
Key Points
-
Two decades of clinical experience with the immunomodulatory treatment of multiple sclerosis points to distinct immunological pathways that drive disease relapses and progression. Although the immunomodulatory drugs reduce the frequency of relapses, the trade-off of efficacy is a range of side effects, and the long-standing drugs approved for multiple sclerosis do not ultimately halt neurodegeneration.
-
Dissecting the distinct roles of the immune system in multiple sclerosis is complicated by one, the multicellular pathophysiology that involves infiltrating adaptive and innate immune cells, as well as central nervous system (CNS)-resident innate cells with inflammatory capacity; and two, the chronic nature of the disease that unfolds over a period of many decades.
-
Multiple sclerosis is associated with more than 100 different genetic variants that promote disease predisposition and with environmental influences that alter disease penetrance and stochastic occurrences, although the exact triggering events may vary from one patient to the next. Despite the progress in identifying the genetic determinants of the disease, their phenotypic consequences remain to be elucidated, and a substantial understanding of environmental contributors is lacking.
-
Dysregulation of immune effector–suppressor cell interactions occurs in multiple sclerosis, ultimately resulting in autoreactive adaptive immune cells that are capable of infiltrating and promoting damage within the CNS. However, these cells may not be the main drivers of more chronic, progressive neurodegeneration.
-
Chronic inflammation in multiple sclerosis may reflect a long-term stress response to homeostatic dysregulation in the CNS by tissue-resident innate cells that exceedingly burdens the system, leading to progressive and irreversible neurodegenerative decline.
-
The most imminent goal for future treatment is the concomitant improved targeting of relapses and progression, potentially through combinatorial therapies that modulate both arms of the disease. Improved disease prognosis and potential patient stratification for more directed healthcare provision are also much-anticipated prospects and may become tangible as we move into the immune informatics era and as large-scale, organized health resources become increasingly accessible.
Abstract
Two decades of clinical experience with immunomodulatory treatments for multiple sclerosis point to distinct immunological pathways that drive disease relapses and progression. In light of this, we discuss our current understanding of multiple sclerosis immunopathology, evaluate long-standing hypotheses regarding the role of the immune system in the disease and delineate key questions that are still unanswered. Recent and anticipated advances in the field of immunology, and the increasing recognition of inflammation as an important component of neurodegeneration, are shaping our conceptualization of disease pathophysiology, and we explore the potential implications for improved healthcare provision to patients in the future.
Similar content being viewed by others

Main
Approximately 2.5 million people worldwide are afflicted with multiple sclerosis, a chronic neuroinflammatory disease of the brain and spinal cord that is a common cause of serious physical disability in young adults1, especially women. Multiple sclerosis poses a major personal and socioeconomic burden: the average age of disease onset is 30 years — a time that is decisive for work and family planning — and 25 years after diagnosis, approximately 50% of patients require permanent use of a wheelchair. The condition has a heterogeneous presentation (Box 1) that can include sensory and visual disturbances, motor impairments, fatigue, pain and cognitive deficits1. The variation in clinical manifestations correlates with the spatiotemporal dissemination of lesional sites of pathology within the central nervous system (CNS)2. These lesions are a hallmark of multiple sclerosis (Box 2) and are caused by immune cell infiltration across the blood–brain barrier (BBB) that promotes inflammation, demyelination, gliosis and neuroaxonal degeneration, leading to disruption of neuronal signalling3. T cells appear early in lesion formation, and the disease is considered to be autoimmune, initiated by autoreactive lymphocytes that mount aberrant responses against CNS autoantigens, the precise nature of which, however, remains enigmatic.
Infiltration of immune cells from the periphery — which is particularly prominent in the common, relapsing-remitting form of the disease — has been the main target of currently available therapies for multiple sclerosis (see Supplementary information S1 (table)). Although these broad-spectrum immunomodulatory drugs reduce immune cell activity and entry into the CNS and decrease relapse frequency, they are often associated with side effects. These range from flu-like symptoms and the development of other autoimmune disorders to malignancies and even fatal opportunistic infections such as progressive multifocal leukoencephalopathy4 (see Supplementary information S1 (table)), indicating the need to identify more specific therapeutic targets that can be efficaciously modulated but without inducing such significant adverse reactions.
Concomitantly, it has been increasingly acknowledged that although the long-standing treatments approved for multiple sclerosis can reduce relapses, they do not substantially halt the disease4,5, and neuroaxonal damage — with ensuing physical disability — continues to accumulate and become permanent. This supports the concept that there is some degree of discord between the processes driving overt relapses and those driving chronic progression. Indeed, secondary progressive disease may not be a temporally distinct phase of the condition arising as a direct consequence of the relapsing-remitting disease but may instead be the outcome of other underlying pathophysiological mechanisms. This is also in keeping with the existence of the relapse-free, primary progressive form of multiple sclerosis (Box 1).
The inferred uncoupling of relapses and disability progression has considerable ramifications for our understanding of disease pathways and for therapeutic design, as there are currently no drugs approved to specifically treat primary or secondary progressive multiple sclerosis5. Although disease progression is not greatly influenced by the available immunomodulatory therapies, which target peripheral immune cell activation and entry into the CNS, immunological involvement is implicated in this process: there is an additional inflammatory component residing in the CNS that is only marginally influenced by peripheral immune control and that contributes to gradual neuroaxonal loss and demise of myelin-producing oligodendrocytes6,7,8. This CNS-resident inflammatory arm of the disease is less well defined but is likely to involve continuous activation of innate immune cells; these cells have been found to predominate in demyelinated areas, but they are also present diffusely throughout normal-appearing white matter, and their numbers correlate with tissue damage9.
Dissecting the distinct roles of the immune system in the events that trigger multiple sclerosis development and those that contribute to disease progression is thus complicated by the multicellular pathophysiology associated with infiltrating adaptive and innate immune cells, as well as CNS-resident innate immune cells with inflammatory capacity and by the chronic nature of the disease that unfolds over a period of many decades.
In this Review, we evaluate how our understanding of the involvement of the immune system in driving the development of multiple sclerosis is being shaped by the ongoing interrogation of genetic predisposition and environmental influences. We discuss the changing role of peripheral immune cells — including effector and regulatory lymphocytes and innate immune cells — in promoting pathogenesis as the disease takes its course, and we point to CNS-resident innate cells as emerging key contributors to chronic inflammation. In considering our current view of multiple sclerosis immunopathology, we highlight the outstanding clinical needs and the imminent biomedical challenges for the future.


What causes multiple sclerosis?
The exact cause of multiple sclerosis, and whether this varies from one patient to the next, still remains elusive, but the disease is thought to arise in genetically susceptible individuals, with stochastic events and environmental factors influencing disease penetrance. Genetic variation accounts for approximately 30% of the overall disease risk, and with the advent of genome-wide association studies (GWASs), more than 100 distinct genetic regions have been identified as being associated with multiple sclerosis, collectively explaining approximately one-third of the genetic component of the condition10. Despite the fact that non-genetic factors have a proportionately larger contribution than genetic factors to immunological heterogeneity in general11, comparatively less progress has been made in elucidating environmental determinants of multiple sclerosis, perhaps reflecting the difficulty of accurately interpreting complex, and sometimes confounding, epidemiological data12.
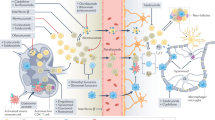
Without a known predominant exogenous risk factor, it is an open question whether multiple sclerosis is triggered in the periphery or in the CNS. In the CNS-extrinsic (peripheral) model, autoreactive T cells that are activated at peripheral sites — potentially through molecular mimicry13,14,15, bystander activation or the co-expression of T cell receptors (TCRs) with different specificities16 — traffic to the CNS along with activated B cells and monocytes (Fig. 1). This model is consistent with the method used to induce the multiple sclerosis-like disease experimental autoimmune encephalomyelitis (EAE) in animals: emulsified CNS antigen is administered along with immune stimulants, resulting in the generation of pathogenic CD4+ T helper 1 (TH1) cells and TH17 cells in the draining lymph nodes. These cells then enter the circulation and ultimately exert their effector functions within the CNS, having crossed the BBB or the blood–cerebrospinal fluid (CSF) barrier at the choroid plexus (Box 3).

