
Key Points
-
A narrow definition of dysbiosis is as a stable microbial community state that functionally contributes to the aetiology, diagnosis or treatment of a disease.
-
Dysbiosis is often driven by infection and inflammation, diet and xenobiotics, host genetics or the host's environment.
-
Innate and adaptive immunity control the colonization niche of the intestinal microbiota through mechanisms including the production of antimicrobial peptides and IgA antibodies.
-
A dysbiotic microbiota may actively influence its colonization niche by altering the functions of innate and adaptive intestinal immunity.
-
Dysbiosis has been associated with many immune-related human diseases, but in many cases it remains to be established whether dysbiosis is a cause or consequence of the disease.
-
Personalized nutrition and metabolite-based 'postbiotic' therapy may present ways in which to harness the increasing knowledge about dysbiosis in disease for the design of new therapies.
Abstract
Throughout the past century, we have seen the emergence of a large number of multifactorial diseases, including inflammatory, autoimmune, metabolic, neoplastic and neurodegenerative diseases, many of which have been recently associated with intestinal dysbiosis — that is, compositional and functional alterations of the gut microbiome. In linking the pathogenesis of common diseases to dysbiosis, the microbiome field is challenged to decipher the mechanisms involved in the de novo generation and the persistence of dysbiotic microbiome configurations, and to differentiate causal host–microbiome associations from secondary microbial changes that accompany disease course. In this Review, we categorize dysbiosis in conceptual terms and provide an overview of immunological associations; the causes and consequences of bacterial dysbiosis, and their involvement in the molecular aetiology of common diseases; and implications for the rational design of new therapeutic approaches. A molecular- level understanding of the origins of dysbiosis, its endogenous and environmental regulatory processes, and its downstream effects may enable us to develop microbiome-targeting therapies for a multitude of common immune-mediated diseases.
Similar content being viewed by others


Main
The incidence of many common multifactorial human diseases, such as diabetes and obesity, allergy and asthma, neurodegeneration and inflammatory bowel disease (IBD), has substantially increased during the past two centuries. The short duration of this period, which encompasses only a limited number of human generations, makes it unlikely that these disorders can be explained by genetic factors alone1. Instead, changes in lifestyle and environmental factors, which are broadly adopted by post-industrial revolution societies, compared with the conditions prevalent during the preceding evolution of the human gene pool are probably associated with the increasing incidence of these autoimmune, inflammatory and metabolic diseases2. These lifestyle and environmental factors include alterations in diet, physical activity, hygiene, longevity, exposure to xenobiotics and a newly acquired human ability to control light and temperature. In the quest to better understand the origin of these pandemics, it has recently been recognized that another gene pool needs to be considered when evaluating the impact of such environmental factors on human health, namely the metagenome of the entirety of microorganisms that colonize the human body, which is collectively termed the microbiome3. The microbiome has co-evolved with the eukaryotic genome of its host and colonizes the host's interfaces with the outside world, including the gastrointestinal tract, skin, respiratory tract and urogenital tract. Both the human and microbial genomes have been subject to dietary and environmental pressures, including the rapid environmental changes that characterized the industrial revolution that has occurred in the past two centuries. The substantially shorter generation times of commensal microorganisms, relative to humans, make the microbiome amenable to rapid evolutionary changes on a much shorter timescale and may suggest that adaptation of the metagenome to changes in environmental conditions is more rapid than that of the host genome. In recent years, many of the modern multifactorial diseases that show an increasing incidence have been associated with an abnormal microbiome structure, termed dysbiosis, which affects the taxonomical composition as well as the metagenomic function of the microbial community. The microbiome consists of complex bacterial, archaeal, fungal, viral and protozoan communities that colonize multiple body sites. In this Review, we focus primarily on the bacterial part of the gastrointestinal tract microbiome, and its effects on immune homeostasis and the risk of immune-mediated and immune-associated diseases.
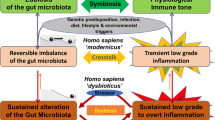
The healthy intestinal microbial community can be characterized in terms of diversity, stability and resistance, and resilience4, which are defined, respectively, as the richness of the ecosystem, its amenability to perturbation and its ability to return to the pre-perturbation state. Data from large human cohort studies suggest that multiple stable states of the microbial ecosystem can colonize a host in the absence of overt signs of disease5,6,7 (Fig. 1). A common definition of dysbiosis describes it as a compositional and functional alteration in the microbiota that is driven by a set of environmental and host-related factors that perturb the microbial ecosystem to an extent that exceeds its resistance and resilience capabilities. Once the microbiota configuration is shifted, dysbiosis likewise persists as a stable state and can assume various compositional manifestations depending on the trigger8. Thus, the stability of the intestinal microbial community can be viewed on a conceptual energy landscape (Fig. 1), in which both healthy and dysbiotic states can exist in several different configurations, but the transition between them requires external forces that are stronger than the stability properties of the system9.

The transition between the healthy state and the dysbiotic state requires stimuli such as diet, host genetics, infection or inflammation.
There are a number of limitations to the basic definition of dysbiosis as an altered state of the intestinal bacterial community. First, the enormous interindividual variability in the taxonomic microbiota composition between healthy individuals across geography, age and dietary habits5,6,7,9 raises the question of what can be considered a reference population, and allows for almost any given gut microbial configuration to be considered 'dysbiotic' when compared with a particular control. Similarly, the microbiota of laboratory mice is dramatically influenced by vivaria (rearing conditions) and the rodent diet10. It is therefore crucial that studies in both humans and animal models are very carefully controlled to avoid conclusions being drawn from 'spurious' dysbiosis caused by interindividual variability, vertical transmission, housing effects, variations in pathogen screening in animal facilities and other factors accounting for incidental deviations of microbiome composition from a given reference population10. By contrast, in the case of true phenotype-associated dysbiosis, the inflammatory, genetic or dietary causes are sufficient to provoke the de novo manifestation of dysbiosis, and the dysbiotic microbiota is sufficient to cause disease in experimental models10. Second, adaptations of the microbiome to altered environmental conditions or changes in the state of the host — which result in abnormal community composition and function — may generally have beneficial, neutral or harmful consequences for the host. As with host tissue deviations from homeostasis, adaptive changes in the microbiome in response to perturbations of the steady state might become detrimental in those cases in which the microbial community does not return to the previous state after normalization of the environmental conditions, but instead persists chronically in a 'maladaptive' state that has detrimental consequences for the host11,12. In this Review, we therefore suggest the use of a narrow definition of dysbiosis, namely a microbial community state that is not only statistically associated with a disease, but also functionally contributes to the aetiology, diagnosis or treatment of the disease. Thereby, a dysbiotic microbiome configuration should fulfil Koch's postulates for the definition of a disease-causing microbial agent, with the exception of the requirements for cultivability and absence from a healthy host.
Types of dysbiosis
Dysbiosis typically features one or more of the following non-mutually exclusive characteristics.
Bloom of pathobionts. Members of the commensal microbiota that have the potential to cause pathology have been termed pathobionts13. Such bacteria are typically present at low relative abundances but proliferate when aberrations occur in the intestinal ecosystem. A prototypical example of such population expansion is the outgrowth of the bacterial family Enterobacteriaceae, which is frequently observed in enteric infection and inflammation14. Importantly, this bloom of Enterobacteriaceae is consistently observed in both patients with IBD15 and mouse models of IBD16, which suggests that conserved and robust mechanisms underlie this phenomenon. However, the bloom of Enterobacteriaceae may represent a consequence rather than a cause of the inflammation-induced remodelling of the intestinal ecosystem.
Loss of commensals. Conversely to the outgrowth of pathobionts, dysbiosis frequently features the reduction or complete loss of normally residing members of the microbiota, which can be the consequence of microbial killing or diminished bacterial proliferation17. Such a loss of commensals can be functionally important, and restoration of the abolished bacteria or their metabolites has the potential to reverse dysbiosis-associated phenotypes. This has been demonstrated, for instance, in two mouse models of autism spectrum disorder in which reconstitution of Lactobacillus reuteri in a diet-induced model18 and of Bacteroides fragilis in a maternally transmitted model19 reduced disease severity. Replenishment of diminished commensal bacteria has also proved effective against enteric infection, as in the case of Clostridium difficile-induced inflammation, which was ameliorated by colonization with Clostridium scindens20. Such studies suggest that targeted microbiota reconstitution could be an effective way to harness our understanding of the functional importance of disease-associated dysbiosis21. Knowledge about particular microbiome-derived metabolites can further enhance the power of this approach, as has been demonstrated, for instance, for the impact of the microbiota on microglia function22, intestinal cytokine production23 and neurodegeneration24.
Loss of diversity. A recurrent characteristic of disease-associated dysbiosis is a reduction in alpha diversity. The richness of the intestinal microbiota increases during the first years of life7, can be influenced by dietary patterns25 and is associated with metabolic health26. By contrast, low bacterial diversity has been documented in the context of dysbiosis induced by abnormal dietary composition21, IBD27, AIDS28 and type 1 diabetes (T1D)29, among many other conditions30.
Origins of dysbiosis
Given the above definition of dysbiosis as a distinct microbial ecological state that is causally linked to the manifestation, diagnosis or treatment of a particular disease, it is crucial to consider the mechanisms that contribute to the development and maintenance of a dysbiotic state. In this section, we focus on the most prevailing categories of factors that influence the composition of the intestinal microbial community (Fig. 1).
