
Key Points
-
Lysosomes are the primary catabolic compartments of eukaryotic cells. They degrade extracellular material that has been internalized by endocytosis and intracellular components that have been sequestered by autophagy.
-
Most lysosomal hydrolases acquire a mannose-6-phosphate (M6P) tag in the Golgi complex. This tag is recognized in the trans-Golgi network (TGN) by M6P receptors (M6PRs) that target the lysosomal hydrolases to the endo-lysosomal pathway. M6PR-independent pathways for the transport of lysosomal hydrolases do exist but, like the pathways for the transport of lysosomal membrane proteins (LMPs), they are mostly undetermined.
-
Recently, the LMP lysosome integral membrane protein 2 was found to be required for the M6PR-independent transport of the lysosomal hydrolase β-glucocerebrosidase to the lysosome. This finding indicates a novel role for LMPs in intracellular protein transport and links the lysosomal targeting of an LMP to a lysosomal hydrolase, albeit by an as yet unidentified pathway.
-
Recent studies provide evidence for both clathrin- and non-clathrin-mediated exits for LMPs from the TGN, which would allow for a timely and targeted delivery of LMPs to distinct endosomal intermediates. This is further supported by the presence of multiple sorting signals in the cytosolic tails of some LMPs, the observation that domains other than the cytosolic tails can be involved in lysosomal targeting and the occurrence of post-translational modifications that can provide fine-tuning of the sorting signals.
-
The significance of understanding LMP trafficking is further illustrated by the ongoing discovery of new and unexpected roles for LMPs in cellular physiology and of mutations in LMPs that lead to lysosomal dysfunction and disease. An important emerging theme is that the absence or presence of LMPs can essentially change the properties of the target compartment; for example, its acidity, fusogenicity, catabolic capacity, dynamics and drug resistance and the availability of cytoplasmic substrates.
-
Elucidating alternative pathways for the delivery of lysosomal proteins is a major challenge for future studies and is of key importance to expand our knowledge of lysosome biogenesis and to understand the pathologies that are associated with lysosomal dysfunctioning.
Abstract
Lysosomes are the primary catabolic compartments of eukaryotic cells. They degrade extracellular material that has been internalized by endocytosis and intracellular components that have been sequestered by autophagy. In addition, specialized cells contain lysosome-related organelles that store and secrete proteins for cell-type-specific functions. The functioning of a healthy cell is dependent on the proper targeting of newly synthesized lysosomal proteins. Accumulating evidence suggests that there are multiple lysosomal delivery pathways that together allow the regulated and sequential deposition of lysosomal components. The importance of lysosomal trafficking pathways is emphasized by recent findings that reveal new roles for lysosomal membrane proteins in cellular physiology and in an increasing number of diseases that are characterized by defects in lysosome biogenesis.
Similar content being viewed by others
Main
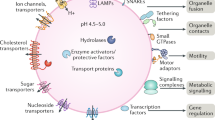
Lysosomes are ubiquitous organelles that constitute the primary degradative compartments of the cell. They receive their substrates through endocytosis, phagocytosis or autophagy (Figs 1,2). The catabolic function of lysosomes is complemented by lysosome-related organelles (LROs), such as melanosomes, lytic granules, major histocompatibility complex (MHC) class II compartments and platelet-dense granules1. LROs share many properties with lysosomes, but they also contain cell-type-specific proteins and might require additional cellular machinery for their biogenesis2,3. Lysosomes and LROs are involved in various physiological processes, such as cholesterol homeostasis, plasma membrane repair, bone and tissue remodelling, pathogen defence, cell death and cell signalling (Fig. 1). These complex functions make the lysosome a central and dynamic organelle and not simply the dead end of the endocytic pathway.

The lysosome is a central, acidic organelle that is involved in the degradation of macromolecules through the activity of lysosomal hydrolases. Lysosomes are crucial for the maturation of phagosomes to phagolysosomes in phagocytosis, which is important for cellular pathogen defence. The macroautophagy pathway mediates the turnover of cytoplasmic components, such as organelles and large complexes, and is involved in cell death and proliferation. Macroautophagy depends on the fusion of lysosomes with autophagosomes to create autolysosomes, in which degradation occurs. Macroautophagy and chaperone-mediated autophagy, a direct lysosomal transport process for cytosolic proteins, are regulated by lysosome-associated membrane proteins (LAMPs). Lysosomal exocytosis and plasma membrane repair are Ca2+ and synaptotagmin 7 (SYT7)-dependent fusion events, which are possibly involved in pathogen entry, autoimmunity and neurite outgrowth. The lysosomal cell death pathway is triggered by a release of lysosomal cathepsins through an unknown mechanism. Cellular cholesterol homeostasis is controlled by lysosomal cholesterol efflux through Niemann–Pick C1 protein (NPC1). Major histocompatibility complex (MHC) class II-dependent antigen presentation requires lysosomal proteases and membrane proteins. The release of exosomes is thought to be involved in adaptive immune responses. Lysosomal membrane proteins are also involved in the transport of newly synthesized hydrolases to the lysosome (for example, lysosomal integral membrane protein 2 (LIMP2)) and across the lysosomal membrane (for example, the V-type H+-ATPase complex and chloride channel protein 7 (CLC7)) .

Lysosome biogenesis requires the concerted involvement of biosynthetic and endocytic pathways. Lysosomes receive cargo for degradation as well as newly synthesized lysosomal proteins by the endocytic pathway (green arrows). Lysosomal proteins are synthesized in the endoplasmic reticulum (ER) and transported through the Golgi complex to the trans-Golgi network (TGN). From the TGN, they can follow the constitutive secretory pathway (blue arrows) to the plasma membrane and subsequently reach lysosomes by endocytosis. In addition, lysosomal proteins can follow a direct intracellular pathway (red arrows) to the endo-lysosomal system. The best-characterized direct intracellular pathway is the clathrin-dependent transport of lysosomal hydrolases by mannose-6-phosphate receptors (M6PRs). The available literature suggests that there are multiple pathways for both lysosomal hydrolases and lysosomal membrane proteins (for example, lysosomal integral membrane protein 2-mediated transport of β-glucocerebrosidase), which may enter the endo-lysosomal pathways at distinct stages of maturation (grey arrows). The black arrows represent multiple retrograde pathways from endosomes. For more information on the molecular machinery involved in these pathways, see the main text. ETC, endosome-to-TGN carrier; ILV, intraluminal vesicle.
Two classes of proteins are essential for the function of lysosomes: soluble lysosomal hydrolases (also referred to as acid hydrolases) and integral lysosomal membrane proteins (LMPs). Each of the 50 known lysosomal hydrolases targets specific substrates for degradation, and their collective action is responsible for the total catabolic capacity of the lysosome. In addition to bulk degradation and pro-protein processing, lysosomal hydrolases are involved in antigen processing, degradation of the extracellular matrix and initiation of apoptosis4. The mammalian lysosome contains ∼25 LMPs5, but additional LMPs are being revealed5,6,7. LMPs reside mainly in the lysosomal limiting membrane and have diverse functions, including acidification of the lysosomal lumen, protein import from the cytosol, membrane fusion and transport of degradation products to the cytoplasm8 (Fig. 1). The most abundant LMPs are lysosome-associated membrane protein 1 (LAMP1), LAMP2, lysosome integral membrane protein 2 (LIMP2; also known as SCARB2) and the tetraspanin CD63 (see Supplementary information S1 (table)).
Lysosome biogenesis requires integration of the endocytic and biosynthetic pathways of the cell (Fig. 2). Lysosomal targeting of newly synthesized lysosomal proteins can be direct, from the trans-Golgi network (TGN) to the endosomal system, or indirect, involving transport to the plasma membrane and subsequent endocytosis. The best understood direct pathway is the mannose-6-phosphate receptor (M6PR)-mediated transport of lysosomal hydrolases9,10. By contrast, remarkably little is known about the structural and molecular machinery for the transport of LMPs to lysosomes. The significance of tightly regulated LMP trafficking is illustrated by recent findings that describe new and unexpected roles for LMPs in cellular physiology. It is becoming apparent that LMPs can impose specific functions onto the organelles through which they are transported or in which they reside, such as the endoplasmic reticulum (ER), lysosomes and the plasma membrane. Their importance is further highlighted by the discovery of an increasing number of gene mutations that lead to lysosomal dysfunction and disease11 (Table 1). In addition, various knockout mice and non-mammalian model organisms have highlighted the role of LMPs in cell physiology (see Supplementary information S1 (table)).
Here, we give an overview of the cellular pathways involved in lysosome biogenesis, with a focus on the biosynthetic pathways that are independent of M6PR functioning. In addition, we discuss the putative and emerging roles of LMPs in the transport of proteins and organelles and the consequences of their impaired trafficking for human health.
Endocytic pathways to the lysosome
The degradative endocytic pathway starts at the plasma membrane and ends in lysosomes. Between these two 'stations', endocytosed cargo passes through a range of endosomal intermediates (Fig. 2) that are distinguished by their content, molecular make-up, morphology and pH and by the kinetics by which endocytic tracers reach them12 (Table 2). A constant exchange of incoming and outgoing membranes and multiple fusion events result in the gradual remodelling of an endosomal intermediate into a later-stage endosome13,14,15,16, a process called maturation17. In addition, endosomes can exchange content through vesicular transport carriers, tubular connections and kiss-and-run fusion events18,19,20.
The widely used distinction between early endosomes (EEs) and late endosomes (LEs)12 is based on functional and biological characteristics, but oversimplifies the complexity of the endocytic pathway. This was exemplified by a recent immunoelectron microscopy (IEM) study linking the molecular make-up of endosomes with their ultrastructural characteristics21. Distinct EE marker proteins showed different distributions, ranging from a restricted localization on early-stage EEs (for example, early endosome antigen 1 (EEA1)) to a more widespread distribution on other EEs and on early-stage LEs (for example, the Rab proteins RAB11 and RAB4, and HRS (also known as ESCRT0)). These observations indicate that functionally different intermediates of EEs and LEs can be distinguished and that the transition between EEs and LEs is gradual. The endocytic pathway is therefore best regarded as a spatiotemporal continuum of intermediates, which continuously exchange their content while undergoing gradual molecular and structural remodelling and functional transformation (Table 2).
Sorting events in early endocytic compartments. EEs are the main recipients of incoming endocytic vesicles from the plasma membrane. They receive cargo that cycles back to the plasma membrane, as well as material that is transported further along the endocytic pathway. In addition, EEs receive endogenous proteins from the TGN, such as M6PRs carrying lysosomal hydrolases22,23 (Fig. 2; Table 2). The main function of EEs is to sort this cargo for recycling or degradation. EEs contain a vacuolar part (also referred to as the sorting endosome) from which a reticulum of multi-branching tubules emerges24,25 (referred to as the tubular sorting endosome (TSE)26,27 or tubular endosomal network)27,28 (Fig. 2). Generally, cargo destined for lysosomes remains in the EE vacuole, whereas cargo to be recycled enters the TSE. Proteins without a specific targeting signal enter the TSE by default29,30. In the EE vacuole, a few intraluminal vesicles (ILVs) are present that form by inward budding of the EE limiting membrane.