During the establishment of central tolerance in the thymus, most autoreactive T cells are deleted; however, this process is imperfect, and some autoreactive T cells are released into the periphery. In health, peripheral tolerance mechanisms keep these cells in check. If this tolerance is broken — through the reduced function of regulatory T (TReg) cells and/or the increased resistance of effector B cells and T cells to suppressive mechanisms — central nervous system (CNS)-directed autoreactive B cells and T cells can be activated in the periphery to become aggressive effector cells by molecular mimicry, novel autoantigen presentation, recognition of sequestered CNS antigen released into the periphery or bystander activation. Genetic and environmental factors, including infectious agents and smoke constituents, contribute to these events. Once activated, CD8+ T cells, differentiated CD4+ T helper 1 (TH1) and TH17 cells, B cells and innate immune cells can infiltrate the CNS, leading to inflammation and tissue damage. B cells trafficking out of the CNS can undergo affinity maturation in the lymph nodes before re-entering the target organ and promoting further damage. Dashed arrows indicate differentiation. BCR, B cell receptor; CD8+ MAIT cell, CD8+ mucosa-associated invariant T cell; TCR, T cell receptor.
Alternatively, CNS-intrinsic events may trigger disease development, with the infiltration of autoreactive lymphocytes occurring as a secondary phenomenon. It is unclear what these CNS-intrinsic events might be, although postulated mechanisms include inflammatory responses to an as yet unknown CNS viral infection — a hypothesis based partly on the emerging appreciation of CNS immune surveillance17,18 (Box 3) — or to processes leading to primary neurodegeneration, similar to those that have been implicated in Alzheimer disease or Parkinson disease19. However, drawing support for either model of multiple sclerosis aetiology from other diseases warrants a closer consideration of how known multiple sclerosis risk factors compare to those for other common autoimmune and neurodegenerative conditions.
Genetic predisposition. The majority of multiple sclerosis-associated candidate genes are thought to be immunological. Consequently, the notable overlap in associated genomic regions between multiple sclerosis and other autoimmune diseases is not surprising10 and may suggest some sharing of predisposing immunological processes, thereby supporting the peripheral model of multiple sclerosis initiation. However, in some cases this is only an apparent overlap: for instance, the same variant in the gene region encoding tumour necrosis factor receptor 1 (TNFR1) confers susceptibility to multiple sclerosis but promotes protection against ankylosing spondylitis, consistent with the opposing effects of drugs targeting the TNFR1 pathway, which exacerbate multiple sclerosis relapses but show efficacy in ankylosing spondylitis20. Despite this caveat, efforts to obtain a more comprehensive interpretation of the genetic data have led to the construction of interactome networks21 using the presumed candidate genes assigned to each associated region. For multiple sclerosis, such analyses implicate the involvement of interleukin-2 (IL-2), interferons (IFNs) and nuclear factor-κB signalling, among numerous other immunological pathways, in disease predisposition22.
These findings are consistent with pre-GWAS concepts regarding immunological mechanisms in multiple sclerosis, but the more substantial use of GWAS data to dissect disease pathways requires more in-depth investigations. Epigenetic23,24, transcriptomic25 and immunoprofiling26,27 analyses are just beginning to shed light on how the variants correlate with immune cell subset-specific differences in the regulation of gene expression, as most associated genetic variants are non-coding and many colocalize with gene enhancers or repressors in immune cells23. However, correlations do not necessarily reflect causality. To date, a more detailed, but not definitive, understanding of genetically determined disease pathways has been attained for only a handful of associated loci, such as the HLA-A*02:01 and HLA-DRB1*15:01 variants28,29, and the genes encoding the α-chains of the IL-2 and IL-7 receptors (IL-2Rα and IL-7Rα, respectively)30,31,32,33. The data implicate central tolerance mechanisms, as well as peripheral differences in effector T cell function due to altered cytokine responsiveness, cytokine production and homeostatic proliferation, in multiple sclerosis predisposition.
Although still limited, the present view regarding the functional implications of multiple sclerosis-associated genetic polymorphisms is that the HLA variants primarily define the CNS specificity of the disease by affecting the T cell repertoire, whereas the non-HLA variants more broadly influence the threshold of immune cell activation, thereby ultimately altering the likelihood of a CNS-directed autoimmune response being mounted.
Overall, strikingly few genetic associations are shared between multiple sclerosis and other neurodegenerative conditions such as Alzheimer disease34 and Parkinson disease35. This indicates that non-immunological, primary neurodegenerative processes are less likely to promote the initiation of multiple sclerosis, although genetic contributions to disease severity or subphenotypes of the disease may yet reveal a role for neurological genes. Intriguingly, however, disease risk associations in the HLA region have also been observed for the other neurodegenerative conditions34,35, even though T cell infiltration is uncharacteristic for these diseases, and thus further investigation is needed to determine the significance of these findings.
The genetic architecture of multiple sclerosis emphasizes the prominent role of the immune system in disease predisposition. The clinical relevance of determining the specific phenotypic consequences of multiple sclerosis-associated polymorphisms has now begun to be recognized20, and this plethora of variants can serve as a platform for interrogating human immune system diversity: to help to fine-tune our understanding of disease immunopathogenesis, to identify more targeted treatment approaches and to even uncover novel immunological pathways that can be harnessed for therapeutic benefit.
Environmental factors. In line with the perceived distinct roles of multiple sclerosis genetic risk factors in the direct triggering of autoreactivity and in the broader modulation of thresholds of immune cell activation, the environmental factors that contribute to disease development may also fall into two similar categories.
Those environmental factors more directly involved in the triggering of autoreactive T cells are often postulated to be viral or microbial in nature and mediate their effects through molecular mimicry13,14,15. Tolerance breakdown may also arise through the environmental factor-driven generation of novel autoantigens36. In addition to directly providing or modifying relevant antigens, environmental determinants such as CNS-tropic infectious agents may also promote the release of sequestered CNS antigens into the periphery, as has been observed in a model of Theiler's murine encephalomyelitis virus infection14,37 (Fig. 1).
Environmental influences with a more modulatory role may indirectly alter the activation thresholds of autoreactive T cells by triggering a pro-inflammatory milieu. Intriguingly, peripheral inflammation due to infection may also have a direct influence on the CNS: locally secreted cytokines can activate afferent nerve endings, circumventricular organ and choroid plexus innate immune cells can respond to circulating pathogen-associated molecular patterns38,39, and pro-inflammatory cytokines at high concentrations in the circulation can be transported across the BBB and can induce signalling in perivascular macrophages40. The outcome of this immune system-to-CNS communication seems to typically involve the pro-inflammatory activation of microglial cells. This raises the provocative question of whether, in some cases, multiple sclerosis can result indirectly from peripheral inflammation that drives microglia-dependent neurodegeneration, without the need for a CNS-directed autoreactive response to be mounted.
Considering that the numerous non-HLA genetic risk factors for multiple sclerosis probably affect a multitude of immunological pathways, environmental factors that influence any one of these different pathways may also contribute to disease development. Based on this parallel, there may be just as many different environmental determinants of multiple sclerosis as there are genetic risk variants. To date, the reported environmental factors implicated in multiple sclerosis variably, but not exclusively, include vitamin D, human cytomegalovirus infection12 and circadian disruption41. However, smoking and Epstein–Barr virus (EBV) infection remain the best-confirmed environmental contributors12, although it is notable that the modest impact of their individual effects on overall multiple sclerosis risk is comparable to that of any single associated genetic variant.
