
Key Points
-
Recognizing the presence of invading pathogens is key to mounting an effective innate immune defence. Mammalian cells express different classes of germline-encoded pattern recognition receptors (PRRs) that monitor the extracellular and the intracellular compartments of host cells for signs of infection and that initiate several conserved signalling pathways.
-
Recent advances have identified several new extracellular and intracellular PRRs and have shed light on the complex interplay of innate immune signalling pathways during pathogen infection.
-
Although the function of most Toll-like receptors (TLRs) has been determined in the past, the functions of orphan mouse TLR11, TLR12 and TLR13, which are not found in humans, have so far eluded researchers. The ligands for these receptors have recently been characterized, which has revealed key differences between the human and mouse innate immune system.
-
Cytoplasmic DNA is a strong activator of immune responses and induces type I interferon (IFN) production through the signalling adaptor stimulator of IFN genes protein (STING). Several candidate proteins have been proposed to bind to double-stranded DNA and to induce type I IFN production. More recently, a previously unrecognized role for cyclic dinucleotides in this signalling pathway has been described.
-
NOD-like receptors are the largest group of intracellular receptors and are known for their ability to induce the assembly of inflammasome complexes. Recent reports have identified new inflammasome complexes and have characterized their function in the recognition of various bacterial pathogens.
-
Canonical inflammasomes have been defined as macromolecular complexes that activate the cysteine protease caspase 1. However, recent findings have highlighted the role of caspase 8 and caspase 11 in inducing pro-inflammatory cytokine maturation and host cell death.
Abstract
Recognizing the presence of invading pathogens is key to mounting an effective innate immune response. Mammalian cells express different classes of germline-encoded pattern recognition receptors that monitor the extracellular and intracellular compartments of host cells for signs of infection and that activate several conserved signalling pathways. An efficient immune response often requires the sequential detection of a pathogen by different receptors in different subcellular compartments, which results in a complex interplay of downstream signalling pathways. In this Review, we discuss the recent identification of previously unknown pattern recognition receptors and how they complement the repertoire of established receptors.
Similar content being viewed by others
Main
The mammalian innate immune system provides a first line of defence against microbial attack through phagocytosis and the induction of inflammation. These responses are stimulated by several classes of germline-encoded pattern recognition receptors (PRRs) that primarily recognize conserved microbial molecules termed pathogen-associated molecular patterns (PAMPs) but that also recognize host-derived danger signals, which are released in response to stress, tissue damage and necrotic cell death1. Bacterial PAMPs are diverse and include various molecules ranging from lipoproteins, lipopolysaccharide (LPS), flagellin and peptidoglycan to unique bacterial nucleic acid structures, such as cyclic dinucleotides (CDNs). By contrast, viruses are mainly recognized through viral fusion glycoproteins and through unique nucleic acids, such as double-stranded RNA (dsRNA), uncapped single-stranded RNA (ssRNA) and viral DNA. Comparably little is known about the recognition of intracellular parasites. However, similar to other microorganisms, parasite recognition is dependent on the detection of unique molecules2.
PRRs initiate antimicrobial defence mechanisms through several conserved signalling pathways. The activation of transcription factors such as nuclear factor-κB (NF-κB) and interferon-regulatory factors (IRFs) promotes the production of inflammatory cytokines and type I interferons (IFNs), respectively. Other PRRs initiate the assembly of cytoplasmic signalling complexes, termed inflammasomes, which activate inflammatory caspases3,4. Active caspase 1 controls the maturation and the secretion of leaderless cytokines such as interleukin-1β (IL-1β) and IL-18, and induces pyroptosis, which is a lytic form of cell death that can restrict pathogen replication5. The inflammatory response that is induced by PRR activation recruits and activates circulating immune cells and is essential for priming adaptive immune responses.
Two main classes of PRRs have been described in mammalian cells: membrane-bound receptors, such as Toll-like receptors (TLRs) and C-type lectin receptors (CLRs), and cytoplasmic sensors, including NOD-like receptors (NLRs), pyrin and HIN domain-containing (PYHIN) family members, RIG-I-like receptors (RLRs) and an increasing range of cytosolic nucleic acid sensors. TLRs were the first group of PRRs to be characterized and they recognize PAMPs in the extracellular compartment or within endosomes6. Following ligation with their ligands, TLRs interact with different combinations of the adaptor proteins TIR domain-containing adaptor protein (TIRAP; also known as MAL), myeloid differentiation primary-response protein 88 (MYD88), TIR-domain-containing adaptor protein inducing IFNβ (TRIF; also known as TICAM1) and TRIF-related adaptor molecule (TRAM; also known as TICAM2)7. The MYD88-dependent pathway controls the activation of mitogen-activated protein kinases (MAPKs) and the transcription factor NF-κB, whereas the TRIF-dependent pathway mainly mediates type I IFN production. Plasmacytoid dendritic cells (pDCs) have an unusual network of signalling pathways that links MYD88 to IRF7 and that enables these cells to produce large quantities of IFNα in response to TLR7 and TLR9 ligands8.
NLRs constitute the largest group of cytoplasmic receptors. The first members of this group that were identified — nucleotide-binding oligomerization domain-containing protein 1 (NOD1) and NOD2 — recognize peptidoglycan fragments and initiate both NF-κB activation and IFNβ production9. Some reports have also linked NOD2 to the recognition of RNA10. In addition, other members of the NLR family drive the assembly of inflammasome complexes in response to various danger signals and PAMPs4.
Nucleic acids and their derivatives are one of the most important groups of PAMPs, particularly in the innate immune response against viruses that otherwise present few conserved PAMPs11. Microbial nucleic acids can be discriminated from self nucleic acids using various parameters, such as their sequence, their tertiary structure, their molecular modifications and their localization. In addition, mislocalized DNA and RNA can be an indicator of cellular damage and infection. Different classes of PRRs recognize cytoplasmic nucleic acids and initiate several distinct immune responses. RLRs — comprising retinoic acid-inducible gene I (RIG-I), melanoma differentiation-associated protein 5 (MDA5) and LGP2 (also known as DHX58) — detect several different ssRNA and dsRNA viruses and induce type I IFN production through mitochondrial antiviral signalling protein (MAVS) and IRF3 (Refs 12, 13, 14, 15, 16, 17). The response to cytoplasmic DNA is not as well characterized, but can lead to type I IFN production, to inflammasome activation and even to the induction of a newly described cell death pathway that is linked to immunity, termed necroptosis18 (Box 1). A surprisingly large number of cytoplasmic DNA receptors have been identified in recent years, but many still await definitive validation.
In this Review, we discuss recently identified PRRs, their ligands, their modes of signalling and their interactions with other mammalian PRRs.
New functions for orphan TLRs
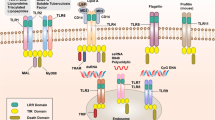
TLRs are arguably the best-studied group of PRRs. So far, 10 members of the TLR family have been identified in humans and 12 in mice, and a number of genetic studies have revealed their respective ligands and their modes of signalling (for a review see Refs 6, 19). TLR-mediated recognition of PAMPs can occur at the plasma membrane or at endosomal and endolysosomal membranes. TLR1, TLR2, TLR4, TLR5 and TLR6 primarily, but not exclusively, localize to the plasma membrane and recognize microbial components such as lipids, lipoproteins, LPS and proteins. Conversely, TLR3, TLR7, TLR8 and TLR9 localize to intracellular vesicular compartments and are involved in the recognition of nucleic acids (Table 1). Nevertheless, the ligands for several TLRs including TLR10, which is only found in humans, and TLR11, TLR12 and TLR13, which are present in mice but not humans, have remained unknown so far (Fig. 1).

Two Toll-like receptors (TLRs) — TLR5 and TLR11 — recognize bacterial flagellin and induce nuclear factor-κB (NF-κB) signalling through the adaptor molecules myeloid differentiation primary-response protein 88 (MYD88) and TIR domain-containing adaptor protein (TIRAP; also known as MAL). TLR5 localizes to the cell membrane, whereas TLR11 is thought to localize from the endoplasmic reticulum (ER) to the endolysosomal compartments (indicated by the dashed arrow), as it requires protein unc-93 homolog B1 (UNC93B1; a protein that is necessary for the ER–endosome trafficking of TLRs) for its function. TLR11 also forms a heterodimer in endosomes with TLR12, which recognizes profilin-like proteins from apicomplexan parasites, such as Toxoplasma gondii and Plasmodium falciparum. The correct function of TLR12 also requires UNC93B1. In plasmacytoid dendritic cells (pDCs), TLR12 has the ability to form homodimers that recognize profilin and that induce MYD88 signalling. Finally, mouse TLR13 recognizes the CGGAAAGACC motif of bacterial 23S ribosomal RNA (rRNA) in the endosomal compartment and induces NF-κB signalling via MYD88.