In the mildly acidic environment of EEs, lysosomal hydrolases dissociate from M6PRs and remain in the endosomal lumen, and the M6PRs return to the TGN for other rounds of transport. M6PRs can enter specialized recycling carriers — the endosome-to-TGN carriers that form directly from endosomal vacuoles21 (Fig. 2) — or can enter the TSEs, where additional recycling carriers exit28. Recycling of M6PRs by endosome-to-TGN carriers seems to be independent of clathrin, but requires the retromer subunit sorting nexin 1 (SNX1) and/or SNX2 (Refs 21, 31,32,33,34,35,36). Association of the retromer with EEs is regulated by the sequential action of RAB5 and RAB7 (Ref. 37), which are exchanged during the maturation of EEs to LEs38. Interference with RAB7 function causes dissociation of the retromer vacuolar protein sorting-associated protein 26 (VPS26)–VPS29–VPS35 trimer and also interferes with M6PR recycling. Notably, the retromer component VPS26 colocalizes with clathrin on TSEs39,40, suggesting that there are two retromer-dependent pathways for EE-to-TGN recycling of M6PRs: through endosome-to-TGN carriers directly from the EE vacuole21 and from TSEs39, presumably in a clathrin-dependent manner (Fig. 2).
A TSE can contain proteins for recycling to the plasma membrane or to the TGN, as well as low levels of LMPs (for example, LAMP1, LAMP2 and CD63) that are destined for the lysosome26. TSEs have multiple membrane buds that can contain the adaptor complex proteins AP1 and AP3, with or without the association of clathrin26. AP1 has been implicated in the recycling of M6PRs to the TGN41. AP3 is required for the efficient transport of LMPs from TSEs to lysosomes and LROs26,42. The importance of this pathway is illustrated by the pigmentation and bleeding disorder Hermansky–Pudlak syndrome 2, in which patients lack a functional AP3 complex, and this results in a redistribution of LMPs to the plasma membrane and impaired biogenesis, particularly of melanosomes and platelet-dense granules43,44,45. Therefore, following the initial sorting in EE vacuoles, TSEs further sort proteins to the plasma membrane, TGN or lysosomes. The perinuclear endosomal recycling compartment46 can be thought of as a subdomain of the TSE28 or as a separate compartment that emanates from the TSE after the removal of proteins that are targeted for destinations other than the plasma membrane26,27.
Sorting events in late endosomal compartments. Cargo proteins that are sorted into ILVs thereby remain in the endosomal lumen and are routed for lysosomal degradation. The ESCRT machinery has been identified as an important regulator for ubiquitin-dependent cargo sorting into ILVs as well as for ILV formation47,48,49,50,51,52. A well-established cargo for the ESCRT pathway is epidermal growth factor receptor (EGFR)53,54,55,56. Prior to sorting into ILVs, EGFR and growth hormone receptor are concentrated in a subdomain of the limiting membrane of the EE vacuole that bears a clathrin coat55,57 comprising two layers57 — a bilayered coat (Fig. 2). The bilayered coat contains concentrated levels of HRS, which binds to both ubiquitylated cargo and clathrin, providing a link between cargo selection and coat formation55,56,58. At the edges of the coat, small membrane invaginations bud off into the endosomal lumen to generate the ILVs59. Notably, however, impairment of ILV formation by the phosphoinositide 3-kinase (PI3K) inhibitor wortmannin60 or ablation of clathrin by RNA interference61 does not abolish EGFR degradation in lysosomes, suggesting that there are additional endosomal retention mechanisms.
CD63 and other tetraspanins62 are efficiently sorted into ILVs (Fig. 3). In LEs, this leads to a ∼7-fold enrichment of CD63 in the ILVs relative to the endosomal limiting membrane63. Interestingly, in oligodendroglial cells, which secrete CD63-positive exosomes, CD63 sorting into exosomes and into their ILV precursors was found to be independent of the ESCRT machinery, but required the sphingolipid ceramide64. Recent studies in dendritic cells revealed that the mechanism by which the LMP MHC class II is sorted into ILVs varies with the activation state of the cells. In immature dendritic cells, endocytosis of MHC class II from the plasma membrane as well as sorting of MHC class II into ILVs requires ubiquitylation and leads to lysosomal degradation. Maturation of the cells leads to a decrease in the ubiquitylation of MHC class II and an increase in the plasma membrane levels of MHC class II65,66. In these maturated cells, endosome-associated MHC class II is still sorted into ILVs, not for degradation but for secretion by exosomes (Fig. 1). These exosomes also contain the tetraspanin CD9 (Ref. 67), which had previously been shown to be enriched on exosomes together with CD63 (Refs 62,68). Because sorting to ILVs and exosomes in these conditions seems to be independent of ubiquitin, an ESCRT-independent mechanism of sorting tetraspanins into ILVs is implicated69. The possible involvement of other factors in CD63 transport, as well as the emerging roles of CD63 as a transport regulator of other proteins and as a determinant of kidney physiology, was recently described62,70 and will not be discussed further here. In contrast to CD63, LAMP1, LAMP2 and LIMP2 are predominantly localized at the endosomal limiting membrane63,71 (Fig. 3), but their relative intra-endosomal distribution can vary (Fig. 3). These observations indicate that the intra-endosomal distributions of LMPs are subject to regulation, which might have major implications for their functioning.

Electron micrographs of ultrathin cryosections immunogold labelled for the indicated lysosomal membrane proteins. a | Endogenous lysosome-associated membrane protein 1 (LAMP1) localization in human HepG2 cells. LAMP1 (labelled by 10 nm gold particles) is predominantly found at the limiting membrane of the lysosome (LYS). b | Endogenous LAMP1 localization in human HepG2 cells can vary between distinct endo-lysosomal intermediates. The thin arrow points to an inward-budding vesicle of the late endosome (LE) or lysosome. The nucleus (N) is indicated. c | Lysosomal integral membrane protein 2 (LIMP2)-deficient fibroblasts co-transfected with LIMP2–green fluorescent protein (GFP) and β-glucocerebrosidase (βGC). LIMP2 (labelled by 15 nm gold particles) is predominantly found in the endo-lysosomal limiting membrane, whereas βGC (labelled by 10 nm gold particles) is found associated with the limiting membrane as well as in the endo-lysosomal lumen. d | Endogenous CD63 localization in human HepG2 cells. CD63 (labelled by 10 nm gold particles) is almost exclusively found in the endo-lysosomal lumen, where it associates with intraluminal vesicles (ILVs). Scale bars represent 200 nm. Thick arrows point to BSA-coated 5 nm gold particles that were internalized for 3 hours.
LE intermediates13,38, often referred to as multivesicular bodies (MVBs), are globular vacuoles with numerous ILVs (Table 2). LEs no longer contain significant amounts of cargo for recycling to the plasma membrane, but instead have elevated levels of proteins that are destined for lysosomes and substantial levels of lysosomal hydrolases72,73. LEs form fewer and less extensive tubular extensions than EEs and communicate with the TGN, other LEs and lysosomes rather than with the plasma membrane20. In addition to the retromer-dependent pathways for retrograde transport of M6PRs from the EE vacuoles and TSEs, other transport proteins have been implicated in M6PR retrieval from endosomes74: RAB9 (Ref. 75), the target (t)-SNARE syntaxin 16 (Ref. 76), phosphofurin acidic cluster sorting protein 1 (PACS1)77, EpsinR (also known as clathrin-interactor 1)78, the recently identified GARP complex79 and tail-interacting protein of 47 kD (TIP47; also known as M6PRBP1)75, although the functioning of TIP47 protein in M6PR trafficking was recently debated80. It is not clear how these proteins relate to each other and where they act in the endosomal system. Because TSEs exhibit AP1- and clathrin-coated buds26 and PACS1 and EpsinR can interact with AP1–clathrin, these pathways might be active in TSEs. RAB9 mainly mediates M6PR recycling from LEs81, suggesting that there are multiple retrograde pathways for M6PR recycling at different places in the endocytic system, including both EEs and LEs.
The sorting events in LEs eventually result in an increase in the degradative capacity of these compartments38, as well as an increased ability for homotypic fusion with other LEs and heterotypic fusion with pre-existing lysosomes15,20,69,82,83,84. Lysosomes are generally defined as organelles of heterogeneous size and content that are LMP-rich but lack M6PRs and have a pH below five12,15 (Table 2). The specific functional properties of lysosomes are discussed below.
Direct transport of lysosomal hydrolases
In addition to the endocytic pathway, lysosome biogenesis requires input from the biosynthetic pathway for the delivery of newly synthesized lysosomal proteins. The complexity of the endosomal system allows for multiple sites at which the biosynthetic pathway can intersect with endosomal intermediates (Fig. 2). The best understood pathways are those for the delivery of lysosomal hydrolases.
M6PR-dependent transport. Most lysosomal hydrolases acquire an M6P tag during transport through the Golgi complex85, which is recognized by M6PRs in the TGN9,10. There are two types of M6PR, 300 kD cation-independent M6PR (CI-M6PR; also known as IGF2R) and 46 kD cation-dependent M6PR (CD-M6PR), both of which are ubiquitously expressed. The M6PR pathway is the major pathway for the lysosomal targeting of lysosomal hydrolases and is fairly well understood. A recent review74 effectively summarizes our knowledge of M6PR-dependent transport and we refer to this paper for detailed reading. In the TGN, M6PRs bind to AP1 and/or to GGA proteins86,87,88,89,90,91,92, resulting in the formation of 60–100nm clathrin-coated vesicles22,27,89,92. In addition, live-cell imaging has revealed the existence of larger, pleiomorphic carriers that are positive for GGA1, AP1, clathrin or the cytosolic tail of CI-M6PR23,91,93 that originate from the TGN and move towards the cell periphery23,94. It is not known exactly where in the endocytic pathway the M6PRs deliver their bound ligands, but live-cell imaging23 as well as the relatively equal distribution of M6PRs on EEs and LEs22 suggests that EEs are the predominant site for entry. Most M6PRs travel through this direct, clathrin-dependent pathway to endosomes. It is not yet clear whether GGA proteins and AP1 generate distinct types of M6PR carriers, or whether GGA proteins facilitate the entry of M6PR into clathrin-coated vesicles by interacting with AP1 (Refs 89,90).
