Key Points
-
Notable heterogeneity exists in the neuropathological substrates that underlie dementia in the setting of Parkinson's disease dementia (PDD). Nevertheless, the presumptive caudal-to-rostral spread of Lewy body and neurite pathology from the lower brainstem to telencephalic regions, which culminates in a heavy burden of this α-synuclein (α-syn) pathology in limbic and neocortical structures, is the most characteristic pathological finding in most PDD cases.
-
Up to 50% of patients with PDD can have sufficient amyloid-β (Aβ) plaque and tau neurofibrillary tangle (NFT) pathology for the diagnosis of a second neurodegenerative dementia — that is, Alzheimer's disease (AD) — and this co-morbid pathology is more common in PDD than PD.
-
Tau NFT and Aβ plaque pathology may act synergistically with Lewy body and neurite pathology to confer a worse prognosis and a higher burden of cortical Lewy body and neurite pathology in PDD.
-
Clinical phenotypes of PD may help to identify neuropathological subtypes of PD that exhibit differing propensities for developing dementia. For example, non-tremor-dominant or postural gait instability PD phenotypes may often be associated with greater Aβ plaque pathology and shorter times to dementia in patients with PDD than in patients with PD exhibiting less co-morbid AD neuropathology and a tremor-dominant phenotype.
-
Genetic variations may contribute to the heterogeneity in the neuropathology and the time-of-onset of dementia in PD. For example, the apolipoprotein E (APOE) ε4 genotype may increase both Lewy body and neurite pathology and AD neuropathology and result in an increased risk of dementia in PD. Moreover, heterozygous mutations in β-glucocerebrosidase (GBA) may increase the severity of Lewy body and neurite patholgy in 'pure' synucleinopathies.
-
Further biomarker and detailed clinicopathological correlation studies of prospective patients will help to further elucidate the inter-relationships of AD and α-syn pathology in PD and the development of dementia in patients with PD. These discoveries will be crucial in the development of meaningful disease-modifying therapies for PDD.
Abstract
Dementia is increasingly being recognized in cases of Parkinson's disease (PD); such cases are termed PD dementia (PDD). The spread of fibrillar α-synuclein (α-syn) pathology from the brainstem to limbic and neocortical structures seems to be the strongest neuropathological correlate of emerging dementia in PD. In addition, up to 50% of patients with PDD also develop sufficient numbers of amyloid-β plaques and tau-containing neurofibrillary tangles for a secondary diagnosis of Alzheimer's disease, and these pathologies may act synergistically with α-syn pathology to confer a worse prognosis. An understanding of the relationships between these three distinct pathologies and their resultant clinical phenotypes is crucial for the development of effective disease-modifying treatments for PD and PDD.
Similar content being viewed by others


Main
Parkinson's disease (PD) is the most common neurodegenerative movement disorder. The clinical syndrome of PD is typically associated with neuronal loss in the substantia nigra and inclusions containing the synaptic protein α-synuclein (α-syn) in the cell bodies and processes of surviving neurons (known as Lewy bodies and Lewy neurites, respectively) in this region. However, PD is now recognized as being a more complex clinicopathological entity. Many individuals with PD have widespread α-syn pathology in the brain. Moreover, most patients with this condition exhibit some level of cognitive dysfunction — including dementia (PD dementia (PDD)) — during the natural course of their illness (see Ref. 1 for a detailed review on the history of the major developments in PD research).
Given this expanded view of PD as both a movement and cognitive disorder, here we examine the complex connections between the neuropathological aetiologies that underlie the variable expression of the cognitive deficits in PD. By doing so, we aim to further the understanding of the pathobiology of PDD and to distinguish genetic, clinical and neuropathological subtypes of this condition, which will be important for developing and administering appropriate disease-modifying treatments.
Clinical features of PDD
Epidemiology and clinical impact of PDD. Cognitive impairment (that is, cognitive dysfunction that occurs in the absence of a functional deficit that is sufficient for the diagnosis of dementia) is a common feature of PD and may be a natural consequence of end-stage disease2,3,4, as the overall prevalence of cognitive impairment in PD is roughly 30%3,5, and approximately 80% of longitudinally followed patients with PD develop dementia over the course of their disease2,3,6. It is estimated that the incidence rate of dementia in patients with PD is at least fourfold higher than it is in the general population3,5,7,8. In addition, cognitive dysfunction that does not meet the threshold for dementia can be present early in the disease course, with cognitive deficits seen in up to 24% of patients at the initial diagnosis of PD9. Furthermore, patients with PDD experience dementia for an average of 3–4 years during their disease course4. Thus, including the period of cognitive dysfunction before progression to PDD, many patients with PD show cognitive deficits for the majority of their illness duration. This fact has implications for patient quality of life, public health and the cost of care.
Cognitive difficulties can exacerbate the disabilities caused by motor symptoms in PD. Indeed, the presence of cognitive impairment or dementia in patients with PD is associated with a loss of independence, a lower quality of life and a reduction in survival time10,11. Furthermore, individuals with end-stage PD who develop dementia often exhibit a decline in function that follows a stereotypical pattern, indicating that dementia onset may herald impending residential care and mortality4. Thus, the onset and progression of cognitive impairment and dementia in PD have an important influence on patient management and prognosis, and understanding the pathophysiology underlying cognitive dysfunction in PD is crucial for therapeutic development.
Cognitive features of MCI and dementia in PD. The specific cognitive symptoms associated with PD vary12, probably because of the heterogeneous nature of the underlying neuropathology. The most common cognitive impairments in PD are deficits in attention, executive functioning and visuospatial processing, although patients may also exhibit varying degrees of memory loss, which, in some cases, may be related more to a frontally mediated retrieval deficit (that is, a deficit in executive functioning) than to an intrinsic encoding problem (see Ref. 5 for a thorough review of cognitive studies in PDD). Similarly, PDD is not characterized by an intrinsic core language deficit5, but it can be associated with difficulties in sentence processing, which are also related to executive dysfunction13. Some researchers propose that executive difficulties in PD are an early phenomenon of altered dopaminergic tone in the frontal cortex, whereas deficits in visuospatial functioning and semantic memory correspond to the later neuropathological involvement of the temporal and parietal cortices, and that these 'posterior symptoms' confer an increased risk of PDD8. The recognition of cognitive symptoms in PD has led to the recent development of clinical criteria for PDD5 and mild cognitive impairment (MCI) in PD (PD-MCI)12 (Box 1). These formalized criteria will facilitate the earlier detection and better characterization of cognitive impairment in PD and will allow detailed comparative studies to be conducted in the future.
There is some similarity between the cognitive symptoms of PDD and Alzheimer's disease (AD), but patients with PDD may be differentiated from individuals with AD by the presence of hallucinations, cognitive fluctuations, depression and sleep disturbance14. By contrast, the cognitive features of PDD are similar to and often indistinguishable from the clinical syndrome of dementia with Lewy bodies (DLB)5,14,15. The research criteria for DLB make a somewhat arbitrary distinction between these two entities, with a diagnosis of DLB being assigned to patients with an onset of dementia within 1 year after the onset of motor symptoms and PDD being assigned to individuals when dementia occurs more than 1 year after PD onset16. The underlying pathophysiology responsible for the different chronological patterns for the emergence of dementia in these clinical syndromes is a matter of intense research and continued debate14.
Patients with PD may have neuropsychiatric symptoms, including depression, anxiety, apathy, hallucinations and delusions5. Furthermore, some individuals may exhibit an impulse control disorder that is characterized by compulsive gambling, eating, purchasing, sexual behaviour and/or a dopamine dysregulation syndrome17. The aetiology of impulse control disorders in PD is thought to be due to stimulation of hypersensitive ventral striatal–frontal connections by dopaminergic therapy rather than a direct consequence of the neurodegenerative process that is specific to PD and PDD17,18.
Clinical risk factors for PDD. Identification of clinical features that may predict impending cognitive decline in PD is of interest for clinical practice and disease management. Age is the most prominent risk factor for PDD4,7,8,19. The increased risk of dementia with age has been dissociated from the potential influence of age of PD onset20; in other words, patients with PDD have a similar age of dementia onset regardless of when they first present with motor symptoms4. Studies show that cognitive impairment in PD also correlates with the severity of motor disability21 and that the effects of ageing on cognition may be additive to the severity of the motor disturbance7. Other factors and disease features linked to PDD include male sex22, low education22, visual hallucinations3,4,15 and prominent axial rigidity and bradykinesia relative to tremor3,5,23. The presence of MCI in PD12, as revealed by poor performance on specific neuropsychological measures, including semantic fluency8, constructional praxis8, verbal memory24 and executive function24, has also been associated with an increased risk of developing dementia.