Infection and inflammation. Dysbiosis caused by enteric infection was first observed in mouse models of infection with Citrobacter rodentium31 and Salmonella enterica subsp. enterica serovar Typhimurium32, in which inflammation compromises the microbiota's ability to provide colonization resistance against invading microorganisms. Inflammation induced by dextran sodium sulfate or genetic deficiency of interleukin-10 (Il10) in mice led to similar changes in the microbial community and favoured the growth of enteric pathogens31,32. In addition to intestinal infection, inflammation-induced outgrowth of members of the Enterobacteriaceae family can promote the development of colorectal cancer33 and sepsis34. The molecular mechanisms leading to the establishment of Enterobacteriaceae in the inflamed gut are manifold, and include the release of nutrients35, the use of metal ions36, intermicrobial competition and horizontal gene transfer37, the exploitation of antimicrobial peptides38, as well as the harnessing of aerobic and anaerobic cellular respiration39,40.
Diet and xenobiotics. Diet has a considerable short-term41 and long-term6 influence on the composition of the intestinal microbiota. In mice fed a low-fibre diet, microbial diversity is progressively reduced across consecutive generations21. Similarly, a high-fat diet reduces microbial diversity in mice42. In addition to the nutritional content of food, dietary xenobiotics have the potential to alter homeostatic commensal colonization. This is most intuitive in the case of antibiotics43, but has also been described for non-caloric artificial sweeteners44 and dietary emulsifiers45, although the mechanisms by which the latter two examples shape the microbiome remain to be determined. Diet-induced and xenobiotic-induced dysbiosis may be strong drivers of disease manifestations, as has been documented in mice46 and, in certain cases, even in humans47.
Genetics. In addition to the above-mentioned environmental factors, host genetics are involved in shaping the composition of the intestinal microbiota48. A twin study identified the abundance of multiple taxa of the intestinal microbiota influenced by host genetics49, such as the association of the Bifidobacterium genus and the human gene locus that encodes lactase. This association, among several others, was also found by genome-wide association studies that linked genetic loci with microbial taxa and functional pathways50,51. In addition, the locus encoding the human vitamin D receptor, and several other human loci involved in immune and metabolic functions, were highlighted as potential drivers of microbial control through host genetics52. In mice, genomic studies have likewise identified an impact of host genetics on colonization with particular taxa53. In certain cases, the genetic influence on microbial composition may be involved in the manifestation of certain phenotypes, as demonstrated for Christensenellaceae and low body mass index51, thereby meeting the narrow definition of dysbiosis. The relative contributions of diet versus host genetics in humans await further elucidation. However, the impact of diet seems to outweigh the genetic background of the host in mouse models54, which suggests that a particular diet may compensate for the genetic predisposition of the host for intestinal colonization with a particular microorganism.
Familial transmission. The early succession of intestinal colonization after birth is determined by the maternal microbiota55 and, in particular, by the mode of delivery56. Thus, transmission across generations is an important contributor that shapes individual microbiomes, although studies in both germ-free mice and human neonates have demonstrated that maternal factors alone do not suffice to explain an individual's microbiota assembly55,57. Environmental transmission seems to be of additional importance, as households feature characteristic microbiome signatures, and the microbiomes of members of a particular household are more similar to one another than to the microbiomes of members of other households58. In laboratory mice, the effects of coprophagy and isolated housing conditions potentiate this effect, leading to the establishment of mouse line-specific or even vivarium-specific microbiomes59. Both familial and environmental microbiome transmission may be of phenotypic importance in some disorders, by introducing a transmissible microbiota component to non-infectious diseases, but they can also result in incidental 'spurious' dysbiosis10.
Other causes. Several other factors have been suggested as potential instigators of dysbiosis that is causally involved in the manifestation of host phenotypes, including circadian disruption60,61, maternal high-fat diet18, pregnancy62 and physical injury63. Given the importance of the microbiome in influencing host physiology and the microbiome's high degree of amenability to change by environmental conditions, it is likely that this list will be further expanded by future studies.
Immune control of microbial homeostasis
The immune system is considered to be one of the most important forces by which the host shapes the configuration of the normal and dysbiotic microbiome. As such, understanding immune system–microbiome crosstalk is crucial in defining the direct and indirect effects of host immunity on dysbiosis-driven diseases (Fig. 2).

Innate mechanisms of regulation include nucleotide-binding oligomerization domain-containing protein 2 (NOD2)-mediated recognition of microbial peptidoglycan, which contributes to intestinal homeostasis by signalling through the kinase receptor-interacting protein 2 (RIP2; also known as RIPK2) and nuclear factor-κB (NF-κB), and by inducing the production of antimicrobial peptides (AMPs) and mucin. Other microbial products such as flagellin and lipoproteins stimulate Toll-like receptor 5 (TLR5) in dendritic cells (DCs) and epithelial cells to enhance the epithelial expression of AMPs. The NOD-, LRR- and pyrin domain-containing 6 (NLRP6) inflammasome is activated by microbial metabolites, resulting in the secretion of interleukin-18 (IL-18) and AMPs. Adaptive mechanisms of microbial regulation include the production of secretory IgA (sIgA) — which is mediated by T follicular helper (TFH) cells — as well as CD1d-mediated activation of invariant natural killer T (iNKT) cells and secretion of anti-inflammatory cytokines. Likewise, the microbiota modulates the activity of γδ intraepithelial lymphocytes (IELs). IL, interleukin; ILC, innate lymphoid cell; PD1, programmed cell death protein 1; PDL1, PD1 ligand 1; TCR, T cell receptor.
The innate immune system in the regulation of microbial composition. Microbial sensing through germline-encoded pattern recognition receptors (PRRs) was suggested to influence microbial colonization in mice64. For instance, mice deficient in the Toll-like receptor (TLR) signalling adaptor myeloid differentiation primary response protein 88 (MYD88) harbour a distinct intestinal microbiota65. Furthermore, loss of MYD88 signalling specifically in epithelial cells results in increased numbers of mucosa-associated bacteria and increased translocation of bacteria to the mesenteric lymph nodes, as well as altered bacterial composition66. One particular TLR that was suggested to be involved in the prevention of dysbiosis is the flagellin sensor TLR5. Tlr5−/− mice develop altered intestinal microbiota compared with littermate wild-type controls, and this alteration leads to the manifestation of hyperphagia (excessive eating) and metabolic syndrome, whereas microbiota depletion using antibiotics corrected the metabolic phenotype67. Furthermore, Tlr5−/− mice feature high levels of Enterobacteriaceae in close proximity to the intestinal epithelium68. By contrast, Myd88−/− mice did not develop metabolic syndrome, which suggests the existence of additional compensatory mechanisms67. Despite the reported differences in microbial composition, the role of TLR signalling in the control of intestinal microbial ecology remains unresolved, as follow-up studies have suggested that maternal transmission, rather than genetic deficiency, might explain the microbial differences observed in TLR-deficient mice59.
Additional PRRs with a suggested link to microbial dysbiosis are the NOD-like receptors (NLRs). In the absence of nucleotide-binding oligomerization domain-containing protein 1 (NOD1), which recognizes peptidoglycan from Gram-negative bacteria, the bacterial population is expanded, and this includes an increase in commensal Clostridiales, Bacteroides spp., segmented filamentous bacteria (SFB) and Enterobacteriaceae69. Similarly, Nod2−/− mice have an altered microbiota composition that is characterized by an increased burden of commensals as well as an increased proportion of mucosa-associated bacteria, thereby predisposing the mice to intestinal inflammation and colorectal cancer70,71,72. Similarly to these observations made in mice, human NOD2 polymorphisms are also associated with dysbiosis in Crohn disease73. Interestingly, NOD2 expression is dependent on the presence of commensal bacteria, therefore suggesting a negative-feedback relationship between commensal bacteria and NOD2. Consequently, NOD2 deficiency breaks this homeostatic interaction, leading to dysbiosis development71. However, as in the case of TLRs, a study using littermate breeding failed to observe gross alterations in the structure of the intestinal microbiota of mice lacking Nod1 or Nod2, and this raised the possibility that these divergent observations could be explained by different experimental designs and environmental sources of variation74. These examples highlight the importance of well-designed experimental controls in the study of dysbiosis in mice that have innate immune defects.
Aside from NOD1 and NOD2, some NLR proteins assemble into multiprotein complexes known as inflammasomes, leading to the activation of caspase 1, which then processes the cytokines interleukin-1β (IL-1β) and IL-18 (Ref. 75). NOD-, LRR- and pyrin domain-containing 6 (NLRP6) is one such protein that induces or affects intestinal epithelial inflammasome formation. NLRP6 inflammasome formation was shown in vitro, and its deficiency in vivo led to impaired caspase 1 activation and IL-18 secretion; however, it has yet to be associated with ASC speck formation23. NLRP6 was shown to have a role in the maintenance of a stable microbial community in the intestine. Mice deficient in NLRP6 feature a dysbiotic microbiota that confers transmissible susceptibility to colitis, features of metabolic syndrome, pathogenic infection, and colitis-associated colorectal cancer23,76,77,78,79. Mechanistically, commensal microbiota-derived metabolites activate the NLRP6-associated inflammasome, thereby maintaining a homeostatic environment of mucus and antimicrobial peptides that act to control the microbiota composition23,79,80. Similarly, deficiencies in other inflammasome components — namely, ASC and caspase 1 — have been associated with a dysbiotic microbiota and increased susceptibility to the development of dextran sodium sulfate-induced colitis76.
Although in vivo NLRP6 inflammasome formation has not been structurally proved to date, a similarly altered microbiota composition and metagenomic function has been found across animal facilities in mice deficient in NLRP6 and downstream inflammasome components; however, the local wild-type mice differed considerably between vivaria23. The greater functional than compositional congruency of the microbiomes associated with NLRP6 deficiency across facilities23 is in line with findings in humans, in which core metagenomic functions are shared across a wide variability of taxonomic assortments5. This finding also illustrates the importance of careful interpretation of dysbiosis, as this crucially depends on the nature of the reference population, which in the case of both laboratory mice and human populations underlies global variability7,81. This has recently been exemplified by the discovery that a commensal protist, Tritrichomonas musculis, elicits inflammasome activation in epithelial cells that results in the secretion of IL-18 and the downstream activation of the intestinal immune system, including changes in myeloid cells and innate lymphoid cells (ILCs), and the expansion of T helper 1 (TH1) and TH17 cells82. Thus, protozoan colonization markedly alters epithelial inflammasome signalling and the activation state of the immune system, and is a potential source of inter-facility variation.