There is robust evidence that high levels of EBV-specific antibodies correlate with increased multiple sclerosis risk, as does a history of infectious mononucleosis12,42. Several mechanisms for the role of EBV infection in multiple sclerosis development have been proposed. One hypothesis is that inadequate regulation of latent EBV infection leads to viral reactivation in the CNS, resulting in EBV-transformed B cells in the meningeal and perivascular space expressing viral proteins that could activate effector T cells43. Furthermore, chronic viral infection can lead to an increase in the numbers of virus-specific memory T cells44, and this increase may be accentuated in multiple sclerosis; indeed, homeostatic peripheral T cell proliferation in response to an accelerated thymic involution has been observed in patients with relapsing-remitting disease45. However, there is conflicting evidence regarding whether EBV RNA or protein is present in the CNS of patients with multiple sclerosis43,46, and this hypothesis thus remains controversial. A second hypothesis suggests that EBV may instead have a more general role in immune system dysregulation, which is in keeping with the correlation of EBV infection with the risk of developing other autoimmune diseases, such as systemic lupus erythematosus14.
As the vastness of the human virome is just beginning to be appreciated47, our understanding of viral involvement in multiple sclerosis is still in its infancy. This is equally true for the bacterial microbiome, the genome of which is approximately 100-times larger than the human genome, and which fluctuates in composition based on environmental factors such as diet48. EAE studies have demonstrated that changes to the gut microbiota, for example, can alter the incidence and severity of CNS inflammation and ensuing disease49. However, a direct link between the microbiota and multiple sclerosis in humans has yet to be demonstrated.
Although identifying the many environmental factors that may alter multiple sclerosis risk and comprehending their mode of action poses a particularly significant challenge, the putative ease of modifying exogenous influences and human behaviour to reduce disease risk or severity is an attractive prospect for future medical intervention.

Chronic multicellular disease development
The multifactorial nature of multiple sclerosis — involving a potential deluge of different gene–environment interactions at its inception — unfolds through a complex, highly multicellular pathophysiological process that evolves throughout the duration of the disease course (Figs 1,2).

Immune cell infiltration from the periphery is a prominent feature of early-stage multiple sclerosis (top panel) and can occur from the meningeal blood vessels by direct crossing of the blood–brain barrier (denoted '1' in the figure) or the subarachnoid space (SAS; denoted '2'), or from the choroid plexus across the blood–cerebrospinal fluid (CSF) barrier (denoted '3'). Peripheral innate and adaptive immune cells can accumulate in perivascular spaces and enter the central nervous system (CNS) parenchyma. These cells, along with activated CNS-resident microglia and astrocytes, promote demyelination and oligodendrocyte (ODC) and neuroaxonal injury through direct cell contact-dependent mechanisms and through the action of soluble inflammatory and neurotoxic mediators. Later on in the disease (bottom panel), immune cell infiltration wanes, perhaps due to adaptive immune cell exhaustion from chronic antigen exposure. However, chronic CNS-intrinsic inflammation and neurodegeneration continue. Meningeal tertiary lymphoid-like structures, which have specifically been documented in secondary progressive disease, may contribute to late-stage inflammation in patients with this form of multiple sclerosis. The action of the CNS-resident innate cells may contribute to chronic inflammation irrespective of the precise disease subtype. Stimulated by the microglia, astrocytes can produce CC-chemokine ligand 2 (CCL2) and granulocyte–macrophage colony-stimulating factor (GM-CSF), leading to even further microglial recruitment and activation, and the astrocytes can prevent remyelination at sites of neuroaxonal injury by inhibiting progenitor cells from developing into mature ODCs. APC, antigen-presenting cell; CD8+ MAIT cell, CD8+ mucosa-associated invariant T cell; FDC, follicular dendritic cell; IFNγ, interferon-γ; IL-17, interleukin-17; NO, nitric oxide; RNS, reactive nitrogen species; ROS, reactive oxygen species; TH1 cell, T helper 1 cell.
Autoreactive T cells. The presence of T cells within CNS lesions is detectable in the early stages of multiple sclerosis9, and the long-appreciated HLA associations with the disease are thought to reflect the presentation of specific CNS autoantigens to autoreactive T cells. As demyelination is a key feature of multiple sclerosis neuropathology, myelin protein-derived antigens have been hypothesized to be the main autoreactive targets. Myelin basic protein (MBP), proteolipid protein and myelin oligodendrocyte glycoprotein (MOG), for example, have been demonstrated to be recognized by circulating CD4+ T cells in patients with multiple sclerosis but also in healthy individuals, and there is conflicting evidence regarding potential differences in the frequency and avidity of these cells between the two groups50,51. This controversy, as well as the absence of a dominant T cell autoantigen in multiple sclerosis, may be attributed to technical limitations in detecting such autoantigens, to inter-patient variation, or to epitope spreading52,53, but unbiased combinatorial library screening methods54 and antigen-tolerizing approaches55 may help to further elucidate anti-myelin immune responses in the disease.
In EAE, infiltrating CD4+ T cells are re-activated in the CNS by antigen-presenting cells (APCs), including CD11c+ dendritic cells (DCs), with the resulting inflammatory response leading to monocyte recruitment into the CNS, as well as naive CD4+ T cell activation through epitope spreading that further fuels the inflammation52. TH1 cells and TH17 cells are the main CD4+ T cell subsets implicated in disease, and thus skewing of T cell differentiation away from these subsets and towards a TH2 cell phenotype has been a prominent therapeutic concept and is considered to be a mechanism of action of the first-line, disease-modifying therapies IFNβ56, glatiramer acetate (Copaxone; Teva and Sanofi–Aventis)57 and dimethyl fumarate (Tecfidera; Biogen)58.
However, the relative importance of TH1 cells versus TH17 cells in multiple sclerosis pathogenesis is contentious: conflicting studies variably report the predominance of one cell type over the other at initial diagnosis and during subsequent relapses and progression59,60, and compared with controls, patient myelin-reactive peripheral CD4+ T cells expressing CC-chemokine receptor 6 (CCR6) show enhanced expression of both the respective TH1 and TH17 cell signature cytokines IFNγ and IL-17A61. Furthermore, some lesional CD4+ T cells have an intermediate phenotype, expressing IFNγ and IL-17A simultaneously62. Despite these inconsistencies, the failure of a Phase II clinical trial in patients with relapsing-remitting multiple sclerosis following the administration of ustekinumab (Stelara; Janssen)63 — an antibody that targets the p40 subunit that is shared by the IL-12 and IL-23 cytokines, which are involved in TH1 and TH17 cell differentiation, respectively — was not anticipated. Suggested explanations have included a putative inability of the drug to cross the BBB and exert an effect directly in the CNS, and a diminished importance for IL-12 and/or IL-23 at later stages of disease63. The very premise for the ustekinumab trial, based partly on EAE studies, has also been questioned; although EAE models (see Supplementary information S2 (table)) are indispensable for studying disease mechanisms in vivo, interspecies immunological differences have been recognized (Box 4), including the essential requirement for IL-23 in TH17 cell induction in mice but not in humans64,65. In addition, the function of TH17 cells seems to differ between mice and humans. TH17 cell-mediated granulocyte–macrophage colony-stimulating factor (GM-CSF) production contributes to chronic inflammation in EAE66, whereas TH1 cells and other cell subsets are the primary producers of this cytokine in humans67.
CD8+ T cells are found in higher frequency than CD4+ T cells in the white matter and in grey matter cortical demyelinating lesions, and their numbers closely correlate with axonal damage3. Consistent with a key role for these cells in disease pathogenesis, myelin-specific CD8+ T cells are readily activated by epitope spreading, even in a CD4+ T cell-driven EAE model, with this being aided by antigen cross-presentation by monocyte-derived DCs in the CNS53. Non-myelin astrocyte-derived antigen can also trigger spontaneous relapsing-remitting disease in mice by driving the establishment of non-recirculating resident memory-like CD8+ T cells within the CNS68. The disease course and pathology in this model was modulated by B cells and by viral triggering, suggesting that complex multicellular and environmental interactions can contribute to disease heterogeneity. In humans, up to a quarter of CD8+ T cells in the active lesions of patients with multiple sclerosis can produce IL-17 and are thus thought to be mucosa-associated invariant T cells (MAIT cells)69. Efficacious autologous haematopoietic stem cell transplantation in patients with highly active disease results in the long-lasting depletion of these cells, suggesting that they have an important role in disease pathogenesis70. The precise contribution of CD8+ T cells, compared with that of CD4+ T cells and other cell types, following autologous haematopoietic stem cell transplantation and the relative importance of targeting these cells in the therapeutic efficacy of broad-spectrum drugs — such as natalizumab (Tysabri; Biogen and Elan), alemtuzumab (Campath-1H; Genzyme) or fingolimod (FTY720 and Gilenya; Novartis)4 — is not entirely clear and requires further study; however, based on evidence published to date, the exploration of CD8+ T cell-specific therapies in the future is warranted.