The pathogenesis of PD
Role of Lewy bodies and Lewy neurites. α-syn is a 140-amino-acid presynaptic protein that is involved in vesicular transport. This protein was implicated in the pathogenesis of PD when pathogenic mutations in SNCA (which encodes α-syn)25,26 and, later, multiplications of this gene27 were linked to hereditary forms of this disease. Some of these genetic abnormalities may increase the propensity of α-syn to fibrillize in vitro28. Thus, molecular changes in the α-syn protein that increase protein misfolding and aggregation have a direct role in disease pathogenesis. The initial discovery of SNCA mutations was closely followed by evidence from post-mortem studies of PD and DLB brains that the characteristic Lewy bodies and Lewy neurites visualized through immunohistochemical methods using α-syn-specific antibodies were composed mainly of insoluble, fibrillar forms of α-syn29. Several experimental models support a role for α-syn aggregation in neurodegeneration. For example, overexpression of the Ala53Thr mutant variant of α-syn in transgenic animals30,31 led to the formation of α-syn immunoreactive deposits that resembled Lewy body and neuritic pathology as well as a cognitive and motor clinical phenotype and reduced survival time. Furthermore, suppression of Ala53Thr α-syn expression resulted in decreases in neuropathology and clinical symptoms31 (see below for further discussion of the role of fibrillized α-syn in the progression of PD and Ref. 32 for a comprehensive review of murine models of PD).
A detailed neuropathological examination of a large number of PD and control cases by Braak and colleagues33,34 revealed a stereotypical caudal-to-rostral ascending progression of α-syn pathology from brainstem structures in early stages of the disease to limbic and neocortical areas in the later stages. Staging systems of α-syn pathology based on these observations have been proposed for PD33 and DLB16. These hypothetical systems are supported by some independent studies35,36,37, although not all studies demonstrate a similar topography of disease spread37,38, and α-syn staging may be more applicable to patients with younger-onset PD6. Indeed, up to 30% of elderly patients with α-syn pathology at autopsy show no clinical signs of dementia or a movement disorder, thereby prompting such cases to be termed incidental Lewy body disease (ILBD)34,36,39,40,41,42. These observations have led some researchers to infer that α-syn pathology may be neuroprotective rather than a cause of neuronal cell death37,43. In addition, quantitative studies of Lewy bodies and Lewy neurites suggest a dissociation between α-syn pathology and neuron loss in PD44,45. The explanation for this discrepancy is unclear. It is possible that individuals with ILBD could be in a prodromal state of PD in which the level of α-syn pathology does not cross the threshold to manifest clinical symptoms46, and that they would develop full blown PD if they survived for a longer period34. This would be consistent with findings showing that the locations of most incidental α-syn pathology match the areas affected in early stages of PD34,36.
α-syn pathology is also found in up to 50% of patients with AD and has a distribution that is largely restricted to the amygdala in many cases35,36,39,47,48,49. The amygdala seems to be exceptionally vulnerable to other neurodegenerative inclusions, such as tau and TAR DNA-binding protein 43 (TDP43) inclusions, and 'amygdala-only' (Ref. 50) patterns of α-syn pathology in AD cases may reflect a separate disease process from the pathogenic spread of these inclusions in PD and DLB, as has been proposed by Braak and colleagues35,49 (for a detailed review of the Braak staging system and proposed revisions to this system, see Refs 43,51).
Despite the exceptions discussed above to the staging systems proposed by Braak and colleagues, the stereotypical non-random progression of α-syn pathology in most patients with PD suggests that α-syn undergoes cell-to-cell transmission within individuals. In addition, α-syn pathology discovered in fetal dopaminergic neurons that were implanted in patients with PD52,53,54 as well as recent in vitro cell studies55,56 and in vivo animal studies57,58 indicate that pathogenic α-syn can transfer between cells, leading to neurodegeneration. These and other transmission studies59,60,61,62 are reviewed in depth elsewhere63,64, and Fig. 1 summarizes the proposed mechanisms by which the aggregation and spread of α-syn leads to deleterious consequences for the affected neurons65. The evidence for α-syn transmission in synucleinopathies prompts speculation that PD and other neurodegenerative proteinopathies could spread between individuals in a similar way to the spread of pathological prion proteins in human transmissible spongiform encephalopathies, but there is currently no definitive evidence of human-to-human transmission of clinical PD66 (Box 2).

Under physiological conditions, α-synuclein (α-syn) exists in a soluble random coil state. Under pathological conditions, however, the native protein undergoes misfolding into pathogenic species of α-syn (dimers, trimers and oligomers) that further aggregate into higher-order structures (protofibrils, other intermediates and amyloid fibrils). Ultimately, these higher-order structures are the building blocks for the pathological inclusions of α-syn — that is, Lewy bodies and Lewy neurites — that are visualized under light microscopy at autopsy65. Genetic abnormalities and environmental factors may accelerate this process26,27,28,65,114,116. Normal quality-control systems (chaperones, ubiquitin proteosomes and phagosome–lysosome systems) that prevent or reverse protein misfolding or eliminate misfolded proteins are overwhelmed by oligomeric species of α-syn (indicated by dashed lines)65. Remarkably, recent data suggest that the progression of PD and related disorders may be linked to the cell-to-cell spread of pathological species of α-syn56,57,58,63. α-syn transmission is thought to have various, inter-related toxic consequences65. It is unclear which species of pathogenic α-syn is directly toxic to neurons; however, recent animal studies show that synthetic α-syn fibrils alone are sufficient to transmit disease (that is, α-syn pathology) between neurons and cause clinical disease57,58. UPS, ubiquitin proteasome system.
The data reviewed here — including recent transmission studies showing that injections of synthetic α-syn pre-formed fibrils into the dorsal striatum are followed first by Lewy body formation in the substantia nigra pars compacta and then later by neuron loss57,58 — suggest that α-syn misfolding and aggregation within substantia nigra neurons result in neuron loss and the clinical syndrome of idiopathic PD. Although the exact pathogenic species of α-syn (for example, dimers, oligomers, protofibrils or fibrils) that are neurotoxic and that can spread from neuron to neuron are not yet known, α-syn pathologies are likely to be the proximal cause of neuron dysfunction and death in the cortex of patients with PDD, as discussed in more detail below.
Neuropathology underlying PDD
Clinicopathological correlations in PDD. The neuropathology underlying PDD is heterogeneous in nature (Fig. 2) and has been challenging to discern, with several factors making it difficult to conduct accurate clinicopathological correlation studies for this condition. First, human post-mortem studies are limited to visualization of neuropathology at the time of death, which may occur from one of many potential non-neurological aetiologies and precede end-stage PD as well as the full progression of its neuropathology. Although patients with PDD may have been followed longitudinally, autopsy data are by definition cross-sectional in nature, which makes it difficult to directly correlate such data with antecedent clinical deficits that manifest years before death. Second, interpretations of the neuropathological substrates of cognitive status in PD are limited by the accuracy of clinical diagnosis and by the ability to perform adequate follow-up studies in the end stages of disease2. There also may be several years between a patient's last clinic visit and autopsy because the reduced mobility of patients with end-stage PD renders office visits infeasible, and thus large numbers of well-annotated cases are needed to make meaningful observations. Last, criteria for neuropathological diagnoses have evolved over time, and advanced immunochemical techniques have revealed higher burdens of cortical α-syn pathology in cases of PDD than were previously recognized67,68. Thus, for these reasons, there has been considerable variation in results from earlier clinicopathological studies, as reviewed elsewhere69.

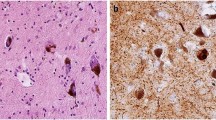
The main neuropathological features of PDD70 include a severe burden of Lewy bodies (asterisks) and Lewy neurites (arrows) (image from the mid-frontal cortex) (panel a), moderate tau-reactive diffuse threads (arrows) and neurofibrillary tangles (asterisk) (image from the anterior cingulate gyrus) (panel b) and extensive diffuse amyloid-β-reactive plaque pathology (asterisks) (image from the superior temporal cortex) (panel c). The colocalization (shown in yellow; panel f) of pathological α-synuclein (α-syn) in Lewy bodies (shown in red) (panel d) and the amyloid-binding dye thioflavin S (ThS; shown in green) (panel e) reveal the fibrillar nature of the majority of α-syn pathology (tissue stained in panels e and f was from the anterior cingulate gyrus).
In general, cortical Lewy body and neuritic pathology is more extensive and severe in PDD than in PD without dementia, and several lines of evidence from studies using α-syn immunohistochemistry implicate cortical α-syn pathology as the strongest correlate of dementia in PDD. First, most large studies have found that PDD cases are almost exclusively of the limbic-predominant stage (transitional) or neocortical-predominant stage16 (diffuse) of α-syn pathology3,21,69,70,71,72 and have higher levels of cortical α-syn pathology than do cases of PD4,67,69,70,71,72,73,74. Indeed, many studies find that the level of global cortical and limbic α-syn pathology can be used to discriminate between PD and PDD21,69,70. However, some studies suggest that increased levels of α-syn pathology in PDD in specific brain regions, including the parahippocampal gyrus74,75 or anterior cingulate gyrus, can be used to differentiate this condition from PD75,76. Second, strong correlations of diminishing performance on several cognitive measures with advancing α-syn pathology stages or increasing cortical α-syn pathology burden with have been described3,21,38,75,76. Last, multivariate regression analyses that included multiple neuropathological and genetic variables implicated in PDD found that the burden of cortical and limbic Lewy bodies and neurites was the strongest correlate of dementia in a large, well-annotated cohort of patients with PD or PDD70.