TLR11: a new flagellin receptor. Previous work had shown that TLR11 recognizes profilin20, which is a protein from the apicomplexan parasite Toxoplasma gondii20 and is an unknown proteinaceous component of uropathogenic Escherichia coli (UPEC)21. TLR11 is highly expressed in the intestinal epithelium and therefore its role in the recognition of enteropathogenic bacteria has recently been investigated22. TLR11-knockout mice infected with Salmonella enterica subsp. enterica serovar Typhimurium showed signs of increased intestinal invasion and enhanced bacterial dissemination to systemic organs, which indicates that TLR11 detects an S. Typhimurium ligand. Fractionation of heat-killed S. Typhimurium and UPEC extracts showed that TLR11 recognized flagellin21, which is also a TLR5 ligand23. Further analysis showed that TLR11 induces immune responses independently of TLR5 and that TLR5-knockout mice had higher levels of expression of TLR11 (Ref. 22), which might be an explanation for the previously reported increased resistance of Tlr5−/− animals to S. Typhimurium infection24.
Although both TLR5 and TLR11 recognize flagellin, they function in different subcellular compartments. TLR5 is reportedly localized at the cell membrane, whereas TLR11 probably localizes to the endolysosomes as its function requires protein unc-93 homolog B1 (UNC93B1) (Ref. 25), which is a protein that is necessary for the trafficking of TLRs from the endoplasmic reticulum (ER) to the endosomes26. Interestingly, the presence of TLR11 correlates with the resistance of mice to Salmonella enterica subsp. enterica serovar Typhi infection, which has in the past hindered the development of a small-animal model of typhoid fever. Mathur et al.22 found that Tlr11−/− mice were susceptible to S. Typhi infections, that they showed signs of febrile illness with features of human typhoid fever and that they could be efficiently immunized against S. Typhi. Thus, TLR11 alone mediates the resistance of mice to S. Typhi infections, which suggests that Tlr11−/− mice might be a suitable mouse model of human typhoid fever.
However, several questions remain unanswered; for example, why can S. Typhimurium overcome the intestinal TLR11 barrier whereas S. Typhi cannot? A reduced arsenal of virulence factors in S. Typhi might be a possible explanation. Alternatively, motility might be involved: it seems to be important for S. Typhi infections, as aflagellated S. Typhi are non-virulent, whereas flagella are less important for S. Typhimurium pathogenesis22. S. Typhi infection of Tlr11−/− mice does not fully recapitulate the pathological and immunological features of human typhoid fever. In particular, the severe intestinal destruction and IL-12 production seen in these mice are not typically seen in S. Typhi-infected humans22. Nevertheless, the availability of a small-animal model for the study of S. Typhi infections is an important step in understanding typhoid infections and might also contribute to the study of other human enteric diseases.
Recognition of apicomplexan parasites by TLR12. T. gondii is an obligate intracellular apicomplexan parasite that infects a wide range of warm-blooded hosts. In infected mice, survival requires IL-12 production, which is dependent on MYD88 (Ref. 27). However, although TLR2, TLR4 and TLR11 induce a cytokine response following the recognition of T. gondii glycosylphosphatidylinositol (GPI)-anchored proteins and T. gondii profilin, deletions of these TLRs do not recapitulate the lethality observed in MYD88-deficient mice, which indicates that additional TLRs recognize T. gondii20,27,28.
Koblansky et al.29 have now shown that TLR12 has a crucial role in the control of T. gondii infections. TLR12 is highly homologous to TLR11 and, although their expression overlaps in macrophages and DCs, TLR12 is predominantly expressed in myeloid cells, whereas TLR11 is mostly expressed in epithelial tissue29. Macrophages and conventional DCs deficient for both TLR11 and TLR12 failed to respond to T. gondii profilin, which indicates that these TLRs function as a heterodimer in these cell types. Interestingly, TLR11 and TLR12 also recognize profilin from Plasmodium falciparum, but only TLR11 responds to UPEC flagellin29. Nevertheless, unlike Tlr11−/− animals, Tlr12−/− mice rapidly succumb to T. gondii infection. A possible explanation for this was provided by the observation that TLR12 alone was necessary and sufficient to induce IL-12 and IFNα expression in pDCs in response to profilin, which leads to the production of IFNγ by natural killer (NK) cells and to host resistance against T. gondii infection29. Thus, in addition to TLR11, TLR12 is involved in the recognition of profilin from apicomplexan parasites. TLR12 can function either alone or as a heterodimer with TLR11. It is unclear why mice and rats but not humans have evolved such an efficient system to recognize apicomplexan parasites, but it is possible that resistance to these parasites might be more important in rodents, as they are an intermediate host in the T. gondii life cycle.
Mouse TLR13 and human TLR8 recognize bacterial RNA. TLR2 is generally thought to be a central detector of Gram-positive bacteria. However, the activation of host immune responses by group A streptococcus was shown to occur by a MYD88-dependent but TLR2-, TLR4- and TLR9-independent pathway30, which suggests that other TLRs might be important for the recognition of this pathogen. Mice lacking UNC93B1 were similarly unresponsive31. Surprisingly, macrophages lacking multiple TLRs (specifically TLR2, TLR3, TLR4, TLR7 and TLR9) were still responsive to heat-inactivated Streptococcus aureus, but not if the preparations had been treated with ribonuclease A (RNase A)32, which suggests that an RNA-sensing pathway is involved.
Using DC subsets with distinct TLR expression profiles, a recent study showed that the TLR2-independent sensing of heat-inactivated S. aureus only requires TLR13. Large bacterial ribosomal RNAs (rRNAs) — specifically the conserved CGGAAAGACC motif of 23S rRNA — were identified as the ligand for TLR13. Notably, this immunostimulatory sequence is targeted by the macrolide, lincosamide and streptogramin B (MLS) group of antibiotics. Importantly, the modification of 23S rRNA in certain MLS-resistant clinical isolates of S. aureus abolished their immunostimulatory activity32. This motif is highly conserved among Gram-negative and Gram-positive bacteria: the 23S rRNA of E. coli was also shown to induce a transcriptional response that was dependent on TLR13, which resulted in the induction of pro-IL-1β33. Finally, a third study confirmed the importance of these results, showing that both live and heat-killed Streptococcus pyogenes are recognized by this TLR13-dependent pathway34. Given the importance of TLR13 in sensing bacteria, it is surprising that this TLR is not present in humans. It is possible that a related RNA-sensing PRR has evolved in humans to recognize species of bacteria that have modified their 23S rRNA.
Although the studies mentioned above excluded an involvement of mouse TLR8 in the recognition of bacterial RNA32, human TLR8 has a different specificity to both physiological and synthetic TLR8 ligands. Indeed, several reports indicate that human TLR8 responds to total bacterial RNA35, as well as to infections with several bacterial pathogens, by inducing the expression of pro-inflammatory cytokines and type I IFNs36. However, the exact ligands for TLR8 and the possible redundancies between TLR8 and TLR13 still need to be determined. In addition, links and cooperations between RNA sensing by endosomal TLRs and by cytoplasmic RNA receptors remain uncharacterized.
Sensing cytosolic DNA and CDNs
The cytosolic responses to RNA and RNA viruses have been fairly extensively characterized. Members of the DExD/H box helicase (Asp–Glu–x–Asp/His box) family — RIG-I, MDA5 and LGP2 — are involved in the recognition of cytosolic ssRNA and dsRNA and signal through MAVS, which activates IRF1, IRF3, IRF7 and NF-κB; this ultimately leads to the expression of type I IFNs and pro-inflammatory cytokines11 (Fig. 2).

a | Microbial pathogens release different types of nucleic acids into the cytosol. Viruses are mainly detected by their single-stranded RNA (ssRNA), double-stranded RNA (dsRNA) or DNA. In addition, virulence-associated secretion systems might leak cyclic-dinucleotides (CDNs) into the host cell cytosol that can be sensed by pattern recognition receptors. The lysis of bacteria in the cytosol can release DNA and CDNs, as has been shown for Francisella novicida and Listeria monocytogenes. Bacterial RNA is thought to reach the cytosol by leakage from the lysosomes and endolysosomes. b | Various cytosolic receptors detect these different types of nucleic acids. Retinoic acid-inducible gene I (RIG-I) and melanoma differentiation-associated protein 5 (MDA5) detect ssRNA and dsRNA and induce type I interferons (IFNs) through mitochondrial antiviral-signalling protein (MAVS), TANK-binding kinase 1 (TBK1) and IFN-regulatory factor 3 (IRF3). RNA polymerase III can convert AT-rich DNA into dsRNA that is recognized by RIG-I. DNA is detected by DDX41, IFNγ-inducible protein 16 (IFI16) and maybe another as yet unknown receptor (depicted as a question mark), and all of these receptors signal through the endoplasmic reticulum (ER)-resident protein stimulator of IFN genes protein (STING), which acts upstream of TBK1. DNA-dependent activator of IFN-regulatory factors (DAI) is another sensor that has been proposed to interact with STING (dashed arrow), but which has recently been implicated in the induction of necroptosis through its direct interaction with receptor-interacting protein 3 (RIP3). STING not only functions as a signalling adaptor for the cytosolic DNA response but has also been shown to directly bind to CDNs and to activate type I IFN signalling. DDX41 also detects CDNs and activates STING. In response to DNA, cyclic GMP–AMP synthase (cGAS) synthesizes CDNs, which function as endogenous danger signals. A dsRNA-sensing and DNA-sensing pathway that is controlled by leucine-rich repeat flightless-interacting protein 1 (LRRFIP1) functions as a co-activator of type I IFN signalling by activating β-catenin, which enhances IFNβ production. CBP, CREB-binding protein.
