Base excision repair intermediates induce p53-independent cytotoxic and genotoxic responses. J. Biol. Chem. 278, 39951–39959 (2003).
Whitehouse, C. J. et al. XRCC1 stimulates human polynucleotide kinase activity at damaged DNA termini and accelerates DNA single-strand break repair. Cell 104, 107–117 (2001). These authors describe the first indication of the most important role identified for XRCC1 so far — promoting the processing of damaged DNA termini.
Karimi-Busheri, F. et al. Molecular characterization of a human DNA kinase. J. Biol. Chem. 274, 24187–24194 (1999).
Jilani, A. et al. Molecular cloning of the human gene, PNKP, encoding a polynucleotide kinase 3′-phosphatase and evidence for its role in repair of DNA strand breaks caused by oxidative damage. J. Biol. Chem. 274, 24176–24186 (1999).
Winters, T. A., Weinfeld, M. & Jorgensen, T. J. Human HeLa cell enzymes that remove phosphoglycolate 3′-end groups from DNA. Nucleic Acids Res. 20, 2573–2580 (1992).
Winters, T. A., Henner, W. D., Russell, P. S., McCullough, A. & Jorgensen, T. J. Removal of 3′-phosphoglycolate from DNA strand-break damage in an oligonucleotide substrate by recombinant human apurinic/apyrimidinic endonuclease 1. Nucleic Acids Res. 22, 1866–1873 (1994).
Chen, D. S., Herman, T. & Demple, B. Two distinct human DNA diesterases that hydrolyze 3′-blocking deoxyribose fragments from oxidized DNA. Nucleic Acids Res. 19, 5907–5914 (1991).
Izumi, T. et al. Requirement for human AP endonuclease 1 for repair of 3′-blocking damage at DNA single-strand breaks induced by reactive oxygen species. Carcinogenesis 21, 1329–1334 (2000).
Parsons, J. L., Dianova, I. I. & Dianov, G. L. APE1 is the major 3′-phosphoglycolate activity in human cell extracts. Nucleic Acids Res. 32, 3531–3536 (2004).
Matsumoto, Y. & Kim, K. Excision of deoxyribose phosphate residues by DNA polymerase beta during DNA repair. Science 269, 699–702 (1995).
Sung, J. S. & Demple, B. Roles of base excision repair subpathways in correcting oxidized abasic sites in DNA. Febs J. 273, 1620–1629 (2006).
Wiederhold, L. et al. AP endonuclease-independent DNA base excision repair in human cells. Mol. Cell 15, 209–220 (2004).
Yang, S. W. et al. A eukaryotic enzyme that can disjoin dead-end covalent complexes between DNA and type I topoisomerases. Proc. Natl. Acad. Sci. USA 93, 11534–11539 (1996).
Pouliot, J. J., Yao, K. C., Robertson, C. A. & Nash, H. A. Yeast gene for a Tyr-DNA phosphodiesterase that repairs topoisomerase I complexes. Science 286, 552–555 (1999). This paper contains the seminal finding that TDP1 is an end-processing factor.
Ahel, I. et al. The neurodegenerative disease protein aprataxin resolves abortive DNA ligation intermediates. Nature 443, 713–716 (2006). These authors describe the seminal finding that 5′-AMP strand breaks are the likely physiological substrate for APTX.
Rass, U., Ahel, I. & West, S. C. Actions of aprataxin in multiple DNA repair pathways. J. Biol. Chem. 282, 9469–9474 (2007).
Loizou, J. I. et al. The protein kinase CK2 facilitates repair of chromosomal DNA single-strand breaks. Cell 117, 17–28 (2004). This paper contains the seminal finding that CK2 is a DNA repair protein and is required to assemble XRCC1 end-processing complexes.
Clements, P. M. et al. The ataxia-oculomotor apraxia 1 gene product has a role distinct from ATM and interacts with the DNA strand break repair proteins XRCC1 and XRCC4. DNA Repair (Amst.) 3, 1493–1502 (2004). This paper and references 75–77 establish APTX as a component of the SSBR machinery.
Gueven, N. et al. Aprataxin, a novel protein that protects against genotoxic stress. Hum. Mol. Genet. 13, 1081–1093 (2004).