A further example of the centrality of microbial control through epithelial cell secretion of antimicrobial peptides is provided by α-defensins, which are expressed by Paneth cells and are essential regulators of intestinal ecology. Mice that lack α-defensins display an altered microbial composition compared with wild-type controls, albeit with normal bacterial numbers83. Likewise, Paneth cells secrete the antimicrobial lectin REGIIIγ, which targets Gram-positive bacteria, is expressed in response to bacterial colonization and is important for the maintenance of separation between the microbiota and the epithelial surface84.
In addition to epithelial cells, ILCs were recently suggested to have a role in the regulation of homeostatic microbial colonization85,86. For example, RORγt+ ILCs are the dominant source of IL-22, which was shown to be important for epithelial production of antimicrobial peptides such as REGIIIγ, REGIIIβ, S100A8 and S100A9 (Ref. 87). Reduction of IL-22 levels results in the expansion of SFB populations88 and systemic colonization with commensals89. In addition, T-bet+ ILCs are a source of interferon-γ (IFNγ) and tumour necrosis factor (TNF), and may have an important role in regulating the composition of the microbiota, as mice deficient in T-bet in the innate immune system develop transmissible colitis and dysbiosis characterized by the outgrowth of Helicobacter typhlonius16,90.
The adaptive immune system in the regulation of microbial composition. Similarly to the role of the innate immune system in the regulation of a healthy microbiota, accumulating evidence suggests a role for the adaptive immune system in microbiome control (Fig. 2). In particular, B cells are crucial players in the maintenance of intestinal homeostasis through the production of secretory IgA. Secretory IgA antibodies can be targeted to specific bacteria91 and even specific bacterial functions such as flagella production92. Interestingly, the bacteria most preferentially targeted by secretory IgA are those that are associated with mucosal-proximal colonization and colitogenic potential93. In germ-free mice, the number of IgA-secreting B cells is reduced, although the total number of B cells is comparable to the number of B cells found in colonized mice94. In the absence of secretory IgA, the total amount of luminal microbial DNA is normal; however, serum lipopolysaccharide (LPS) concentrations are higher than they are in the presence of secretory IgA, and moderate changes in bacterial composition are observed95. In the case of activation-induced cytidine deaminase deficiency, the absence of somatic hypermutation results in increased SFB colonization in the small intestine and the expansion of anaerobes, whereas reconstitution of IgA reverses these microbial abnormalities96,97.
Furthermore, a subset of CD4+ T cells known as T follicular helper (TFH) cells, which promote IgA selection, is associated with microbiome control in the intestine. TFH cells express high levels of the inhibitory receptor programmed cell death protein 1 (PD1). Bacterial communities and IgA production are regulated by PD1 signalling, and PD1 deficiency results in a reduced frequency of faecal bacteria coated with IgA, and in an altered microbial composition characterized by a reduced frequency of Bifidobacterium species and an increased frequency of Enterobacteriaceae, although the total number of bacteria is comparable98.
Invariant natural killer T (iNKT) cells comprise an additional class of immune cells involved in the regulation of bacterial composition. These cells respond to a wide range of microbial glycolipids. Mice deficient in the MHC class I-like molecule CD1d have an altered faecal microbiota composition characterized by an increased frequency of adherent bacteria, SFB localization in close proximity to epithelial cells and enhanced colonization by pathogens99. Furthermore, intraepithelial lymphocytes that express γδ T cell receptors prevent mucosal dissemination of bacteria through the secretion of cytokines and antimicrobial molecules following mucosal injury. In the absence of γδ intraepithelial lymphocytes, the control of invasive bacteria is compromised and invasive bacteria populations are expanded100.
Impact of dysbiosis on the host immune system
Although the above mechanisms are involved in preventing the development of dysbiosis, a dysbiotic microbial community, once established, substantially affects both the local mucosal and systemic landscape of immune cells, thereby creating a feedback loop in which the host immune system and its microbiota cross-regulate each other101. The microbiota features a large repertoire of signals and mechanisms by which it can affect immune activation, including epigenetic remodelling and altered gene expression (summarized in Box 1). Intriguingly, in the context of dysbiosis, microbial signalling to the immune system can be important for the maintenance of the dysbiotic state, which is achieved by at least two phenomena. First, pathobionts arising under inflammatory conditions contribute to the perpetuation of inflammation, thereby preserving the conditions that favour their own growth. Second, a dysbiotic microbiota can in some cases be dominantly transferred to a new host, in which immune system hijacking alters the microbial colonization niche (Fig. 3).

A dysbiotic microbiota can hijack the host immune system through various mechanisms that collectively contribute to the stabilization of the dysbiotic configuration. These mechanisms include the modulation of inflammasome signalling through microbial metabolites, the modulation of Toll-like receptor (TLR) signalling and the degradation of secretory IgA (sIgA). AMPs, antimicrobial peptides; IL, interleukin; MYD88, myeloid differentiation primary response protein 88; NF-κB, nuclear factor-κB; NLRP6, NOD-, LRR- and pyrin domain-containing 6.
Signalling to innate immunity. A healthy or dysbiotic microbiota can influence the host innate immune system via two types of signal: microbial cell components and metabolites. In a dysbiotic state, alterations in the signature of microbial molecules sensed by the host can lead to a different activation state of the immune system. This is exemplified by a recent study of three infant cohorts whose microbiomes differed in terms of the immunogenicity of LPS, and thereby in their ability to stimulate TLR4, activate nuclear factor-κB and tolerate endotoxin102. Importantly, the less immunogenic LPS was found to be produced by bacterial species in children from countries in which there is a high prevalence of autoimmune disease, which suggests a direct link between microbiota structure, immune activation and susceptibility to disease.
An additional TLR that is modulated by bacteria is TLR2 (Ref. 103). The oral anaerobic bacterium Porphyromonas gingivalis transforms the oral microbiota into a dysbiotic community and contributes to inflammation104. P. gingivalis has the capacity to manipulate the host immune response by promoting the degradation of MYD88, and thereby inhibiting the antimicrobial response while maintaining inflammation through crosstalk between TLR2 and the complement receptor C5aR103. The uncoupling of bacterial eradication from tissue inflammation is an example in which a single commensal can disrupt host–microbiota homeostasis to cause inflammation and maintain persistent dysbiosis. Similarly, the microbiota perpetuates abnormal tissue immunity after infection with Yersinia pseudotuberculosis, inducing lymphatic leakage into the mesenteric adipose tissue12.
Similarly to TLR signalling, NLR signalling is controlled by signals derived from the microbiota. NLRP6-associated inflammasome activation in the gut leads to the secretion of IL-18, thereby regulating intestinal inflammation, epithelial repair and host defence against infections. As briefly described above, NLRP6 signalling is suggested to be involved in constructing the intestinal colonization niche through the secretion of mucus and antimicrobial peptides, and the absence of these mechanisms facilitates dysbiosis development23. Notably, the dysbiotic configuration can be stably transferred to wild-type mice and promotes disease manifestations in the new host23,76,77,78,79. NLRP6 activity is influenced by the concentrations of microbe-modulated metabolites, including the bile acid conjugate taurine, the amino acid histamine and the polyamine spermine23. In the absence of NLRP6, the metabolite profile changes into one that has an inflammasome-suppressing capability, such that on transfer to a new host, the dysbiotic configuration modulates the antimicrobial peptide landscape in a way that favours its preferential colonization over that of the invaded wild-type microbiome configuration.
A similar phenomenon can be observed in the case of IL-22. The absence of IL-22 leads to an altered and transmissible disease-promoting microbiota, and cohousing of wild-type mice with IL-22-deficient mice reduced their expression of IL-22-induced antimicrobial proteins to the levels found in IL-22-deficient mice105. These examples represent strategies by which an altered microbiota composition may contribute to its own maintenance by regulating specific factors involved in the orchestration of mucosal immunity.
Signalling to adaptive immune cells. Similarly sophisticated mechanisms of microbial modulation have been suggested to involve the adaptive arm of the immune system. One remarkable mechanism by which the microbiota can affect the colonization niche is through the degradation of secretory IgA106. Sutterella species are associated with low secretory IgA levels, as they can degrade both IgA and the associated stabilizing peptide106. Transfer of microbiota from mice that have low faecal secretory IgA levels by cohousing or faecal transplantation can change the intestinal environment in the new host from high to low secretory IgA levels, thereby also transferring the susceptibility to chemically induced intestinal inflammation106. Collectively, these observations in both innate and adaptive immunity provide evidence for the intriguing notion that intestinal microorganisms shape the very same mechanisms that organize their colonization conditions — namely, the production of antimicrobial peptides, mucus and secretory IgA. As such, commensals are not mere bystanders but are active participants in intestinal niche construction. In the case of dysbiosis, these microbe-controlled mechanisms contribute to the perpetuation of a stably altered community (Fig. 3).

Dysbiosis and immunological diseases
Despite the recent surge in associations of immune-mediated diseases with dysbiosis, it remains unclear for many associations whether dysbiosis is a direct cause of the disease manifestation or whether changes in the microbial communities in individuals with disease are a result of a change in the host's immune system, diet or metabolism. A causal contribution of a dysbiotic microbiome to disease pathogenesis can be demonstrated in a number of ways, ranging from prospective cohort studies to interventional trials in humans and preclinical studies involving microbiome transfers from gnotobiotic mice to germ-free mice. Through the manifold impact of the microbiota on host immunity, dysbiosis may directly influence immune-mediated diseases, as discussed in this section and summarized in Fig. 4.