By contrast, the response to cytosolic DNA, which leads to the induction of type I IFNs and/or inflammasome activation, has not been characterized to the same extent. The first cytosolic DNA sensors to be identified were DNA-dependent activator of IFN regulatory factors (DAI; also known as ZBP1)37 and absent in melanoma 2 (AIM2)38,39.
However, subsequent studies have shown that DAI is not essential for the IFN response to DNA40 and instead DAI was linked to the recognition of murine cytomegalovirus and to the induction of necroptosis18 (Box 1). In addition, AIM2 assembles inflammasome complexes and does not promote an IFN response38,39,41. So, how is IFN induced by cytosolic DNA? RNA polymerase III can also function as a sensor of B-form DNA (poly(dA:dT)) by converting it into dsRNA that is recognized by RIG-I (Refs 42, 43). However, as other forms of DNA induce type I IFN independently of RNA polymerase III, there must be additional cytoplasmic DNA sensors. The search for these elusive receptors has led to the recent identification of several different candidate proteins that seem to be involved in cytosolic nucleic acid sensing, either as receptors or as signalling adaptors (Table 2).
STING: a PRR and a signalling adaptor protein? A role for stimulator of IFN genes protein (STING) in the cytosolic response to nucleic acids was independently reported by several groups that screened for proteins that activate the IFNβ promoter44,45. STING was subsequently found to predominantly reside in the ER and to have a crucial role in the response to transfected dsDNA, as well as to viral, bacterial and eukaryotic intracellular pathogens in bone marrow-derived macrophages (BMDMs) and in bone marrow-derived DCs (BMDCs)46. Further analysis indicated that STING was also essential for the IFN response to CDNs47. However, for both the sensing of DNA and CDNs, STING was thought to function as an adaptor protein, linking upstream PRRs to IRF3 activation.
In an attempt to identify host components that are upstream of STING in the CDN-sensing pathway, Burdette et al.48 found that the expression of STING alone is sufficient to reconstitute IFNβ production following cyclic diguanylate monophosphate (c-di-GMP) and cyclic diadenylate monophosphate (c-di-AMP) treatment of HEK293T cells, which lack endogenous STING expression. STING directly binds to CDNs, and an Arg231Ala mutant of STING does not respond to CDNs but still responds to DNA when expressed in Sting−/− BMDMs. These findings, taken together with the observation that STING alone is not sufficient to restore the responsiveness of HEK293T cells to transfected DNA, indicate that STING can function both as a direct sensor of CDNs and as a signalling adaptor molecule in response to cytosolic DNA48.
Five groups have recently published the crystal structure of STING alone or in a complex with c-di-GMP49,50,51,52,53. These studies show that the cytoplasmic domain (CTD) of STING adopts a new α/β-fold that has some structural similarity to the RAS family of small G proteins. In addition, these studies suggest that the STING CTD forms a V-shaped dimer, even when it is not bound to its ligand, and that it binds to one molecule of c-di-GMP at the interface of the dimer. This indicates that ligand-induced dimerization is not the mechanism by which STING is activated. As only one part of STING has been crystallized, it remains to be investigated how ligand binding affects other domains of STING, which could be involved in downstream signalling.
Recent reports have confirmed the central role of STING in the type I IFN response to DNA and CDNs. Sensing of the bacterial secondary messengers c-di-GMP and c-di-AMP by STING was initially thought to be a mechanism for the detection of intracellular bacteria, such as Listeria monocytogenes, Legionella pneumophila and Pseudomonas aeruginosa54, but recently it has been shown that CDNs could also be an endogenous secondary messenger or a danger signal55,56. A first study showed that cyclic GMP–AMP (cGAMP) was synthesized from GTP and ATP in cytosolic extracts treated with DNA, or in cells transfected with DNA or infected with a DNA virus56. Interestingly, cGAMP could activate IRF3 by binding to STING, indicating that it might be a danger signal56. A second study identified cGAMP synthase (cGAS) as a cytosolic DNA sensor55. Knockdown of cGAS reduced IRF3 activation and IFNβ induction in response to DNA and to a DNA virus55. Purified cGAS catalysed the synthesis of cGAMP in the presence of different forms of DNA, but not RNA, which indicates that cGAS activity is only stimulated by DNA55. In addition, cGAS was shown to directly bind immunostimulatory DNA but not RNA. Finally, subcellular fractionation and immunofluorescence studies confirmed that cGAS primarily localizes to the cytosol and not to the nucleus, where it can sense cytosolic DNA55.
In conclusion, these studies have established STING as an essential component of cytosolic nucleic acid sensing, functioning as a PRR as well as a signalling adaptor protein. In addition, CDNs (either host-derived or from intracellular bacterial pathogens) are important immunostimulatory compounds that might be valuable as immunotherapeutics or as adjuvants57,58.
DDX41 recognizes DNA and CDNs. Several reports suggest the existence of at least one putative DNA sensor upstream of STING. One possible candidate is DDX41, which is a member of the DExD/H box helicases. DDX41 was identified in a small interfering RNA screen in a mouse DC line (the D2SC cell line), and knockdown of DDX41 affected the IFN response to B-form DNA (poly(dA:dT)), to Z-form DNA (poly(dG:dC)) and to the DNA virus herpes simplex virus 1 (HSV1)59. Interestingly, despite its homology to other DExD/H box helicases, the knockdown of Ddx41 does not affect the response to polyinosinic–polycytidylic acid (polyI:C) or to RNA viruses. Similar effects of Ddx41 knockdown were observed in BMDCs and in the THP1 monocytic cell line59. Biochemical analysis indicates that DDX41 specifically binds DNA via its DExD/H box domain, that it interacts with endogenous STING and TANK-binding kinase 1 (TBK1) and that it colocalizes with STING in the ER. These results suggest that DDX41 is a cytosolic DNA sensor, but it remains to be definitively shown whether DDX41 activates STING to induce an IFN response. Although these knockdown studies show a role for DDX41 in sensing cytosolic DNA, it remains possible that there are additional cytosolic DNA sensors that recognize other forms of DNA and that induce IFN.
In addition, DDX41 might have a role in the recognition of CDNs. Knockdown of DDX41 by short hairpin RNA (shRNA) in D2SC cells and THP1 cells abolishes the IFN response to CDN transfection and L. monocytogenes60, which is a bacterial pathogen that is known to induce IFN via the release of c-di-AMP54. Binding assays using biotinylated c-di-GMP showed that the central DExD/H box domain, but not the helicase domain, of DDX41 is required for CDN binding60. The mechanism of DDX41 signalling remains unclear, but CDN transfection led to the co-immunoprecipitation of DDX41 with STING, and the CDN-dependent STING–TBK1 association was reduced in the presence of DDX41-specific shRNA. This indicates that DDX41 and STING might form a CDN-sensing complex, in which STING functions downstream of DDX41 or as a cofactor. This model is supported by results showing that DDX41 has a higher affinity for c-di-GMP than STING does60 and that Ddx41 knockdown reduces the association of STING with c-di-GMP60. Further crystallographic analysis of DDX41 in complex with c-di-GMP, and ideally also with STING, will be necessary to fully understand the mechanism underlying CDN recognition.