M6PR-independent transport. In patients with I-cell disease (also known as mucolipidosis type II), lysosomal hydrolases do not acquire M6P tags because of a deficiency in N-acetylglucosamine (GlcNAc)-phosphotransferase activity95,96,97. Nevertheless, in some I-cell-diseased cells, such as hepatocytes, Kupffer cells and lymphocytes, a significant portion of newly synthesized lysosomal hydrolases do reach the lysosome96,98,99. Similar observations were made in GlcNAc phosphotransferase-knockout mice, a model system for I-cell disease100, and in mice deficient for both CI-M6PR and CD-M6PR, inferring a complete ablation of the M6PR pathway101. The M6PR-independent pathways of lysosomal hydrolase transport are mostly unknown, with the exception of β-glucocerebrosidase (βGC) transport (Fig. 4). In the absence of a functional M6PR pathway, newly synthesized lysosomal hydrolases can follow the constitutive secretory pathway to the plasma membrane and after secretion might be taken up by fluid-phase endocytosis. The multiligand-binding mannose receptor that is preferentially expressed on subsets of dentritic cells, on liver sinusoidal endothelial cells and on tissue macrophages binds lysosomal hydrolases102 and could mediate endocytosis in these cell types103. Mannose receptor-mediated uptake is also important for the successful delivery of some recombinant enzymes for the treatment of lysosomal storage disorders.

a | In wild-type cells, most lysosomal hydrolases are transported to lysosomes by mannose-6-phosphate receptors (M6PRs) that bind their ligands in the trans-Golgi network (TGN). β-Glucocerebrosidase (βGC), however, does not follow the classical M6PR pathway but instead binds to lysosomal integral membrane protein 2 (LIMP2) in the endoplasmic reticulum (ER), which directs it to the lysosome. b | In cells that lack M6PRs (or in I-cell-diseased cells, which lack the phosphotransferase necessary to make the M6P tag), most lysosomal hydrolases are secreted, but βGC continues to be targeted to the lysosomes. c | In LIMP2-deficient cells, most lysosomal acid hydrolases are delivered to lysosomes, whereas βGC is secreted.
The VPS10 receptor family . Recently, members of the mammalian vacuolar protein sorting (VPS10) domain-containing protein family of receptors have emerged as candidates for mediating direct transport of lysosomal hydrolases to the lysosome104. VPS10 domain-containing proteins share homology with the luminal part of yeast Vps10, which mediates sorting of the soluble hydrolase carboxypeptidase Y to the vacuole105. In mammals, the VPS10 domain-containing family presently consists of the multiligand receptors sortilin, SorLA (also known as sortilin-related receptor) and SORCS1–SORCS3 (Refs 106,107). Sortilin is likely to be responsible for the direct lysosomal targeting of the acid sphingomyelinase and sphingolipid activator proteins (SAPs)108,109, which are non-enzymatic cofactors that are required for glycosphingolipid degradation in lysosomes110. In addition, a substantial portion of the SAP precursor protein prosaposin is secreted, after which re-uptake can be carried out by the low-density lipoprotein receptor-related proteins, the M6PRs and, as seen in macrophages, the mannose receptors111.
The carboxyl terminus of sortilin contains AP1- and GGA-binding motifs that are essential for trafficking. As shown by IEM, sortilin colocalizes with CI-M6PR in the TGN21,112,113, indicating that these non-related proteins are transported by the same clathrin-dependent pathway to the endo-lysosomal system. Moreover, sortilin and CI-M6PR share a retromer-dependent pathway that recycles them to the TGN21,39,114. Interestingly, antibodies to the lysosomal hydrolases cathepsin D and cathepsin H can co-immunoprecipitate sortilin115, but the implication of this finding and a role for sortilin in lysosomal hydrolase sorting remain to be established. The crystal structure of human sortilin in complex with neurotensin revealed that sortilin binds its ligand in a tunnel formed by a β-propeller domain116. Hence, sortilin — similar to LIMP2 (see below) — binds its ligands through proteinaceous interactions that, unlike the M6PR pathway, do not require glycosylation.
LIMP2-dependent transport of β GC. Unlike most soluble lysosomal hydrolases, βGC does not obtain an M6P tag, and in I-cell-diseased cells or in cells lacking M6PR βGC is normally transported to lysosomes (Fig. 4). For a long time it was not understood how βGC, of which mutation causes the most common lysosomal storage disorder, Type I Gaucher disease, is transported to lysosomes. In a recent study, LIMP2 was unexpectedly found to be a selective and specific binding partner for βGC117. LIMP2 is a heavily glycosylated LMP that spans the membrane twice, with both the N and C termini facing the cytosol (Fig. 1) and with a putative sorting signal in the C terminus (see Supplementary information S1 (table)). Binding between LIMP2 and βGC is pH dependent, enabling these proteins to associate in the ER and all the way to the lysosome, where they dissociate because of the acidic pH. In mice lacking LIMP2 (Ref. 118), βGC is no longer sorted to lysosomes but is instead secreted (Fig. 4). The site of interaction between βGC and LIMP2 was mapped to a highly conserved coiled-coil motif in the luminal loop of LIMP2, but is still unresolved in βGC119. It is not yet known whether LIMP2 only carries βGC or if it also sorts other lysosomal hydrolases. The observations that βGC and LIMP2 associate, that they colocalize in lysosomes (Figs 3,4) and that the activity, levels and localization of βGC correlate with the expression of LIMP2 imply that M6PR-independent sorting of βGC requires βGC to bind to LIMP2 (Ref. 117). Furthermore, these data suggest that the transport of LMPs can be linked to transport pathways of lysosomal hydrolases. Interestingly, deletion of the C-terminal tail of LIMP2 did not affect its lysosomal localization. Moreover, the steady-state localization of LIMP2 and βGC remained unaltered in AP3-deficient mocha cells, suggesting that there are additional sorting motifs in the transmembrane or luminal domain of LIMP2 that do not involve interactions with AP3.
Transport of βGC is probably not the only function of LIMP2. Overexpression of LIMP2 leads to enlarged EEs, LEs and lysosomes with impaired membrane trafficking out of the endo-lysosomal compartments, accumulation of free cholesterol in the limiting membranes and impaired transferrin receptor recycling120. In mice, LIMP2 deficiency causes ureteric pelvic junction obstruction, peripheral neuropathy and deafness118, which is probably associated with impaired apical trafficking and distribution of K+ channels and megalin, a low-density lipoprotein-like endocytic receptor121. Recently, LIMP2-null mutations were found in patients suffering from action myoclonus-renal failure syndrome (AMRF)122,123, a severe, autosomal-recessive multisystem disorder. It still needs to be determined whether possible truncated forms of LIMP2 are associated with the disease and to what extent they can bind βGC. Patient fibroblasts show a severe enzymatic deficiency of βGC, whereas the βGC activity in patient leukocytes seems unaffected, suggesting an additional βGC targeting mechanism123. Finally, LIMP2 was shown to have an extralysosomal role in intercalated discs of cardiac myocytes124.
TGN sorting of lysosomal membrane proteins
The studies on LIMP2 and βGC underline the central role of LMPs in lysosome biogenesis and lysosome functioning, and show that the lysosomal targeting pathways of an LMP and a soluble lysosomal hydrolase can overlap. Our general understanding of how LMPs are sorted to lysosomes and other cellular compartments is still in its infancy, but accumulating evidence suggests that multiple pathways might exist that can be independent of clathrin.
Direct and indirect transport pathways. A substantial fraction of the LMPs that exit the TGN travel to the plasma membrane along the default secretory pathway and subsequently reach lysosomes through the endocytic pathway125. In addition, LMPs can travel directly from the TGN to the endo-lysosomal pathway. Increasing evidence suggests that there are multiple TGN exits for LMPs; this would allow for a targeted delivery to defined endosomal intermediates, including LEs (J.K., unpublished observations). The relative contributions of each transport pathway might differ depending on the cell type, the LMP26,125,126,127 and its expression level128 and the cellular conditions. A particular LMP can contain sorting motifs for distinct trafficking pathways. For example, the LMP mucolipin 1 (MCOLN1) is a putative ion channel with six predicted transmembrane domains that is defective in the lysosomal storage disorder mucolipidosis type IV129 (Table 1; see Supplementary information S1 (table)). A chimeric protein containing the C-terminal tail of MCOLN1, fused to the luminal and transmembrane domains of the plasma membrane glycoprotein TAC, is transported to the plasma membrane and subsequently endocytosed. By contrast, a chimaera containing the N-terminal tail of MCOLN1 fused to TAC travels directly from the TGN to the endo-lysosomal system130.
Multiple TGN exits. Many LMPs contain di-leucine or tyrosine-based sorting motifs131 (see Supplementary information S1 (table)), which equip them to use the AP1–clathrin- and GGA–clathrin-dependent exits from the TGN. Indeed, LAMP1 was found in AP1–clathrin-positive TGN membranes by both biochemical and IEM studies125,132,133. In addition, TGN exit and efficient lysosomal targeting of the LMP lysosomal-associated protein transmembrane 5 (LAPTM5), which is specifically expressed in haematopoietic cells, requires GGA3, the same component involved in the exit of M6PRs from the TGN134. However, in vitro studies indicated that LAMPs can be sorted into non-coated TGN-derived vesicles, which are different from those containing the M6PRs135. Moreover, in mice lacking a functional AP1 complex, LAMP1 is still found in lysosomes without increased transport through the plasma membrane41, and when AP1 or clathrin is depleted from HeLa cells125 LAMP1 still travels to lysosomes. The transport of MHC class II to MHC class II compartments also seems to involve sorting into non-coated TGN-derived vesicles136 (Fig. 1). Biochemical and IEM studies in B lymphoblasts revealed that MHC class II enters TGN exit carriers that are devoid of AP1, clathrin and CD-M6PR137. Together, these data indicate that LMPs can exit the TGN in AP1–clathrin-coated vesicles, but that additional TGN exit carriers must exist for direct TGN-to-lysosome transport.
Lysosomal targeting of multiprotein complexes, such as ion channels, exhibits an additional level of complexity in that different subunits may be targeted independently. A recent study showed that a mutation in the gene encoding VMA21, an assembly chaperone of the V-type H+ ATPase complex, causes X-linked myopathy with excessive autophagy (XMEA). The V-type H+ ATPase complex is required for the import of protons into the lysosomal lumen. In patients carrying a mutation in VMA21, only reduced levels of the V-type H+ ATPase complex reach the lysosomes. This in turn results in a general increase in endo-lysosomal pH and a subsequent decrease of autophagic degradation, resulting in the accumulation of autolysosomes and autophagosomes. This study illustrates the importance of tightly linked regulation of LMP assembly and transport138. Other examples of ion and protein transport over the lysosomal limiting membrane through channels or by transporters include chloride transport through chloride channel protein 7 (ClC7), cystine transport through cystinosin, sialic acid transport through sialin, cobalamin transfer by the cobalamin transporter and cholesterol and lipid transport through Niemann–Pick C1 protein (NPC1)11,139 (see Supplementary information S1 (table)). Mutations in the genes encoding these proteins cause a block or retardation of the respective transport events and can cause various lysosomal storage diseases, neurodegeneration and osteopetrosis11 (Table 1).