Despite these observations, some non-demented individuals with PD21,67,74 and some patients with ILBD37,40 have a marked burden of cortical and limbic α-syn pathology. Furthermore, a minority of patients with PDD were found to have minimal cortical α-syn pathology at autopsy15,21,67,70,77. These findings suggest that cortical and limbic α-syn pathology is not exclusively required for the development of dementia in PD. Thus, the presence of cortical α-syn pathology at autopsy should be correlated with clinical information on the subject's cognitive status before death for the neuropathological diagnosis of PDD. These discrepancies might be explained by methodological difficulties in ascertainment of clinical symptoms across studies and/or by cognitive reserve, brain plasticity or other factors that could make some individuals more resistant to the detrimental effects of α-syn pathology, but further research is needed to clarify these issues.
Dementia in patients with PD who have limited cortical and limbic α-syn pathology could also result from subcortical involvement or the presence of co-morbid neuropathologies. Cholinergic deficits occur in patients with PDD78,79, with highest loss in those with the longest duration of parkinsonism before dementia and who also have the lowest cortical and limbic Lewy body and neuritic burden78. These neurochemical changes have been ascribed to neuronal loss in basal forebrain cholinergic nuclei79,80. Of note, neuronal loss in these regions is associated with the transition of α-syn pathology into limbic and neocortical regions21,69. Moreover, it has been found that the burden of α-syn pathology68,72 and diffuse amyloid-β (Aβ) plaques (a feature of AD)81 in the striatum is higher in PDD than in PD without dementia, and this striatal pathology has been considered to be a possible substrate for cognitive impairment. Indeed, a study of 92 patients with PDD found 4 individuals with minimal cortical Lewy pathology, and these cases all had notable cerebrovascular disease (CVD) or featured subcortical Lewy pathology, which could have explained the cognitive deficits70.
Despite the central role of α-syn pathology in PDD, some studies have also found that the levels of Aβ plaques and tau neurofibrillary tangles (NFTs) — the hallmark pathologies of AD — inversely correlate with cognitive status in cases of PDD71,73,75,82,83 or in a subset of patients with this condition70,76 (Fig. 2). Indeed, two studies have shown that AD neuropathology seems to be a more specific correlate of dementia in PD than α-syn pathology, as the majority of PD patients with sufficient numbers of NFTs and Aβ plaques for a diagnosis of co-morbid AD have clinical dementia and hence could be assigned a diagnosis of PDD plus AD (PDD+AD)70,71. One of these studies found, however, that a regression model incorporating cortical α-syn, tau and Aβ pathologies together more accurately predicted dementia than any single marker alone71. Similarly, the other study found that a regression model that considered the apolipoprotein E (APOE) ε4 genotype status alongside cortical α-syn pathology improved the diagnostic accuracy for PDD compared with a model in which only the severity of cortical α-syn pathology was considered70. Interestingly, the association of the APOE ε4 genotype with PDD was independent of measures of AD neuropathology70. Overall, patients with PDD tend to have a higher cortical Aβ plaque burden4,21,69,70,71,72,73,84 and, to a lesser degree, a higher NFT burden4,70,71,72,73,82 than patients with PD without dementia. The percentage of patients with PDD who have levels of NFTs and plaques that meet the threshold for a second diagnosis of AD varies between studies and depends on the criteria used; however, up to 50% of patients with PDD may have co-morbid AD (that is, PDD+AD)39,70,76,82,85,86. Thus, AD neuropathology, especially Aβ plaque pathology, appears to have an important role in the pathogenesis of PDD for a significant proportion of patients.
Interestingly, patients with PDD+AD seem to have higher levels of cortical and limbic α-syn pathology than patients with PDD without co-morbid AD70,77, and increased severity of cortical Aβ plaque and NFT burden is associated with increased cortical α-syn pathology density70,71,74,76,82,87. These data imply a potential synergy between α-syn pathology and AD-related pathology. In support of this assertion, transgenic mice overexpressing human wild-type α-syn88 or human Ala53Thr mutant α-syn89 with human mutant forms of tau89 and/or the amyloid precursor protein (which leads to an increase in Aβ production)88,89 show greater neurodegenerative pathology and a more severe clinical phenotype than do mice overexpressing AD-associated proteins alone. In addition, patients with PD who carry the pathogenic Ala53Thr SNCA mutation exhibit marked tau pathology90, and in vitro experiments show that tau and α-syn can each cross-seed and enhance each other's polymerization into fibrils91,92.
This putative additive effect of α-syn pathology, Aβ plaques and NFTs could potentially influence the clinical features of PDD. Indeed, patients with PDD+AD may have a shorter disease duration than those with PDD39,70,82,83,86, and PDD+AD is also associated with an older age-at-onset of motor symptoms70,83,86. In addition, a higher Aβ plaque burden alone has been associated with shortened survival times in patients with PD6,71,84,93 and an older age of PD onset6,71. Thus, the presence of AD neuropathology in patients with PD could lead to an older age-of-onset PD subtype that has a more malignant course6,71. These prognostic findings for PDD+AD have important implications for patient care and emphasize the need for reliable biomarkers of the underlying neuropathology in PDD.
A few other potential neuropathologies that contribute to PDD have been examined. One study found that argyrophilic grain disease was a common feature of PDD94; however, it did not seem to correlate well with PDD in two large autopsy series21,70. TDP43 inclusions, which are characteristic of frontotemporal lobar degeneration and amyotrophic lateral sclerosis, have also been found in limbic regions in a small number of patients with PD and PDD95, but they do not seem to influence the outcome of dementia in PD70. Finally, CVD, as defined by varying methods and criteria, has been described in a minority (roughly 10–15% or less) of individuals with PDD2,3,70,82,85, although the presence of cerebrovascular lesions (including AD-associated amyloid angiopathy) was reported in 94% of patients with PDD versus roughly 50% of individuals with PD in one cohort73. Thus, systematic evaluation of CVD with validated neuropathological criteria is needed in PDD, but overall CVD does not seem to contribute notably to dementia in most patients with PDD.
In summary, these data implicate the progression of Lewy body and neurite pathology from subcortical areas into limbic and cortical structures as the major driving force behind the development of dementia in most individuals with PDD, although there is notable heterogeneity in the underlying dementia-linked neuropathology of PDD.
Clinical subtypes of PD and DLB. In addition to the aforementioned clinical similarity between PDD and DLB, there is neuropathological continuity between these syndromes, as the level and distribution of the underlying α-syn pathology in both of these patient groups are often indistinguishable36,49,74,78,82,96,97, although some studies have suggested that DLB and PDD can be differentiated between on the basis of an increased number of striatal Aβ plaques in DLB98,99. Dual pathology may contribute to the observed low diagnostic accuracy for clinical DLB82,100,101,102: co-morbid AD neuropathology can hinder the diagnosis of DLB100,101,103, as the clinical features of DLB, such as hallucinations, fluctuations and executive impairments, are less prominent in cases of this disorder in which there are high levels of AD neuropathology100,101,104. These findings suggest that the clinical phenotype of disorders featuring Lewy body and neurite pathology could be masked in some cases by concomitant AD14,100. Thus, the aetiology of the differing pattern of emergence of cognitive deficits across PDD and DLB is unclear but may be partly due to varying degrees of co-morbid AD neuropathology.
The effects of ageing97 also seem to have a role in the expression of dementia in PDD and DLB. The lack of correlation between α-syn pathology and disease duration38 and the variability of nigral neuronal cell loss in PD45 suggest that there is considerable heterogeneity in the rate of disease progression in PD. Indeed, patients who develop PD at an early age have been shown to have a longer course of disease on average6,19,105,106 than patients who are older at onset. Those older patients with a shorter interval (<10 years) of PD before dementia onset tend to have a higher degree of both cortical and limbic α-syn pathology and AD pathology6,70,71,78. In addition, the age-of-onset of PD itself does not incur a greater risk of dementia20 but instead influences the interval to dementia4, as patients with early-onset PD (which is associated with a long PD-to-dementia interval) and patients with late-onset PD (which is associated with a short PD-to-dementia interval) have similar ages of dementia onset4,70,78. This may be partly related to the presence of AD neuropathology6,70,71,87. Of note, the age of dementia onset in DLB and PDD is similar despite differences between the age-of-onset of motor features in these syndromes2,78.