IFI16: an unusual PYHIN member. IFNγ-inducible protein 16 (IFI16), which is a member of the PYHIN protein family, is another putative DNA sensor. Transfection of cells with either viral or synthetic DNA has long been known to induce a type I IFN response. Even though IFI16 is predominantly a nuclear protein, immunofluorescence analysis showed that IFI16 could colocalize with immunostimulatory DNA in the cytoplasm. Consistent with this observation, knockdown of IFI16 reproducibly resulted in a reduced IFN response to cytosolic DNA or to HSV1 (Ref. 61). The signalling mechanism of IFI16 is likely to involve STING, as STING was shown to co-immunoprecipitate with IFI16 from DNA-treated cells; however, it remains unclear whether this interaction is direct or whether it involves other proteins61. Interestingly, a recent report indicated that IFI16-mediated HSV1 sensing occurs in the nucleus62, but the authors did not see a relocalization of IFI16 from the nucleus to the cytoplasm where STING is located. Thus, additional factors might mediate the interaction of IFI16 with STING62. Intriguingly, IFI16 might also be involved in the inflammasome response to Kaposi's sarcoma-associated herpesvirus and in the inflammasome response that restricts human cytomegalovirus replication independently of type I IFN63. In conclusion, more work is required to understand in which subcellular compartment IFI16 functions and how IFI16 can not only function as an activator of the STING signalling pathway but also as an initiator of inflammasome assembly.
LRRFIP1: a co-activator of the cytoplasmic DNA response. The leucine-rich repeat (LRR) protein LRRFIP1 (leucine-rich repeat flightless-interacting protein 1) was recently identified in a screen that investigated the role of LRR-containing and LRR-interacting proteins in the IFN response to L. monocytogenes64. Knockdown of Lrrfip1 reduces IFNβ production in response to vesicular stomatitis virus (VSV), synthetic RNA and synthetic B-form and Z-form DNA, which confirms the previous reports that LRRFIP1 binds to both dsRNA and dsDNA64. Interestingly, Lrrfip1 knockdown does not abolish the activation of IRF3, NF-κB and MAPKs in L. monocytogenes-infected cells, but rather it reduces the phosphorylation of β-catenin, which is thought to function as a transcriptional co-activator at the Ifnb1 promoter64. Thus, these results have identified LRRFIP1 as an essential component of the type I IFN response to VSV and to L. monocytogenes infections.
As L. monocytogenes is known to induce the type I IFN response through CDNs54, it will be interesting to determine the role of LRRFIP1 in CDN sensing. In addition, the generation of LRRFIP1-deficient mice is required to further understand the physiological role of LRRFIP1 in vivo and to delineate its role in the complex network of nucleic acid-sensing pathways.
Other cytoplasmic nucleic acid sensors. In addition to the sensors discussed above, a multitude of other proteins have been implicated in the sensing of cytoplasmic nucleic acids (Table 2); for example, NOD2 (Ref. 10), NLRP3 (NOD-, LRR- and pyrin domain-containing 3)65,66 and KU70 (also known as XRCC6; a DNA sensor that induces type III IFN)67 can recognize nucleic acids.
Members of the DExD/H box helicases seem to have a very prominent role in RNA sensing. DDX3 was proposed to interact with RIG-I, MDA5 and MAVS to promote IFN production in response to viral RNA68. Another study proposed that DDX1, DDX21 and DDX36 form a complex that activates TRIF in response to polyI:C in DCs69. DDX60 associates with RIG-I, MDA5 and LGP2 and enhances type I IFN production in response to RNA and DNA viruses70. It has been suggested that DHX9 and DHX36 sense oligodeoxynucleotides and induce MYD88 signalling71. However, these results must be treated with caution, as the DExD/H box helicase family members also have an important role in RNA metabolism72.
Another gene family that has a role in antiviral defence is the family of IFN-induced proteins with tetratricopeptide repeats (IFITs). Some of the IFITs recognize viral ssRNA that has a 5′-triphosphate group (PPP–ssRNA), which distinguishes it from the host RNA73. The crystal structures of IFIT5 and a fragment of IFIT1 showed there to be a previously unidentified domain with a positively charged cavity that specifically facilitates the binding of PPP–ssRNA, as well as providing the structural basis for the selective recognition of PPP–ssRNA that is distinct from the recognition of PPP–dsRNA by RIG-I (Ref. 74). The mode of action of IFIT proteins is unclear, but it has been suggested to involve either the disruption of protein–protein interactions in the host translation-initiation machinery or the binding of viral RNA, thus preventing viral replication or packaging into new viral particles.
Recent progress has led to the identification of a surprisingly large variety of cytoplasmic nucleic acid sensors and has raised a number of questions. How do all of these newly identified PRRs cooperate in cytosolic nucleic acid sensing? Are there redundant pathways and cell type-specific differences? How do these sensors contribute to immunity, vaccination and autoimmune diseases in vivo? A better understanding will be gained from a rigorous validation of the function of these newly identified PRRs, as their identification and characterization has so far relied on gene knockdown and overexpression studies in non-physiological cell lines. Given the complexity of the cytoplasmic nucleic acid response, future studies need to move away from analysing each sensor and pathway individually and to take a more holistic approach, such as systems analysis, to understand the response as a whole.
Orphan NLRs assemble new inflammasomes
The recognition of intracellular pathogens is not restricted to the detection of nucleic acids but involves, in analogy to TLR sensing, the recognition of various PAMPs. This aspect of immune recognition is mainly carried out by the family of NLRs. The human genome encodes 23 NLR family members and more than 34 NLRs have been identified in mice. Some of the NLRs, such as NOD1, NOD2 and class II transactivator (CIITA), are involved in NF-κB signalling and transcriptional activation; however, the majority of these family members are thought to initiate the assembly of inflammasomes. Inflammasomes that are assembled by NLRs usually activate caspase 1 and are sometimes referred to as canonical inflammasomes. Other inflammasome complexes have been identified and are sometimes referred to as non-canonical inflammasomes because they activate other caspases that lead to pro-inflammatory cell death or to the release of pro-inflammatory cytokines (Box 2). However, until recently only three NLRs — NLRC4 (NOD-, LRR- and CARD-containing 4), NLRP1 (or murine NLRP1B) and NLRP3 — were definitely known to initiate inflammasome assembly. In addition, the PYHIN member AIM2 was shown to assemble inflammasomes in response to cytoplasmic DNA38,39,41. The ligands and mode of signalling of these receptors have been extensively reviewed4 and will not be discussed in this Review. As inflammasomes also induce pyroptosis of the infected cell, their activation is very tightly controlled; for example, the activation of TLRs and/or of the type I IFN response is required for the expression of pro-IL-1β, NLRP3, AIM2 and pro-caspase 1 (Refs 4, 41, 75, 76), which shows the importance of PRR crosstalk in the response to intracellular pathogens. In the section below, we highlight recent work that has characterized the function of orphan NLRs in inflammasome signalling in response to intracellular pathogens (Fig. 3). Although there are some differences in the inflammasome recognition of certain pathogens between mice and humans77, many of the mechanisms of inflammasome activation are similar.

a | Neuronal apoptosis inhibitory proteins (NAIPs) function as upstream direct receptors for NLRC4 (NOD-, LRR- and CARD-containing 4). NAIP2 binds the type III secretion system (T3SS) rod subunit; NAIP5 or NAIP6 bind flagellin; and human NAIP binds the T3SS needle subunit. The binding of the ligand facilitates an interaction with NLRC4. NLRC4 function also requires the receptor to become phosphorylated, which is mediated by the protein kinase Cδ (PKCδ). b | Human NLRP7 (NOD-, LRR- and pyrin domain-containing 7) recognizes acylated lipopeptides from several bacteria. The activation of the NLRP7 inflammasome promotes cytokine maturation and restricts pathogen replication. c | Lipopolysaccharide (LPS) from Yersinia spp. induces NLRP12 and pro-interleukin-1β (pro-IL-1β) expression via Toll-like receptor 4 (TLR4). The activation of NLRP12 requires the T3SS of Yersinia spp., YopJ, and possibly the injection of an additional ligand (indicated by a question mark). d | In the presence of NLRP6, basal levels of IL-18 production maintain gut homeostasis and a normal gut microbiota. Possible mechanisms involve the production of antimicrobial peptides and epithelial cell regeneration. e | Absence of NLRP6 promotes an altered, colitogenic microbiota and reduced epithelial cell repair in response to damage. The colitogenic microbiota stimulates the secretion of CC-chemokine ligand 5 (CCL5) by epithelial cells, which results in the recruitment of immune cells, triggering a chronic inflammatory response. NF-κB, nuclear factor-κB; PAMP, pattern-associated molecular pattern.
The NAIP–NLRC4 inflammasome. One of the first NLRs that was shown to mediate the assembly of inflammasome complexes was NLRC4 (Ref. 78). Previous work had shown that NLRC4 responds to both bacterial flagellin and to type III secretion system (T3SS) rod proteins from different bacterial pathogens79,80,81,82, but it was unclear whether these proteins were directly sensed by NLRC4 or whether other factors were involved. Neuronal apoptosis inhibitory protein 5 (NAIP5) was implicated in NLRC4 activation in mouse macrophages in response to L. pneumophila, but NAIP5 only partially contributed to NLRC4 activation during S. Typhimurium and P. aeruginosa infections83. These puzzling observations were clarified by two recent studies that showed that NAIPs — four of which (NAIP1, NAIP2, NAIP5 and NAIP6) are expressed in C57BL/6 mice — function upstream of mouse NLRC4 as receptors for flagellin and T3SS rod subunits84,85.