The pathways used by LE and lysosomal SNARE proteins — that is, syntaxin 7, syntaxin 8, vesicle transport through interaction with t-SNAREs homologue 1B (VTI1B), vesicle-associated membrane protein 8 (VAMP8) and VAMP7 (Ref. 20) — are virtually unknown. Although the cytoplasmic domains of SNAREs are essential for sorting140, their transport is complicated by their association in multiple cis- or trans-SNARE complexes, which may also be required for sorting141. VAMP7 depends on its so-called longin domain for delivery to lysosomes. Interestingly, this domain can bind AP3, linking the lysosomal targeting of VAMP7 to LMP transport rather than to the M6PR pathway142. However, significant levels of VAMP7 are found at the plasma membrane, where endocytosis is mediated by a specific adaptor, HIV-1 Rev-binding protein (HRB; also known as AGFG1)143. It therefore remains to be established whether VAMP7 reaches AP3-positive TSE tubules through a direct intracellular pathway, by passage over the plasma membrane, or both.
Luminal sorting determinants. An entirely different clue to the mechanism of sorting comes from recent studies in melanocytes. In addition to lysosomes, these cells contain melanosomes — LROs that store melanin pigment. The melanosomal LMP tyrosinase-related protein 1 (TYRP1) follows a direct TGN-to-melanosome pathway in melanocytes, but uses an indirect pathway via the cell surface in cells that lack glucosylceramide127. Unexpectedly, LAMP1 and LAMP2 behave in the opposite manner and are transported indirectly in wild-type cells and directly in the absence of glucosylceramide. Because the level of glucosylceramide is in part regulated through the activity of βGC, which is transported to lysosomes with the help of LIMP2 (see earlier), it is conceivable that LIMP2 also indirectly controls the transport of proteins to melanosomes. A chimeric protein comprising the luminal domain of TYRP1 and the cytosolic tail of LAMP1 was transported directly to melanosomes, indicating that the luminal domain of TYRP1 contains sorting information to guide proteins into a direct TGN-to-melanosome pathway127. Another melanosomal LMP, PMEL17 (also known as SILV), is efficiently sorted into ILVs by a mechanism that does not require the cytosolic domain of PMEL17 and is independent of ubiquitylation and the ESCRT machinery. Instead, PMEL17 requires its luminal domain for an unknown sorting mechanism that is conserved in non-pigmented cells144.
LMP trafficking and function
Because LMPs mediate a wide range of functions (Table 1; see Supplementary information S1 (table)), the presence, absence or mutation of a given LMP in a membrane can significantly alter the functional properties of the underlying compartment or transport intermediate. This emphasizes the need to understand how LMPs are sorted to various cellular compartments, and how their intracellular transport and distribution relates to their functioning.
LAMPs and lysosomal integrity. Although most LMPs predominantly reside in lysosomes, their subcellular distributions can change and are more dynamic than so far appreciated. This has become especially apparent for LAMP1 and LAMP2. LAMPs are type-1 transmembrane proteins with considerable sequence homology that contain a large, heavily glycosylated luminal domain and a short cytosolic tail. For example, there are three LAMP2 isoforms with different transmembrane and cytosolic domains, which show a preferential localization in either endosomes and lysosomes or the plasma membrane145. More generally, the cell surface expression of LAMPs is increased in the activation of platelets, peripheral blood monocytes and cytotoxic T cells146 and in highly malignant tumour cells147.
We are only beginning to understand the significance of local LAMP concentrations. The plasma membrane levels of CD63 are of major consequence for other local protein concentrations62, but elevated plasma membrane levels of LAMP1 and LAMP2 have not yet been linked to a specific phenotype. An important clue to the significance of sustained LAMP levels in lysosomes came from a recent study which showed that LAMP proteins are involved in sensitizing tumour cells to lysosomal cell death (LCD) (Fig. 1). Oncogenic transformation of fibroblasts on the one hand leads to a decrease in the levels of LAMP proteins in lysosomes and on the other hand increases the susceptibility of these cells to the LCD pathway148. Likewise, decreased levels of LAMP1 and LAMP2 also contribute to an enhanced sensitivity of transformed cells to anti-cancer drugs that trigger LCD. Overexpression of LAMPs had the opposite effect, indicating that LAMPs can protect cells from the LCD pathway. In addition, LAMP-depleted cells showed a redistribution of lysosomes to the cell periphery, pointing to a role for LAMPs in lysosomal dynamics. Together with earlier studies correlating surface expression of LAMP proteins to metastatic potential of carcinoma cells147,149, these findings exemplify the importance of LAMP targeting in maintaining lysosomal integrity and in regulating LCD pathways. They also underscore the need to understand the relationship between LAMP trafficking and successful anti-cancer treatment.
LAMPs and lysosomal dynamics. The significance of the intracellular localization of LAMPs is further exemplified by the observation that in LAMP-deficient cells the distribution of lysosomes is more dispersed and peripheral, suggesting that lysosome migration towards the microtubule organization centre is delayed150. These findings resemble observations in AP3-deficient cytotoxic T lymphocytes, in which minus-end-directed, microtubule-mediated movement of lytic granules towards the immunological synapse was impaired, resulting in a loss of cytotoxicity151. Similar to the lysosomes in LAMP-knockout cells, lytic granules in AP3-deficient cytotoxic T lymphocytes remained at the cell periphery. These findings suggest that LAMPs are required to move lysosomes because a reduction in LAMP, by genetic downregulation or impaired transport as a result of AP3 deficiency, leads to their redistribution. It is not yet clear whether and how the short (11 amino acids) C-terminal tails of LAMP proteins can interact, directly or indirectly, with the dynein-mediated centripetal migration system of lysosomes.
In addition, a recent study has shown that LAMP1 participates in the regulation of lysosomal exocytosis152. Unexpectedly, lysosomal exocytosis is increased in macrophages and fibroblasts that are defective in neuraminidase, a model for the lysosomal disorder sialidosis, which results in over-sialylation of LAMP1. Lysosomal exocytosis, which is involved in plasma membrane repair, is Ca2+ dependent and requires the ubiquitously expressed LMP synaptotagmin 7 (SYT7)153, a Ca2+ sensor that cooperates with SNARE proteins to promote the fusion of the lysosomal and plasma membranes153. The lysosomal vesicle SNARE VAMP7 is also required for Ca2+-triggered lysosomal exocytosis154 and forms complexes with syntaxin 4 and synaptosomal-associated protein 23 (SNAP23), two plasma membrane t-SNAREs that interact in a Ca2+-dependent manner with SYT7 (Ref. 154). VAMP7 and SYT7 colocalize in LEs and lysosomal compartments that exocytose after an increase in the intracellular Ca2+ concentration. Interestingly, genetic ablation of SYT7 leads to a defect in neurite outgrowth, suggesting that SYT7-regulated exocytosis of LEs and lysosomes plays a part in the addition of new membrane to developing neurites155. How LAMP1 fits into this picture and how luminal alterations in LAMP1 give rise to altered lysosome dynamics need to be resolved.
LAMPs150, VAMP7 (Ref. 156) and SYT7 are also required for Ca2+-dependent phagosome–lysosome fusion, a process that is necessary for limiting the intracellular growth of pathogenic bacteria. Bacterial type III secretion systems can permeabilize membranes and cause a Ca2+ influx in mammalian cells, thereby promoting lysosomal exocytosis. Phagolysosome fusion is also a Ca2+-dependent process. Analysis of SYT7-deficient cells revealed that the intracellular survival of bacteria that have to evade lysosomes in order to replicate is dependent on SYT7. Most likely, shortly after invasion phagosomes are permeabilized, Ca2+ influx into the cytosol is triggered and SYT7-dependent phagolysosomal fusion and bacterial killing is initiated. Therefore, the lysosomal repair response is an evolutionarily conserved protection mechanism against pathogens157. Also, invasion of the protozoan Trypanosoma cruzi, which causes the debilitating human Chagas disease, requires host cell lysosomes that are recruited to the site of parasite entry and gradually fuse with the plasma membrane, thereby providing the membrane for formation of the parasitophorous vacuole158.
LAMPs in autophagy and phagocytosis. Apart from the documented role of lysosomal hydrolases in defence against bacteria, there is an increasing body of evidence that suggests that LAMPs are of key importance for the maturation of autophagosomes and phagosomes (Fig. 1). Autophagosomes mediate autophagy, a conserved and strictly regulated lysosomal pathway that degrades cytoplasmic material and organelles159 and is activated during stress conditions, such as during amino acid starvation, the unfolded protein response or viral infection. Phagocytosis is also an evolutionarily conserved mechanism that mediates the lysosome-mediated degradation of large particles, such as internalized pathogens.
Deficiency of LAMP2 in mice160 recapitulates human Danon disease, a rare disorder characterized by LAMP2 mutations that lead to a clinical triad of hypertrophic cardiomyopathy, skeletal myopathy and mental retardation161 (Table 1). The pathological hallmark of LAMP2 deficiency in both mice and humans is an accumulation of autophagic compartments, which is most pronounced in muscle cells. The cellular pathology is explained by a role of LAMP2, and possibly also of LAMP1 (Ref. 162), in mediating the fusion of lysosomes with autophagosomes. The lack, or large reduction, of these fusion events would explain the abnormal accumulation of autophagosomes and autolysosomes. Interestingly, analysis of LAMP-deficient cells revealed that RAB7 recruitment to autophagosomes and their fusion with lysosomes was severely retarded150,163,164. A similar lysosome–phagosome fusion defect is observed in neutrophils that lack LAMP2, possibly explaining the reduced ability of these cells to kill pathogens, which in LAMP2-knockout mice leads to periodontis165.
The C-terminal tail of LAMP2A is also implicated in the transport of cytosolic substrates across the lysosomal limiting membrane, a process referred to as chaperone-mediated autophagy (CMA)166. CMA is important for different biological processes, such as the presentation of cytoplasmic antigens by MHC class II molecules167, cellular ageing168 and neurodegeneration169. How the recognition of CMA-targeting sequences and the translocation into the lysosome occurs at the molecular level is not yet understood.
Conclusion and perspectives
Each protein that enters the endocytic pathway passes a number of decision stations that determine the rest of its journey. The exact sorting decisions that need to be taken depend on where the cargo enters the endosomal continuum. Apart from the M6PR pathway, only little is known about the biosynthetic pathways to the lysosome. Identification of alternative receptors and/or sorting mechanisms for direct targeting of lysosomal hydrolases or LMPs to the lysosome is one of the major challenges in the lysosomal trafficking field. The recent availability of a mouse model system for I cell disease100 provides an exciting new opportunity to study M6PR-independent mechanisms of lysosomal hydrolase sorting and may lead to new clues to the role of Vps10 domain-containing receptors. Moreover, the LIMP2-dependent transport of βGC indicates that the M6PR-independent delivery of lysosomal hydrolases might be linked to the lysosomal targeting pathways of LMPs.