Luo, H. et al. A new XRCC1-containing complex and its role in cellular survival of methyl methanesulfonate treatment. Mol. Cell Biol. 24, 8356–8365 (2004).
Sano, Y. et al. Aprataxin, the causative protein for EAOH is a nuclear protein with a potential role as a DNA repair protein. Ann. Neurol. 55, 241–249 (2004).
Kubota, Y. et al. Reconstitution of DNA base excision-repair with purified human proteins: interaction between DNA polymerase beta and the XRCC1 protein. EMBO J. 15, 6662–6670 (1996).
Plo, I. et al. Association of XRCC1 and tyrosyl DNA phosphodiesterase (Tdp1) for the repair of topoisomerase I-mediated DNA lesions. DNA Repair (Amst.) 2, 1087–1100 (2003).
Sossou, M. et al. APE1 overexpression in XRCC1-deficient cells complements the defective repair of oxidative single strand breaks but increases genomic instability. Nucleic Acids Res. 33, 298–306 (2005).
Mani, R. S. et al. XRCC1 stimulates polynucleotide kinase by enhancing its damage discrimination and displacement from DNA repair intermediates. J. Biol. Chem. 282, 28004–28013 (2007).
Hirano, M. et al. DNA single-strand break repair is impaired in aprataxin-related ataxia. Ann. Neurol. 61, 162–174 (2007).
Fortini, P., Pascucci, B., Belisario, F. & Dogliotti, E. DNA polymerase beta is required for efficient DNA strand break repair induced by methyl methanesulfonate but not by hydrogen peroxide. Nucleic Acids Res. 28, 3040–3046 (2000).
Pascucci, B., Russo, M. T., Crescenzi, M., Bignami, M. & Dogliotti, E. The accumulation of MMS-induced single strand breaks in G1 phase is recombinogenic in DNA polymerase beta defective mammalian cells. Nucleic Acids Res. 33, 280–288 (2005).
Vermeulen, C., Verwijs-Janssen, M., Cramers, P., Begg, A. C. & Vens, C. Role for DNA polymerase beta in response to ionizing radiation. DNA Repair (Amst.) 6, 202–212 (2007).
Braithwaite, E. K. et al. DNA polymerase lambda protects mouse fibroblasts against oxidative DNA damage and is recruited to sites of DNA damage/repair. J. Biol. Chem. 280, 31641–31647 (2005).
Garcia-Diaz, M., Bebenek, K., Kunkel, T. A. & Blanco, L. Identification of an intrinsic 5′-deoxyribose-5-phosphate lyase activity in human DNA polymerase lambda: a possible role in base excision repair. J. Biol. Chem. 276, 34659–34663 (2001).
Bebenek, K. et al. 5′-Deoxyribose phosphate lyase activity of human DNA polymerase iota in vitro. Science 291, 2156–2159 (2001).
Prasad, R., Dianov, G. L., Bohr, V. A. & Wilson, S. H. FEN1 stimulation of DNA polymerase beta mediates an excision step in mammalian long patch base excision repair. J. Biol. Chem. 275, 4460–4466 (2000).
Hashiguchi, K., Matsumoto, Y. & Yasui, A. Recruitment of DNA repair synthesis machinery to sites of DNA damage/repair in living human cells. Nucleic Acids Res. 35, 2913–2923 (2007).
Frosina, G. et al. Two pathways for base excision repair in mammalian cells. J. Biol. Chem. 271, 9573–9578 (1996). These data underpin the concept of long-patch and short-patch BER.
Klungland, A. & Lindahl, T. Second pathway for completion of human DNA base excision-repair: reconstitution with purified proteins and requirement for DNase IV (FEN1). Embo J. 16, 3341–3348 (1997).
Fan, J., Otterlei, M., Wong, H. K., Tomkinson, A. E. & Wilson, D. M. 3rd. XRCC1 co-localizes and physically interacts with PCNA. Nucleic Acids Res. 32, 2193–2201 (2004).
Uchiyama, Y., Suzuki, Y. & Sakaguchi, K. Characterization of plant XRCC1 and its interaction with proliferating cell nuclear antigen. Planta 227, 1233–1241 (2008).