Autoreactive B cells. Compared with T cells, infiltrating B cell numbers in the CNS vary more throughout disease progression. Clonally expanded B cells can be found in the meninges, parenchyma and CSF, and intrathecal B cells produce antibodies that are detectable in the CSF and are of diagnostic value. Numbers of antibody-secreting plasma cells are increased with age in patients with primary or secondary progressive multiple sclerosis3. The meninges of patients with secondary progressive disease often contain tertiary lymphoid structures of aggregated plasma cells, B cells, T cells and follicular DCs (FDCs)71, which are a product of long-term inflammation as observed in other chronic inflammatory or infectious diseases72. By contrast, primary progressive disease is characterized by diffuse meningeal infiltration without such structures73. Despite initial reports that certain autoantigens — including MOG, neurofascin, contactin and the ATP-dependent inwardly rectifying potassium channel KIR4.1 — are recognized by pathogenic B cells in subgroups of patients, these findings still await verification74. Moreover, other antibody-mediated neurological diseases, such as myasthenia gravis, neuromyelitis optica and autoimmune encephalitis, show a clinical uniformity75 that is absent in the subgroup of patients with antibody-positive multiple sclerosis.
In the absence of known autoantigens, the mechanisms controlling B cell activation, selection and affinity maturation have been a matter of speculation. However, the recent application of next-generation sequencing technologies to analyse B cell receptor diversity has allowed for the characterization of B cell clonotypes in the peripheral compartments and the CSF of patients with multiple sclerosis, and such studies indicate that antigen-experienced B cells can undergo maturation in draining cervical lymph nodes before transmigration to the CNS76,77. These data imply a therapeutic potential for the peripheral modulation of specific B cell subtypes76,77. Currently, Phase II clinical trials have shown that CD20-specific monoclonal antibodies rituximab (MabThera; Roche)78 or ocrelizumab (Roche and Biogen)79 are efficacious in reducing relapse rates. These drugs deplete the majority of B cell subsets but not autoantibody-producing terminally differentiated plasma cells, and they may thus serve to effectively reduce B cell-mediated antigen presentation and other non-autoantibody-associated pathogenic contributions such as pro-inflammatory IL-6 production80.
Defective regulatory cells. The emergence and action of autoreactive B cells and T cells in multiple sclerosis may be due to the defective functions of regulatory cells, such as forkhead box P3 (FOXP3)-expressing CD4+ regulatory T (TReg) cells81 and IL-10-producing T regulatory type 1 (TR1) cells82. Although few such cells are present in the CNS of patients83, disease-associated HLA class II variants could skew thymic selection such that the regulatory T cells that are released into the periphery inadequately suppress autoreactive effector T cells81. Alternatively, dysfunction of peripheral suppressor cells could be indirectly driven by the dysregulation of tolerogenic APCs, as shown in EAE84. Non-HLA genetic associations, such as variation in the BACH2 gene region10, may also be implicated in altering TReg cell function, as the transcription factor BACH2 has an essential role in the development of these cells85 and acts as a super-enhancer for T cell identity86. However, patients with immunodysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX), who have a FOXP3 deficiency, do not develop CNS-directed autoimmunity87, and therefore TReg cell dysfunction in patients with multiple sclerosis may be an acquired rather than a primary defect.
Studies have variably reported a decreased frequency and/or suppressive capacity of TReg cells88,89, as well as an altered frequency of specific TReg cell subsets (such as CD39+ cells90), in the periphery of patients with multiple sclerosis compared with controls. Such defects have been attributed to reduced frequencies of naive circulating TReg cells of recent thymic origin (identified as CD45RA+CD31+), along with the compensatory but ineffective expansion of the memory TReg cell population88. Another possibility is the skewing of TReg cells towards an IFNγ-secreting TH1 cell-like phenotype in patients, which is reversible upon IFNβ therapy91. An alternative but non-mutually exclusive explanation for the action of autoreactive effector T cells in multiple sclerosis is that, rather than being the outcome of inadequate peripheral suppression, the effector T cells themselves are actively resistant to suppressive mechanisms, with the suggestion that IL-6-induced signal transducer and activator of transcription 3 (STAT3)-mediated signalling contributes to this resistance92,93. Such resistance mechanisms emphasize the putative caveat of studies that document patient TReg cell dysfunction using autologous effector T cells, as these reports may in fact reflect increased effector T cell resistance.
In addition to CD4+ TReg cells, CD8+ regulatory T cells have been implicated in EAE94. These cells have also been found in patients with multiple sclerosis in whom HLA-E-restricted CD8+ T cells display a less regulatory phenotype than those in healthy individuals95, and neuroantigen-specific CD8+ T cells may have less suppressive capacity during relapses96. Enhanced cytotoxic CD8+ regulatory T cell function has also been observed in patients following glatiramer acetate therapy97, and expansion of a putative regulatory CD103+CD8+ T cell subset has been reported in some patients treated with natalizumab98. Drug administration may also influence regulatory B cells: IFNβ therapy correlates with an increase in the numbers of IL-10-producing regulatory CD19+CD24hiCD38hi transitional B cells in treated patients with multiple sclerosis99. Other regulatory B cell subsets, such as those secreting IL-35, have also been implicated in recovery from EAE100.
Collectively, dysregulation of effector–regulatory cell interactions occurs in multiple sclerosis, ultimately resulting in the emergence of autoreactive adaptive immune cells that are capable of infiltrating and promoting damage within the CNS. The skewing of effector–regulatory cell interactions may provide some therapeutic benefit but may not be sufficient to prevent neurodegeneration.
Inflammation in progressive neurodegeneration
As currently available immunomodulatory therapies decrease relapse rates but not necessarily long-term multiple sclerosis progression, it has been suggested that autoimmune response-instigated neuroaxonal injury triggers a potentially self-sustaining chronic neurodegenerative process. This proceeds even in the absence of continued immune cell infiltration from the periphery, which eventually wanes regardless of therapy, possibly due to immune cell exhaustion associated with chronic antigenic exposure101. Although neurodegeneration in multiple sclerosis is thought to be the culmination of a cascade of events occurring in axons and neurons — including oxidative stress responses, energy deficiencies, ionic imbalances, and the failure of neuroprotective and regenerative mechanisms6 (Box 5) — chronic CNS inflammation may fuel these processes through the action of cells that have become or are already resident within the CNS (Fig. 2).
Previously infiltrating adaptive immune cells may contribute to long-term inflammation in multiple sclerosis through the eventual establishment of tertiary lymphoid structures within the CNS71. However, it is becoming increasingly apparent that CNS-resident cells that sense homeostatic disturbances, mainly microglia and astrocytes, can also produce a range of neurotoxic inflammatory mediators (such as cytokines, chemokines and reactive oxygen species) that promote and sustain neuroaxonal damage and thus neurodegeneration6,19 (Fig. 2).
Moreover, these cells are likely to have a role in multiple sclerosis-associated CNS inflammation not only during the later stages of the disease when immune cell infiltration from the periphery subsides but also from the outset (Fig. 2). Even after the very first manifestation of disease, increases in the numbers and activation status of microglia and macrophages can be observed in lesions and in the normal-appearing white matter9,102. In addition, as neuroaxonal degeneration disseminates (Box 5), microglia in the vicinity of axons emanating from distally damaged neurons may become activated; these cells may hence serve as the nucleus of new lesion formation103 and might also contribute to the general brain atrophy that is observed in early disease104. Notably, the relative role of microglia versus monocyte-derived macrophages throughout the course of multiple sclerosis has not been fully elucidated owing to the difficulty in distinguishing these two morphologically and functionally similar cell types.