An important emerging theme is that the delivery of LMPs can essentially change the properties of the target compartment; for example, acidity can be altered by the delivery of the V-type H+ ATPase complex, fusogenicity can be altered by the delivery of VAMP7, catabolic capacity can be altered by the delivery of LIMP2, dynamics and drug resistance can be altered by the delivery of LAMP1 and LAMP2, and the availability of cytoplasmic substrates can be altered by the delivery of LAMP2A. Moreover, LMPs are increasingly being implicated as the causative genes for a range of human diseases. The increasing number of functions ascribed to the LAMP cytosolic tails and the presence of enlarged, more peripheral lysosomes in LAMP-deficient cells raise the notion that LAMPs might be incorporated in functional microdomains at the lysosomal limiting membrane (allowing, for example, association with microtubules, lysosomal membrane transporters or sorting determinants to the lysosomal pathway) that can act in events as diverse as lysosome exocytosis, dynamics and protein translocation.
A timely and targeted delivery of LMPs to endosomal intermediates at distinct stages of the endocytic pathway might be an important mechanism for the gradual delivery of essential building blocks to assemble a functional lysosome. This scenario is supported by the presence of multiple sorting signals in the cytosolic tails of some LMPs, the evidence that domains other than the cytosolic tails can be involved in lysosomal targeting117,127,144 and the occurrence of post-translational modifications (glycosylation, phosphorylation, palmitoylation and ubiquitylation) that can fine-tune the sorting signals170.
Multiple pathways for the delivery of newly synthesized proteins to the endo-lysosomal system would require multiple TGN exits. Current data suggest the existence of clathrin-independent TGN exit pathways that could be used by LMPs and even, to a minor extent, by lysosomal hydrolases. Additional evidence for multiple pathways comes from yeast, in which the lysosomal targeting of the lysosomal hydrolase carboxypeptidase Y and the LMP alkaline phosphatase requires distinct pathways and machinery. In Drosophila melanogaster, the granule group protein Deep Orange (DOR) is involved in the lysosomal targeting of lysosomal hydrolases, but not of LMPs171. Because the mammalian endosomal system is far more complex than in yeast, the presence of multiple TGN-to-endosome pathways seems a realistic possibility. Elucidating these pathways is a major challenge for future studies and is of key importance to expand our knowledge of lysosome biogenesis and to understand the pathologies associated with lysosomal dysfunctioning.
References
Dell'Angelica, E. C., Mullins, C., Caplan, S. & Bonifacino, J. S. Lysosome-related organelles. FASEB J. 14, 1265–1278 (2000).
Bonifacino, J. S. Insights into the biogenesis of lysosome-related organelles from the study of the Hermansky–Pudlak syndrome. Ann. NY Acad. Sci. 1038, 103–114 (2004).
Dell'Angelica, E. C. The building BLOC(k)s of lysosomes and related organelles. Curr. Opin. Cell Biol. 16, 458–464 (2004).
Conus, S. & Simon, H. U. Cathepsins: key modulators of cell death and inflammatory responses. Biochem. Pharmacol. 76, 1374–1382 (2008).
Lübke, T., Lobel, P. & Sleat, D. E. Proteomics of the lysosome. Biochim. Biophys. Acta 1793, 625–635 (2009).
Schroder, B. et al. Integral and associated lysosomal membrane proteins. Traffic 8, 1676–1686 (2007).
Callahan, J. W., Bagshaw, R. D. & Mahuran, D. J. The integral membrane of lysosomes: its proteins and their roles in disease. J. Proteomics 72, 23–33 (2009). References 6 and 7 present new proteomic experiments that reveal a greater than expected number of LMPs of largely unknown function.
Eskelinen, E. L., Tanaka, Y. & Saftig, P. At the acidic edge: emerging functions for lysosomal membrane proteins. Trends Cell Biol. 13, 137–145 (2003).
Kornfeld, S. & Mellman, I. The biogenesis of lysosomes. Annu. Rev. Cell Biol. 5, 483–525 (1989).
Figura, K. V. & Hasilik, A. Lysosomal enzymes and their receptors. 55, 167–193 (1986).
Ruivo, R., Anne, C., Sagne, C. & Gasnier, B. Molecular and cellular basis of lysosomal transmembrane protein dysfunction. Biochim. Biophys. Acta 1793, 636–649 (2009).
Sachse, M., Ramm, G., Strous, G. & Klumperman, J. Endosomes: multipurpose designs for integrating housekeeping and specialized tasks. Histochem. Cell Biol. 117, 91–104 (2002).
Stoorvogel, W., Strous, G. J., Geuze, H. J., Oorschot, V. & Schwartz, A. L. Late endosomes derive from early endosomes by maturation. Cell 65, 417–427 (1991).
Dunn, K. W. & Maxfield, F. R. Delivery of ligands from sorting endosomes to late endosomes occurs by maturation of sorting endosomes. J. Cell Biol. 117, 301–310 (1992).
Futter, C. E., Pearse, A., Hewlett, L. J. & Hopkins, C. R. Multivesicular endosomes containing internalized EGF–EGF receptor complexes mature and then fuse directly with lysosomes. J. Cell Biol. 132, 1011–1023 (1996).
van Deurs, B., Holm, P. K., Kayser, L., Sandvig, K. & Hansen, S. H. Multivesicular bodies in HEp-2 cells are maturing endosomes. Eur. J. Cell Biol. 61, 208–224 (1993).
Murphy, R. F. Maturation models for endosome and lysosome biogenesis. Trends Cell Biol. 1, 77–82 (1991).
Bright, N. A., Gratian, M. J. & Luzio, J. P. Endocytic delivery to lysosomes mediated by concurrent fusion and kissing events in living cells. Curr. Biol. 15, 360–365 (2005).
Gruenberg, J. & Stenmark, H. The biogenesis of multivesicular endosomes. Nature Rev. Mol. Cell Biol. 5, 317–323 (2004).
Luzio, J. P., Pryor, P. R. & Bright, N. A. Lysosomes: fusion and function. Nature Rev. Mol. Cell Biol. 8, 622–632 (2007).
Mari, M. et al. SNX1 defines an early endosomal recycling exit for sortilin and mannose 6-phosphate receptors. Traffic 9, 380–393 (2008). Describes a new SNX1- andSNX2-dependent, clathrin-independent exit for M6PRs from EE vacuoles and provides detailed molecular and ultrastructural characterization of endosomal intermediates.
Klumperman, J. et al. Differences in the endosomal distributions of the two mannose 6-phosphate receptors. J. Cell Biol. 121, 997–1010 (1993).
Waguri, S. et al. Visualization of TGN to endosome trafficking through fluorescently labeled MPR and AP-1 in living cells. Mol. Biol. Cell 14, 142–155 (2003).
Tooze, J. & Hollinshead, M. Tubular early endosomal networks in AtT20 and other cells. J. Cell Biol. 115, 635–653 (1991).
Stoorvogel, W., Oorschot, V. & Geuze, H. J. A novel class of clathrin-coated vesicles budding from endosomes. J. Cell Biol. 132, 21–33 (1996).
Peden, A. A. et al. Localization of the AP-3 adaptor complex defines a novel endosomal exit site for lysosomal membrane proteins. J. Cell Biol. 164, 1065–1076 (2004). Functional analysis of AP3 function in the transport of LMPs out of TSEs.
van Meel, E. & Klumperman, J. Imaging and imagination: understanding the endo-lysosomal system. Histochem. Cell Biol. 129, 253–266 (2008).
Bonifacino, J. S. & Rojas, R. Retrograde transport from endosomes to the trans-Golgi network. Nature Rev. Mol. Cell Biol. 7, 568–579 (2006).
Draye, J. P., Quintart, J., Courtoy, P. J. & Baudhuin, P. Relations between plasma membrane and lysosomal membrane. 1. Fate of covalently labelled plasma membrane protein. Eur. J. Biochem. 170, 395–403 (1987).
Yamashiro, D. J., Tycko, B., Fluss, S. R. & Maxfield, F. R. Segregation of transferrin to a mildly acidic (pH 6.5) para-Golgi compartment in the recycling pathway. Cell 37, 789–800 (1984).
Carlton, J. G. et al. Sorting nexin-2 is associated with tubular elements of the early endosome, but is not essential for retromer-mediated endosome-to-TGN transport. J. Cell Sci. 118, 4527–4539 (2005).
Carlton, J. et al. Sorting nexin-1 mediates tubular endosome-to-TGN transport through coincidence sensing of high-curvature membranes and 3-phosphoinositides. Curr. Biol. 14, 1791–1800 (2004).
Seaman, M. N. J. Cargo-selective endosomal sorting for retrieval to the Golgi requires retromer. J. Cell Biol. 165, 111–122 (2004).
Seaman, M. N. Recycle your receptors with retromer. Trends Cell Biol. 15, 68–75 (2005).
Bonifacino, J. S. & Hurley, J. H. Retromer. Curr. Opin. Cell Biol. 20, 427–436 (2008).
Rojas, R., Kametaka, S., Haft, C. R. & Bonifacino, J. S. Interchangeable but essential functions of SNX1 and SNX2 in the association of retromer with endosomes and the trafficking of mannose 6-phosphate receptors. Mol. Cell. Biol. 27, 1112–1124 (2007).
Rojas, R. et al. Regulation of retromer recruitment to endosomes by sequential action of Rab5 and Rab7. J. Cell Biol. 183, 513–526 (2008).
Rink, J., Ghigo, E., Kalaidzidis, Y. & Zerial, M. Rab conversion as a mechanism of progression from early to late endosomes. Cell 122, 735–749 (2005).
Arighi, C. N., Hartnell, L. M., Aguilar, R. C., Haft, C. R. & Bonifacino, J. S. Role of the mammalian retromer in sorting of the cation-independent mannose 6-phosphate receptor. J. Cell Biol. 165, 123–133 (2004).
Johannes, L. & Popoff, V. Tracing the retrograde route in protein trafficking. Cell 135, 1175–1187 (2008).
Meyer, C. et al. μ1A-adaptin-deficient mice: lethality, loss of AP-1 binding and rerouting of mannose 6-phosphate receptors. EMBO J. 19, 2193–2203 (2000).
Dell'Angelica, E. C., Klumperman, J., Stoorvogel, W. & Bonifacino, J. S. Association of the AP-3 adaptor complex with clathrin. Science 280, 431–434 (1998). First study to show AP3 localization on EE-associated recycling tubules.
Dell'Angelica, E. C., Shotelersuk, V., Aguilar, R. C., Gahl, W. A. & Bonifacino, J. S. Altered trafficking of lysosomal proteins in Hermansky–Pudlak syndrome due to mutations in the β3A subunit of the AP-3 adaptor. Mol. Cell 3, 11–21 (1999).
Huizing, M. & Gahl, W. A. Disorders of vesicles of lysosomal lineage: the Hermansky–Pudlak syndromes. Curr. Mol. Med. 2, 451–467 (2002).
Starcevic, M., Nazarian, R. & Dell'Angelica, E. C. The molecular machinery for the biogenesis of lysosome-related organelles: lessons from Hermansky–Pudlak syndrome. Semin. Cell Dev. Biol. 13, 271–278 (2002).
Maxfield, F. R. & McGraw, T. E. Endocytic recycling. Nature Rev. Mol. Cell Biol. 5, 121–132 (2004).