These studies showed that NAIP2 binds to the S. Typhimurium rod protein PrgJ, which facilitates an NAIP2–NLRC4 interaction and results in the assembly of this inflammasome, and that NAIP5 and NAIP6 bind to flagellin. Interestingly, although S. Typhimurium is known to strongly induce caspase 1 activation in human cells, human NAIP does not bind flagellin or PrgJ85. Further analysis of human NAIP showed that it interacts with the T3SS needle subunit of Citrobacterium violaceum to activate NLRC4. Consistent with these findings, transfection of homologous subunits of enterohaemorrhagic E. coli, Brucella thailandensis, P. aeruginosa, Shigella flexneri and S. Typhimurium also activated the NAIP–NLRC4 inflammasome85. Thus, although human NAIP has a different substrate specificity to mouse NAIP2, NAIP5 and NAIP6, it can also recognize T3SS rod proteins and functions upstream of NLRC4.
The mechanism by which NAIPs activate NLRC4 remains to be investigated, but electron microscopic analysis of a complex containing an S. Typhimurium flagellin fragment, mouse NAIP5 and human NLRC4 showed there to be disc-shaped structures in which NAIP5 and NLRC4 occupied an equivalent position, which suggests that both proteins are part of a larger complex86. The generation of NAIP-deficient mice is still necessary to validate the above findings and to define the activation mechanism of NLRC4; this is particularly important given the recent observation that protein kinase Cδ (PKCδ)-mediated phosphorylation of NLRC4 is also required to 'license' the receptor for inflammasome activation during S. Typhimurium and L. pneumophila infection87.
In the future it will be interesting to determine whether upstream sensors are also important for the activation of other inflammasomes, especially the NLRP3 inflammasome, which recognizes a panoply of chemically and structurally different stimuli.
NLRP6 inflammasome. Intestinal homeostasis depends on complex interactions between the microbiota, the intestinal epithelium and the host immune system. Previous studies have firmly established a role for the NLRP3 inflammasome in acute dextran sodium sulphate (DSS)-induced colitis, partly because of a defect in the repair of the intestinal mucosa in Nlrp3−/− mice90. Similarly, Nlrp6−/− mice are more susceptible to chemically induced colitis and colitis-induced tumourigenesis than wild-type mice88,89; this has been attributed to impaired self-renewal and proliferation of mucosal epithelial cells mediated by alterations in the intestinal stem cell niche88, or alternatively to an impaired NLRP6 function in haematopoietic cells89. Consistent with these results, another study showed that Nlrp6−/− mice were characterized by spontaneous intestinal hyperplasia, by inflammatory cell recruitment and by an exacerbation of chemical colitis induced by exposure to DSS90. Surprisingly, 16S rRNA-based analysis of the faecal microbiota showed that Nlrp6−/− mice had an altered microbiota, which was characterized by an increased representation of bacteria from the phyla Bacteroidetes (the Prevotellaceae family) and TM7 (Ref. 90). Further investigation showed that NLRP6 deficiency in colonic epithelial cells and reduced basal IL-18 secretion from epithelial cells caused this altered microbiota. Importantly, this study showed that co-housing wild-type mice with Nlrp6−/− mice increased the susceptibility of the wild-type mice to the development of DSS-induced colitis, which indicates that the microbiota that is associated with NLRP6 deficiency is colitogenic. The microbiota from Nlrp6−/− mice was associated with an increased production of CC-chemokine ligand 5 (CCL5), which might increase inflammation following epithelial damage by DSS, leading to the recruitment of immune cells, such as neutrophils, that induce a chronic inflammatory response and that exacerbate the DSS response.
IL-18 production is key to maintaining intestinal homeostasis and its loss is responsible for the increased severity of DSS-induced colitis in caspase 1-deficient mice91. The mechanism by which IL-18 exerts this protective effect is unclear, but the studies discussed above88,89,90 suggest that IL-18 has a dual role in gut homeostasis, stimulating the release of antimicrobial peptides that control the gut microbiota92 and controlling epithelial cell regeneration93. Thus, a deficiency in IL-18 production could result in increased severity of DSS-induced colitis through reduced epithelial repair88 as well as through an altered colitogenic microbiota90. Whether the NLRP6 inflammasome functions in one or several intestinal cell types (epithelial or haematopoietic cells) remains to be determined. Nevertheless, several studies88,89,90 have established a central role for the NLRP6 inflammasome in maintaining intestinal homeostasis. Further work is required to define to what extent the functions of the NLRP3 and NLRP6 inflammasomes in the gut overlap and to identify the bacterial or the host-derived ligands that are recognized by NLRP6.
The NLRP7 inflammasome detects bacterial lipopeptides. The characterization of the inflammasome response to Mycoplasma spp. led to the identification of an NLRP7 inflammasome in human macrophages and THP1 cells, which is induced in response to microbial acylated lipopeptides such as Pam3Cys-Ser-Lys4-trihydrochloride (Pam3CSK4)94. The activation of NLRP7 resulted in caspase 1 activation mediated by the adaptor protein ASC and the subsequent release of IL-1β and IL-18, but it did not result in pyroptosis94. Knockdown of NLRP7 led to the increased replication of S. aureus and L. monocytogenes in THP1 cells94, which was similar to NLRP3 silencing94; this indicates that both inflammasomes restrict pathogen growth. Interestingly, this study shows that there must be important differences in the sensing of acylated lipopeptides between the human and the mouse systems, as NLRP7 is only found in humans and not in mice. Furthermore, Pam3CSK4 is generally used for inflammasome priming and the addition of exogenous ATP is required for a robust inflammasome response in BMDMs, whereas human cells respond to Pam3CSK4 even in the absence of exogenous ATP94. This observation shows that there is a fundamental and important difference in the requirement for inflammasome priming between the human and the mouse systems. Future studies are necessary to investigate whether there is a cytoplasmic acylated lipopeptide sensor in mice, as well as to define the ligand range and the binding mechanism of acylated lipopeptides to human NLRP7.
Triggering NLRP12. NLRP12 was initially identified as a negative regulator of non-canonical NF-κB signalling95,96; however, the role of NLRP12 in the inflammasome response to infections remained unclear. A recent report has linked NLRP12 to caspase 1 activation during Yersinia spp. infections97. Neutrophils and BMDMs from Nlrp12−/− mice infected with Yersinia pestis had partially reduced levels of active caspase 1 and mature IL-1β and IL-18 compared with cells from infected wild-type mice97. This phenotype was less severe than that of mice with a deficiency in ASC or caspase 1, which completely ablated cytokine maturation, but comparable to those with a deficiency in NLRP3. Activation of the NLRP12 inflammasome was also dependent on the Y. pestis T3SS and the effector molecule YopJ, which is similar to NLRP3 inflammasome activation during Yersinia spp. infections98,99 and could indicate that NLRP12 functions in conjunction with NLRP3. Similarly to Nlrp3 induction, NLRP12 expression was dependent on a preceding priming signal in the form of TLR4 signalling, which again shows the close connection between extracellular and intracellular pathogen recognition. Consistent with the in vitro data, Nlrp12−/− mice were more susceptible to Y. pestis infections and had reduced levels of IL-1β, IL-18 and IFNγ, which indicates that there is an important role for the NLRP12 inflammasome in host defence against Yersinia spp. infection.
Although the first evidence of a role for NLRP12 in inflammasome activation has now been shown97, further work — particularly the generation of multi-gene-deficient mice — is necessary to clarify whether NLRP12 functions alone or together with NLRP3 and NLRC4, which are the two other NLRs known to be activated by Yersinia spp. infection98,99. In addition, it is unknown whether NLRP12 activation is restricted to Yersinia spp. infections or whether other pathogens also activate this pathway.
Analysis of the function of orphan NLRs has substantially increased our understanding of the inflammasome complex itself and its function in pathogen recognition as well as in tissue homeostasis. An important emerging theme is that several NLRs can engage in the assembly of the same inflammasome complex by providing specificity for different types of ligands, as has been elegantly shown for the NAIP–NLRC4 inflammasome. In addition, the discovery of the NLRP6 inflammasome has highlighted the importance of studying the role of inflammasomes in different cell types and not just in haematopoietic cells. Nevertheless, many questions regarding the new canonical and non-canonical inflammasomes remain unanswered; in particular, the nature of their ligands and their mode of activation will be active areas of research in the future.