Various factors can contribute to the development and maintenance of a dysbiotic state. The dysbiotic microbiota, through metabolites and toxins, can influence disease development in the intestine as well as in distal organs. 4-EPS, 4-ethylphenylsulfate; AHR, aryl hydrocarbon receptor; IBD, inflammatory bowel disease; NAFLD, nonalcoholic fatty liver disease; NASH, nonalcoholic steatohepatitis; SCFAs, short-chain fatty acids; TMAO, trimethylamine-N-oxide.
IBD. IBD is a group of multifactorial disorders that are characterized by chronic relapsing inflammation of the intestinal mucosa and extra-intestinal organs. The microbiota is central in the pathogenesis of IBD, and IBD is associated with decreased microbial richness15,107. Several attempts have been made to identify a single bacterium that could be a mono-associated cause of IBD, among them pathogenic Escherichia coli, C. difficile and Fusobacterium nucleatum108. However, inconsistent observations regarding the microbial compositions of patients with IBD have hindered efforts to assess the aetiological role of specific bacterial species in the pathophysiology of IBD, and a causal relationship has yet to be established. Moreover, it is possible that functional analysis of the microbiome is more relevant to the pathogenesis of IBD than is compositional analysis. For instance, it has been suggested that dysbiosis in IBD involves a decrease in the frequency of butyrate-producing bacteria along with an increase in sulfate reduction, which results in reduced butyrate levels and increased epithelial permeability and bacterial translocation109. Multiple additional mechanisms have been suggested to contribute to the pathogenesis of IBD, such as microbial sensing, antigen processing and oxygen levels110. Nonetheless, it is still unclear whether dysbiosis is one of the causes of inflammation in patients with IBD or is merely the result of a disturbed intestinal environment.
Coeliac disease. Coeliac disease is an autoimmune intestinal disease that is triggered by an immune response to peptides found in dietary gluten, and it is accompanied by dysbiotic changes. However, no consistent microbial signature has been determined in patients with coeliac disease to date111. The pathogenesis of coeliac disease involves the induction of a gluten-specific inflammatory TH1 and TH17 cell response, as well as the targeted killing of intestinal epithelial cells by T cells112. Several studies have suggested that the composition and function of the intestinal microbiota may contribute to the development of coeliac disease in several ways113, yet no studies have been performed to elucidate the mechanisms by which dysbiosis could drive disease susceptibility. The levels of short-chain fatty acids (SCFAs) are modified in patients with coeliac disease113, which potentially indicates a mechanism by which the microbiota modulates oral tolerance. Establishing whether dysbiosis is a cause or consequence of coeliac disease remains a challenge for the future.
Rheumatoid arthritis. Rheumatoid arthritis is a systemic inflammatory disease that results in joint destruction. Germ-free mice are protected from the development of experimental arthritis114, which indicates a fundamental role of intestinal commensal bacteria in the development and progression of the disease. In humans, the presence of Prevotella copri correlated with disease in a cohort of patients with new-onset rheumatoid arthritis115. A recent metagenome-wide association study also highlighted Lactobacillus salivarius as a marker of rheumatoid arthritis116. The intestinal community in patients with rheumatoid arthritis featured deviations in several metagenomic functions, including metal ion metabolism, redox functions and arginine metabolism116; however, the contribution of the microbiome to the pathogenesis of human rheumatoid arthritis merits further study.
T1D. T1D is an autoimmune disease that originates from the T cell-mediated destruction of insulin-producing cells in the pancreas. Although approximately two-thirds of all patients with T1D have been found to have HLA risk alleles, less than 10% of all individuals carrying these alleles actually develop the disease, which suggests an important contribution of non-genetic factors in determining the proportion of individuals that ultimately develop this type of autoimmunity117. In a mouse model of T1D, the microbiota has been implicated as an important contributor to disease pathogenesis65. A recent study in humans concluded that intestinal community alterations, including loss of bacterial diversity, occur after the seroconversion of patients with T1D but precede the onset of diabetes symptoms29, which raises the possibility that the microbiota causally contributes to the instigation of autoimmunity.
Asthma. Early microbial colonization of mucosal tissues during infancy has long-lasting influences, among them the development of disease later in life. Germ-free conditions and early-life antibiotic exposure are associated with increased susceptibility to allergy and asthma118. Studies in animal models of asthma suggest that neonatal colonization influences the education of the immune system. For instance, the frequency of intestinal regulatory T (Treg) cells is reduced in vancomycin-treated mice, and IgE levels are concomitantly elevated119. In an additional study, antibiotic treatment elevated lung inflammation, IgE titres and circulating basophil numbers. It was postulated that the microbiota, through B cell-intrinsic MYD88 signalling, limits serum IgE levels and basophil abundance120, and that in the absence of microbiota, B cells preferentially undergo isotype switching to IgE (rather than IgA), which supports allergic inflammation121. iNKT cells represent an additional cell type that has a role in microbiota-driven asthma. Germ-free mice have elevated numbers of iNKT cells in the colonic lamina propria and in the lungs, which leads to a higher susceptibility of these mice to the development of asthma122. Early exposure to intestinal microbiota reduces iNKT cell abundance, partly through epigenetic modification of the gene that encodes CXC-chemokine ligand 16 (Ref. 122). In human studies, intestinal and lung dysbiosis characterized by reduced bacterial diversity correlated with asthma123. The intestinal microbiota of infants at risk of asthma exhibited microbial dysbiosis accompanied by reduced levels of faecal acetate, and restoration of four bacterial genera that have a decreased abundance in children at risk of asthma ameliorated airway inflammation in germ-free mice124. However, additional studies are needed to provide a causal link between dysbiosis and the inflammatory response that includes increased numbers of iNKT cells, elevated IgE levels and reduced Treg cell numbers in asthma.
Multiple sclerosis. Multiple sclerosis is an immune-mediated disease that affects the central nervous system, and both genetic and environmental factors contribute to its pathogenesis. The intestinal microbiome profile of human patients with multiple sclerosis is distinct from that of healthy controls, with decreased species richness in patients who have active disease125. In a mouse model of relapsing-remitting multiple sclerosis, spontaneous experimental autoimmune encephalomyelitis (EAE) in TCR-transgenic mice, it was suggested that the commensal microbiota, in addition to self-antigen recognition, is required for the induction of an autoimmune response126. Germ-free mice did not develop disease symptoms, whereas microbial colonization was sufficient to induce EAE development through the activation of autoreactive CD4+ T cells126. Further elucidation of microbial contributions to the pathogenesis of multiple sclerosis will constitute an exciting area of future research.
Thus, through the alteration of normal immune system reactivity, states of dysbiosis may directly influence immunological disease. In addition, dysbiosis can contribute to the large range of modern human diseases that are not classically considered as immune mediated, but feature an inflammatory component that often contributes to disease development, progression and clinical manifestations (see Box 2).
Dysbiosis in diagnostics and therapy
Given the association of dysbiosis with the aetiology of numerous diseases, the possibility of using information on the state and function of the microbiota for the diagnosis and therapy of human immune-mediated or immune-associated diseases is enthralling. Indeed, we have recently witnessed a revolution in analysis tools for surveying the microbiome, including DNA sequencing for the identification of strains and their genomes, RNA sequencing for the determination of microbial gene activity, and metabolome analysis. Several diseases are associated with the early development of dysbiosis, such that it occurs before the development of overt clinical manifestations. In a large cohort of untreated patients with newly diagnosed Crohn disease, a 'dysbiosis index' was proposed and shown to be associated with clinical parameters107. Inflammatory conditions strongly correlated with an overall decrease in microbiota species richness and an alteration in the abundance of several taxa, with mucosal samples correlating better with disease severity than did luminal faecal samples, while antibiotic use exacerbated microbial dysbiosis associated with disease.
This demonstrates the potential of shotgun metagenomic sequencing and non-targeted metabolomics to characterize microbial community function in IBD, which may enable the identification of microbial biomarkers127. It remains possible that the difficulty in identifying a clear disease-associated dysbiotic profile in IBD has its origin in the complexity of the disease, and in the fact that several different manifestations of intestinal inflammation and extra-intestinal involvement are jointly classified as IBD. A more individualized approach that links microbial community structure and disease phenotype might therefore prove more effective with regard to diagnosing early disease progression on the basis of microbial biomarkers. As such, the diagnostic value of the microbiome may lie in differentiating subtypes of IBD that share common clinical symptoms, and microbiome signatures that differentiate between IBD and other intestinal inflammatory diseases might be more valuable than those that merely distinguish between individuals with disease and individuals who are healthy128.
The potential for the microbiome as a diagnostic tool for immune-mediated diseases reaches far beyond the intestine. For example, in Parkinson disease, chronic constipation and changes in microbiota composition precede motor symptoms by years, and might be promising biomarkers for screening tests to aid early disease detection among individuals at risk of developing this disorder129. Similarly, the microbiota has been implicated in the development of Alzheimer disease130, which emphasizes the notion that microbiome-based diagnosis might provide opportunities for the early detection of neurodegenerative diseases. Further well-powered and appropriately analysed studies should take into consideration all layers of microbiome complexity, including profiling of the metagenome, metatranscriptome and metabolome signatures of the microbiota, to maximize the repertoire of microbial biomarkers that is available for early disease detection. Furthermore, as medications can affect and are metabolized by the microbiome, the identification of disease-associated signature microbiomes is most informative before patients undergo any treatment. When patients are sampled during medical therapy, potential medication-induced dysbiosis should be taken into consideration131.
Targeting of dysbiosis for therapy. Owing to our increasing understanding of the drivers and consequences of dysbiosis, an intensifying effort is being made to engineer or reconstitute the microbiota to prevent or treat disease. The most dramatic reconstitution is achieved by faecal microbiota transplantation (FMT), in which the entire intestinal community of a patient is replaced by the microbiota of a healthy donor. FMT achieved spectacular results in patients with pseudomembranous colitis caused by recurrent infection with antibiotic-resistant C. difficile132. Understanding the specific mechanisms of intermicrobial competition and the host-stimulatory activity of particular microorganisms may allow the development of more targeted interventions against pseudomembranous colitis in the future20.