Saksena, S., Sun, J., Chu, T. & Emr, S. D. ESCRTing proteins in the endocytic pathway. Trends Biochem. Sci. 32, 561–573 (2007).
Seaman, M. N. Endosome protein sorting: motifs and machinery. Cell. Mol. Life Sci. 65, 2842–2858 (2008).
Raiborg, C. & Stenmark, H. The ESCRT machinery in endosomal sorting of ubiquitylated membrane proteins. Nature 458, 445–452 (2009).
Hanson, P. I., Roth, R., Lin, Y. & Heuser, J. E. Plasma membrane deformation by circular arrays of ESCRT-III protein filaments. J. Cell Biol. 180, 389–402 (2008).
Malerod, L. & Stenmark, H. ESCRTing membrane deformation. Cell 136, 15–17 (2009).
Saksena, S., Wahlman, J., Teis, D., Johnson, A. E. & Emr, S. D. Functional reconstitution of ESCRT-III assembly and disassembly. Cell 136, 97–109 (2009).
Bache, K. G., Brech, A., Mehlum, A. & Stenmark, H. Hrs regulates multivesicular body formation via ESCRT recruitment to endosomes. J. Cell Biol. 162, 435–442 (2003).
Raiborg, C. et al. Hrs sorts ubiquitinated proteins into clathrin-coated microdomains of early endosomes. Nature Cell Biol. 4, 394–398 (2002).
Raiborg, C., Bache, K. G., Mehlum, A., Stang, E. & Stenmark, H. Hrs recruits clathrin to early endosomes. EMBO J. 20, 5008–5021 (2001).
Urbe, S. et al. The UIM domain of Hrs couples receptor sorting to vesicle formation. J. Cell Sci. 116, 4169–4179 (2003).
Sachse, M., Urbe, S., Oorschot, V., Strous, G. J. & Klumperman, J. Bilayered clathrin coats on endosomal vacuoles are involved in protein sorting toward lysosomes. Mol. Biol. Cell 13, 1313–1328 (2002).
Raiborg, C., Wesche, J., Malerod, L. & Stenmark, H. Flat clathrin coats on endosomes mediate degradative protein sorting by scaffolding Hrs in dynamic microdomains. J. Cell Sci. 119, 2414–2424 (2006).
Murk, J. L. et al. Endosomal compartmentalization in three dimensions: implications for membrane fusion. Proc. Natl Acad. Sci. USA 100, 13332–13337 (2003).
Futter, C. E., Collinson, L. M., Backer, J. M. & Hopkins, C. R. Human VPS34 is required for internal vesicle formation within multivesicular endosomes. J. Cell Biol. 155, 1251–1264 (2001).
Sigismund, S. et al. Clathrin-mediated internalization is essential for sustained EGFR signaling but dispensable for degradation. Dev. Cell 15, 209–219 (2008).
Pols, M. S. & Klumperman, J. Trafficking and function of the tetraspanin CD63. Exp. Cell Res. 315, 1584–1592 (2008).
Escola, J. M. et al. Selective enrichment of tetraspan proteins on the internal vesicles of multivesicular endosomes and on exosomes secreted by human B-lymphocytes. J. Biol. Chem. 273, 20121–20127 (1998).
Trajkovic, K. et al. Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science 319, 1244–1247 (2008). First demonstration that the formation of ILVs containing CD63 that are destined for secretion as exosomes is ESCRT independent and requires ceramide.
Shin, J. S. et al. Surface expression of MHC class II in dendritic cells is controlled by regulated ubiquitination. Nature 444, 115–118 (2006).
van Niel, G. et al. Dendritic cells regulate exposure of MHC class II at their plasma membrane by oligoubiquitination. Immunity 25, 885–894 (2006).
Buschow, S. I. et al. MHC II in dendritic cells is targeted to lysosomes or T cell-induced exosomes via distinct multivesicular body pathways. Traffic 14 Jul 2009 (doi:10.1111/j.1600-0854.2009.00963.x).
Thery, C. et al. Molecular characterization of dendritic cell-derived exosomes. Selective accumulation of the heat shock protein hsc73. J. Cell Biol. 147, 599–610 (1999).
Woodman, P. G. & Futter, C. E. Multivesicular bodies: co-ordinated progression to maturity. Curr. Opin. Cell Biol. 20, 408–414 (2008).
Schroder, J. et al. Deficiency of the tetraspanin CD63 associated with kidney pathology but normal lysosomal function. Mol. Cell. Biol. 29, 1083–1094 (2009).
Kuronita, T. et al. A role for the lysosomal membrane protein LGP85 in the biogenesis and maintenance of endosomal and lysosomal morphology. J. Cell Sci. 115, 4117–4131 (2002). Shows that overexpression of LIMP2 causes an enlargement of EEs and lysosomes, probably in a RAB5-dependent manner, suggesting that LIMP2 participates in the organization of endosomal and lysosomal compartments.
Geuze, H. J. et al. Sorting of mannose 6-phosphate receptors and lysosomal membrane proteins in endocytic vesicles. J. Cell Biol. 107, 2491–2501 (1988).
Griffiths, G., Hoflack, B., Simons, K., Mellman, I. & Kornfeld, S. The mannose 6-phosphate receptor and the biogenesis of lysosomes. Cell 52, 329–341 (1988).
Braulke, T. & Bonifacino, J. S. Sorting of lysosomal proteins. Biochim. Biophys. Acta 1793, 605–614 (2009).
Diaz, E., Schimmoller, F. & Pfeffer, S. R. A novel Rab9 effector required for endosome-to-TGN transport. J. Cell Biol. 138, 283–290 (1997).
Amessou, M. et al. Syntaxin 16 and syntaxin 5 are required for efficient retrograde transport of several exogenous and endogenous cargo proteins. J. Cell Sci. 120, 1457–1468 (2007).
Crump, C. M. et al. PACS-1 binding to adaptors is required for acidic cluster motif-mediated protein traffic. EMBO J. 20, 2191–2201 (2001).
Saint-Pol, A. et al. Clathrin adaptor epsinR is required for retrograde sorting on early endosomal membranes. Dev. Cell 6, 525–538 (2004).
Perez-Victoria, F. J., Mardones, G. A. & Bonifacino, J. S. Requirement of the human GARP complex for mannose 6-phosphate-receptor-dependent sorting of cathepsin D to lysosomes. Mol. Biol. Cell 19, 2350–2362 (2008).
Bulankina, A. V. et al. TIP47 functions in the biogenesis of lipid droplets. J. Cell Biol. 185, 641–655 (2009).
Diaz, E. & Pfeffer, S. R. TIP47: a cargo selection device for mannose 6-phosphate receptor trafficking. Cell 93, 433–443 (1998).
Bakker, A. C., Webster, P., Jacob, W. A. & Andrews, N. W. Homotypic fusion between aggregated lysosomes triggered by elevated [Ca2+]i in fibroblasts. J. Cell Sci. 110, 2227–2238 (1997).
Ward, D. M., Leslie, J. D. & Kaplan, J. Homotypic lysosome fusion in macrophages: analysis using an in vitro assay. J. Cell Biol. 139, 665–673 (1997).
Mullock, B. M., Bright, N. A., Fearon, C. W., Gray, S. R. & Luzio, J. P. Fusion of lysosomes with late endosomes produces a hybrid organelle of intermediate density and is NSF dependent. J. Cell Biol. 140, 591–601 (1998).
Sleat, D. E. et al. The human brain mannose 6-phosphate glycoproteome: a complex mixture composed of multiple isoforms of many soluble lysosomal proteins. Proteomics 5, 1520–1532 (2005).
Boman, A. L., Zhang, C., Zhu, X. & Kahn, R. A. A family of ADP-ribosylation factor effectors that can alter membrane transport through the trans-Golgi. Mol. Biol. Cell 11, 1241–1255 (2000).
Dell'Angelica, E. C. et al. GGAs: a family of ADP ribosylation factor-binding proteins related to adaptors and associated with the Golgi complex. J. Cell Biol. 149, 81–94 (2000).
Hirst, J. et al. A family of proteins with γ-adaptin and VHS domains that facilitate trafficking between the trans-Golgi network and the vacuole/lysosome. J. Cell Biol. 149, 67–80 (2000).
Doray, B., Ghosh, P., Griffith, J., Geuze, H. J. & Kornfeld, S. Cooperation of GGAs and AP-1 in packaging MPRs at the trans-Golgi network. Science 297, 1700–1703 (2002).
Puertollano, R., Aguilar, R. C., Gorshkova, I., Crouch, R. J. & Bonifacino, J. S. Sorting of mannose 6-phosphate receptors mediated by the GGAs. Science 292, 1712–1716 (2001).
Puertollano, R. et al. Morphology and dynamics of clathrin/GGA1-coated carriers budding from the trans-Golgi network. Mol. Biol. Cell 14, 1545–1557 (2003).
Geuze, H. J., Slot, J. W., Strous, G. J., Hasilik, A. & von Figura, K. Possible pathways for lysosomal enzyme delivery. J. Cell Biol. 101, 2253–2262 (1985).
Polishchuk, R. S., San Pietro, E., Di Pentima, A., Tete, S. & Bonifacino, J. S. Ultrastructure of long-range transport carriers moving from the trans Golgi network to peripheral endosomes. Traffic 7, 1092–1103 (2006).
Bonifacino, J. S. & Lippincott-Schwartz, J. Coat proteins: shaping membrane transport. Nature Rev. Mol. Cell Biol. 4, 409–414 (2003).
Hasilik, A., Waheed, A. & von Figura, K. Enzymatic phosphorylation of lysosomal enzymes in the presence of UDP-N-acetylglucosamine. Absence of the activity in I-cell fibroblasts. Biochem. Biophys. Res. Commun. 98, 761–767 (1981).
Waheed, A. et al. Deficiency of UDP-N-acetylglucosamine: lysosomal enzyme N-acetylglucosamine-1-phosphotransferase in organs of I-cell patients. Biochem. Biophys. Res. Commun. 105, 1052–1058 (1982).
Reitman, M. L., Varki, A. & Kornfeld, S. Fibroblasts from patients with I-cell disease and pseudo-Hurler polydystrophy are deficient in uridine 5′-diphosphate-N-acetylglucosamine: glycoprotein N-acetylglucosaminylphosphotransferase activity. J. Clin. Invest. 67, 1574–1579 (1981).
Little, L. et al. Properties of N-acetylglucosamine 1-phosphotransferase from human lymphoblasts. Biochem. J. 248, 151–159 (1987).
Owada, M. & Neufeld, E. F. Is there a mechanism for introducing acid hydrolases into liver lysosomes that is independent of mannose 6-phosphate recognition? Evidence from I-cell disease. Biochem. Biophys. Res. Commun. 105, 814–820 (1982).
Gelfman, C. M. et al. Mice lacking α/β subunits of GlcNAc-1-phosphotransferase exhibit growth retardation, retinal degeneration, and secretory cell lesions. Invest. Ophthalmol. Vis. Sci. 48, 5221–5228 (2007).