Some insights have been gained from transgenic EAE models, which have enabled these cell types to be studied in distinction: these studies have shown that at disease onset monocyte-derived macrophages initiate demyelination, whereas microglia may be more involved in debris clearing105. This suggests that microglia may have some neuroprotective capacity by helping to resolve inflammation105, as well as by actively displacing specific neuronal synapses to maintain CNS homeostasis106 and by producing neurotrophic factors (such as brain-derived neurotrophic factor) to aid the repair of neuroaxonal damage. Primary neurodegeneration in conditions such as Alzheimer disease is partly attributed to inadequate neuroprotective microglial action19, and this functional failure may also be relevant to multiple sclerosis development.
Conversely, healthy neurons constitutively express inhibitors that block the phagocytic capacity of microglia107, implying that if left unchecked, microglia may promote tissue injury, driving a feedback loop of progressive neuroaxonal damage. Moreover, specific transgenic targeting of microglia has also been reported to reduce EAE-associated CNS inflammation108. In addition, activated microglia can promote astrocyte dysfunction. Similarly to microglia, astrocytes can display both pro-inflammatory and anti-inflammatory properties, and they have a crucial CNS barrier function by forming the glia limitans that lines the neuronal tissue. Their dysfunction can permit and even facilitate peripheral immune cell infiltration early in multiple sclerosis through the production of chemokines. Moreover, upon activation by stimulated microglia, astrocytes can produce CC-chemokine ligand 2 (CCL2) and GM-CSF, leading to even further microglial recruitment and activation, and they can prevent remyelination at sites of neuroaxonal injury by inhibiting the generation of mature oligodendrocytes8 (Fig. 2). Therefore, targeting pro-inflammatory mediators produced by astrocytes may serve to inhibit both peripheral immune cell infiltration and the continuous inflammation within the CNS, and may thus be of therapeutic value.
The incompletely resolved role of CNS-resident innate-like immune cells in multiple sclerosis immunopathology — in dampening down inflammation and/or actively contributing to it — may reflect our only partial understanding of how the function of these cells varies across different regions of the CNS and throughout the course of the disease. Notably, the pro-inflammatory action of CNS-resident innate-like immune cells in progressive neurodegeneration may be intrinsically linked to multiple sclerosis chronicity. Inflammation in the CNS may be viewed as a stress response to maintain tissue homeostasis109 — particularly as both the innate immune and neuronal compartments of the CNS are specialized to sense an array of stressors including ions, low pH110, temperature changes, hormones and cytokines — and even in the absence of disease, CNS-derived inflammatory processes increase as a function of time, eventually promoting ageing-associated neurodegeneration111.
In multiple sclerosis, the additional inflammation induced and/or enhanced by peripheral immune cell filtration and by CNS-resident innate-like immune cells may effectively contribute to the acceleration of the inevitable ageing processes in the CNS, and as the subjugating burden of the stress response is too great for homeostasis to be adequately maintained, pronounced progressive neurodegenerative decline follows.

Concluding remarks
Our understanding of multiple sclerosis immunopathology has been consistently modified since the approval of the first immunomodulatory therapy for the condition, and therefore questions regarding the mechanisms underscoring the triggers and long-term development of the disease remain to be definitively answered, although these questions are now better defined.
Perhaps most significantly, the appreciation of multiple sclerosis as the pathophysiological intersection between interlinked but not entirely interdependent autoimmune and neurodegenerative processes has set imminent research challenges. There is a dire need to meaningfully integrate rapidly emerging technologies and data with existing neuroimmunological clinical concepts in order to interrogate the multicellular interplay that unfolds within the CNS throughout disease progression. Underscoring this is the requirement to further decompartmentalize the study of immunology and neurology in multiple sclerosis through the investigation of neuroinflammation as a whole in order to better delineate which inflammatory and neurodegenerative mechanisms are truly distinct but occur in parallel and which are inextricably associated, so as to aid the design of more effective therapeutic strategies.
A goal for future treatment of multiple sclerosis may thus be the simultaneous, early targeting of peripheral immune cell function and of CNS-intrinsic inflammation, potentially through combinatorial therapies designed to effectively and specifically modulate these two immunological arms of the disease, along with the provision of neuroprotective or neuroregenerative drugs. Improved disease prognosis and potential patient stratification for more directed healthcare provision are also much-anticipated prospects and may become tangible as we move into the 'immune informatics' era and as large-scale, organized health resources become increasingly accessible.
References
Compston, A. & Coles, A. Multiple sclerosis. Lancet 372, 1502–1517 (2008).
Kearney, H. et al. Cervical cord lesion load is associated with disability independently from atrophy in MS. Neurology 84, 367–373 (2015).
Frischer, J. M. et al. The relation between inflammation and neurodegeneration in multiple sclerosis brains. Brain 132, 1175–1189 (2009).
Haghikia, A., Hohlfeld, R., Gold, R. & Fugger, L. Therapies for multiple sclerosis: translational achievements and outstanding needs. Trends Mol. Med. 19, 309–319 (2013).
Feinstein, A., Freeman, J. & Lo, A. C. Treatment of progressive multiple sclerosis: what works, what does not, and what is needed. Lancet Neurol. 14, 194–207 (2015).
Friese, M. A., Schattling, B. & Fugger, L. Mechanisms of neurodegeneration and axonal dysfunction in multiple sclerosis. Nat. Rev. Neurol. 10, 225–238 (2014).
Schattling, B. et al. TRPM4 cation channel mediates axonal and neuronal degeneration in experimental autoimmune encephalomyelitis and multiple sclerosis. Nat. Med. 18, 1805–1811 (2012).
Mayo, L. et al. Regulation of astrocyte activation by glycolipids drives chronic CNS inflammation. Nat. Med. 20, 1147–1156 (2014).
Popescu, B. F. & Lucchinetti, C. F. Pathology of demyelinating diseases. Annu. Rev. Pathol. 7, 185–217 (2012).
Beecham, A. H. et al. Analysis of immune-related loci identifies 48 new susceptibility variants for multiple sclerosis. Nat. Genet. 45, 1353–1360 (2013).
Brodin, P. et al. Variation in the human immune system is largely driven by non-heritable influences. Cell 160, 37–47 (2015). A comprehensive study of the contribution of non-genetic factors to the variation of more than 200 immunological parameters.
Belbasis, L., Bellou, V., Evangelou, E., Ioannidis, J. P. A. & Tzoulaki, I. Environmental risk factors and multiple sclerosis: an umbrella review of systematic reviews and meta-analyses. Lancet Neurol. 14, 263–273 (2015).
Harkiolaki, M. et al. T cell-mediated autoimmune disease due to low-affinity crossreactivity to common microbial peptides. Immunity 30, 348–357 (2009).
Muenz, C., Luenemann, J. D., Getts, M. T. & Miller, S. D. Antiviral immune responses: triggers of or triggered by autoimmunity? Nat. Rev. Immunol. 9, 246–258 (2009).
Olson, J. K., Croxford, J. L., Calenoff, M. A., Dal Canto, M. C. & Miller, S. D. A virus-induced molecular mimicry model of multiple sclerosis. J. Clin. Invest. 108, 311–318 (2001).
Ji, Q., Perchellet, A. & Goverman, J. M. Viral infection triggers central nervous system autoimmunity via activation of CD8+ T cells expressing dual TCRs. Nat. Immunol. 11, 628–634 (2010).
Ransohoff, R. M. & Engelhardt, B. The anatomical and cellular basis of immune surveillance in the central nervous system. Nat. Rev. Immunol. 12, 623–635 (2012).
Louveau, A. et al. Structural and functional features of central nervous system lymphatic vessels. Nature 523, 337–341 (2015).
Heneka, M. T., Kummer, M. P. & Latz, E. Innate immune activation in neurodegenerative disease. Nat. Rev. Immunol. 14, 463–477 (2014).