Patients with PD can also be clinically subdivided according to differences in motor phenotypes. Individuals with prominent postural instability and gait disorder symptoms or a non-tremor-dominant presentation seem to have a shorter survival time and are more likely to develop dementia than those with a tremor-dominant presentation3,23,83,106. A subset of rapidly progressive tremor-dominant patients with PD who do not develop dementia has also been described106. Furthermore, the emergence of the postural gait-instability phenotype in patients with an initial presentation of tremor-dominant PD was associated with an accelerated rate of cognitive decline107. In addition, autopsy data106, cerebrospinal fluid (CSF) biomarker data108,109,110 and in vivo Aβ neuroimaging data111 suggest that underlying AD Aβ pathology may be more common in non-tremor-dominant patients. Patients with a non-tremor-dominant phenotype have higher cortical and limbic Lewy body burden as well106. Interestingly, the non-tremor-dominant phenotype is reminiscent of the motor features of DLB102, and it is tempting to hypothesize that AD Aβ pathology contributes to the variable and overlapping natures of the clinical presentations of PDD and DLB. One caveat to this interpretation is the accelerated progression of cortical and limbic α-syn pathology without the influence of AD neuropathology that causes early dementia in 'pure' DLB: that is, DLB without marked co-morbid AD neuropathology. This observation is in contrast to the slow progression of cortical and limbic α-syn pathology in patients with PD who have a long PD–dementia interval and who, in most cases, are also thought to have a 'pure' synucleinopathy; that is, they have negligible AD neuropathology6. These findings suggest that a complex interaction of genetic and neuropathological factors result in the varying expression of clinical phenotypes across the Lewy body and neurite pathology spectrum (Box 3); thus, we feel that PDD and DLB are most accurately viewed as clinicopathological entities on one continuous spectrum of disease (that is, synulceinopathies). For a detailed review on the relationships between clinical subtypes across the PDD–DLB spectrum and underlying neuropathology, see Ref. 112. Further detailed longitudinal clinical and biomarker studies with autopsy confirmation are needed to elucidate the relationship between PDD and DLB. The data described above suggest that such studies may yield important prognostic and perhaps diagnostic information for PDD and DLB.
Genetic associations. Genetic factors may also play an important part in the expression of cognitive deficits in PDD and DLB. Indeed, some hereditary forms of PD have been associated with dementia, most notably those arising from a pathogenic mutation in or triplication of SNCA, or a mutation in β-glucocerebrosidase (GBA; also known as acid β-glucosidase)26. By contrast, mutations in leucine-rich repeat kinase 2 (LRRK2) and other less common genetic aetiologies of PD do not seem to be as strongly linked to PDD and DLB26,113.
Homozygous mutations in GBA result in the lysosomal storage disorder Gaucher's disease, whereas heterozygous mutations in this gene are associated with an increased risk of PD114 or DLB115. Moreover, GBA-linked PD is associated with a higher risk of dementia and an earlier age of dementia onset than non-GBA-linked PD116. Recently, an international multicentre study found that individuals with DLB were more than eight times likely to harbour a GBA mutation than were controls, and these cases had a more aggressive disease course than patients with DLB who did not carry the mutation117. Furthermore, autopsy studies showed that GBA mutation-carrying patients with PD had higher levels of cortical and limbic α-syn pathology than non-carrier patients with PD116 and that there was an increased rate of carriers in cases of 'pure' DLB compared with DLB cases with a high degree of AD neuropathology (DLB+AD)115,118. Given that the protein encoded by GBA is functionally involved in lysosomal pathways, the findings described above suggest that mutations in GBA could contribute to increased α-syn deposition and influence the cortical spreading of α-syn pathology and the resultant dementia.
Genetic changes other than those linked to hereditary forms of PD have also been implicated in PDD. Indeed, the APOE ε4 allele has been extensively studied as a risk factor for AD and may confer an increased risk of dementia in PD70,119,120. However, no association was found between APOE ε4 carrier status and the rate of cognitive decline in a population-based longitudinally followed PD cohort, and a meta-analysis of previous studies revealed a potential influence of publication bias and heterogeneity in odds ratios as an alternative explanation for the association between APOE ε4 and dementia in PDD121. The presence of one or more copies of the APOE ε4 allele may affect cognition in the later stages of PD119, which could also explain the discrepancies mentioned above. Interestingly, APOE ε4 status is associated with both an increased number of Aβ plaques87,122 and a high cortical and limbic α-syn pathology burden in PD42,76,120,122. Furthermore, in one study, the association between APOE ε4 status and cognitive impairment was lost after adjusting for CSF levels of the 42-amino-acid form of Aβ (Aβ1–42), suggesting that the observed influence of APOE ε4 on cognition in PD was mediated by associated AD neuropathological changes123. By contrast, as mentioned above, the APOE ε4 genotype was predictive of PDD, independently of AD-related pathology or α-syn pathology, in a multivariate analysis of a large PD–PDD autopsy cohort70. Finally, the APOE ε4 allele frequency was higher in 'pure' synucleinopathies than in cognitively normal elderly controls, but it was less frequent in these pure cases than in DLB+AD and AD cases120. Thus, the APOE ε4 genotype may contribute to neurodegeneration in PDD through pathways that are shared with and diverge from those involved in AD.
The gene that encodes tau, MAPT, contains two major haplotypes in humans: H1 and H2. The H1/H1 haplotype has been associated with an increased risk of some tauopathies that clinically resemble PD but lack α-syn pathology (for example, progressive supranuclear palsy)124. Interestingly, this variation in MAPT has been associated with PD as well125, and hence provides another link between α-syn and tau pathologies in PD. The cognitive phenotypes that are associated with the H1/H1 haplotype in PD have been less well studied, although one study found that this haplotype is associated with poor memory performance in PD (but does not influence cognitive decline)119, and two others found that the H1/H1 haplotype in PD is an independent predictor of PDD8,126. In addition, the H1/H1 haplotype may have a synergistic effect on the risk of PDD when it occurs with a polymorphism in SNCA126. Furthermore, a polymorphism in MAPT may influence the degree of co-morbid AD pathology in PD, as shown by higher CSF levels of tau in a subgroup of PDD risk allele carriers with low CSF Aβ1–42 levels127, further suggesting an influence of genetic factors on the variable expression of AD and α-syn pathology in PDD. By contrast, one large autopsy study found no association of the H1/H1 haplotype and PDD, as individuals with PD and patients with PDD had nearly the same proportion of H1 carriers70. The discrepancies outlined above may be due to differences in sample size or other poorly understood issues.
Further prospective studies of large cohorts of patients who meet the modern criteria for PDD (which can be later examined at autopsy) will help to confirm these observations and clarify the impact of genetic factors on the emergence of PDD. Therapeutic trial designs may benefit from taking account of genetic subgroups into their analyses.
Biomarker studies. Biofluid biomarkers with a strong predictive value for cognitive impairment in PD could be useful in clinical trials. Indeed, a recent biomarker exploratory study with an immune-based multiplex approach found that epidermal growth factor (EGF) plasma levels were lower in PDD than in PD and could predict progression from PD to PDD with 79% accuracy in a mixed PD–PDD cohort128. Furthermore, patients with PD who had the lowest EGF levels were at greater risk of developing PDD in longitudinal follow-up128. These findings were replicated in drug-naive, newly diagnosed patients with PD: here, EGF levels were inversely correlated with cognitive functions, including semantic fluency and executive function, at the 2-year follow-up129. Thus, these results suggest that EGF could be used as a biofluid biomarker for predicting cognitive decline in PD, although this possibility requires further validation.
CSF analytes are other biomarker candidates for PDD, as proteins characteristic of inclusions in neurodegenerative disease, such as tau and Aβ1–42, can be readily detected in CSF. Indeed, PDD was found to be associated with higher CSF levels of total tau (t-tau)130 and phosphorylated tau (p-tau)130,131 than was PD, although these levels were not as high as in AD131. Patients with PDD have also been described as having lower levels of Aβ1–42 in CSF than those found in individuals with PD and intermediate to those in patients with AD and healthy controls130,132. Furthermore, an 'AD CSF biomarker signature' (that is, increased levels of t-tau and decreased levels of Aβ1–42) was found in a higher percentage of PDD cases than PD cases133. In addition, low Aβ1–42 levels in PDD were associated with poor performances on a memory task132 and executive tasks130,134. High t-tau levels in PDD were associated with a poor performance on a memory task130 and, when combined in a ratio to Aβ1–42 levels, with poor performances in executive tasks134. Finally, a prospective study found that low CSF Aβ1–42 levels that are indicative of AD predict cognitive decline in PD across several cognitive domains123. Thus, the levels of CSF tau and Aβ1–42 that are associated with cognitive decline may have predictive value for PDD and implicate AD neuropathology in cognitive decline in PD.