The success of FMT in C. difficile infection has given rise to the hope that this procedure might have the potential to be similarly effective in treating other diseases that involve dysbiosis, such as IBD and colorectal cancer. Nevertheless, the chronic stability of the transferred microbiome in FMT remains elusive, as does the long-term efficacy of FMT when repeatedly performed133. As such, whether FMT can be an efficient alternative to current treatment protocols awaits the generation of robust data in future studies. Likewise, the application of antibiotics has been widely practised in IBD, as several randomized controlled studies suggested this approach to be of benefit. However, further studies have deemed these results inconsistent, thereby precluding clear conclusions about the effectiveness of antibiotic treatment in IBD134.
Another form of microbiome engineering consists of the administration of probiotics, such as Lactobacillus and Bifidobacterium species, to support the expansion of a healthy microbiota. Nevertheless, there is very limited evidence that supports the efficacy of probiotics in microbiome-associated disorders135. For example, a large number of studies have been performed on the use of probiotics in Crohn disease, but there is currently no overall evidence to support their widespread use. The reason for inconsistencies between studies with regard to antibiotics and probiotics for the treatment of dysbiosis in IBD might lie in the strong interindividual variability in the susceptibility of the intestinal microbial community to biotic intervention. For instance, a recent study examining the kinetics of effective Bifidobacterium longum engraftment in humans found that low pretreatment levels of both B. longum and microbial carbohydrate utilization gene expression are a requirement for efficient probiotic colonization136. Improved knowledge regarding the prerequisites for effective probiotic engraftment and the range of interindividual variability in the susceptibility of the microbiota to probiotic intervention may thus be pivotal for efficient and personalized approaches.
In contrast to the probiotic administration of specific strains, dietary prebiotics aim to modify the composition of the intestinal ecosystem through nutritional changes. Given the rapid and reproducible responsiveness of the microbiota to dietary intervention41, a promising microbiome-modulating approach consists of the rational design of personalized diets137. In the case of metabolic disease, knowledge about the microbiota composition aids in predicting individual responses to dietary intervention138. Deciphering the amenability of an individual patient's microbiome to change through dietary or biotic intervention may similarly offer the chance to better tailor a therapeutic intervention to the specific characteristics of a particular microbial ecosystem. A prime example of how a mechanistic understanding of the contribution of diet and the microbiome to immune-mediated disease can facilitate the design of new treatment options relates to atherosclerosis. The metabolism of the dietary lipid phosphatidylcholine, as well as that of l-carnitine, a red meat component, by commensal bacteria results in the accumulation of trimethylamine-N-oxide (TMAO), which promotes atherosclerosis and thrombosis47,139,140. Targeted inhibition of this reaction can reverse TMAO accumulation and ameliorate disease development141. Further understanding of which bacteria lead to the production of high amounts of TMAO will allow the identification of groups of individuals who are at higher risk of developing disease, and these individuals could potentially undergo microbiome correction before disease develops.
Intriguingly, in certain clinical contexts, inducing a shift in the composition of the intestinal microbiota may have positive effects on the host. The administration of the anticancer drug cyclophosphamide alters the intestinal microbial community, in particular inducing the outgrowth of Gram-positive bacteria and their translocation to secondary lymphoid organs. This drives TH1 and TH17 cell responses, which help to potentiate the anticancer effect of the drug142. Similarly, cancer treatment with antibodies against cytotoxic T lymphocyte antigen 4 (CTLA4) was associated with the outgrowth of B. fragilis, which in turn promoted immunostimulatory anticancer effects143. Furthermore, colonization with bifidobacteria was found to contribute to PD1 ligand 1 (PDL1)-targeted cancer immunotherapy through effects on CD8+ T cells144.
Challenges and future avenues
Although the primary goal of the human microbiome project was to establish the normality of intestinal microbial composition and function145, subsequent efforts have aimed to define and understand dysbiotic states associated with human disease146. This has resulted in a surge of recent associations of aberrant microbial composition with host phenotypic manifestations in both mice and humans. Although several of these new associations have brought about promising implications for future diagnostic and therapeutic approaches, various challenges need to be overcome by the field to harness the new wealth of information on different states of the microbial ecosystem and their role in disease development.
First, given the wide range of composition that the intestinal microbiota can assume in the absence of overt disease, the importance of appropriate controls for defining a microbial ecosystem as dysbiotic is imminent10. Thus, for studies of human disease-associated microbiomes, it might not be sufficient to compare individuals with a disease-free control cohort, but it may rather necessitate the analysis of control samples from the same living environment, ideally corrected for dietary habits7, sampling time60 and localization6. Furthermore, rather than simply comparing healthy individuals with those with disease, it might be more insightful to compare the microbiomes of patients across different diseases, and particularly across different manifestations of the same disease, to use the information provided by the microbiome to better define and stratify patients according to disease subtypes. A similar approach applies to studies in mice. The microbiota of wild-type mice varies considerably between vivaria and between commercial vendors23,147, thus diminishing the universality of local compositional comparisons of the microbial taxa present in different mice.
Second, as host–microbiome co-evolution probably selected for microbial function rather than microbial composition5, the concept of dysbiosis likewise deserves a functional rather than a taxonomic interpretation. Relative to microbial composition, microbial functionalities and metabolite profiles associated with a particular condition or genotype might be not only more consistent across populations, geography and animal facilities23, but also of much higher causative relevance for the associated disease manifestation. As such, the field should strive to achieve an understanding of dysbiosis, its triggers, and the maintenance of its different stable states at the level of metagenomic gene expression and metabolite abundance (Fig. 1).
Third, the extent and manifestation of dysbiosis seem to be highly context dependent. The evolution of the microbiota has occurred on the background of several thousands of host genes, and the roles of complementarity and redundancy need to be taken into consideration when evaluating phenome–biome interactions. For instance, the effect of a particular genomic mutation on disease susceptibility might only become apparent in the context of a particular microbial configuration148. Similarly, susceptibility to dysbiosis development might only become relevant in the combinatorial context of host genotype and environmental microbial 'repertoire', including diet and household (or animal vivarium).
Finally, an improved understanding of the precise microbiota-derived or microbiota-modulated molecules that mediate the impact of dysbiosis on disease development may allow the design of metabolite-based interventions that act directly on the physiology of the host, thereby bypassing the vast complexity of interindividual variability in microbiota composition and disease associations. Such interventions, aptly termed 'postbiotics', have proved efficacious in mouse models of IBD23,149 and allergic inflammation150, among other models of microbiome-associated conditions151. The potential superiority of the postbiotic approach lies in its ease of application and its reduced complexity compared with interventions that aim to modulate the entire microbial ecosystem that resides in the gut. As such, the advancement of insights into the molecular mechanisms that drive dysbiosis-associated pathologies may enable the development of individualized dietary, probiotic and postbiotic interventions that could control the development, progression and variable manifestations of immune-mediated and immune-associated diseases.
References
Bach, J. F. The effect of infections on susceptibility to autoimmune and allergic diseases. N. Engl. J. Med. 347, 911–920 (2002).
Egger, G. & Dixon, J. Beyond obesity and lifestyle: a review of 21st century chronic disease determinants. Biomed Res. Int. 2014, 731685 (2014).
Kelsen, J. R. & Wu, G. D. The gut microbiota, environment and diseases of modern society. Gut Microbes 3, 374–382 (2012).
Lozupone, C. A., Stombaugh, J. I., Gordon, J. I., Jansson, J. K. & Knight, R. Diversity, stability and resilience of the human gut microbiota. Nature 489, 220–230 (2012).
Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature 486, 207–214 (2012). This study highlights extensive interindividual variability in the composition of the microbiome of healthy individuals.
Wu, G. D. et al. Linking long-term dietary patterns with gut microbial enterotypes. Science 334, 105–108 (2011).
Yatsunenko, T. et al. Human gut microbiome viewed across age and geography. Nature 486, 222–227 (2012).
David, L. A. et al. Host lifestyle affects human microbiota on daily timescales. Genome Biol. 15, R89 (2014).
Lloyd-Price, J., Abu-Ali, G. & Huttenhower, C. The healthy human microbiome. Genome Med. 8, 51 (2016).
Stappenbeck, T. S. & Virgin, H. W. Accounting for reciprocal host−microbiome interactions in experimental science. Nature 534, 191–199 (2016).
Thaiss, C. A. et al. Persistent microbiome alterations modulate the rate of post-dieting weight regain. Nature 540, 544–551 (2016).
Fonseca, D. M. et al. Microbiota-dependent sequelae of acute infection compromise tissue-specific immunity. Cell 163, 354–366 (2015). This paper describes a role for the microbiome in mediating post-infection immune system aberrations.
Chow, J. & Mazmanian, S. K. A pathobiont of the microbiota balances host colonization and intestinal inflammation. Cell Host Microbe 7, 265–276 (2010).
Stecher, B., Maier, L. & Hardt, W. D. 'Blooming' in the gut: how dysbiosis might contribute to pathogen evolution. Nat. Rev. Microbiol. 11, 277–284 (2013).
Frank, D. N. et al. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc. Natl Acad. Sci. USA 104, 13780–13785 (2007).
Garrett, W. S. et al. Communicable ulcerative colitis induced by T-bet deficiency in the innate immune system. Cell 131, 33–45 (2007). This study describes microbially transmissible colitis in mice that lack T-bet in the innate immune system.
Korem, T. et al. Growth dynamics of gut microbiota in health and disease inferred from single metagenomic samples. Science 349, 1101–1106 (2015).
Buffington, S. A. et al. Microbial reconstitution reverses maternal diet-induced social and synaptic deficits in offspring. Cell 165, 1762–1775 (2016).
Hsiao, E. Y. et al. Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell 155, 1451–1463 (2013).