Conclusions and perspectives
The concept of PRRs, as formulated by Charles Janeway Jr over two decades ago1, has profoundly shaped our understanding of how pathogens are recognized and how innate immune responses are initiated. Research in the last couple of years has led to the identification and the characterization of an increasing number of extracellular and intracellular PRRs, including a growing number of cytoplasmic nucleic acid sensors. Interestingly, there seem to be several intracellular receptors for the same kind of ligands, as exemplified by cytoplasmic DNA sensors. A possible explanation for this could be that, depending on their source (bacterial, viral or endogenous), the ligands might have different modifications, thus enabling the host to use a range of receptors to specifically recognize these types of ligands and, accordingly, to tune the response to pathogens or to tissue damage. The compartmentalization and the activation kinetics of each of these receptors are probably different and might influence how they bind to and respond to ligands. In addition, there might be differences in terms of the tissue-specific and the cell type-specific expression, as well as the downstream signalling pathways, and this will require thorough validation. The generation of single-knockout and multi-knockout mice will be an essential tool to consolidate this wealth of information into broad models, in order to define the physiological significance of individual PRRs.
The mechanisms by which these pathways are regulated and whether there is crosstalk between PRRs that recognize the same PAMPs or pathogens will be an active area of research in the future. Only recently has it become apparent that these signalling pathways can interact to initiate appropriate and robust host responses, as exemplified by the strict requirement of prior NF-κB and type I IFN signalling for the activation of certain types of inflammasome complexes41,75,76,100. However, whether this cooperation extends further, for example, to the initiation of adaptive immune responses, remains to be determined. Finally, as research on pattern recognition continues, it is probable that the knowledge gained about these processes will yield new approaches for the selective therapeutic manipulation of innate immune signalling pathways during infectious and inflammatory diseases.
References
Janeway, C. A. Jr. Approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harb. Symp. Quant. Biol. 54, 1–13 (1989).
McGuinness, D. H., Dehal, P. K. & Pleass, R. J. Pattern recognition molecules and innate immunity to parasites. Trends Parasitol. 19, 312–319 (2003).
Martinon, F., Burns, K. & Tschopp, J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-β. Mol. Cell 10, 417–426 (2002).
Schroder, K. & Tschopp, J. The inflammasomes. Cell 140, 821–832 (2010).
Miao, E. A. et al. Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nature Immunol. 11, 1136–1142 (2010).
Kawai, T. & Akira, S. TLR signaling. Cell Death Differ. 13, 816–825 (2006).
O'Neill, L. A. & Bowie, A. G. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nature Rev. Immunol. 7, 353–364 (2007).
Barbalat, R., Ewald, S. E., Mouchess, M. L. & Barton, G. M. Nucleic acid recognition by the innate immune system. Annu. Rev. Immunol. 29, 185–214 (2011).
Strober, W., Murray, P. J., Kitani, A. & Watanabe, T. Signalling pathways and molecular interactions of NOD1 and NOD2. Nature Rev. Immunol. 6, 9–20 (2006).
Sabbah, A. et al. Activation of innate immune antiviral responses by Nod2. Nature Immunol. 10, 1073–1080 (2009).
Desmet, C. J. & Ishii, K. J. Nucleic acid sensing at the interface between innate and adaptive immunity in vaccination. Nature Rev. Immunol. 12, 479–491 (2012).
Kang, D. C. et al. mda-5: An interferon-inducible putative RNA helicase with double-stranded RNA-dependent ATPase activity and melanoma growth-suppressive properties. Proc. Natl Acad. Sci. USA 99, 637–642 (2002).
Kato, H. et al. Cell type-specific involvement of RIG-I in antiviral response. Immunity 23, 19–28 (2005).
Yoneyama, M. et al. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nature Immunol. 5, 730–737 (2004).
Rothenfusser, S. et al. The RNA helicase Lgp2 inhibits TLR-independent sensing of viral replication by retinoic acid-inducible gene-I. J. Immunol. 175, 5260–5268 (2005).
Satoh, T. et al. LGP2 is a positive regulator of RIG-I- and MDA5-mediated antiviral responses. Proc. Natl Acad. Sci. USA 107, 1512–1517 (2010).
Saito, T. et al. Regulation of innate antiviral defenses through a shared repressor domain in RIG-I and LGP2. Proc. Natl Acad. Sci. USA 104, 582–587 (2007).
Upton, J. W., Kaiser, W. J. & Mocarski, E. S. DAI/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe 11, 290–297 (2012).
Leulier, F. & Lemaitre, B. Toll-like receptors--taking an evolutionary approach. Nature Rev. Genet. 9, 165–178 (2008).
Yarovinsky, F. et al. TLR11 activation of dendritic cells by a protozoan profilin-like protein. Science 308, 1626–1629 (2005).
Zhang, D. et al. A Toll-like receptor that prevents infection by uropathogenic bacteria. Science 303, 1522–1526 (2004).
Mathur, R. et al. A mouse model of Salmonella typhi infection. Cell 151, 590–602 (2012). This study reports that TLR11 is a novel flagellin receptor, which functions independently of TLR5.
Hayashi, F. et al. The innate immune response to bacterial flagellin is mediated by Toll-like receptor 5. Nature 410, 1099–1103 (2001).
Uematsu, S. et al. Detection of pathogenic intestinal bacteria by Toll-like receptor 5 on intestinal CD11c+ lamina propria cells. Nature Immunol. 7, 868–874 (2006).
Pifer, R., Benson, A., Sturge, C. R. & Yarovinsky, F. UNC93B1 is essential for TLR11 activation and IL-12-dependent host resistance to Toxoplasma gondii. J. Biol. Chem. 286, 3307–3314 (2011).
Kim, Y. M., Brinkmann, M. M., Paquet, M. E. & Ploegh, H. L. UNC93B1 delivers nucleotide-sensing Toll-like receptors to endolysosomes. Nature 452, 234–238 (2008).
Scanga, C. A. et al. Cutting edge: MyD88 is required for resistance to Toxoplasma gondii infection and regulates parasite-induced IL-12 production by dendritic cells. J. Immunol. 168, 5997–6001 (2002).
Debierre-Grockiego, F. et al. Fatty acids from Plasmodium falciparum down-regulate the toxic activity of malaria glycosylphosphatidylinositols. Infect. Immun. 74, 5487–5496 (2006).
Koblansky, A. A. et al. Recognition of profilin by Toll-like receptor 12 is critical for host resistance to Toxoplasma gondii. Immunity 30, 119–130 (2012).
Gratz, N. et al. Group A streptococcus activates type I interferon production and MyD88-dependent signaling without involvement of TLR2, TLR4, and TLR9. J. Biol. Chem. 283, 19879–19887 (2008).
Deshmukh, S. D. et al. Macrophages recognize streptococci through bacterial single-stranded RNA. EMBO Rep. 12, 71–76 (2011).
Oldenburg, M. et al. TLR13 recognizes bacterial 23S rRNA devoid of erythromycin resistance-forming modification. Science 337, 1111–1115 (2012). This study describes a role for mouse TLR13 in the recognition of a conserved CGGAAAGACC motif in S. aureus 23S rRNA, which is also targeted by the MLS group of antibiotics. Mutations in this sequence, which are found in certain MLS-resistant strains of S. aureus , not only promote antibiotic resistance but also abolish innate immune recognition.
Li, X. D. & Chen, Z. J. Sequence specific detection of bacterial 23S ribosomal RNA by TLR13. Elife 1, e00102 (2012).
Hidmark, A., von Saint Paul, A. & Dalpke, A. H. Cutting edge: TLR13 is a receptor for bacterial RNA. J. Immunol. 189, 2717–2721 (2012).
Kariko, K., Buckstein, M., Ni, H. & Weissman, D. Suppression of RNA recognition by Toll-like receptors: the impact of nucleoside modification and the evolutionary origin of RNA. Immunity 23, 165–175 (2005).
Cervantes, J. L., Weinerman, B., Basole, C. & Salazar, J. C. TLR8: the forgotten relative revindicated. Cell. Mol. Immunol. 9, 434–438 (2012).
Takaoka, A. et al. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature 448, 501–505 (2007).
Hornung, V. et al. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 458, 514–518 (2009).
Fernandes-Alnemri, T., Yu, J. W., Datta, P., Wu, J. & Alnemri, E. S. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature 458, 509–513 (2009).
Ishii, K. J. et al. TANK-binding kinase-1 delineates innate and adaptive immune responses to DNA vaccines. Nature 451, 725–729 (2008).
Jones, J. W. et al. Absent in melanoma 2 is required for innate immune recognition of Francisella tularensis. Proc. Natl Acad. Sci. USA 107, 9771–9776 (2010).
Chiu, Y. H., Macmillan, J. B. & Chen, Z. J. RNA polymerase III detects cytosolic DNA and induces type I interferons through the RIG-I pathway. Cell 138, 576–591 (2009).
Ablasser, A. et al. RIG-I-dependent sensing of poly(dA:dT) through the induction of an RNA polymerase III-transcribed RNA intermediate. Nature Immunol. 10, 1065–1072 (2009).