Immunoassays have been developed to detect forms of α-syn135,136,137 and proteins involved in inflammatory and oxidative stress pathways131,138,139 in CSF; however, the relationships between these potential biomarkers, PDD and cognitive status have not been systematically evaluated. Interestingly, drug-naive patients with early-stage PD without dementia have lower levels of CSF t-tau than do controls, and these low levels of tau correlate with lower CSF levels of α-syn110. Longitudinal data on CSF tau levels in PD are lacking, but one possibility is that CSF tau levels may increase over the course of the disease140 and could predict incipient dementia, as indicated by the aforementioned associations between cognitive dysfunction and increased CSF tau levels in PDD.
Neuroimaging studies provide a non-invasive method for determining risk of dementia in PD and, possibly, identifying underlying neuropathology. A volumetric MRI study using a specialized algorithm to score an individual's 'AD-like pattern' of atrophy found that this spatial pattern of atrophy predicted cognitive decline in PD141. Nuclear imaging using positron-emission scanning (PET) with a radioligand specific for Aβ deposition in the brain (Pittsburgh compound B (PIB)) provides in vivo evidence of AD neuropathology. Indeed, higher PIB retention, which reflects a higher burden of cortical Aβ plaques, corresponded to a higher likelihood that patients with PD would convert to a cognitively worse diagnosis (that is, PD to PD-MCI or PD-MCI to PDD)142 and was associated with cognitive dysfunction142,143. Finally, functional PET imaging with 2-deoxy-2-[18F] fluoro-d-glucose (18F-FDG PET) showed that patients with PD who eventually progressed to PDD showed hypometabolism in the visual association and posterior cingulate cortex144. Multimodal analyses find that CSF measures of tau and Aβ are associated with structural changes on MRI scans in PD and PDD140,145. Note that in addition to these predictive studies, numerous comparative neuroimaging studies have been conducted in PD and PDD, and these have been reviewed in detail in Refs 5,113.
Prospective multimodal studies of clinical, genetic, biochemical and neuroimaging biomarkers in initially drug-naive patients with PD, such as the Parkinson's Progression Marker Initiative (PPMI)146, are ongoing, and they will provide crucial insight into the dynamic changes that occur in these markers over time and their relationships with emerging dementia.
Therapeutic strategies and drug development
The current pharmacological strategy for the treatment of PDD includes augmenting neurotransmitter deficits, similar to AD treatment. Acetylcholinesterase inhibitors have been shown to improve cognition and improve the ability to perform activities of daily living in PDD147, with the largest body of supportive data for rivastigmine148. There is inconsistent evidence for the glutaminergic agent memantine having a beneficial effect in PDD, but further investigation is warranted113,148.
Increasing cholinergic tone with acetylcholinesterase inhibitors has the potential to worsen motor symptoms and, conversely, dopamine agonists used to treat motor symptoms may worsen cognition, complicating therapeutic options in PDD113. Furthermore, all of these agents do not target the underlying pathobiology of PDD and thus do not affect the progression of disease. The aforementioned studies on cell-to-cell transmission suggest that prevention of α-syn aggregation may help to arrest motor and cognitive difficulties in PD. Concepts of drug development for prevention of α-syn-mediated neurodegeneration are reviewed in detail elsewhere149 but broadly include decreasing synthesis and aggregation of α-syn and promoting clearance of pathological inclusions. Furthermore, the role of tau and Aβ pathology in the development of cognitive deficits in PD suggests that emerging disease-modifying therapies for AD may be beneficial in a subset of PDD cases. Immune-based therapies targeting α-syn are one potential approach for disease-modifying therapies in PD and PDD150. Timing the initiation of potential disease-modifying therapies is also a critical issue, as it is unclear if halting the spread of α-syn aggregations in symptomatic patients with PD would affect cognitive and motor symptoms or if preventive strategies through treatment of at-risk individuals or early-onset patients would be more efficacious150. Thus, biomarkers of incipient cognitive decline and pre-motor PD would be instrumental in the implementation and evaluation of α-syn-directed therapies in a protective manner in highly susceptible patients.
Conclusions
Dementia in PD is a common occurrence and has a significant impact on patient well-being. Although most evidence suggests a role for α-syn pathology in the development of clinical symptoms in PD, including the expression of cognitive dysfunction, notable heterogeneity exists between cases of PDD in terms of symptomatology and timing of dementia. This highlights the fact that complex interactions between multiple factors — including age, genetics and cognitive reserve — contribute to the clinicopathological expression of symptoms in this condition. Furthermore, AD neuropathology appears to contribute to the emergence of dementia in PD and may indeed be the underlying basis for dementia in a subset of older patients with PD by acting synergistically with α-syn pathology to promote the spread of α-syn inclusions. These findings illustrate the need to consider PD, and all neurodegenerative diseases, as clinicopathological entities rather than purely clinical syndromes. Indeed, this view has emerged with new clinical and pathological criteria for AD151,152 that incorporate biomarker evidence of Aβ plaque and NFT pathology to support clinical criteria. This approach has the benefit of providing refined prognostic information for different PD subtypes and could be useful in the selection of homogenous patient populations to more effectively study possible disease-modifying treatments. For example, patients with clinical phenotype and biomarker evidence of AD neuropathology may be predicted to have shorter disease duration and a more rapid time to dementia, and such patients could be administered Aβ- or tau-directed therapies as they are developed. Thus, coordinated efforts between researchers from multiple backgrounds to further elucidate these relationships, such as in the PPMI study, will be crucial for the advancement of meaningful disease-modifying treatments that could help to preserve patient independence and improve the quality of life in patients with PD.
References
Goedert, M., Spillantini, M. G., Del Tredici, K. & Braak, H. 100 years of Lewy pathology. Nature Rev. Neurol. 9, 13–24 (2012).
Hely, M. A., Reid, W. G., Adena, M. A., Halliday, G. M. & Morris, J. G. The Sydney multicenter study of Parkinson's disease: the inevitability of dementia at 20 years. Mov. Disord. 23, 837–844 (2008).
Aarsland, D., Andersen, K., Larsen, J. P., Lolk, A. & Kragh-Sorensen, P. Prevalence and characteristics of dementia in Parkinson disease: an 8-year prospective study. Arch. Neurol. 60, 387–392 (2003).
Kempster, P. A., O'Sullivan, S. S., Holton, J. L., Revesz, T. & Lees, A. J. Relationships between age and late progression of Parkinson's disease: a clinico-pathological study. Brain 133, 1755–1762 (2010).
Emre, M. et al. Clinical diagnostic criteria for dementia associated with Parkinson's disease. Mov. Disord. 22, 1689–1707 (2007). The Movement Disorder Society Task Force criteria for PDD.
Halliday, G., Hely, M., Reid, W. & Morris, J. The progression of pathology in longitudinally followed patients with Parkinson's disease. Acta Neuropathol. 115, 409–415 (2008). This is one of the largest prospectively followed PD autopsy series; it found that 80% of patients with PD develop dementia after 20 years.
Levy, G. et al. Combined effect of age and severity on the risk of dementia in Parkinson's disease. Ann. Neurol. 51, 722–729 (2002).
Williams-Gray, C. H. et al. The distinct cognitive syndromes of Parkinson's disease: 5 year follow-up of the CamPaIGN cohort. Brain 132, 2958–2969 (2009).
Muslimovic, D., Post, B., Speelman, J. D. & Schmand, B. Cognitive profile of patients with newly diagnosed Parkinson disease. Neurology 65, 1239–1245 (2005).
Rosenthal, E. et al. Association between cognition and function in patients with Parkinson disease with and without dementia. Mov. Disord. 25, 1170–1176 (2010).
Lo, R. Y. et al. Clinical features in early Parkinson disease and survival. Arch. Neurol. 66, 1353–1358 (2009).
Litvan, I. et al. Diagnostic criteria for mild cognitive impairment in Parkinson's disease: Movement Disorder Society Task Force guidelines. Mov. Disord. 27, 349–356 (2012). The Movement Disorder Society Task Force criteria for PD-MCI.
Grossman, M. et al. Difficulty processing temporary syntactic ambiguities in Lewy body spectrum disorder. Brain Lang. 120, 52–60 (2012).
Lippa, C. F. et al. DLB and PDD boundary issues: diagnosis, treatment, molecular pathology, and biomarkers. Neurology 68, 812–819 (2007). An in-depth discussion of the clinicopathological overlap between PDD and DLB.
Galvin, J. E., Pollack, J. & Morris, J. C. Clinical phenotype of Parkinson disease dementia. Neurology 67, 1605–1611 (2006).
McKeith, I. G. Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): report of the Consortium on DLB International Workshop. J. Alzheimers Dis. 9, 417–423 (2006). In this paper, the DLB Consortium outlines its clinical and neuropathological diagnostic criteria for DLB.
Weintraub, D. Dopamine and impulse control disorders in Parkinson's disease. Ann. Neurol. 64 (Suppl. 2), 93–100 (2008).
Weintraub, D., Papay, K., Siderowf, A. & Parkinson's Progression Markers Initiative. Screening for impulse control symptoms in patients with de novo Parkinson disease: a case-control study. Neurology 80, 176–180 (2013).