Buffie, C. G. et al. Precision microbiome reconstitution restores bile acid mediated resistance to Clostridium difficile. Nature 517, 205–208 (2015). References 18–20 describe how microbiome reconstitution approaches can be used to treat dysbiosis-associated phenotypes.
Sonnenburg, E. D. et al. Diet-induced extinctions in the gut microbiota compound over generations. Nature 529, 212–215 (2016).
Erny, D. et al. Host microbiota constantly control maturation and function of microglia in the CNS. Nat. Neurosci. 18, 965–977 (2015).
Levy, M. et al. Microbiota-modulated metabolites shape the intestinal microenvironment by regulating NLRP6 inflammasome signaling. Cell 163, 1428–1443 (2015). This study demonstrates that microbial hijacking of the innate immune system contributes to the establishment of dysbiosis.
Sampson, T. R. et al. Gut microbiota regulate motor deficits and neuroinflammation in a model of Parkinson's disease. Cell 167, 1469–1480.e12 (2016).
Cotillard, A. et al. Dietary intervention impact on gut microbial gene richness. Nature 500, 585–588 (2013).
Le Chatelier, E. et al. Richness of human gut microbiome correlates with metabolic markers. Nature 500, 541–546 (2013). References 25 and 26 report that the decrease in microbiome richness predisposes patients to dysmetabolism and low-grade inflammation.
Norman, J. M. et al. Disease-specific alterations in the enteric virome in inflammatory bowel disease. Cell 160, 447–460 (2015).
Monaco, C. L. et al. Altered virome and bacterial microbiome in human immunodeficiency virus-associated acquired immunodeficiency syndrome. Cell Host Microbe 19, 311–322 (2016).
Kostic, A. D. et al. The dynamics of the human infant gut microbiome in development and in progression toward type 1 diabetes. Cell Host Microbe 17, 260–273 (2015).
Mosca, A., Leclerc, M. & Hugot, J. P. Gut microbiota diversity and human diseases: should we reintroduce key predators in our ecosystem? Front. Microbiol. 7, 455 (2016).
Lupp, C. et al. Host-mediated inflammation disrupts the intestinal microbiota and promotes the overgrowth of Enterobacteriaceae. Cell Host Microbe 2, 119–129 (2007).
Stecher, B. et al. Salmonella enterica serovar typhimurium exploits inflammation to compete with the intestinal microbiota. PLoS Biol. 5, 2177–2189 (2007).
Arthur, J. C. et al. Intestinal inflammation targets cancer-inducing activity of the microbiota. Science 338, 120–123 (2012).
Ayres, J. S., Trinidad, N. J. & Vance, R. E. Lethal inflammasome activation by a multidrug-resistant pathobiont upon antibiotic disruption of the microbiota. Nat. Med. 18, 799–806 (2012).
Ng, K. M. et al. Microbiota-liberated host sugars facilitate post-antibiotic expansion of enteric pathogens. Nature 502, 96–99 (2013).
Deriu, E. et al. Probiotic bacteria reduce Salmonella Typhimurium intestinal colonization by competing for iron. Cell Host Microbe 14, 26–37 (2013).
Stecher, B. et al. Gut inflammation can boost horizontal gene transfer between pathogenic and commensal Enterobacteriaceae. Proc. Natl Acad. Sci. USA 109, 1269–1274 (2012).
Behnsen, J. et al. The cytokine IL-22 promotes pathogen colonization by suppressing related commensal bacteria. Immunity 40, 262–273 (2014).
Lopez, C. A. et al. Virulence factors enhance Citrobacter rodentium expansion through aerobic respiration. Science 353, 1249–1253 (2016).
Winter, S. E. et al. Host-derived nitrate boosts growth of E. coli in the inflamed gut. Science 339, 708–711 (2013).
David, L. A. et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 505, 559–563 (2014).
Denou, E., Marcinko, K., Surette, M. G., Steinberg, G. R. & Schertzer, J. D. High-intensity exercise training increases the diversity and metabolic capacity of the mouse distal gut microbiota during diet-induced obesity. Am. J. Physiol. Endocrinol. Metab. 310, E982–E993 (2016).
Cho, I. et al. Antibiotics in early life alter the murine colonic microbiome and adiposity. Nature 488, 621–626 (2012).
Suez, J. et al. Artificial sweeteners induce glucose intolerance by altering the gut microbiota. Nature 514, 181–186 (2014).
Chassaing, B. et al. Dietary emulsifiers impact the mouse gut microbiota promoting colitis and metabolic syndrome. Nature 519, 92–96 (2015).
Yoshimoto, S. et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 499, 97–101 (2013).
Zhu, W. et al. Gut microbial metabolite TMAO enhances platelet hyperreactivity and thrombosis risk. Cell 165, 111–124 (2016).
Levy, M., Thaiss, C. A. & Elinav, E. Metagenomic cross-talk: the regulatory interplay between immunogenomics and the microbiome. Genome Med. 7, 120 (2015).
Goodrich, J. K. et al. Human genetics shape the gut microbiome. Cell 159, 789–799 (2014).
Bonder, M. J. et al. The effect of host genetics on the gut microbiome. Nat. Genet. 48, 1407–1412 (2016).
Turpin, W. et al. Association of host genome with intestinal microbial composition in a large healthy cohort. Nat. Genet. 48, 1413–1417 (2016).
Wang, J. et al. Genome-wide association analysis identifies variation in vitamin D receptor and other host factors influencing the gut microbiota. Nat. Genet. 48, 1396–1406 (2016). References 49–52 highlight the influence of host genetics on microbiota composition.
Benson, A. K. et al. Individuality in gut microbiota composition is a complex polygenic trait shaped by multiple environmental and host genetic factors. Proc. Natl Acad. Sci. USA 107, 18933–18938 (2010).
Carmody, R. N. et al. Diet dominates host genotype in shaping the murine gut microbiota. Cell Host Microbe 17, 72–84 (2015).
Koenig, J. E. et al. Succession of microbial consortia in the developing infant gut microbiome. Proc. Natl Acad. Sci. USA 108 (Suppl. 1), 4578–4585 (2011).
Dominguez-Bello, M. G. et al. Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proc. Natl Acad. Sci. USA 107, 11971–11975 (2010).
McCafferty, J. et al. Stochastic changes over time and not founder effects drive cage effects in microbial community assembly in a mouse model. ISME J. 7, 2116–2125 (2013).
Lax, S. et al. Longitudinal analysis of microbial interaction between humans and the indoor environment. Science 345, 1048–1052 (2014).
Ubeda, C. et al. Familial transmission rather than defective innate immunity shapes the distinct intestinal microbiota of TLR-deficient mice. J. Exp. Med. 209, 1445–1456 (2012).
Thaiss, C. A. et al. Transkingdom control of microbiota diurnal oscillations promotes metabolic homeostasis. Cell 159, 514–529 (2014).
Voigt, R. M. et al. The circadian clock mutation promotes intestinal dysbiosis. Alcohol. Clin. Exp. Res. 40, 335–347 (2016).
Koren, O. et al. Host remodeling of the gut microbiome and metabolic changes during pregnancy. Cell 150, 470–480 (2012).
Kigerl, K. A. et al. Gut dysbiosis impairs recovery after spinal cord injury. J. Exp. Med. 213, 2603–2620 (2016).
Thaiss, C. A., Levy, M., Suez, J. & Elinav, E. The interplay between the innate immune system and the microbiota. Curr. Opin. Immunol. 26, 41–48 (2014).
Wen, L. et al. Innate immunity and intestinal microbiota in the development of type 1 diabetes. Nature 455, 1109–1113 (2008).
Frantz, A. L. et al. Targeted deletion of MyD88 in intestinal epithelial cells results in compromised antibacterial immunity associated with downregulation of polymeric immunoglobulin receptor, mucin-2, and antibacterial peptides. Mucosal Immunol. 5, 501–512 (2012).
Vijay-Kumar, M. et al. Metabolic syndrome and altered gut microbiota in mice lacking Toll-like receptor 5. Science 328, 228–231 (2010). The authors of this study report microbial transmissible metabolic syndrome in mice that lack TLR5.
Carvalho, F. A. et al. Transient inability to manage proteobacteria promotes chronic gut inflammation in TLR5-deficient mice. Cell Host Microbe 12, 139–152 (2012).
Bouskra, D. et al. Lymphoid tissue genesis induced by commensals through NOD1 regulates intestinal homeostasis. Nature 456, 507–510 (2008).
Couturier-Maillard, A. et al. NOD2-mediated dysbiosis predisposes mice to transmissible colitis and colorectal cancer. J. Clin. Invest. 123, 700–711 (2013).
Petnicki-Ocwieja, T. et al. Nod2 is required for the regulation of commensal microbiota in the intestine. Proc. Natl Acad. Sci. USA 106, 15813–15818 (2009).
Rehman, A. et al. Nod2 is essential for temporal development of intestinal microbial communities. Gut 60, 1354–1362 (2011).
Li, E. et al. Inflammatory bowel diseases phenotype. C. difficile and NOD2 genotype are associated with shifts in human ileum associated microbial composition. PLoS ONE 7, e26284 (2012).
Robertson, S. J. et al. Nod1 and Nod2 signaling does not alter the composition of intestinal bacterial communities at homeostasis. Gut Microbes 4, 222–231 (2013).
Henao-Mejia, J., Elinav, E., Thaiss, C. A. & Flavell, R. A. Inflammasomes and metabolic disease. Annu. Rev. Physiol. 76, 57–78 (2014).
Elinav, E. et al. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell 145, 745–757 (2011).
Henao-Mejia, J. et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature 482, 179–185 (2012).
Hu, B. et al. Microbiota-induced activation of epithelial IL-6 signaling links inflammasome-driven inflammation with transmissible cancer. Proc. Natl Acad. Sci. USA 110, 9862–9867 (2013).