Cotner-Gohara, E., Kim, I. K., Tomkinson, A. E. & Ellenberger, T. Two DNA binding and nick recognition modules in human DNA ligase III. J. Biol. Chem. 283, 10764–10772 (2008).
Lakshmipathy, U. & Campbell, C. Antisense-mediated decrease in DNA ligase III expression results in reduced mitochondrial DNA integrity. Nucleic Acids Res. 29, 668–676 (2001).
De, A. & Campbell, C. A novel interaction between DNA ligase III and DNA polymerase gamma plays an essential role in mitochondrial DNA stability. Biochem. J. 402, 175–186 (2007).
Mortusewicz, O., Rothbauer, U., Cardoso, M. C. & Leonhardt, H. Differential recruitment of DNA ligase I and III to DNA repair sites. Nucleic Acids Res. 34, 3523–3532 (2006).
Caldecott, K. W., Tucker, J. D., Stanker, L. H. & Thompson, L. H. Characterization of the XRCC1-DNA ligase III complex in vitro and its absence from mutant hamster cells. Nucleic Acids Res. 23, 4836–4843 (1995).
Wei, Y. F. et al. Molecular cloning and expression of human cDNAs encoding a novel DNA ligase IV and DNA ligase III, an enzyme active in DNA repair and recombination. Mol. Cell Biol. 15, 3206–3216 (1995).
Taylor, R. M., Whitehouse, C. J. & Caldecott, K. W. The DNA ligase III zinc finger stimulates binding to DNA secondary structure and promotes end joining. Nucleic Acids Res. 28, 3558–3563 (2000).
Taylor, R. M., Whitehouse, J., Cappelli, E., Frosina, G. & Caldecott, K. W. Role of the DNA ligase III zinc finger in polynucleotide binding and ligation. Nucleic Acids Res. 26, 4804–4810 (1998).
Mackey, Z. B. et al. DNA ligase III is recruited to DNA strand breaks by a zinc finger motif homologous to that of poly(ADP-ribose) polymerase. Identification of two functionally distinct DNA binding regions within DNA ligase III. J. Biol. Chem. 274, 21679–21687 (1999).
Wang, H. et al. DNA ligase III as a candidate component of backup pathways of nonhomologous end joining. Cancer Res. 65, 4020–4030 (2005).
Audebert, M., Salles, B. & Calsou, P. Involvement of poly(ADP-ribose) polymerase-1 and XRCC1/DNA ligase III in an alternative route for DNA double-strand breaks rejoining. J. Biol. Chem. 279, 55117–55126 (2004).
Wong, H. K., Kim, D., Hogue, B. A., McNeill, D. R. & Wilson, D. M. 3rd. DNA damage levels and biochemical repair capacities associated with XRCC1 deficiency. Biochemistry 44, 14335–14343 (2005).
Bekker-Jensen, S. et al. Human Xip1 (C2orf13) is a novel regulator of cellular responses to DNA strand breaks. J. Biol. Chem. 282, 19638–19643 (2007).
Iles, N., Rulten, S., El-Khamisy, S. F. & Caldecott, K. W. APLF (C2orf13) is a novel human protein involved in the cellular response to chromosomal dna strand breaks. Mol. Cell Biol. 27, 3793–3803 (2007).
Schreiber, V. et al. Poly(ADP-ribose) polymerase-2 (PARP-2) is required for efficient base excision DNA repair in association with PARP-1 and XRCC1. J. Biol. Chem. 277, 23028–23036 (2002).
Tan, Y., Raychaudhuri, P. & Costa, R. H. Chk2 mediates stabilization of the FoxM1 transcription factor to stimulate expression of DNA repair genes. Mol. Cell Biol. 27, 1007–1016 (2007).
Li, N., Wu, H., Yang, S. & Chen, D. Ischemic preconditioning induces XRCC1, DNA polymerase-beta, and DNA ligase III and correlates with enhanced base excision repair. DNA Repair (Amst.) 6, 1297–306 (2007).
Fritz, G., Grosch, S., Tomicic, M. & Kaina, B. APE/Ref-1 and the mammalian response to genotoxic stress. Toxicology 193, 67–78 (2003).