Dittmer, F. et al. Alternative mechanisms for trafficking of lysosomal enzymes in mannose 6-phosphate receptor-deficient mice are cell type-specific. J. Cell Sci. 112, 1591–1597 (1999). Shows that the lack of both M6PRs in mice leads to an I-cell-disease-like phenotype and that cathepsin D, although following different routes, is targeted independently of M6PRs in hepatocytes and thymocytes but not in fibroblasts.
Allavena, P., Chieppa, M., Monti, P. & Piemonti, L. From pattern recognition receptor to regulator of homeostasis: the double-faced macrophage mannose receptor. Crit. Rev. Immunol. 24, 179–192 (2004).
Elvevold, K. et al. Liver sinusoidal endothelial cells depend on mannose receptor-mediated recruitment of lysosomal enzymes for normal degradation capacity. Hepatology 48, 2007–2015 (2008).
Canuel, M., Libin, Y. & Morales, C. R. The interactomics of sortilin: an ancient lysosomal receptor evolving new functions. Histol. Histopathol. 24, 481–492 (2009).
Marcusson, E. G., Horazdovsky, B. F., Cereghino, J. L., Gharakhanian, E. & Emr, S. D. The sorting receptor for yeast vacuolar carboxypeptidase Y is encoded by the VPS10 gene. Cell 77, 579–586 (1994).
Hampe, W., Rezgaoui, M., Hermans-Borgmeyer, I. & Schaller, H. C. The genes for the human VPS10 domain-containing receptors are large and contain many small exons. Hum. Genet. 108, 529–536 (2001).
Willnow, T. E., Petersen, C. M. & Nykjaer, A. VPS10P-domain receptors — regulators of neuronal viability and function. Nature Rev. Neurosci. 9, 899–909 (2008).
Ni, X. & Morales, C. R. The lysosomal trafficking of acid sphingomyelinase is mediated by sortilin and mannose 6-phosphate receptor. Traffic 7, 889–902 (2006).
Petersen, C. M. et al. Molecular identification of a novel candidate sorting receptor purified from human brain by receptor-associated protein affinity chromatography. J. Biol. Chem. 272, 3599–3605 (1997).
Lefrancois, S., Zeng, J., Hassan, A. J., Canuel, M. & Morales, C. R. The lysosomal trafficking of sphingolipid activator proteins (SAPs) is mediated by sortilin. EMBO J. 22, 6430–6437 (2003). Provides evidence that direct trafficking of both prosaposin and sphingosin activator protein from the TGN to the lysosomal compartment occurs independently of M6PRs by a sortilin-mediated mechanism.
Hiesberger, T. et al. Cellular uptake of saposin (SAP) precursor and lysosomal delivery by the low density lipoprotein receptor-related protein (LRP). EMBO J. 17, 4617–4625 (1998).
Nielsen, M. S. et al. The sortilin cytoplasmic tail conveys Golgi-endosome transport and binds the VHS domain of the GGA2 sorting protein. EMBO J. 20, 2180–2190 (2001).
Lefrancois, S., Zeng, J., Hassan, A. J., Canuel, M. & Morales, C. R. The lysomal trafficking of sphingolipid activator proteins (SAPs) is mediated by sortilin. EMBO J. 23, 1680 (2004).
Seaman, M. N. Identification of a novel conserved sorting motif required for retromer-mediated endosome-to-TGN retrieval. J. Cell Sci. 120, 2378–2389 (2007).
Canuel, M., Korkidakis, A., Konnyu, K. & Morales, C. R. Sortilin mediates the lysosomal targeting of cathepsins D and H. Biochem. Biophys. Res. Commun. 373, 292–297 (2008).
Quistgaard, E. M. et al. Ligands bind to Sortilin in the tunnel of a ten-bladed β-propeller domain. Nature Struct. Mol. Biol. 16, 96–98 (2009).
Reczek, D. et al. LIMP-2 is a receptor for lysosomal mannose-6-phosphate-independent targeting of β-glucocerebrosidase. Cell 131, 770–783 (2007). Demonstrates that LIMP2 functions as a transport chaperone for M6PR-independent transport of the lysosomal hydrolase βGC to lysosomes.
Gamp, A. C. et al. LIMP-2/LGP85 deficiency causes ureteric pelvic junction obstruction, deafness and peripheral neuropathy in mice. Hum. Mol. Genet. 12, 631–646 (2003).
Dvir, H. et al. X-ray structure of human acid-β-glucosidase, the defective enzyme in Gaucher disease. EMBO Rep. 4, 704–709 (2003).
Kuronita, T. et al. The NH2-terminal transmembrane and lumenal domains of LGP85 are needed for the formation of enlarged endosomes/lysosomes. Traffic 6, 895–906 (2005).
Knipper, M. et al. Deafness in LIMP2-deficient mice due to early loss of the potassium channel KCNQ1/KCNE1 in marginal cells of the stria vascularis. J. Physiol. 576, 73–86 (2006).
Berkovic, S. F. et al. Array-based gene discovery with three unrelated subjects shows SCARB2/LIMP-2 deficiency causes myoclonus epilepsy and glomerulosclerosis. Am. J. Hum. Genet. 82, 673–684 (2008).
Balreira, A. et al. A nonsense mutation in the LIMP-2 gene associated with progressive myoclonic epilepsy and nephrotic syndrome. Hum. Mol. Genet. 17, 2238–2243 (2008). References 122 and 123 report the first discoveredhuman mutations in the βGC trafficking receptor LIMP2 that lead to central nervous system and kidney disease.
Schroen, B. et al. Lysosomal integral membrane protein 2 is a novel component of the cardiac intercalated disc and vital for load-induced cardiac myocyte hypertrophy. J. Exp. Med. 204, 1227–1235 (2007).
Janvier, K. & Bonifacino, J. S. Role of the endocytic machinery in the sorting of lysosome-associated membrane proteins. Mol. Biol. Cell 16, 4231–4242 (2005).
Carlsson, S. R. & Fukuda, M. The lysosomal membrane glycoprotein lamp-1 is transported to lysosomes by two alternative pathways. Arch. Biochem. Biophys. 296, 630–639 (1992).
Groux-Degroote, S. et al. Glycolipid-dependent sorting of melanosomal from lysosomal membrane proteins by lumenal determinants. Traffic 9, 951–963 (2008).
Harter, C. & Mellman, I. Transport of the lysosomal membrane glycoprotein lgp120 (lgp-A) to lysosomes does not require appearance on the plasma membrane. J. Cell Biol. 117, 311–325 (1992).
Luzio, J. P. et al. Membrane dynamics and the biogenesis of lysosomes. Mol. Membr. Biol. 20, 141–154 (2003).
Vergarajauregui, S. & Puertollano, R. Two di-leucine motifs regulate trafficking of mucolipin-1 to lysosomes. Traffic 7, 337–353 (2006).
Bonifacino, J. S. & Traub, L. M. Signals for sorting of transmembrane proteins to endosomes and lysosomes. Annu. Rev. Biochem. 72, 395–447 (2003).
Honing, S., Griffith, J., Geuze, H. J. & Hunziker, W. The tyrosine-based lysosomal targeting signal in lamp-1 mediates sorting into Golgi-derived clathrin-coated vesicles. EMBO J. 15, 5230–5239 (1996).
Hunziker, W. & Geuze, H. J. Intracellular trafficking of lysosomal membrane proteins. Bioessays 18, 379–389 (1996).
Pak, Y., Glowacka, W. K., Bruce, M. C., Pham, N. & Rotin, D. Transport of LAPTM5 to lysosomes requires association with the ubiquitin ligase Nedd4, but not LAPTM5 ubiquitination. J. Cell Biol. 175, 631–645 (2006).
Karlsson, K. & Carlsson, S. R. Sorting of lysosomal membrane glycoproteins lamp-1 and lamp-2 into vesicles distinct from mannose 6-phosphate receptor/γ -adaptin vesicles at the trans-Golgi network. J. Biol. Chem. 273, 18966–18973 (1998).
Kleijmeer, M. J., Morkowski, S., Griffith, J. M., Rudensky, A. Y. & Geuze, H. J. Major histocompatibility complex class II compartments in human and mouse B lymphoblasts represent conventional endocytic compartments. J. Cell Biol. 139, 639–649 (1997).
Glickman, J., Morton, P., Slot, J., Kornfeld, S. & Geuze, H. The biogenesis of the MHC class II compartment in human I-cell disease B lymphoblasts. J. Cell Biol. 132, 769–785 (1996).
Ramachandran, N. et al. VMA21 deficiency causes an autophagic myopathy by compromising V-ATPase activity and lysosomal acidification. Cell 137, 235–246 (2009).
Chang, T. Y., Chang, C. C., Ohgami, N. & Yamauchi, Y. Cholesterol sensing, trafficking, and esterification. Annu. Rev. Cell Dev. Biol. 22, 129–157 (2006).
Jahn, R. & Scheller, R. H. SNAREs — engines for membrane fusion. Nature Rev. Mol. Cell Biol. 7, 631–643 (2006).
Gordon, D. E., Mirz, M., Sahlender, D. A., Jakovleska, J. & Peden, A. A. Coiled-coil interactions are required for post-Golgi R-SNARE trafficking. EMBO Rep. 26Jun 2009 (doi:10.1038/embor.2009.96).
Martinez-Arca, S. et al. A dual mechanism controlling the localization and function of exocytic v-SNAREs. Proc. Natl Acad. Sci. USA 100, 9011–9016 (2003).
Pryor, P. R. et al. Molecular basis for the sorting of the SNARE VAMP7 into endocytic clathrin-coated vesicles by the ArfGAP Hrb. Cell 134, 817–827 (2008).
Theos, A. C. et al. A lumenal domain-dependent pathway for sorting to intralumenal vesicles of multivesicular endosomes involved in organelle morphogenesis. Dev. Cell 10, 343–354 (2006).
Gough, N. R. & Fambrough, D. M. Different steady state subcellular distributions of the three splice variants of lysosome-associated membrane protein LAMP-2 are determined largely by the COOH-terminal amino acid residue. J. Cell Biol. 137, 1161–1169 (1997). LAMP2 splice variants have different transmembrane domains and cytosolic tails. This study revealed that the different C-terminal tails of LAMP2 are crucial for the intracellular distribution of the LAMP2 variants.
Kannan, K. et al. Lysosome-associated membrane proteins h-LAMP1 (CD107a) and h-LAMP2 (CD107b) are activation-dependent cell surface glycoproteins in human peripheral blood mononuclear cells which mediate cell adhesion to vascular endothelium. Cell. Immunol. 171, 10–19 (1996).
Fukuda, M. Lysosomal membrane glycoproteins. Structure, biosynthesis, and intracellular trafficking. J. Biol. Chem. 266, 21327–21330 (1991).
Fehrenbacher, N. et al. Sensitization to the lysosomal cell death pathway by oncogene-induced down-regulation of lysosome-associated membrane proteins 1 and 2. Cancer Res. 68, 6623–6633 (2008). Clear demonstration that malignant transformation of cells is associated with downregulation of LAMP proteins and a sensitization of cells to lysosomal cell death pathways induced by anti-cancer drugs.