Wlodarska, M. et al. NLRP6 inflammasome orchestrates the colonic host–microbial interface by regulating goblet cell mucus secretion. Cell 156, 1045–1059 (2014).
Birchenough, G. M., Nystrom, E. E., Johansson, M. E. & Hansson, G. C. A sentinel goblet cell guards the colonic crypt by triggering Nlrp6-dependent Muc2 secretion. Science 352, 1535–1542 (2016).
Rausch, P. et al. Analysis of factors contributing to variation in the C57BL/6J fecal microbiota across German animal facilities. Int. J. Med. Microbiol. 306, 343–355 (2016).
Chudnovskiy, A. et al. Host–protozoan interactions protect from mucosal infections through activation of the inflammasome. Cell 167, 444–456.e14 (2016).
Salzman, N. H. et al. Enteric defensins are essential regulators of intestinal microbial ecology. Nat. Immunol. 11, 76–83 (2010).
Vaishnava, S. et al. The antibacterial lectin RegIIIγ promotes the spatial segregation of microbiota and host in the intestine. Science 334, 255–258 (2011).
Gury-BenAri, M. et al. The spectrum and regulatory landscape of intestinal innate lymphoid cells are shaped by the microbiome. Cell 166, 1231–1246.e13 (2016).
Sonnenberg, G. F. & Artis, D. Innate lymphoid cell interactions with microbiota: implications for intestinal health and disease. Immunity 37, 601–610 (2012).
Zheng, Y. et al. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat. Med. 14, 282–289 (2008).
Qiu, J. et al. Group 3 innate lymphoid cells inhibit T-cell-mediated intestinal inflammation through aryl hydrocarbon receptor signaling and regulation of microflora. Immunity 39, 386–399 (2013).
Sonnenberg, G. F. et al. Innate lymphoid cells promote anatomical containment of lymphoid-resident commensal bacteria. Science 336, 1321–1325 (2012).
Powell, N. et al. The transcription factor T-bet regulates intestinal inflammation mediated by interleukin-7 receptor+ innate lymphoid cells. Immunity 37, 674–684 (2012).
Peterson, D. A., McNulty, N. P., Guruge, J. L. & Gordon, J. I. IgA response to symbiotic bacteria as a mediator of gut homeostasis. Cell Host Microbe 2, 328–339 (2007).
Cullender, T. C. et al. Innate and adaptive immunity interact to quench microbiome flagellar motility in the gut. Cell Host Microbe 14, 571–581 (2013).
Palm, N. W. et al. Immunoglobulin A coating identifies colitogenic bacteria in inflammatory bowel disease. Cell 158, 1000–1010 (2014).
Hapfelmeier, S. et al. Reversible microbial colonization of germ-free mice reveals the dynamics of IgA immune responses. Science 328, 1705–1709 (2010).
Shulzhenko, N. et al. Crosstalk between B lymphocytes, microbiota and the intestinal epithelium governs immunity versus metabolism in the gut. Nat. Med. 17, 1585–1593 (2011).
Fagarasan, S. et al. Critical roles of activation-induced cytidine deaminase in the homeostasis of gut flora. Science 298, 1424–1427 (2002).
Suzuki, K. et al. Aberrant expansion of segmented filamentous bacteria in IgA-deficient gut. Proc. Natl Acad. Sci. USA 101, 1981–1986 (2004).
Kawamoto, S. et al. The inhibitory receptor PD-1 regulates IgA selection and bacterial composition in the gut. Science 336, 485–489 (2012).
Nieuwenhuis, E. E. et al. Cd1d-dependent regulation of bacterial colonization in the intestine of mice. J. Clin. Invest. 119, 1241–1250 (2009).
Ismail, A. S. et al. γδ intraepithelial lymphocytes are essential mediators of host-microbial homeostasis at the intestinal mucosal surface. Proc. Natl Acad. Sci. USA 108, 8743–8748 (2011).
Thaiss, C. A., Zmora, N., Levy, M. & Elinav, E. The microbiome and innate immunity. Nature 535, 65–74 (2016).
Vatanen, T. et al. Variation in microbiome LPS immunogenicity contributes to autoimmunity in humans. Cell 165, 842–853 (2016). This study demonstrates that variations in the immunostimulatory activity of LPS in different microbiomes are associated with the development of autoimmune diseases in children.
Maekawa, T. et al. Porphyromonas gingivalis manipulates complement and TLR signaling to uncouple bacterial clearance from inflammation and promote dysbiosis. Cell Host Microbe 15, 768–778 (2014).
Hajishengallis, G. et al. Low-abundance biofilm species orchestrates inflammatory periodontal disease through the commensal microbiota and complement. Cell Host Microbe 10, 497–506 (2011).
Zenewicz, L. A. et al. IL-22 deficiency alters colonic microbiota to be transmissible and colitogenic. J. Immunol. 190, 5306–5312 (2013).
Moon, C. et al. Vertically transmitted faecal IgA levels determine extra-chromosomal phenotypic variation. Nature 521, 90–93 (2015). This paper shows that commensal bacteria modulate intestinal antibody levels via secretory IgA degradation.
Gevers, D. et al. The treatment-naive microbiome in new-onset Crohn's disease. Cell Host Microbe 15, 382–392 (2014).
Strauss, J. et al. Invasive potential of gut mucosa-derived Fusobacterium nucleatum positively correlates with IBD status of the host. Inflamm. Bowel Dis. 17, 1971–1978 (2011).
Li, J., Butcher, J., Mack, D. & Stintzi, A. Functional impacts of the intestinal microbiome in the pathogenesis of inflammatory bowel disease. Inflamm. Bowel Dis. 21, 139–153 (2015).
Rigottier-Gois, L. Dysbiosis in inflammatory bowel diseases: the oxygen hypothesis. ISME J. 7, 1256–1261 (2013).
Wacklin, P. et al. The duodenal microbiota composition of adult celiac disease patients is associated with the clinical manifestation of the disease. Inflamm. Bowel Dis. 19, 934–941 (2013).
Jabri, B. & Sollid, L. M. Tissue-mediated control of immunopathology in coeliac disease. Nat. Rev. Immunol. 9, 858–870 (2009).
Di Cagno, R. et al. Duodenal and faecal microbiota of celiac children: molecular, phenotype and metabolome characterization. BMC Microbiol. 11, 219 (2011).
Wu, H. J. et al. Gut-residing segmented filamentous bacteria drive autoimmune arthritis via T helper 17 cells. Immunity 32, 815–827 (2010).
Scher, J. U. et al. Expansion of intestinal Prevotella copri correlates with enhanced susceptibility to arthritis. eLife 2, e01202 (2013).
Zhang, X. et al. The oral and gut microbiomes are perturbed in rheumatoid arthritis and partly normalized after treatment. Nat. Med. 21, 895–905 (2015).
Achenbach, P., Bonifacio, E., Koczwara, K. & Ziegler, A. G. Natural history of type 1 diabetes. Diabetes 54 (Suppl. 2), S25–S31 (2005).
Risnes, K. R., Belanger, K., Murk, W. & Bracken, M. B. Antibiotic exposure by 6 months and asthma and allergy at 6 years: findings in a cohort of 1,401 US children. Am. J. Epidemiol. 173, 310–318 (2011).
Russell, S. L. et al. Early life antibiotic-driven changes in microbiota enhance susceptibility to allergic asthma. EMBO Rep. 13, 440–447 (2012).
Hill, D. A. et al. Commensal bacteria-derived signals regulate basophil hematopoiesis and allergic inflammation. Nat. Med. 18, 538–546 (2012).
Cahenzli, J., Koller, Y., Wyss, M., Geuking, M. B. & McCoy, K. D. Intestinal microbial diversity during early-life colonization shapes long-term IgE levels. Cell Host Microbe 14, 559–570 (2013).
Olszak, T. et al. Microbial exposure during early life has persistent effects on natural killer T cell function. Science 336, 489–493 (2012).
Bisgaard, H. et al. Childhood asthma after bacterial colonization of the airway in neonates. N. Engl. J. Med. 357, 1487–1495 (2007).
Arrieta, M. C. et al. Early infancy microbial and metabolic alterations affect risk of childhood asthma. Sci. Transl Med. 7, 307ra152 (2015).
Chen, J. et al. Multiple sclerosis patients have a distinct gut microbiota compared to healthy controls. Sci. Rep. 6, 28484 (2016).
Berer, K. et al. Commensal microbiota and myelin autoantigen cooperate to trigger autoimmune demyelination. Nature 479, 538–541 (2011).
Marchesi, J. R. et al. Rapid and noninvasive metabonomic characterization of inflammatory bowel disease. J. Proteome Res. 6, 546–551 (2007).
Olesen, S. W. & Alm, E. J. Dysbiosis is not an answer. Nat. Microbiol. 1, 16228 (2016).
Scheperjans, F. et al. Gut microbiota are related to Parkinson's disease and clinical phenotype. Mov. Disord. 30, 350–358 (2015).
Minter, M. R. et al. Antibiotic-induced perturbations in gut microbial diversity influences neuro-inflammation and amyloidosis in a murine model of Alzheimer's disease. Sci. Rep. 6, 30028 (2016).
Forslund, K. et al. Disentangling type 2 diabetes and metformin treatment signatures in the human gut microbiota. Nature 528, 262–266 (2015).
van Nood, E. et al. Duodenal infusion of donor feces for recurrent Clostridium difficile. N. Engl. J. Med. 368, 407–415 (2013).
Angelberger, S. et al. Temporal bacterial community dynamics vary among ulcerative colitis patients after fecal microbiota transplantation. Am. J. Gastroenterol. 108, 1620–1630 (2013).
Khan, K. J. et al. Antibiotic therapy in inflammatory bowel disease: a systematic review and meta-analysis. Am. J. Gastroenterol. 106, 661–673 (2011).