Hasan, S. et al. Acetylation regulates the DNA end-trimming activity of DNA polymerase beta. Mol. Cell 10, 1213–1222 (2002).
Parsons, J. L. et al. CHIP-mediated degradation and DNA damage-dependent stabilization regulate base excision repair proteins. Mol. Cell 29, 477–487 (2008).
Chen, D., Yu, Z., Zhu, Z. & Lopez, C. D. E2F1 regulates the base excision repair gene XRCC1 and promotes DNA repair. J. Biol. Chem. 283, 15381–15389 (2008).
Narciso, L. et al. Terminally differentiated muscle cells are defective in base excision DNA repair and hypersensitive to oxygen injury. Proc. Natl Acad. Sci. USA 104, 17010–17015 (2007).
Otterlei, M. et al. Post-replicative base excision repair in replication foci. EMBO J. 18, 3834–3844 (1999).
Parlanti, E., Locatelli, G., Maga, G. & Dogliotti, E. Human base excision repair complex is physically associated to DNA replication and cell cycle regulatory proteins. Nucleic Acids Res. 35, 1569–1577 (2007).
Moore, D. J., Taylor, R. M., Clements, P. & Caldecott, K. W. Mutation of a BRCT domain selectively disrupts DNA single-strand break repair in noncycling Chinese hamster ovary cells. Proc. Natl Acad. Sci. USA 97, 13649–13654 (2000).
Taylor, R. M., Moore, D. J., Whitehouse, J., Johnson, P. & Caldecott, K. W. A cell cycle-specific requirement for the XRCC1 BRCT II domain during mammalian DNA strand break repair. Mol. Cell Biol. 20, 735–740 (2000).
Caldecott, K. W. Mammalian DNA single-strand break repair: an X-ra(y)ted affair. Bioessays 23, 447–455 (2001).
Caldecott, K. W. DNA single-strand break repair and spinocerebellar ataxia. Cell 112, 7–10 (2003).
Caldecott, K. W. DNA single-strand breaks and neurodegeneration. DNA Repair (Amst.) 3, 875–882 (2004).
Aicardi, J. et al. Ataxia-ocular motor apraxia: a syndrome mimicking ataxia-telangiectasia. Ann. Neurol. 24, 497–502 (1988).
Hannan, M. A., Sigut, D., Waghray, M. & Gascon, G. G. Ataxia-ocular motor apraxia syndrome: an investigation of cellular radiosensitivity of patients and their families. J. Med. Genet. 31, 953–956 (1994).
Barbot, C. et al. Recessive ataxia with ocular apraxia: review of 22 Portuguese patients. Arch. Neurol. 58, 201–205 (2001).
Moreira, M. C. et al. Homozygosity mapping of Portuguese and Japanese forms of ataxia-oculomotor apraxia to 9p13, and evidence for genetic heterogeneity. Am. J. Hum. Genet. 68, 501–508 (2001).
Le Ber, I. et al. Cerebellar ataxia with oculomotor apraxia type 1: clinical and genetic studies. Brain 126, 2761–2772 (2003).
Baba, Y. et al. Aprataxin (APTX) gene mutations resembling multiple system atrophy. Parkinsonism Relat. Disord. 13, 139–142 (2006).
Quinzii, C. M. et al. Coenzyme Q deficiency and cerebellar ataxia associated with an aprataxin mutation. Neurology 64, 539–541 (2005).
Le Ber, I. et al. Muscle coenzyme Q10 deficiencies in ataxia with oculomotor apraxia 1. Neurology 68, 295–297 (2007).
Date, H. et al. Early-onset ataxia with ocular motor apraxia and hypoalbuminemia is caused by mutations in a new HIT superfamily gene. Nature Genet. 29, 184–188 (2001). This paper and reference 133 identified APTX as the protein mutated in AOA1.
Moreira, M. C. et al. The gene mutated in ataxia-ocular apraxia 1 encodes the new HIT/Zn-finger protein aprataxin. Nature Genet. 29, 189–193 (2001).
Habeck, M. et al. Aprataxin mutations are a rare cause of early onset ataxia in Germany. J. Neurol. 251, 591–594 (2004).