Gregory, A. P. et al. TNF receptor 1 genetic risk mirrors outcome of anti-TNF therapy in multiple sclerosis. Nature 488, 508–511 (2012). This study demonstrates how investigating the functional consequences of disease-associated genetic variation can help to predict therapeutic outcome.
Menche, J. et al. Uncovering disease-disease relationships through the incomplete interactome. Science 347, 1257601 (2015).
Tasan, M. et al. Selecting causal genes from genome-wide association studies via functionally coherent subnetworks. Nat. Methods 12, 154–159 (2015).
Farh, K. K. et al. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature 518, 337–343 (2014).
Kundaje, A. et al. Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330 (2015).
Raj, T. et al. Polarization of the effects of autoimmune and neurodegenerative risk alleles in leukocytes. Science 344, 519–523 (2014).
Ye, C. J. et al. Intersection of population variation and autoimmunity genetics in human T cell activation. Science 345, 1254665 (2014).
Roederer, M. et al. The genetic architecture of the human immune system: a bioresource for autoimmunity and disease pathogenesis. Cell 161, 387–403 (2015). An extensive immunophenotyping study that provides a bioresource for linking genetic control elements associated with normal immunological traits to common autoimmune and infectious diseases.
Friese, M. A. et al. Opposing effects of HLA class I molecules in tuning autoreactive CD8+ T cells in multiple sclerosis. Nat. Med. 14, 1227–1235 (2008).
Gregersen, J. W. et al. Functional epistasis on a common MHC haplotype associated with multiple sclerosis. Nature 443, 574–577 (2006).
Hartmann, F. J. et al. Multiple sclerosis-associated IL2RA polymorphism controls GM-CSF production in human TH cells. Nat. Commun. 5, 5056 (2014).
Dendrou, C. A. et al. Cell-specific protein phenotypes for the autoimmune locus IL2RA using a genotype-selectable human bioresource. Nat. Genet. 41, 1011–1015 (2009).
Gregory, S. G. et al. Interleukin 7 receptor α chain (IL7R) shows allelic and functional association with multiple sclerosis. Nat. Genet. 39, 1083–1091 (2007).
Lundstrom, W. et al. Soluble IL7Rα potentiates IL-7 bioactivity and promotes autoimmunity. Proc. Natl Acad. Sci. USA 110, E1761–E1770 (2013).
Lambert, J.-C. et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer's disease. Nat. Genet. 45, 1452–1458 (2013).
Nalls, M. A. et al. Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson's disease. Nat. Genet. 46, 989–993 (2014).
Scally, S. W. et al. A molecular basis for the association of the HLA-DRB1 locus, citrullination, and rheumatoid arthritis. J. Exp. Med. 210, 2569–2582 (2013).
Miller, S. D. et al. Persistent infection with Theiler's virus leads to CNS autoimmunity via epitope spreading. Nat. Med. 3, 1133–1136 (1997).
Quan, N., Whiteside, M. & Herkenham, M. Time course and localization patterns of interleukin-1β messenger RNA expression in brain and pituitary after peripheral administration of lipopolysaccharide. Neuroscience 83, 281–293 (1998).
Vitkovic, L. et al. Cytokine signals propagate through the brain. Mol. Psychiatry 5, 604–615 (2000).
Dantzer, R., O'Connor, J. C., Freund, G. G., Johnson, R. W. & Kelley, K. W. From inflammation to sickness and depression: when the immune system subjugates the brain. Nat. Rev. Neurosci. 9, 46–57 (2008).
Hedstrom, A. K., Akerstedt, T., Hillert, J., Olsson, T. & Alfredsson, L. Shift work at young age is associated with increased risk for multiple sclerosis. Ann. Neurol. 70, 733–741 (2011).
Operskalski, E. A., Visscher, B. R., Malmgren, R. M. & Detels, R. A case-control study of multiple sclerosis. Neurology 39, 825–829 (1989).
Lossius, A. et al. High-throughput sequencing of TCR repertoires in multiple sclerosis reveals intrathecal enrichment of EBV-reactive CD8+ T cells. Eur. J. Immunol. 44, 3439–3452 (2014).
Snyder, C. M. et al. Memory inflation during chronic viral infection is maintained by continuous production of short-lived, functional T cells. Immunity 29, 650–659 (2008).
Duszczyszyn, D. A. et al. Thymic involution and proliferative T-cell responses in multiple sclerosis. J. Neuroimmunol. 221, 73–80 (2010).
Sargsyan, S. A. et al. Absence of Epstein-Barr virus in the brain and CSF of patients with multiple sclerosis. Neurology 74, 1127–1135 (2010).
Virgin, H. W. The virome in mammalian physiology and disease. Cell 157, 142–150 (2014).
Yatsunenko, T. et al. Human gut microbiome viewed across age and geography. Nature 486, 222–227 (2012).
Berer, K. et al. Commensal microbiota and myelin autoantigen cooperate to trigger autoimmune demyelination. Nature 479, 538–541 (2011).
Bielekova, B. et al. Expansion and functional relevance of high-avidity myelin-specific CD4+ T cells in multiple sclerosis. J. Immunol. 172, 3893–3904 (2004).
Hellings, N. et al. T-cell reactivity to multiple myelin antigens in multiple sclerosis patients and healthy controls. J. Neurosci. Res. 63, 290–302 (2001).
McMahon, E. J., Bailey, S. L., Castenada, C. V., Waldner, H. & Miller, S. D. Epitope spreading initiates in the CNS in two mouse models of multiple sclerosis. Nat. Med. 11, 335–339 (2005).
Ji, Q., Castelli, L. & Goverman, J. M. MHC class I restricted myelin epitopes are cross-presented by Tip-DCs that promote determinant spreading to CD8+ T cells. Nat. Immunol. 14, 254–261 (2013).
Siewert, K. et al. Unbiased identification of target antigens of CD8+ T cells with combinatorial libraries coding for short peptides. Nat. Med. 18, 824–828 (2012).
Lutterotti, A. et al. Antigen-specific tolerance by autologous myelin peptide-coupled cells: a phase 1 trial in multiple sclerosis. Sci. Transl. Med. 5, 188ra175 (2013).
Kozovska, M. E. et al. Interferon beta induces T-helper 2 immune deviation in MS. Neurology 53, 1692–1697 (1999).
Miller, A. et al. Treatment of multiple sclerosis with copolymer-1 (Copaxone): implicating mechanisms of Th1 to Th2/Th3 immune-deviation. J. Neuroimmunol. 92, 113–121 (1998).
Zoghi, S. et al. Cytokine secretion pattern in treatment of lymphocytes of multiple sclerosis patients with fumaric acid esters. Immunol. Invest. 40, 581–596 (2011).
Frisullo, G. et al. IL17 and IFNγ production by peripheral blood mononuclear cells from clinically isolated syndrome to secondary progressive multiple sclerosis. Cytokine 44, 22–25 (2008).
Tzartos, J. S. et al. Interleukin-17 production in central nervous system-infiltrating T cells and glial cells is associated with active disease in multiple sclerosis. Am. J. Pathol. 172, 146–155 (2008).
Cao, Y. et al. Functional inflammatory profiles distinguish myelin-reactive T cells from patients with multiple sclerosis. Sci. Transl. Med. 7, 287ra274 (2015).
Kebir, H. et al. Preferential recruitment of interferon-γ-expressing TH17 cells in multiple sclerosis. Ann. Neurol. 66, 390–402 (2009).
Segal, B. M. et al. Repeated subcutaneous injections of IL12/23 P40 neutralising antibody, ustekinumab, in patients with relapsing-remitting multiple sclerosis: a phase II, double-blind, placebo-controlled, randomised, dose-ranging study. Lancet Neurol. 7, 796–804 (2008).
Ghoreschi, K. et al. Generation of pathogenic TH17 cells in the absence of TGF-β signalling. Nature 467, 967–971 (2010).
Zielinski, C. E. et al. Pathogen-induced human TH17 cells produce IFN-γ or IL-10 and are regulated by IL-1β. Nature 484, 514–518 (2012).