Wolvers, D. et al. Guidance for substantiating the evidence for beneficial effects of probiotics: prevention and management of infections by probiotics. J. Nutr. 140, 698S–712S (2010).
Maldonado-Gomez, M. X. et al. Stable engraftment of Bifidobacterium longum AH1206 in the human gut depends on individualized features of the resident microbiome. Cell Host Microbe 20, 515–526 (2016).
Zmora, N., Zeevi, D., Korem, T., Segal, E. & Elinav, E. Taking it personally: personalized utilization of the human microbiome in health and disease. Cell Host Microbe 19, 12–20 (2016).
Zeevi, D. et al. Personalized nutrition by prediction of glycemic responses. Cell 163, 1079–1094 (2015).
Koeth, R. A. et al. Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat. Med. 19, 576–585 (2013).
Wang, Z. et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 472, 57–63 (2011).
Wang, Z. et al. Non-lethal inhibition of gut microbial trimethylamine production for the treatment of atherosclerosis. Cell 163, 1585–1595 (2015).
Viaud, S. et al. The intestinal microbiota modulates the anticancer immune effects of cyclophosphamide. Science 342, 971–976 (2013).
Vétizou, M. et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science 350, 1079–1084 (2015).
Sivan, A. et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science 350, 1084–1089 (2015).
Turnbaugh, P. J. et al. The human microbiome project. Nature 449, 804–810 (2007).
Thaiss, C. A. & Elinav, E. Exploring new horizons in microbiome research. Cell Host Microbe 15, 662–667 (2014).
Ivanov, I. I. et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 139, 485–498 (2009).
Chu, H. et al. Gene–microbiota interactions contribute to the pathogenesis of inflammatory bowel disease. Science 352, 1116–1120 (2016).
Tsilingiri, K. et al. Probiotic and postbiotic activity in health and disease: comparison on a novel polarised ex-vivo organ culture model. Gut 61, 1007–1015 (2012).
Trompette, A. et al. Gut microbiota metabolism of dietary fiber influences allergic airway disease and hematopoiesis. Nat. Med. 20, 159–166 (2014).
Levy, M., Thaiss, C. A. & Elinav, E. Metabolites: messengers between the microbiota and the immune system. Genes Dev. 30, 1589–1597 (2016).
Chang, P. V., Hao, L., Offermanns, S. & Medzhitov, R. The microbial metabolite butyrate regulates intestinal macrophage function via histone deacetylase inhibition. Proc. Natl Acad. Sci. USA 111, 2247–2252 (2014).
Zelante, T. et al. Tryptophan catabolites from microbiota engage aryl hydrocarbon receptor and balance mucosal reactivity via interleukin-22. Immunity 39, 372–385 (2013).
Gomez de Aguero, M. et al. The maternal microbiota drives early postnatal innate immune development. Science 351, 1296–1302 (2016).
Atarashi, K. et al. Th17 cell induction by adhesion of microbes to intestinal epithelial cells. Cell 163, 367–380 (2015).
Sano, T. et al. An IL-23R/IL-22 circuit regulates epithelial serum amyloid A to promote local effector Th17 responses. Cell 163, 381–393 (2015).
Lecuyer, E. et al. Segmented filamentous bacterium uses secondary and tertiary lymphoid tissues to induce gut IgA and specific T helper 17 cell responses. Immunity 40, 608–620 (2014).
Mazmanian, S. K., Liu, C. H., Tzianabos, A. O. & Kasper, D. L. An immunomodulatory molecule of symbiotic bacteria directs maturation of the host immune system. Cell 122, 107–118 (2005).
Sefik, E. et al. Individual intestinal symbionts induce a distinct population of RORγ+ regulatory T cells. Science 349, 993–997 (2015).
Atarashi, K. et al. Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature 500, 232–236 (2013).
Arpaia, N. et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature 504, 451–455 (2013).
Furusawa, Y. et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 504, 446–450 (2013).
Smith, P. M. et al. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science 341, 569–573 (2013).
Bunker, J. J. et al. Innate and adaptive humoral responses coat distinct commensal bacteria with immunoglobulin A. Immunity 43, 541–553 (2015).
Jiang, W. et al. Dysbiosis gut microbiota associated with inflammation and impaired mucosal immune function in intestine of humans with non-alcoholic fatty liver disease. Sci. Rep. 5, 8096 (2015).
Ridaura, V. K. et al. Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science 341, 1241214 (2013).
Vrieze, A. et al. Transfer of intestinal microbiota from lean donors increases insulin sensitivity in individuals with metabolic syndrome. Gastroenterology 143, 913–916.e7 (2012).
Reijnders, D. et al. Effects of gut microbiota manipulation by antibiotics on host metabolism in obese humans: a randomized double-blind placebo-controlled trial. Cell Metab. 24, 63–74 (2016).
Caesar, R., Tremaroli, V., Kovatcheva-Datchary, P., Cani, P. D. & Backhed, F. Crosstalk between gut microbiota and dietary lipids aggravates WAT inflammation through TLR signaling. Cell Metab. 22, 658–668 (2015).
Castellarin, M. et al. Fusobacterium nucleatum infection is prevalent in human colorectal carcinoma. Genome Res. 22, 299–306 (2012).
Kostic, A. D. et al. Genomic analysis identifies association of Fusobacterium with colorectal carcinoma. Genome Res. 22, 292–298 (2012).
Dapito, D. H. et al. Promotion of hepatocellular carcinoma by the intestinal microbiota and TLR4. Cancer Cell 21, 504–516 (2012).
Choi, G. B. et al. The maternal interleukin-17a pathway in mice promotes autism-like phenotypes in offspring. Science 351, 933–939 (2016).
Finegold, S. M. et al. Pyrosequencing study of fecal microflora of autistic and control children. Anaerobe 16, 444–453 (2010).
Sandler, R. H. et al. Short-term benefit from oral vancomycin treatment of regressive-onset autism. J. Child Neurol. 15, 429–435 (2000).
Acknowledgements
The authors thank the members of the Elinav laboratory for fruitful discussions. They apologize to those authors whose work could not be cited owing to space limitations. A.A.K. is supported by a European Molecular Biology Organization postdoctoral fellowship. C.A.T. received a Boehringer Ingelheim Fonds Ph.D. Fellowship. E.E. is supported by Yael and Rami Ungar, Israel; the Leona M. and Harry B. Helmsley Charitable Trust; the Gurwin Family Fund for Scientific Research; the Crown Endowment Fund for Immunological Research; the estate of Jack Gitlitz; the estate of Lydia Hershkovich; the Benoziyo Endowment Fund for the Advancement of Science; the Adelis Foundation; John L. and Vera Schwartz, Pacific Palisades, California, USA; Alan Markovitz, Canada; Cynthia Adelson, Canada; Centre National de la Recherche Scientifique; the estate of Samuel and Alwyn J. Weber; Mr and Mrs Schwarz, Sherman Oaks, California, USA; grants funded by the European Research Council; the German–Israel Binational foundation; the Israel Science Foundation; the Minerva Foundation; the Rising Tide Foundation; and the Alon Foundation scholar award. E.E. is the incumbent of the Rina Gudinski Career Development Chair and a senior fellow of the Canadian Institute For Advanced Research.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Glossary
- Xenobiotics
-
Small chemical compounds that enter an organism unnaturally, such as drugs or pollutants.
- Koch's postulates
-
A list of criteria that a microorganism needs to fulfil to be considered the causative agent of a disease, including its presence in all cases of the disease, the ability to grow the microorganism in pure culture, transmissibility of the disease by inoculation of a healthy organism and the re-isolation of the microorganism from the infected host.
- Alpha diversity
-
Alpha diversity describes species richness within a site, in contrast to beta diversity, which refers to differences in species composition between sites.
- Antimicrobial peptides
-
Host-derived peptides that are part of the innate immune system and function in host defence against microorganisms.
- Metabolic syndrome
-
A syndrome of co-occurring conditions, including elevated levels of plasma glucose, high blood pressure, abdominal obesity, elevated serum triglyceride levels and low serum high-density lipoprotein levels, that collectively increase the risk of diabetes, stroke and heart disease.
- Inflammasomes
-
Multiprotein complexes composed of a NOD-like receptor protein, the adaptor protein ASC and caspase 1. Inflammasomes contribute to the secretion of interleukin-1β (IL-1β) and IL-18 by activating caspase 1.
- Innate lymphoid cells
-
(ILCs). Cells of the lymphoid lineage that do not express antigen-specific receptors, but orchestrate tissue homeostasis and immunity through cytokine secretion.
- Metabolome
-
The entirety of small-molecule metabolites at a particular site.
- Probiotics
-
Microorganisms that are administered to an organism to benefit the host.
- Prebiotics
-
Ingredients of food that promote the growth and metabolism of beneficial microorganisms in the host.
- Human microbiome project
-
A consortium project with the goal of comprehensively describing the commensal microorganisms associated with health and disease in humans.
Rights and permissions
About this article

Cite this article
Levy, M., Kolodziejczyk, A., Thaiss, C. et al. Dysbiosis and the immune system. Nat Rev Immunol 17, 219–232 (2017). https://doi.org/10.1038/nri.2017.7
Published:
Issue Date:
DOI: https://doi.org/10.1038/nri.2017.7
This article is cited by
-
Cross-reactive CD8+ T cell responses to tumor-associated antigens (TAAs) and homologous microbiota-derived antigens (MoAs)
Journal of Experimental & Clinical Cancer Research (2024)
-
Gut dysbiosis induces the development of depression-like behavior through abnormal synapse pruning in microglia-mediated by complement C3
Microbiome (2024)
-
Dissecting the respective roles of microbiota and host genetics in the susceptibility of Card9−/− mice to colitis
Microbiome (2024)
-
Fermented foods and gastrointestinal health: underlying mechanisms
Nature Reviews Gastroenterology & Hepatology (2024)
-
Paternal microbiome perturbations impact offspring fitness
Nature (2024)