Schrag, A., Ben-Shlomo, Y., Brown, R., Marsden, C. D. & Quinn, N. Young-onset Parkinson's disease revisited — clinical features, natural history, and mortality. Mov. Disord. 13, 885–894 (1998).
Aarsland, D. et al. The effect of age of onset of PD on risk of dementia. J. Neurol. 254, 38–45 (2007).
Braak, H., Rub, U., Jansen Steur, E. N., Del Tredici, K. & de Vos, R. A. Cognitive status correlates with neuropathologic stage in Parkinson disease. Neurology 64, 1404–1410 (2005).
Levy, G. et al. Motor impairment in PD: relationship to incident dementia and age. Neurology 55, 539–544 (2000).
Jankovic, J. et al. Variable expression of Parkinson's disease: a base-line analysis of the DATATOP cohort. The Parkinson Study Group. Neurology 40, 1529–1534 (1990).
Levy, G. et al. Memory and executive function impairment predict dementia in Parkinson's disease. Mov. Disord. 17, 1221–1226 (2002).
Polymeropoulos, M. H. et al. Mutation in the α-synuclein gene identified in families with Parkinson's disease. Science 276, 2045–2047 (1997). This study made the landmark discovery of SNCA mutations in PD.
Poulopoulos, M., Levy, O. A. & Alcalay, R. N. The neuropathology of genetic Parkinson's disease. Mov. Disord. 27, 831–842 (2012).
Singleton, A. B. et al. α-synuclein locus triplication causes Parkinson's disease. Science 302, 841 (2003).
Conway, K. A., Harper, J. D. & Lansbury, P. T. Accelerated in vitro fibril formation by a mutant α-synuclein linked to early-onset Parkinson disease. Nature Med. 4, 1318–1320 (1998).
Spillantini, M. G. et al. α-Synuclein in Lewy bodies. Nature 388, 839–840 (1997). This study made the landmark discovery of α-syn as the major component of Lewy pathology.
Giasson, B. I. et al. Neuronal α-synucleinopathy with severe movement disorder in mice expressing A53T human α-synuclein. Neuron 34, 521–533 (2002).
Lim, Y. et al. α-Syn suppression reverses synaptic and memory defects in a mouse model of dementia with Lewy bodies. J. Neurosci. 31, 10076–10087 (2011).
Magen, I. & Chesselet, M. F. Genetic mouse models of Parkinson's disease: the state of the art. Prog. Brain Res. 184, 53–87 (2010).
Braak, H. et al. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol. Aging 24, 197–211 (2003).
Del Tredici, K., Rub, U., De Vos, R. A., Bohl, J. R. & Braak, H. Where does parkinson disease pathology begin in the brain? J. Neuropathol. Exp. Neurol. 61, 413–426 (2002).
Dickson, D. W., Uchikado, H., Fujishiro, H. & Tsuboi, Y. Evidence in favor of Braak staging of Parkinson's disease. Mov. Disord. 25 (Suppl. 1), 78–82 (2010).
Jellinger, K. A. Lewy body-related α-synucleinopathy in the aged human brain. J. Neural Transm. 111, 1219–1235 (2004).
Parkkinen, L., Pirttila, T. & Alafuzoff, I. Applicability of current staging/categorization of α-synuclein pathology and their clinical relevance. Acta Neuropathol. 115, 399–407 (2008).
Beach, T. G. et al. Unified staging system for Lewy body disorders: correlation with nigrostriatal degeneration, cognitive impairment and motor dysfunction. Acta Neuropathol. 117, 613–634 (2009).
Mikolaenko, I. et al. α-synuclein lesions in normal aging, Parkinson disease, and Alzheimer disease: evidence from the Baltimore Longitudinal Study of Aging (BLSA). J. Neuropathol. Exp. Neurol. 64, 156–162 (2005).
Parkkinen, L., Kauppinen, T., Pirttila, T., Autere, J. M. & Alafuzoff, I. α-synuclein pathology does not predict extrapyramidal symptoms or dementia. Ann. Neurol. 57, 82–91 (2005).
Dickson, D. W. et al. Evidence that incidental Lewy body disease is pre-symptomatic Parkinson's disease. Acta Neuropathol. 115, 437–444 (2008).
Saito, Y. et al. Accumulation of phosphorylated α-synuclein in aging human brain. J. Neuropathol. Exp. Neurol. 62, 644–654 (2003).
Wakabayashi, K. et al. The Lewy body in Parkinson's disease and related neurodegenerative disorders. Mol. Neurobiol. 47, 495–508 (2013).
Milber, J. M. et al. Lewy pathology is not the first sign of degeneration in vulnerable neurons in Parkinson disease. Neurology 79, 2307–2314 (2012).
Parkkinen, L. et al. Disentangling the relationship between Lewy bodies and nigral neuronal loss in Parkinson's disease. J. Park Dis. 1, 277–286 (2011).
Braak, H. et al. Pathology associated with sporadic Parkinson's disease — where does it end? J. Neural Transm. Suppl. 70, 89–97 (2006).
Hamilton, R. L. Lewy bodies in Alzheimer's disease: a neuropathological review of 145 cases using α-synuclein immunohistochemistry. Brain Pathol. 10, 378–384 (2000).
Lippa, C. F. et al. Lewy bodies contain altered α-synuclein in brains of many familial Alzheimer's disease patients with mutations in presenilin and amyloid precursor protein genes. Am. J. Pathol. 153, 1365–1370 (1998).
Iseki, E. Dementia with Lewy bodies: reclassification of pathological subtypes and boundary with Parkinson's disease or Alzheimer's disease. Neuropathology 24, 72–78 (2004).
Leverenz, J. B. et al. Empiric refinement of the pathologic assessment of Lewy-related pathology in the dementia patient. Brain Pathol. 18, 220–224 (2008).
Jellinger, K. A. A critical reappraisal of current staging of Lewy-related pathology in human brain. Acta Neuropathol. 116, 1–16 (2008).
Li, J. Y. et al. Lewy bodies in grafted neurons in subjects with Parkinson's disease suggest host-to-graft disease propagation. Nature Med. 14, 501–503 (2008).
Kordower, J. H., Chu, Y., Hauser, R. A., Freeman, T. B. & Olanow, C. W. Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson's disease. Nature Med. 14, 504–506 (2008).
Kordower, J. H., Chu, Y., Hauser, R. A., Olanow, C. W. & Freeman, T. B. Transplanted dopaminergic neurons develop PD pathologic changes: a second case report. Mov. Disord. 23, 2303–2306 (2008).
Luk, K. C. et al. Exogenous α-synuclein fibrils seed the formation of Lewy body-like intracellular inclusions in cultured cells. Proc. Natl Acad. Sci. USA 106, 20051–20056 (2009).
Volpicelli-Daley, L. A. et al. Exogenous α-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron 72, 57–71 (2011).
Luk, K. C. et al. Intracerebral inoculation of pathological α-synuclein initiates a rapidly progressive neurodegenerative α-synucleinopathy in mice. J. Exp. Med. 209, 975–986 (2012).
Luk, K. C. et al. Pathological α-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 338, 949–953 (2012). This study made the landmark discovery of how the transmission of α-syn fibrils alone recapitulates human disease in wild-type animals.
Lee, H. J. et al. Assembly-dependent endocytosis and clearance of extracellular α-synuclein. Int. J. Biochem. Cell Biol. 40, 1835–1849 (2008).
Desplats, P. et al. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of α-synuclein. Proc. Natl Acad. Sci. USA 106, 13010–13015 (2009).
Mougenot, A. L. et al. Prion-like acceleration of a synucleinopathy in a transgenic mouse model. Neurobiol. Aging 33, 2225–2228 (2012).
Masuda-Suzukake, M. et al. Prion-like spreading of pathological α-synuclein in brain. Brain 136, 1128–1138 (2013).
Steiner, J. A., Angot, E. & Brundin, P. A deadly spread: cellular mechanisms of α-synuclein transfer. Cell Death Differ. 18, 1425–1433 (2011).
Olanow, C. W. & Brundin, P. Parkinson's disease and alpha synuclein: is Parkinson's disease a prion-like disorder? Mov. Disord. 28, 31–40 (2013). A timely review of PD model transmission studies.
Lee, V. M. & Trojanowski, J. Q. Mechanisms of Parkinson's disease linked to pathological α-synuclein: new targets for drug discovery. Neuron 52, 33–38 (2006).
Irwin, D. J. et al. Evaluation of potential infectivity of Alzheimer's and Parkinson's disease proteins in recipients of cadaver-derived human growth hormone. JAMA Neurol. 70, 462–468 (2013).
Hurtig, H. I. et al. Alpha-synuclein cortical Lewy bodies correlate with dementia in Parkinson's disease. Neurology 54, 1916–1921 (2000).
Duda, J. E., Giasson, B. I., Mabon, M. E., Lee, V. M. & Trojanowski, J. Q. Novel antibodies to synuclein show abundant striatal pathology in Lewy body diseases. Ann. Neurol. 52, 205–210 (2002).