Ishikawa, H. & Barber, G. N. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 455, 674–678 (2008).
Zhong, B. et al. The adaptor protein MITA links virus-sensing receptors to IRF3 transcription factor activation. Immunity 29, 538–550 (2008).
Ishikawa, H., Ma, Z. & Barber, G. N. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 461, 788–792 (2009).
Sauer, J. D. et al. The N-ethyl-N-nitrosourea-induced Goldenticket mouse mutant reveals an essential function of Sting in the in vivo interferon response to Listeria monocytogenes and cyclic dinucleotides. Infect. Immun. 79, 688–694 (2011).
Burdette, D. L. et al. STING is a direct innate immune sensor of cyclic di-GMP. Nature 478, 515–518 (2011). This report demonstrates the dual function of STING as a sensor for cyclic dinucleotides and as an adaptor for the cytosolic DNA response.
Ouyang, S. et al. Structural analysis of the STING adaptor protein reveals a hydrophobic dimer interface and mode of cyclic di-GMP binding. Immunity 36, 1073–1086 (2012).
Shang, G. et al. Crystal structures of STING protein reveal basis for recognition of cyclic di-GMP. Nature Struct. Mol. Biol. 19, 725–727 (2012).
Shu, C., Yi, G., Watts, T., Kao, C. C. & Li, P. Structure of STING bound to cyclic di-GMP reveals the mechanism of cyclic dinucleotide recognition by the immune system. Nature Struct. Mol. Biol. 19, 722–724 (2012).
Yin, Q. et al. Cyclic di-GMP sensing via the innate immune signaling protein STING. Mol. Cell 46, 735–745 (2012).
Huang, Y. H., Liu, X. Y., Du, X. X., Jiang, Z. F. & Su, X. D. The structural basis for the sensing and binding of cyclic di-GMP by STING. Nature Struct. Mol. Biol. 19, 728–730 (2012).
Woodward, J. J., Iavarone, A. T. & Portnoy, D. A. c-di-AMP secreted by intracellular Listeria monocytogenes activates a host type I interferon response. Science 328, 1703–1705 (2010).
Sun, L., Wu, J., Du, F., Chen, X. & Chen, Z. J. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339, 786–791 (2012).
Wu, J. et al. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science 339, 826–830 (2012). References 55 and 56 describe cGAS as a novel DNA sensor that recognizes cytoplasmic DNA and that produces the newly identified endogenous secondary messenger cyclic GMP–AMP, which signals through STING to activate a type I IFN response.
Karaolis, D. K. et al. Bacterial c-di-GMP is an immunostimulatory molecule. J. Immunol. 178, 2171–2181 (2007).
Hu, D. L. et al. c-di-GMP as a vaccine adjuvant enhances protection against systemic methicillin-resistant Staphylococcus aureus (MRSA) infection. Vaccine 27, 4867–4873 (2009).
Zhang, Z. et al. The helicase DDX41 senses intracellular DNA mediated by the adaptor STING in dendritic cells. Nature Immunol. 12, 959–965 (2011).
Parvatiyar, K. et al. The helicase DDX41 recognizes the bacterial secondary messengers cyclic di-GMP and cyclic di-AMP to activate a type I interferon immune response. Nature Immunol. 13, 1155–1161 (2012).
Unterholzner, L. et al. IFI16 is an innate immune sensor for intracellular DNA. Nature Immunol. 11, 997–1004 (2010).
Orzalli, M. H., DeLuca, N. A. & Knipe, D. M. Nuclear IFI16 induction of IRF-3 signaling during herpesviral infection and degradation of IFI16 by the viral ICP0 protein. Proc. Natl Acad. Sci. USA 109, e3008–e3017 (2012).
Gariano, G. R. et al. The intracellular DNA sensor IFI16 gene acts as restriction factor for human cytomegalovirus replication. PLoS Pathog. 8, e1002498 (2012).
Yang, P. et al. The cytosolic nucleic acid sensor LRRFIP1 mediates the production of type I interferon via a β-catenin-dependent pathway. Nature Immunol. 11, 487–494 (2010).
Kanneganti, T. D. et al. Bacterial RNA and small antiviral compounds activate caspase-1 through cryopyrin/Nalp3. Nature 440, 233–236 (2006).
Sander, L. E. et al. Detection of prokaryotic mRNA signifies microbial viability and promotes immunity. Nature 474, 385–389 (2011).
Zhang, X. et al. Cutting edge: Ku70 is a novel cytosolic DNA sensor that induces type III rather than type I IFN. J. Immunol. 186, 4541–4545 (2011).
Oshiumi, H., Sakai, K., Matsumoto, M. & Seya, T. DEAD/H BOX 3 (DDX3) helicase binds the RIG-I adaptor IPS-1 to up-regulate IFN-β-inducing potential. Eur. J. Immunol. 40, 940–948 (2010).
Zhang, Z. et al. DDX1, DDX21, and DHX36 helicases form a complex with the adaptor molecule TRIF to sense dsRNA in dendritic cells. Immunity 34, 866–878 (2011).
Miyashita, M., Oshiumi, H., Matsumoto, M. & Seya, T. DDX60, a DEXD/H box helicase, is a novel antiviral factor promoting RIG-I-like receptor-mediated signaling. Mol. Cell. Biol. 31, 3802–3819 (2011).
Kim, T. et al. Aspartate-glutamate-alanine-histidine box motif (DEAH)/RNA helicase A helicases sense microbial DNA in human plasmacytoid dendritic cells. Proc. Natl Acad. Sci. USA 107, 15181–15186 (2010).
Fuller-Pace, F. V. DExD/H box RNA helicases: multifunctional proteins with important roles in transcriptional regulation. Nucleic Acids Res. 34, 4206–4215 (2006).
Pichlmair, A. et al. IFIT1 is an antiviral protein that recognizes 5′-triphosphate RNA. Nature Immunol. 12, 624–630 (2011).
Abbas, Y. M., Pichlmair, A., Gorna, M. W., Superti-Furga, G. & Nagar, B. Structural basis for viral 5′-PPP-RNA recognition by human IFIT proteins. Nature 494, 60–64 (2013).
Broz, P. et al. Caspase-11 increases susceptibility to Salmonella infection in the absence of caspase-1. Nature 490, 288–291 (2012).
Rathinam, V. A. et al. TRIF licenses caspase-11-dependent NLRP3 inflammasome activation by Gram-negative bacteria. Cell 150, 606–619 (2012). References 75 and 76 report a link between TRIF-mediated type I IFN production and the activation of the non-canonical caspase 11 inflammasome in response to Gram-negative bacteria.
Gavrilin, M. A. & Wewers, M. D. Francisella recognition by inflammasomes: differences between mice and men. Front. Microbiol. 2, 11 (2011).
Mariathasan, S. et al. Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature 430, 213–218 (2004).
Amer, A. et al. Regulation of Legionella phagosome maturation and infection through flagellin and host Ipaf. J. Biol. Chem. 281, 35217–35223 (2006).
Franchi, L. et al. Cytosolic flagellin requires Ipaf for activation of caspase-1 and interleukin 1β in salmonella-infected macrophages. Nature Immunol. 7, 576–582 (2006).
Miao, E. A. et al. Cytoplasmic flagellin activates caspase-1 and secretion of interleukin-1β via Ipaf. Nature Immunol. 7, 569–575 (2006).
Miao, E. A. et al. Innate immune detection of the type III secretion apparatus through the NLRC4 inflammasome. Proc. Natl Acad. Sci. USA 107, 3076–3080 (2010).
Lightfield, K. L. et al. Critical function for Naip5 in inflammasome activation by a conserved carboxy-terminal domain of flagellin. Nature Immunol. 9, 1171–1178 (2008).
Kofoed, E. M. & Vance, R. E. Innate immune recognition of bacterial ligands by NAIPs determines inflammasome specificity. Nature 477, 592–595 (2011).
Zhao, Y. et al. The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature 447, 596–600 (2011).
Halff, E. F. et al. Formation and structure of a NAIP5–NLRC4 inflammasome induced by direct interactions with conserved N- and C-terminal regions of flagellin. J. Biol. Chem. 287, 38460–38472 (2012).
Qu, Y. et al. Phosphorylation of NLRC4 is critical for inflammasome activation. Nature 490, 539–542 (2012). This report shows that phosphorylation of NLRC4 is crucial for the response to flagellin and identifies PKCδ as the relevant kinase.
Normand, S. et al. Nod-like receptor pyrin domain-containing protein 6 (NLRP6) controls epithelial self-renewal and colorectal carcinogenesis upon injury. Proc. Natl Acad. Sci. USA 108, 9601–9606 (2011).