Codarri, L. et al. RORγt drives production of the cytokine GM-CSF in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation. Nat. Immunol. 12, 560–567 (2011).
Noster, R. et al. IL-17 and GM-CSF expression are antagonistically regulated by human T helper cells. Sci. Transl. Med. 6, 241ra280 (2014).
Sasaki, K. et al. Relapsing–remitting central nervous system autoimmunity mediated by GFAP-specific CD8 T cells. J. Immunol. 192, 3029–3042 (2014).
Willing, A. et al. CD8+ MAIT cells infiltrate into the CNS and alterations in their blood frequencies correlate with IL-18 serum levels in multiple sclerosis. Eur. J. Immunol. 44, 3119–3128 (2014).
Abrahamsson, S. V. et al. Non-myeloablative autologous haematopoietic stem cell transplantation expands regulatory cells and depletes IL-17 producing mucosal-associated invariant T cells in multiple sclerosis. Brain 136, 2888–2903 (2013).
Howell, O. W. et al. Meningeal inflammation is widespread and linked to cortical pathology in multiple sclerosis. Brain 134, 2755–2771 (2011).
Drayton, D. L., Liao, S., Mounzer, R. H. & Ruddle, N. H. Lymphoid organ development: from ontogeny to neogenesis. Nat. Immunol. 7, 344–353 (2006).
Choi, S. R. et al. Meningeal inflammation plays a role in the pathology of primary progressive multiple sclerosis. Brain 135, 2925–2937 (2012).
Brickshawana, A. et al. Investigation of the KIR4.1 potassium channel as a putative antigen in patients with multiple sclerosis: a comparative study. Lancet Neurol. 13, 795–806 (2014).
Leypoldt, F., Armangue, T. & Dalmau, J. Autoimmune encephalopathies. Ann. NY Acad. Sci. 1338, 94–114 (2015).
Palanichamy, A. et al. Immunoglobulin class-switched B cells form an active immune axis between CNS and periphery in multiple sclerosis. Sci. Transl. Med. 6, 248ra106 (2014).
Stern, J. N. H. et al. B cells populating the multiple sclerosis brain mature in the draining cervical lymph nodes. Sci. Transl. Med. 6, 248ra107 (2014). References 76 and 77 use B cell receptor sequencing to investigate the relationship between B cell subsets in the periphery and the CNS, supporting the peripheral targeting of B cells for multiple sclerosis treatment.
Hauser, S. L. et al. B-cell depletion with rituximab in relapsing–remitting multiple sclerosis. N. Engl. J. Med. 358, 676–688 (2008).
Kappos, L. et al. Ocrelizumab in relapsing-remitting multiple sclerosis: a phase 2, randomised, placebo-controlled, multicentre trial. Lancet 378, 1779–1787 (2011).
Barr, T. A. et al. B cell depletion therapy ameliorates autoimmune disease through ablation of IL-6-producing B cells. J. Exp. Med. 209, 1001–1010 (2012).
Venken, K. et al. Natural naive CD4+CD25+CD127low regulatory T cell (Treg) development and function are disturbed in multiple sclerosis patients: recovery of memory Treg homeostasis during disease progression. J. Immunol. 180, 6411–6420 (2008).
Martinez-Forero, I. et al. IL-10 suppressor activity and ex vivo Tr1 cell function are impaired in multiple sclerosis. Eur. J. Immunol. 38, 576–586 (2008).
Fritzsching, B. et al. Intracerebral human regulatory T cells: Analysis of CD4+CD25+FOXP3+ T cells in brain lesions and cerebrospinal fluid of multiple sclerosis patients. PLoS ONE 6, e17988 (2011).
Yogev, N. et al. Dendritic cells ameliorate autoimmunity in the CNS by controlling the homeostasis of PD-1 receptor+ regulatory T cells. Immunity 37, 264–275 (2012).
Roychoudhuri, R. et al. BACH2 represses effector programs to stabilize Treg-mediated immune homeostasis. Nature 498, 506–510 (2013).
Vahedi, G. et al. Super-enhancers delineate disease-associated regulatory nodes in T cells. Nature 520, 558–562 (2015).
Bennett, C. L. et al. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat. Genet. 27, 20–21 (2001).
Venken, K. et al. Compromised CD4+CD25high regulatory T-cell function in patients with relapsing-remitting multiple sclerosis is correlated with a reduced frequency of FOXP3-positive cells and reduced FOXP3 expression at the single-cell level. Immunology 123, 79–89 (2008).
Feger, U. et al. Increased frequency of CD4+CD25+ regulatory T cells in the cerebrospinal fluid but not in the blood of multiple sclerosis patients. Clin. Exp. Immunol. 147, 412–418 (2007).
Fletcher, J. M. et al. CD39+Foxp3+ regulatory T cells suppress pathogenic Th17 cells and are impaired in multiple sclerosis. J. Immunol. 183, 7602–7610 (2009).
Dominguez-Villar, M., Baecher-Allan, C. M. & Hafler, D. A. Identification of T helper type 1-like, Foxp3+ regulatory T cells in human autoimmune disease. Nat. Med. 17, 673–675 (2011).
Schneider, A. et al. In active relapsing-remitting multiple sclerosis, effector T cell resistance to adaptive Tregs involves IL-6-mediated signaling. Sci. Transl. Med. 5, 170ra115 (2013).
Bhela, S. et al. Nonapoptotic and extracellular activity of granzyme B mediates resistance to regulatory T cell (Treg) suppression by HLA-DR−CD25hiCD127lo Tregs in multiple sclerosis and in response to IL-6. J. Immunol. 194, 2180–2189 (2015).
Hu, D. et al. Analysis of regulatory CD8 T cells in Qa-1-deficient mice. Nat. Immunol. 5, 516–523 (2004).
Pannemans, K. et al. HLA-E restricted CD8+ T cell subsets are phenotypically altered in multiple sclerosis patients. Mult. Scler. 20, 790–801 (2014).
Baughman, E. J. et al. Neuroantigen-specific CD8+ regulatory T-cell function is deficient during acute exacerbation of multiple sclerosis. J. Autoimmun. 36, 115–124 (2011).
Tennakoon, D. K. et al. Therapeutic induction of regulatory, cytotoxic CD8+ T cells in multiple sclerosis. J. Immunol. 176, 7119–7129 (2006).
Mattoscio, M. et al. Hematopoietic mobilization: potential biomarker of response to natalizumab in multiple sclerosis. Neurology 84, 1473–1482 (2015).
Schubert, R. D. et al. IFN-β treatment requires B cells for efficacy in neuroautoimmunity. J. Immunol. 194, 2110–2116 (2015).
Shen, P. et al. IL-35-producing B cells are critical regulators of immunity during autoimmune and infectious diseases. Nature 507, 366–370 (2014).
Wherry, E. J. T cell exhaustion. Nat. Immunol. 12, 492–499 (2011).
Giannetti, P. et al. Increased PK11195-PET binding in normal-appearing white matter in clinically isolated syndrome. Brain 138, 110–119 (2015).
Kolasinski, J. et al. A combined post-mortem magnetic resonance imaging and quantitative histological study of multiple sclerosis pathology. Brain 135, 2938–2951 (2012).
Chard, D. T. et al. Brain atrophy in clinically early relapsing-remitting multiple sclerosis. Brain 125, 327–337 (2002).
Yamasaki, R. et al. Differential roles of microglia and monocytes in the inflamed central nervous system. J. Exp. Med. 211, 1533–1549 (2014). A study that used transgenic technology, scanning electron microscopy and gene expression profiling to demonstrate a differential role of microglia and monocytes in EAE.
Chen, Z. et al. Microglial displacement of inhibitory synapses provides neuroprotection in the adult brain. Nat. Commun. 5, 4486 (2014).
Neher, J. J. et al. Inhibition of microglial phagocytosis is sufficient to prevent inflammatory neuronal death. J. Immunol. 186, 4973–4983 (2011).