Apaydin, H., Ahlskog, J. E., Parisi, J. E., Boeve, B. F. & Dickson, D. W. Parkinson disease neuropathology: later-developing dementia and loss of the levodopa response. Arch. Neurol. 59, 102–112 (2002).
Irwin, D. J. et al. Neuropathologic substrates of Parkinson disease dementia. Ann. Neurol. 72, 587–598 (2012). A large autopsy cohort involving multivariate analysis of multiple clinical, genetic and neuropathological variables that implicate Lewy body and neurite pathology as the strongest correlate of PDD.
Compta, Y. et al. Lewy- and Alzheimer-type pathologies in Parkinson's disease dementia: which is more important? Brain. 134, 1493–1505 (2011).
Tsuboi, Y., Uchikado, H. & Dickson, D. W. Neuropathology of Parkinson's disease dementia and dementia with Lewy bodies with reference to striatal pathology. Parkinsonism Relat. Disord. 13 (Suppl. 3), 221–224 (2007).
Jellinger, K. A. & Attems, J. Prevalence and impact of vascular and Alzheimer pathologies in Lewy body disease. Acta Neuropathol. 115, 427–436 (2008).
Harding, A. J. & Halliday, G. M. Cortical Lewy body pathology in the diagnosis of dementia. Acta Neuropathol. 102, 355–363 (2001).
Kovari, E. et al. Lewy body densities in the entorhinal and anterior cingulate cortex predict cognitive deficits in Parkinson's disease. Acta Neuropathol. 106, 83–88 (2003).
Mattila, P. M., Rinne, J. O., Helenius, H., Dickson, D. W. & Roytta, M. α-synuclein-immunoreactive cortical Lewy bodies are associated with cognitive impairment in Parkinson's disease. Acta Neuropathol. 100, 285–290 (2000).
Pletnikova, O. et al. Aβ deposition is associated with enhanced cortical α-synuclein lesions in Lewy body diseases. Neurobiol. Aging 26, 1183–1192 (2005).
Ballard, C. et al. Differences in neuropathologic characteristics across the Lewy body dementia spectrum. Neurology 67, 1931–1934 (2006).
Perry, E. K. et al. Cholinergic correlates of cognitive impairment in Parkinson's disease: comparisons with Alzheimer's disease. J. Neurol. Neurosurg. Psychiatry 48, 413–421 (1985).
Whitehouse, P. J., Hedreen, J. C., White, C. L., & Price, D. L. Basal forebrain neurons in the dementia of Parkinson disease. Ann. Neurol. 13, 243–248 (1983).
Kalaitzakis, M. E., Graeber, M. B., Gentleman, S. M. & Pearce, R. K. Striatal β-amyloid deposition in Parkinson disease with dementia. J. Neuropathol. Exp. Neurol. 67, 155–161 (2008).
Jellinger, K. A. Morphological substrates of parkinsonism with and without dementia: a retrospective clinico-pathological study. J. Neural. Transm. Suppl. 72, 91–104 (2007).
Jellinger, K. A., Seppi, K., Wenning, G. K. & Poewe, W. Impact of coexistent Alzheimer pathology on the natural history of Parkinson's disease. J. Neural Transm. 109, 329–339 (2002).
Kotzbauer, P. T. et al. Pathologic accumulation of α-synuclein and Aβ in Parkinson disease patients with dementia. Arch. Neurol. 69, 1326–1331 (2012).
Hughes, A. J., Daniel, S. E., Blankson, S. & Lees, A. J. A clinicopathologic study of 100 cases of Parkinson's disease. Arch. Neurol. 50, 140–148 (1993).
Sabbagh, M. N. et al. Parkinson disease with dementia: comparing patients with and without Alzheimer pathology. Alzheimer Dis. Assoc. Disord. 23, 295–297 (2009).
Lashley, T. et al. Cortical α-synuclein load is associated with amyloid-β plaque burden in a subset of Parkinson's disease patients. Acta Neuropathol. 115, 417–425 (2008).
Masliah, E. et al. β-amyloid peptides enhance α-synuclein accumulation and neuronal deficits in a transgenic mouse model linking Alzheimer's disease and Parkinson's disease. Proc. Natl Acad. Sci. USA 98, 12245–12250 (2001).
Clinton, L. K., Blurton-Jones, M., Myczek, K., Trojanowski, J. Q. & LaFerla, F. M. Synergistic interactions between Aβ, tau, and α-synuclein: acceleration of neuropathology and cognitive decline. J. Neurosci. 30, 7281–7289 (2010).
Duda, J. E. et al. Concurrence of α-synuclein and tau brain pathology in the Contursi kindred. Acta Neuropathol. 104, 7–11 (2002).
Lee, V. M., Giasson, B. I. & Trojanowski, J. Q. More than just two peas in a pod: common amyloidogenic properties of tau and α-synuclein in neurodegenerative diseases. Trends Neurosci. 27, 129–134 (2004).
Giasson, B. I. et al. Initiation and synergistic fibrillization of tau and α-synuclein. Science 300, 636–640 (2003).
Jellinger, K. A., Wenning, G. K. & Seppi, K. Predictors of survival in dementia with lewy bodies and Parkinson dementia. Neurodegener. Dis. 4, 428–430 (2007).
Sabbagh, M. N. et al. Correlation of clinical features with argyrophilic grains at autopsy. Alzheimer Dis. Assoc. Disord. 23, 229–233 (2009).
Nakashima-Yasuda, H. et al. Co-morbidity of TDP-43 proteinopathy in Lewy body related diseases. Acta Neuropathol. 114, 221–229 (2007).
Colosimo, C., Hughes, A. J., Kilford, L. & Lees, A. J. Lewy body cortical involvement may not always predict dementia in Parkinson's disease. J. Neurol. Neurosurg. Psychiatry 74, 852–856 (2003).
Richard, I. H., Papka, M., Rubio, A. & Kurlan, R. Parkinson's disease and dementia with Lewy bodies: one disease or two? Mov. Disord. 17, 1161–1165 (2002).
Jellinger, K. A. & Attems, J. Does striatal pathology distinguish Parkinson disease with dementia and dementia with Lewy bodies? Acta Neuropathol. 112, 253–260 (2006).
Halliday, G. M., Song, Y. J. & Harding, A. J. Striatal β-amyloid in dementia with Lewy bodies but not Parkinson's disease. J. Neural Transm. 118, 713–719 (2011).
Merdes, A. R. et al. Influence of Alzheimer pathology on clinical diagnostic accuracy in dementia with Lewy bodies. Neurology 60, 1586–1590 (2003).
Verghese, J., Crystal, H. A., Dickson, D. W. & Lipton, R. B. Validity of clinical criteria for the diagnosis of dementia with Lewy bodies. Neurology 53, 1974–1982 (1999).
Litvan, I. et al. Accuracy of the clinical diagnoses of Lewy body disease, Parkinson disease, and dementia with Lewy bodies: a clinicopathologic study. Arch. Neurol. 55, 969–978 (1998).
Nelson, P. T. et al. Low sensitivity in clinical diagnoses of dementia with Lewy bodies. J. Neurol. 257, 359–366 (2010).
Kraybill, M. L. et al. Cognitive differences in dementia patients with autopsy-verified AD, Lewy body pathology, or both. Neurology 64, 2069–2073 (2005).
van Rooden, S. M. et al. The identification of Parkinson's disease subtypes using cluster analysis: a systematic review. Mov. Disord. 25, 969–978 (2010).
Selikhova, M. et al. A clinico-pathological study of subtypes in Parkinson's disease. Brain 132, 2947–2957 (2009).
Alves, G., Larsen, J. P., Emre, M., Wentzel-Larsen, T. & Aarsland, D. Changes in motor subtype and risk for incident dementia in Parkinson's disease. Mov. Disord. 21, 1123–1130 (2006).
Prikrylova Vranova, H. et al. CSF markers of neurodegeneration in Parkinson's disease. J. Neural Transm. 117, 1177–1181 (2010).
Alves, G. et al. Cerebrospinal fluid amyloid-β and phenotypic heterogeneity in de novo Parkinson's disease. J. Neurol. Neurosurg. Psychiatry 85, 537–543 (2012).
Kang, J. H. et al. Association of cerebrospinal fluid Aβ1-42, t-tau, p-tau181 and α-synuclein levels with clinical 1 features of early drug naïve Parkinson's disease patients. JAMA Neurol. (in the press).
Müller, M. L. et al. β-amyloid and postural instability and gait difficulty in Parkinson's disease at risk for dementia. Mov. Disord. 28, 296–301 (2012).
Halliday, G. M., Holton, J. L., Revesz, T. & Dickson, D. W. Neuropathology underlying clinical variability in patients with synucleinopathies. Acta Neuropathol. 122, 187–204 (2011).
Svenningsson, P., Westman, E., Ballard, C. & Aarsland, D. Cognitive impairment in patients with Parkinson's disease: diagnosis, biomarkers, and treatment. Lancet Neurol. 11, 697–707 (2012).