Date, H. et al. The FHA domain of aprataxin interacts with the C-terminal region of XRCC1. Biochem. Biophys. Res. Commun. 325, 1279–1285 (2004).
Kanno, S. et al. A novel human AP endonuclease with conserved zinc-finger-like motifs involved in DNA strand break responses. Embo J. 26, 2094–2103 (2007).
Koch, C. A. et al. Xrcc4 physically links DNA end processing by polynucleotide kinase to DNA ligation by DNA ligase IV. Embo J. 23, 3874–3885 (2004).
Becherel, O. J. et al. Nucleolar localization of aprataxin is dependent on interaction with nucleolin and on active ribosomal DNA transcription. Hum. Mol. Genet. 15, 2239–2249 (2006).
Seidle, H. F., Bieganowski, P. & Brenner, C. Disease-associated mutations inactivate AMP-lysine hydrolase activity of Aprataxin. J. Biol. Chem. 280, 20927–20931 (2005).
Hirano, M. et al. Short half-lives of ataxia-associated aprataxin proteins in neuronal cells. Neurosci. Lett. 419, 184–187 (2007).
Criscuolo, C. et al. Very late onset in ataxia oculomotor apraxia type I. Ann. Neurol. 57, 777 (2005).
Criscuolo, C. et al. Ataxia with oculomotor apraxia type 1 in southern Italy: late onset and variable phenotype. Neurology 63, 2173–2175 (2004).
Tranchant, C., Fleury, M., Moreira, M. C., Koenig, M. & Warter, J. M. Phenotypic variability of aprataxin gene mutations. Neurology 60, 868–870 (2003).
Kijas, A. W., Harris, J. L., Harris, J. M. & Lavin, M. F. Aprataxin forms a discrete branch in the HIT (histidine triad) superfamily of proteins with both DNA/RNA binding and nucleotide hydrolase activities. J. Biol. Chem. 281, 13939–13948 (2006).
Rass, U., Ahel, I. & West, S. C. Defective DNA repair and neurodegenerative disease. Cell 130, 991–1004 (2007).
Takashima, H. et al. Mutation of TDP1, encoding a topoisomerase I-dependent DNA damage repair enzyme, in spinocerebellar ataxia with axonal neuropathy. Nature Genet. 32, 267–272 (2002). This paper identified TDP1 as the protein mutated in SCAN1.
Pourquier, P. et al. Trapping of mammalian topoisomerase I and recombinations induced by damaged DNA containing nicks or gaps. Importance of DNA end phosphorylation and camptothecin effects. J. Biol. Chem. 272, 26441–26447 (1997).
Pourquier, P. et al. Induction of reversible complexes between eukaryotic DNA topoisomerase I and DNA-containing oxidative base damages. 7,8-dihydro-8-oxoguanine and 5-hydroxycytosine. J. Biol. Chem. 274, 8516–8523 (1999).
Pourquier, P. et al. Effects of uracil incorporation, DNA mismatches, and abasic sites on cleavage and religation activities of mammalian topoisomerase I. J. Biol. Chem. 272, 7792–7796 (1997).
Pourquier, P. et al. Topoisomerase I-mediated cytotoxicity of N-methyl-N′-nitro-N-nitrosoguanidine: trapping of topoisomerase I by the O6-methylguanine. Cancer Res. 61, 53–58 (2001).
Pourquier, P. & Pommier, Y. Topoisomerase I-mediated DNA damage. Adv. Cancer Res. 80, 189–216 (2001).
Lebedeva, N., Auffret Vander Kemp, P., Bjornsti, M. A., Lavrik, O. & Boiteux, S. Trapping of DNA topoisomerase I on nick-containing DNA in cell free extracts of Saccharomyces cerevisiae. DNA Repair (Amst.) 5, 799–809 (2006).
Daroui, P., Desai, S. D., Li, T. K., Liu, A. A. & Liu, L. F. Hydrogen peroxide induces topoisomerase I-mediated DNA damage and cell death. J. Biol. Chem. 279, 14587–14594 (2004).
Nitiss, J. L., Nitiss, K. C., Rose, A. & Waltman, J. L. Overexpression of type I topoisomerases sensitizes yeast cells to DNA damage. J. Biol. Chem. 276, 26708–26714 (2001).