Chen, G. Y., Liu, M., Wang, F., Bertin, J. & Nunez, G. A functional role for Nlrp6 in intestinal inflammation and tumorigenesis. J. Immunol. 186, 7187–7194 (2011).
Elinav, E. et al. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell 145, 745–757 (2011). This study provides the first evidence for the existence of an NLRP6 inflammasome in gut epithelial cells and describes its essential role in maintaining gut homeostasis.
Dupaul-Chicoine, J. et al. Control of intestinal homeostasis, colitis, and colitis-associated colorectal cancer by the inflammatory caspases. Immunity 32, 367–378 (2010).
Strowig, T., Henao-Mejia, J., Elinav, E. & Flavell, R. Inflammasomes in health and disease. Nature 481, 278–286 (2012).
Zaki, M. H. et al. The NLRP3 inflammasome protects against loss of epithelial integrity and mortality during experimental colitis. Immunity 32, 379–391 (2010).
Khare, S. et al. An NLRP7-containing inflammasome mediates recognition of microbial lipopeptides in human macrophages. Immunity 36, 464–476 (2012).
Lich, J. D. & Ting, J. P. Monarch-1/PYPAF7 and other CATERPILLER (CLR, NOD, NLR) proteins with negative regulatory functions. Microbes Infect. 9, 672–676 (2007).
Allen, I. C. et al. NLRP12 suppresses colon inflammation and tumorigenesis through the negative regulation of noncanonical NF-κB signaling. Immunity 36, 742–754 (2012).
Vladimer, G. I. et al. The NLRP12 inflammasome recognizes Yersinia pestis. Immunity 37, 96–107 (2012).
Brodsky, I. E. et al. A Yersinia effector protein promotes virulence by preventing inflammasome recognition of the type III secretion system. Cell Host Microbe 7, 376–387 (2010).
Zheng, Y., Lilo, S., Mena, P. & Bliska, J. B. YopJ-induced caspase-1 activation in Yersinia-infected macrophages: independent of apoptosis, linked to necrosis, dispensable for innate host defense. PLoS ONE 7, e36019 (2012).
Henry, T., Brotcke, A., Weiss, D. S., Thompson, L. J. & Monack, D. M. Type I interferon signaling is required for activation of the inflammasome during Francisella infection. J. Exp. Med. 204, 987–994 (2007).
Holler, N. et al. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nature Immunol. 1, 489–495 (2000).
Mocarski, E. S., Upton, J. W. & Kaiser, W. J. Viral infection and the evolution of caspase 8-regulated apoptotic and necrotic death pathways. Nature Rev. Immunol. 12, 79–88 (2012).
Sun, L. et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 148, 213–227 (2012).
Wang, Z., Jiang, H., Chen, S., Du, F. & Wang, X. The mitochondrial phosphatase PGAM5 functions at the convergence point of multiple necrotic death pathways. Cell 148, 228–243 (2012).
Feoktistova, M. et al. cIAPs block ripoptosome formation, a RIP1/caspase-8 containing intracellular cell death complex differentially regulated by cFLIP isoforms. Mol. Cell 43, 449–463 (2011).
Tenev, T. et al. The ripoptosome, a signaling platform that assembles in response to genotoxic stress and loss of IAPs. Mol. Cell 43, 432–448 (2011).
Upton, J. W., Kaiser, W. J. & Mocarski, E. S. Virus inhibition of RIP3-dependent necrosis. Cell Host Microbe 7, 302–313 (2010).
Robinson, N. et al. Type I interferon induces necroptosis in macrophages during infection with Salmonella enterica serovar Typhimurium. Nature Immunol. 13, 954–962 (2012).
Kayagaki, N. et al. Non-canonical inflammasome activation targets caspase-11. Nature 479, 117–121 (2011). This study describes a novel non-canonical caspase 11 inflammasome and shows there to be an essential role for caspase 11 in LPS-induced lethality in a mouse model of septic shock.
Case, C. L. et al. Caspase-11 stimulates rapid flagellin-independent pyroptosis in response to Legionella pneumophila. Proc. Natl Acad. Sci. USA 110, 1851–1856 (2013).
Gringhuis, S. I. et al. Dectin-1 is an extracellular pathogen sensor for the induction and processing of IL-1β via a noncanonical caspase-8 inflammasome. Nature Immunol. 13, 246–254 (2012).
Vince, J. E. et al. Inhibitor of apoptosis proteins limit RIP3 kinase-dependent interleukin-1 activation. Immunity 36, 215–227 (2012).
Pierini, R. et al. AIM2/ASC triggers caspase-8-dependent apoptosis in Francisella-infected caspase-1-deficient macrophages. Cell Death Differ. 19, 1709–1721 (2012).
Acknowledgements
This work was supported by the following grants: PP00P3_139120/1 from the Swiss National Science Foundation to P.B., and AI095396 and AI08972 to D.M.M. from the US National Institute of Allergy and Infectious Diseases. We apologize to investigators whose contributions were not cited more extensively because of space limitations.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Glossary
- Cyclic dinucleotides
-
Small bacterial or host-derived nucleic acids — such as cyclic diguanylate monophosphate (c-di-GMP), cyclic diadenylate monophosphate (c-di-AMP) or cyclic GMP–AMP — that function as secondary messengers and that can induce an innate immune response when present in the cytosol.
- Leaderless cytokines
-
Cytokines that lack a classical amino-terminal secretion signal sequence (also referred to as leader peptide or leader sequence) and that are thought to be secreted by an endoplasmic reticulum- and Golgi-independent mechanism.
- Pyroptosis
-
A lytic pro-inflammatory form of programmed cell death that is initiated by the activation of inflammatory caspases.
- Plasmacytoid dendritic cells
-
(pDCs). A dendritic cell subset that morphologically resembles a plasmablast. pDCs produce large amounts of type I interferons in response to viral infection.
- Necroptosis
-
A form of programmed necrosis that is initiated by the kinases receptor-interacting protein 1 (RIP1) and RIP3 in response to external signals, in conditions in which caspase 8 activity is compromised.
- Ribonuclease A
-
(RNase A). An endoribonuclease that specifically cleaves single-stranded RNA and that is often used to remove RNA from samples.
- Macrolide, lincosamide and streptogramin B
-
(MLS). A group of antibiotics that function as translational inhibitors by targeting the 50S ribosomal subunit, which contains 23S ribosomal RNA.
- DExD/H box helicase
-
An enzyme that can unwind double-stranded RNA using energy derived from ATP hydrolysis. The DExD/H box is a characteristic amino acid signature motif of many RNA-binding proteins.
- Small interfering RNA
-
(siRNA). Short double-stranded RNAs of 19 to 23 nucleotides that induce RNA interference, which is a post-transcriptional process that leads to gene silencing in a sequence-specific manner.
- Short hairpin RNA
-
(shRNA). A sequence of RNA that makes a tight hairpin turn, which can be used to silence target gene expression via RNA interference.
- Leucine-rich repeat
-
(LRR). A protein structural motif composed of repeating stretches of 20 to 30 amino acids that are unusually rich in the hydrophobic amino acid leucine and that form an α/β-horseshoe fold. LRRs are found in many pattern recognition receptors, such as Toll-like receptors and NOD-like receptors, but also in many functionally unrelated proteins.
- β-catenin
-
This protein functions both as a transcriptional activator and as a membrane–cytoskeleton linker protein by binding to E-cadherin. Following detachment from E-cadherin, β-catenin can relocate to the nucleus.
- Type III secretion system
-
(T3SS). A virulence-associated specialized molecular machine present in some bacteria that facilitates the translocation of bacterial proteins into host cells.
- Colitis
-
An inflammatory disease of the colon. In humans, colitis is most commonly classified as ulcerative colitis or as Crohn's disease, which are two inflammatory bowel diseases that have unknown aetiologies. Various hereditary and induced mouse models of human colitis have been developed.
Rights and permissions
About this article
Cite this article
Broz, P., Monack, D. Newly described pattern recognition receptors team up against intracellular pathogens. Nat Rev Immunol 13, 551–565 (2013). https://doi.org/10.1038/nri3479
Published:
Issue Date:
DOI: https://doi.org/10.1038/nri3479
This article is cited by
-
RBP–RNA interactions in the control of autoimmunity and autoinflammation
Cell Research (2023)
-
Microbiota in a long survival discourse with the human host
Archives of Microbiology (2023)
-
Association of rare variants in genes of immune regulation with pediatric autoimmune CNS diseases
Journal of Neurology (2022)
-
Comparative Transcriptome Analysis of Head Kidney of Aeromonas hydrophila-infected Hypoxia-tolerant and Normal Large Yellow Croaker
Marine Biotechnology (2022)
-
Interaction of Salmonella Gallinarum and Salmonella Enteritidis with peripheral leucocytes of hens with different laying performance
Veterinary Research (2021)