Saitoh, O., Wang, W. C., Lotan, R. & Fukuda, M. Differential glycosylation and cell surface expression of lysosomal membrane glycoproteins in sublines of a human colon cancer exhibiting distinct metastatic potentials. J. Biol. Chem. 267, 5700–5711 (1992).
Huynh, K. K. et al. LAMP proteins are required for fusion of lysosomes with phagosomes. EMBO J. 26, 313–324 (2007).
Clark, R. H. et al. Adaptor protein 3-dependent microtubule-mediated movement of lytic granules to the immunological synapse. Nature Immunol. 4, 1111–1120 (2003).
Yogalingam, G. et al. Neuraminidase 1 is a negative regulator of lysosomal exocytosis. Dev. Cell 15, 74–86 (2008). Interesting observation that over-sialylated LAMP1 enhances lysosomal exocytosis.
Idone, V., Tam, C. & Andrews, N. W. Two-way traffic on the road to plasma membrane repair. Trends Cell Biol. 18, 552–559 (2008).
Rao, S. K., Huynh, C., Proux-Gillardeaux, V., Galli, T. & Andrews, N. W. Identification of SNAREs involved in synaptotagmin VII-regulated lysosomal exocytosis. J. Biol. Chem. 279, 20471–20479 (2004).
Arantes, R. M. & Andrews, N. W. A role for synaptotagmin VII-regulated exocytosis of lysosomes in neurite outgrowth from primary sympathetic neurons. J. Neurosci. 26, 4630–4637 (2006).
Braun, V. et al. TI-VAMP/VAMP7 is required for optimal phagocytosis of opsonised particles in macrophages. EMBO J. 23, 4166–4176 (2004).
Roy, D. et al. A process for controlling intracellular bacterial infections induced by membrane injury. Science 304, 1515–1518 (2004).
Tardieux, I. et al. Lysosome recruitment and fusion are early events required for trypanosome invasion of mammalian cells. Cell 71, 1117–1130 (1992).
Reggiori, F. 1. Membrane origin for autophagy. Curr. Top. Dev. Biol. 74, 1–30 (2006).
Tanaka, Y. et al. Accumulation of autophagic vacuoles and cardiomyopathy in LAMP-2-deficient mice. Nature 406, 902–906 (2000).
Nishino, I. et al. Primary LAMP-2 deficiency causes X-linked vacuolar cardiomyopathy and myopathy (Danon disease). Nature 406, 906–910 (2000). First report demonstrating that null mutations within the LAMP2 gene lead to Danon disease and the accumulation of autophagic vacuoles in muscle cells.
Eskelinen, E. L. et al. Disturbed cholesterol traffic but normal proteolytic function in LAMP-1/LAMP-2 double-deficient fibroblasts. Mol. Biol. Cell 15, 3132–3145 (2004).
Binker, M. G. et al. Arrested maturation of Neisseria-containing phagosomes in the absence of the lysosome-associated membrane proteins, LAMP-1and LAMP-2. Cell. Microbiol. 9, 2153–2166 (2007).
Jager, S. et al. Role for Rab7 in maturation of late autophagic vacuoles. J. Cell Sci. 117, 4837–4848 (2004).
Beertsen, W. et al. Impaired phagosomal maturation in neutrophils leads to periodontitis in lysosomal-associated membrane protein-2 knockout mice. J. Immunol. 180, 475–482 (2008).
Cuervo, A. M. & Dice, J. F. A receptor for the selective uptake and degradation of proteins by lysosomes. Science 273, 501–503 (1996).
Zhou, D. et al. Lamp-2a facilitates MHC class II presentation of cytoplasmic antigens. Immunity 22, 571–581 (2005).
Zhang, C. & Cuervo, A. M. Restoration of chaperone-mediated autophagy in aging liver improves cellular maintenance and hepatic function. Nature Med. 14, 959–965 (2008). Interesting observation that transgenic upregulation of the CMA receptor LAMP2A in liver cells improves age-related loss of liver functions.
Cuervo, A. M., Stefanis, L., Fredenburg, R., Lansbury, P. T. & Sulzer, D. Impaired degradation of mutant α-synuclein by chaperone-mediated autophagy. Science 305, 1292–1295 (2004).
De Matteis, M. A. & Luini, A. Exiting the Golgi complex. Nature Rev. Mol. Cell Biol. 9, 273–284 (2008).
Sriram, V., Krishnan, K. S. & Mayor, S. Deep-orange and carnation define distinct stages in late endosomal biogenesis in Drosophila melanogaster. J. Cell Biol. 161, 593–607 (2003).
Acknowledgements
We want to thank our colleagues for their input and many discussions. We express our special thanks to M. van Peski, J. Schröder, M. Schwake, R. Scriwanek and V. Oorschot for help with preparation of the original figures, and H. Geuze, M. Pols and R. Galmes for their comments on the manuscript. J.K. is the recipient of VICI grant 918.56.611 of the Netherlands Organization for Scientific research (NWO). P.S. is the recipient of DFG grants GRK1459 and SA683/5-1 and of the Center of Excellence 'Inflammation at Interfaces' grant. Cells used in figure 3, part c, were courtesy of M. Schwake, Department of Biochemistry, Christian-Albrechts University, Kiel, Germany.
Author information
Authors and Affiliations
Supplementary information
Related links
Related links
DATABASES
OMIM
FURTHER INFORMATION
Glossary
- Lysosome-related organelle
-
(LRO). A cell-type-specific organelle belonging to a family that includes melanosomes, platelet-dense bodies and cytotoxic T cell granules. LROs contain subsets of lysosomal proteins in addition to cell-type-specific proteins.
- Tetraspanin
-
A member of a conserved protein family with four transmembrane domains and two extracellular loops. Tetraspanins act as scaffolding proteins, anchoring multiple proteins to a specific area at the plasma membrane.
- Trans-Golgi network
-
(TGN). A convoluted membrane compartment at the trans side of the Golgi complex that mediates sorting and transport of proteins to various cellular destinations.
- Rab protein
-
A member of a family of small GTPases that, when associated with the cytosolic leaflet of the endosomal limiting membrane, can initiate the formation of functional microdomains.
- HRS
-
(Hepatocyte growth factor-regulated tyrosine kinase substrate; also known as ESCRT0). A cytosolic protein that is involved in the recognition of ubiquitylated cargo at endosomes, which initiates the recruitment of the ESCRT complex.
- Clathrin
-
A protein that forms a coat which has a major role in the formation of transport vesicles. The coat consists of multiple triskelions, which are composed of three clathrin heavy chains and three light chains.
- Retromer
-
A heterotetrameric protein complex that associates with the cytosolic leaflet of the endosomal limiting membrane. The mammalian retromer consists of a sorting nexin dimer composed of a still-undefined combination of SNX1, SNX2, SNX5 or SNX6 and the cargo recognition trimer VPS26–VPS29–VPS35.
- AP1
-
A member of the heterotetrameric family of adaptor proteins involved in membrane trafficking, which also includes AP2, AP3 and AP4.
- ESCRT
-
(Endosomalsorting complexes required for transport). Endosomal sorting machinery consisting of four complexes ESCRT0, ESCRTI, ESCRTII and ESCRTIII plus several accessory components. ESCRT components recognize ubiquitylated cargoes, deform the endosomal membrane and catalyse the formation of ILVs containing the sorted cargo.
- Exosome
-
A small vesicle that initially exists as an ILV in the lumen of LEs or MVBs. They are called exosomes once released, following fusion of LEs and MVBs with the plasma membrane. Exosomes are thought to have key roles in antigen presentation, cell-to-cell communication, the pathogenesis of retroviral infections and prion disease.
- Dendritic cell
-
A potent antigen-presenting cell that is part of the mammalian immune system. Its main function is to process antigen material and present it, in the context of the MHC class II complex, on its surface to other cells of the immune system.
- SNARE
-
(Soluble N-ethylmaleimide-sensitive factor (NSF) attachment protein (SNAP) receptor). A member of a family of membrane-tethered coiled-coil proteins that regulate fusion reactions and target specificity in the vacuolar system. They can be divided into vesicle (v)-SNAREs and target (t)-SNAREs on the basis of their membrane localization.
- GGA protein
-
(Golgi-localized, γ-ear-containing, Arf-binding protein). A member of a family of monomeric adaptor proteins. In mammals there are three different GGA proteins: GGA1, GGA2 and GGA3.
- Autophagosome
-
A double-membrane vesicle that forms at an early stage of the autophagic pathway and can fuse with endosomes and lysosomes for degradation of its contents.
- Lysosomal cell death
-
Apoptosis induced by permeabilization of the lysosomal membrane and the subsequent release of cathepsins into the cytosol. The mechanism of membrane permeabilization is not yet known.
- Immunological synapse
-
The interface between an antigen-presenting cell and a lymphocyte, consisting of a cluster of T cell receptors and a ring of adhesion molecules.
- Bacterial type III secretion system
-
A specialized, needle-like, multiprotein structure in Gram-negative bacteria that is involved in the direct secretion of proteins from the bacterial cell to the host.
- Phagosome
-
A vacuole formed around a particle for example, a pathogenic microorganism absorbed by phagocytosis that can mature into a degradative compartment by fusion with lysosomes.
- Autolysosome
-
A single-membrane vesicle that forms at a late stage of the autophagic pathway by fusion of autophagosomes with endosomes or lysosomes and that contains degradative enzymes obtained after fusion.
- Periodontis
-
A common inflammatory disease of the supporting tissues of the teeth that leads to resorption of alveolar bone and eventually to tooth loss.
- Chaperone-mediated autophagy
-
A direct pathway for transporting cytosolic proteins over the lysosomal limiting membrane and into the lysosome lumen for degradation.
Rights and permissions
About this article
Cite this article
Saftig, P., Klumperman, J. Lysosome biogenesis and lysosomal membrane proteins: trafficking meets function. Nat Rev Mol Cell Biol 10, 623–635 (2009). https://doi.org/10.1038/nrm2745
Published:
Issue Date:
DOI: https://doi.org/10.1038/nrm2745
This article is cited by
-
Carnosine regulation of intracellular pH homeostasis promotes lysosome-dependent tumor immunoevasion
Nature Immunology (2024)
-
Lysosomes as coordinators of cellular catabolism, metabolic signalling and organ physiology
Nature Reviews Molecular Cell Biology (2024)
-
Transcriptome analysis of the immunoprotective effect of Bacillus licheniformis on the intestinal tract of Carassius auratus gibelio infected with Aeromonas hydrophila
Aquaculture International (2024)
-
Kinesin family member 18B activates mTORC1 signaling via actin gamma 1 to promote the recurrence of human hepatocellular carcinoma
Oncogenesis (2023)
-
Loss of the batten disease protein CLN3 leads to mis-trafficking of M6PR and defective autophagic-lysosomal reformation
Nature Communications (2023)