Liu, C., Pouliot, J. J. & Nash, H. A. The role of TDP1 from budding yeast in the repair of DNA damage. DNA Repair (Amst.) 3, 593–601 (2004).
El-Khamisy, S. F., Hartsuiker, E. & Caldecott, K. W. TDP1 facilitates repair of ionizing radiation-induced DNA single-strand breaks. DNA Repair (Amst.) 6, 1485–1495 (2007).
Inamdar, K. V. et al. Conversion of phosphoglycolate to phosphate termini on 3′ overhangs of DNA double strand breaks by the human tyrosyl-DNA phosphodiesterase hTdp1. J. Biol. Chem. 277, 27162–27168 (2002).
Interthal, H., Chen, H. J. & Champoux, J. J. Human Tdp1 cleaves a broad spectrum of substrates, including phosphoamide linkages. J. Biol. Chem. 280, 36518–36528 (2005).
Zhou, T. et al. Deficiency in 3′-phosphoglycolate processing in human cells with a hereditary mutation in tyrosyl-DNA phosphodiesterase (TDP1). Nucleic Acids Res. 33, 289–297 (2005).
Nitiss, K. C., Malik, M., He, X., White, S. W. & Nitiss, J. L. Tyrosyl-DNA phosphodiesterase (Tdp1) participates in the repair of Top2-mediated DNA damage. Proc. Natl Acad. Sci. USA 103, 8953–8958 (2006).
Interthal, H. et al. SCAN1 mutant Tdp1 accumulates the enzyme–DNA intermediate and causes camptothecin hypersensitivity. Embo J. 24, 2224–2233 (2005).
Hirano, R. et al. Spinocerebellar ataxia with axonal neuropathy: consequence of a Tdp1 recessive neomorphic mutation? Embo J. 26, 4732–4743 (2007).
Katyal, S. et al. TDP1 facilitates chromosomal single-strand break repair in neurons and is neuroprotective in vivo. Embo J. 26, 4720–4731 (2007).
Mosesso, P. et al. The novel human gene aprataxin is directly involved in DNA single-strand-break repair. Cell. Mol. Life Sci. 62, 485–491 (2005).
El-Khamisy, S. F. & Caldecott, K. W. TDP1-dependent DNA single-strand break repair and neurodegeneration. Mutagenesis 21, 219–224 (2006).
Sweasy, J. B., Lang, T. & DiMaio, D. Is base excision repair a tumor suppressor mechanism? Cell Cycle 5, 250–259 (2006).
Barzilai, A., Rotman, G. & Shiloh, Y. ATM deficiency and oxidative stress: a new dimension of defective response to DNA damage. DNA Repair (Amst.) 1, 3–25 (2002).
Acknowledgements
I thank the MRC, BBSRC, and EU for financial support, and members of my laboratory for comments and suggestions. I apologize to my colleagues for work I have not referenced in this article, this is due to restrictions in citation numbers.
Author information
Authors and Affiliations
Related links
Glossary
- Alternative non homologous end joining
-
Also known as back-up non homologous end joining. A subpathway of non homologous end joining (NHEJ) that does not require classical NHEJ proteins and which might rejoin DSBs located within short regions of microhomology.
- Clastogenic
-
Capable of causing chromosome damage and/or rearrangements.
Rights and permissions
About this article
Cite this article
Caldecott, K. Single-strand break repair and genetic disease. Nat Rev Genet 9, 619–631 (2008). https://doi.org/10.1038/nrg2380
Issue Date:
DOI: https://doi.org/10.1038/nrg2380
This article is cited by
-
Covalent PARylation of DNA base excision repair proteins regulates DNA demethylation
Nature Communications (2024)
-
Regulation of Rad52-dependent replication fork recovery through serine ADP-ribosylation of PolD3
Nature Communications (2023)
-
Targeting PARP proteins in acute leukemia: DNA damage response inhibition and therapeutic strategies
Journal of Hematology & Oncology (2022)
-
Doxorubicin impacts chromatin binding of HMGB1, Histone H1 and retinoic acid receptor
Scientific Reports (2022)
-
The threat of programmed DNA damage to neuronal genome integrity and plasticity
Nature Genetics (2022)