Goldmann, T. et al. A new type of microglia gene targeting shows TAK1 to be pivotal in CNS autoimmune inflammation. Nat. Neurosci. 16, 1618–1626 (2013).
Kotas, M. E. & Medzhitov, R. Homeostasis, inflammation, and disease susceptibility. Cell 160, 816–827 (2015). An intriguing review on the parallels between homeostatic and inflammatory control mechanisms in health and disease.
Friese, M. A. et al. Acid-sensing ion channel-1 contributes to axonal degeneration in autoimmune inflammation of the central nervous system. Nat. Med. 13, 1483–1489 (2007).
Baruch, K. et al. Aging-induced type I interferon response at the choroid plexus negatively affects brain function. Science 346, 89–93 (2014).
Kira, J. Neuromyelitis optica and Asian phenotype of multiple sclerosis. Ann. NY Acad. Sci. 1142, 58–71 (2008).
Fischer, M. T. et al. NADPH oxidase expression in active multiple sclerosis lesions in relation to oxidative tissue damage and mitochondrial injury. Brain 135, 886–899 (2012).
Shechter, R., London, A. & Schwartz, M. Orchestrated leukocyte recruitment to immune-privileged sites: absolute barriers versus educational gates. Nat. Rev. Immunol. 13, 206–218 (2013).
Perdiguero, E. G. et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature 518, 547–551 (2015).
Yednock, T. A. et al. Prevention of experimental autoimmune encephalomyelitis by antibodies against α4β1 integrin. Nature 356, 63–66 (1992).
Ben-Nun, A. et al. From classic to spontaneous and humanized models of multiple sclerosis: impact on understanding pathogenesis and drug development. J. Autoimmun. 54, 33–50 (2014).
Campbell, G. R. et al. Clonally expanded mitochondrial DNA deletions within the choroid plexus in multiple sclerosis. Acta Neuropathol. 124, 209–220 (2012).
Haider, L. et al. Oxidative damage in multiple sclerosis lesions. Brain 134, 1914–1924 (2011).
Weisfeld-Adams, J. D., Katz Sand, I. B., Honce, J. M. & Lublin, F. D. Differential diagnosis of Mendelian and mitochondrial disorders in patients with suspected multiple sclerosis. Brain 138, 517–539 (2015).
Craner, M. J. et al. Molecular changes in neurons in multiple sclerosis: altered axonal expression of Nav1.2 and Nav1.6 sodium channels and Na+/Ca2+ exchanger. Proc. Natl Acad. Sci. USA 101, 8168–8173 (2004).
Acknowledgements
This work was supported by the Wellcome Trust, the Medical Research Council, the Alan and Babette Sainsbury Charitable Fund, and the Rosetrees Trust (L.F.).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary information S1 (table)
Approved drugs for the treatment of multiple sclerosis (PDF 134 kb)
Supplementary information S2 (table)
Murine EAE models. (PDF 180 kb)
PowerPoint slides
Glossary
- Demyelination
-
Damage to the myelin sheath surrounding nerves in the brain and spinal cord, which affects the function of the nerves involved. Demyelination occurs in multiple sclerosis and in experimental autoimmune encephalomyelitis, which is an animal model of multiple sclerosis.
- Gliosis
-
The proliferation and activation of glial cells (microglia, oligodendrocytes and astrocytes) in response to damage in the central nervous system.
- Tumefactive multiple sclerosis
-
A subtype of multiple sclerosis characterized by atypical, large demyelinated lesions that appear tumour-like and oedematous and can exert pressure on the surrounding central nervous system tissue due to their size.
- Molecular mimicry
-
A mechanism by which a peptide from a foreign antigen that is presented to a T cell closely resembles part of a self-protein, thereby triggering an autoimmune reaction.
- Choroid plexus
-
The site of production of cerebrospinal fluid in the adult brain. It is formed by invagination of ependymal cells into the ventricles, which become highly vascularized.
- Primary neurodegeneration
-
The process of progressive dysfunction and loss of axons and neurons, triggered by mechanisms involving central nervous system-resident cells, as opposed to cells infiltrating from the periphery.
- Candidate genes
-
Genes assumed to be affected by disease-associated genetic polymorphisms, based on their functional relevance and/or their physical proximity to the polymorphisms in question. The determination of whether the assigned candidates are truly affected by the polymorphisms and how they influence disease susceptibility typically requires functional follow-up investigations at the molecular, cellular and systemic levels.
- Interactome networks
-
Maps of molecular interactions, often segregated by cell type, and used as a framework to simplify cellular organization and to help address systems biology questions at the cellular level. These networks may reflect sets of physical intermolecular interactions as well as other molecules that indirectly act together in specific pathways.
- Central tolerance
-
Self-tolerance that is created at the level of the central lymphoid organs. Developing T cells (in the thymus) and B cells (in the bone marrow) that strongly recognize self-antigen must undergo further rearrangement of antigen-receptor genes to become self-tolerant, or they face deletion.
- Immune-privileged site
-
An area in the body with a decreased immune response to foreign antigens, including tissue grafts. These sites include the brain, eye, testis and placenta.
- Dural sinuses
-
Venous channels located between layers of the brain dura mater. These sinuses receive blood from both internal and external brain veins, and cerebrospinal fluid from the subarachnoid space, and empty into the jugular vein.
- Epitope spreading
-
This term is used to describe how a self-directed immune response induced by a single peptide (or epitope) could spread to include other peptides (or epitopes) not only on the same autoantigen (intramolecular spreading) but also on other self-molecules clustered in close vicinity within the target cell (intermolecular spreading).
- Diapedesis
-
The migration of leukocytes across the endothelium, which occurs by leukocytes squeezing through the junctions between adjacent endothelial cells.
- Cross-presentation
-
The initiation of a CD8+ T cell response to an antigen that is not present within antigen-presenting cells (APCs). This exogenous antigen must be taken up by APCs and then re-routed to the MHC class I pathway of antigen presentation.
- Mucosa-associated invariant T cells
-
(MAIT cells). A type of CD8+ T cell that is enriched at mucosal sites and is characterized by the expression of a semi-invariant T cell receptor (a dimer of Vα7.2 in combination with Jα12, Jα20 or Jα33) and is restricted by the non-polymorphic, highly evolutionarily conserved MHC class Ib molecule, MR1.
- Tertiary lymphoid structures
-
Organized lymphocytic aggregates that form in sites of chronic inflammation. Typically, B cell- and T cell-rich zones are segregated, and dendritic cells (DCs), germinal centres with follicular DC networks and specialized endothelial cells are present.
- Super-enhancer
-
A cluster of regulatory elements within a genomic region, often particularly enriched in sites that bind transcriptional co-activators.
- Immunodysregulation, polyendocrinopathy, enteropathy, X-linked syndrome
-
(IPEX). A disease caused by mutations in FOXP3 (which encodes forkhead box P3) and characterized by refractory enteritis and, in some patients, autoimmune endocrinopathies, autoimmune diabetes and thyroiditis.
- Exhaustion
-
Non-responsiveness of the immune system resulting from the deletion of specific thymocytes (central tolerance) and the deletion or functional inactivation of specific T cells in the periphery (peripheral tolerance) in the presence of large quantities of antigen.
Rights and permissions
About this article

Cite this article
Dendrou, C., Fugger, L. & Friese, M. Immunopathology of multiple sclerosis. Nat Rev Immunol 15, 545–558 (2015). https://doi.org/10.1038/nri3871
Published:
Issue Date:
DOI: https://doi.org/10.1038/nri3871
This article is cited by
-
Border-associated macrophages in the central nervous system
Journal of Neuroinflammation (2024)
-
MIF contribution to progressive brain diseases
Journal of Neuroinflammation (2024)
-
Causal relationship between multiple sclerosis and cortical structure: a Mendelian randomization study
Journal of Translational Medicine (2024)
-
Helicobacter pylori infection and risk of multiple sclerosis: an updated meta-analysis
Neurological Sciences (2024)
-
Relapsing–remitting multiple sclerosis patients exhibit differential natural killer functional subpopulations
Acta Neurologica Belgica (2024)