Sidransky, E. et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson's disease. N. Engl. J. Med. 361, 1651–1661 (2009).
Tsuang, D. et al. GBA mutations increase risk for Lewy body disease with and without Alzheimer disease pathology. Neurology 79, 1944–1950 (2012).
Neumann, J. et al. Glucocerebrosidase mutations in clinical and pathologically proven Parkinson's disease. Brain 132, 1783–1794 (2009).
Nalls, M. A. et al. A multicenter study of glucocerebrosidase mutations in dementia with lewy bodies. JAMA Neurol. 70, 727–735 (2013).
Clark, L. N. et al. Association of glucocerebrosidase mutations with dementia with lewy bodies. Arch. Neurol. 66, 578–583 (2009).
Morley, J. F. et al. Genetic influences on cognitive decline in Parkinson's disease. Mov. Disord. 27, 512–518 (2012).
Tsuang, D. et al. APOE ε4 increases risk for dementia in pure synucleinopathies. JAMA Neurol. 70, 223–228 (2013).
Williams-Gray, C. H. et al. Apolipoprotein E genotype as a risk factor for susceptibility to and dementia in Parkinson's disease. J. Neurol. 256, 493–498 (2009).
Wakabayashi, K. et al. Apolipoprotein E ε4 allele and progression of cortical Lewy body pathology in Parkinson's disease. Acta Neuropathol. 95, 450–454 (1998).
Siderowf, A. et al. CSF amyloid β1–42 predicts cognitive decline in Parkinson disease. Neurology 75, 1055–1061 (2010).
Baker, M. et al. Association of an extended haplotype in the tau gene with progressive supranuclear palsy. Hum. Mol. Genet. 8, 711–715 (1999).
Zabetian, C. P. et al. Association analysis of MAPT H1 haplotype and subhaplotypes in Parkinson's disease. Ann. Neurol. 62, 137–144 (2007).
Goris, A. et al. Tau and α-synuclein in susceptibility to, and dementia in, Parkinson's disease. Ann. Neurol. 62, 145–153 (2007).
Compta, Y. et al. High cerebrospinal tau levels are associated with the rs242557 tau gene variant and low cerebrospinal β-amyloid in Parkinson disease. Neurosci. Lett. 487, 169–173 (2011).
Chen-Plotkin, A. S. et al. Plasma epidermal growth factor levels predict cognitive decline in Parkinson disease. Ann. Neurol. 69, 655–663 (2011).
Pellecchia, M. T. et al. Serum epidermal growth factor predicts cognitive functions in early, drug-naive Parkinson's disease patients. J. Neurol. 260, 438–444 (2012).
Compta, Y. et al. Cerebrospinal tau, phospho-tau, and β-amyloid and neuropsychological functions in Parkinson's disease. Mov. Disord. 24, 2203–2210 (2009).
Hall, S. et al. Accuracy of a panel of 5 cerebrospinal fluid biomarkers in the differential diagnosis of patients with dementia and/or parkinsonian disorders. JAMA Neurol. 69, 1445–1452 (2012).
Alves, G. et al. CSF amyloid-β and tau proteins, and cognitive performance, in early and untreated Parkinson's disease: the Norwegian ParkWest study. J. Neurol. Neurosurg. Psychiatry 81, 1080–1086 (2010).
Montine, T. J. et al. CSF Aβ42 and tau in Parkinson's disease with cognitive impairment. Mov. Disord. 25, 2682–2685 (2010).
Leverenz, J. B. et al. Cerebrospinal fluid biomarkers and cognitive performance in non-demented patients with Parkinson's disease. Parkinsonism Relat. Disord. 17, 61–64 (2011).
Mollenhauer, B. et al. Direct quantification of CSF α-synuclein by ELISA and first cross-sectional study in patients with neurodegeneration. Exp. Neurol. 213, 315–325 (2008).
Wang, Y. et al. Phosphorylated α-synuclein in Parkinson's disease. Sci. Transl. Med. 4, 121ra20 (2012).
Tokuda, T. et al. Detection of elevated levels of α-synuclein oligomers in CSF from patients with Parkinson disease. Neurology 75, 1766–1772 (2010).
Hong, Z. et al. DJ-1 and α-synuclein in human cerebrospinal fluid as biomarkers of Parkinson's disease. Brain 133, 713–726 (2010).
Shi, M. et al. Cerebrospinal fluid biomarkers for Parkinson disease diagnosis and progression. Ann. Neurol. 69, 570–580 (2011).
Compta, Y. et al. Grey matter volume correlates of cerebrospinal markers of Alzheimer-pathology in Parkinson's disease and related dementia. Parkinsonism Relat. Disord. 18, 941–947 (2012).
Weintraub, D. et al. Alzheimer's disease pattern of brain atrophy predicts cognitive decline in Parkinson's disease. Brain 135, 170–180 (2011).
Gomperts, S. N. et al. Amyloid is linked to cognitive decline in patients with Parkinson disease without dementia. Neurology 80, 85–91 (2013).
Petrou, M. et al. Aβ-amyloid deposition in patients with Parkinson disease at risk for development of dementia. Neurology 79, 1161–1167 (2012).
Bohnen, N. I. et al. Cerebral glucose metabolic features of Parkinson disease and incident dementia: longitudinal study. J. Nucl. Med. 52, 848–855 (2011).
Beyer, M. K. et al. Cerebrospinal fluid Aβ levels correlate with structural brain changes in Parkinson's disease. Mov. Disord. 28, 302–310 (2013).
Marek, K. et al. The Parkinson Progression Marker Initiative (PPMI). Prog. Neurobiol. 95, 629–635 (2011).
Rolinski, M., Fox, C., Maidment, I. & McShane, R. Cholinesterase inhibitors for dementia with Lewy bodies, Parkinson's disease dementia and cognitive impairment in Parkinson's disease. Cochrane Database Syst. Rev. 3, CD006504 (2012).
Seppi, K. et al. The movement disorder society evidence-based medicine review update: treatments for the non-motor symptoms of Parkinson's disease. Mov. Disord. 26 (Suppl. 3), 42–80 (2011).
Lashuel, H. A., Overk, C. R., Oueslati, A. & Masliah, E. The many faces of α-synuclein: from structure and toxicity to therapeutic target. Nature Rev. Neurosci. 14, 38–48 (2013). A recent review highlighting the pathophysiology of α-syn toxicity in PD and potential therapeutic strategies.
Valera, E. & Masliah, E. Immunotherapy for neurodegenerative diseases: focus on α-synucleinopathies. Pharmacol. Ther. 138, 311–322 (2013).
Sperling, R. A. et al. Toward defining the preclinical stages of Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 7, 280–292 (2011).
Montine, T. J. et al. National Institute on Aging-Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease: a practical approach. Acta Neuropathol. 123, 1–11 (2012).
Ward, C. D. & Gibb, W. R. Research diagnostic criteria for Parkinson's disease. Adv. Neurol. 53, 245–249 (1990).
Prusiner, S. B. Novel proteinaceous infectious particles cause scrapie. Science 216, 136–144 (1982).
Kordower, J. H. & Brundin, P. Propagation of host disease to grafted neurons: accumulating evidence. Exp. Neurol. 220, 224–225 (2009).
Brown, P. et al. Human spongiform encephalopathy: the National Institutes of Health series of 300 cases of experimentally transmitted disease. Ann. Neurol. 35, 513–529 (1994).
Brown, P., Gajdusek, D. C., Gibbs, C. J. Jr & Asher, D. M. Potential epidemic of Creutzfeldt–Jakob disease from human growth hormone therapy. N. Engl. J. Med. 313, 728–731 (1985).
Acknowledgements
We thank the patients and their families who have contributed to the research reviewed here, which has led to meaningful developments in our understanding of Parkinson's disease and related disorders. Funding for our research was provided by the US National Institutes of Health grants P30 AG10124, AG17586, P50 NS53488 and T32-AG000255.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
PowerPoint slides
Glossary
- Executive functioning
-
Abilities in mental flexibility, planning and working memory that are mediated by striatal–frontal networks.
- Semantic memory
-
Memory for the meaning and context of objects and concepts that is mediated by the temporal lobe and its connections throughout the neocortex.
- Mild cognitive impairment
-
(MCI). MCI comprises subjective cognitive complaints with objective findings of cognitive impairment in one or more cognitive domains that does not interfere with the patient's ability to perform tasks of daily living. MCI is thought to represent a prodromal state to Alzheimer's disease and other dementias. Recently, clinical criteria have been defined for MCI in the setting of Parkinson's disease.
- Bradykinesia
-
Symptoms of slowed movement seen in Parkinson's disease and other disorders involving nigral–striatal dopaminergic pathways.
- Constructional praxis
-
The ability to draw or copy a figure (such as clock-drawing or drawing intersecting pentagons), which relies on attention, planning and organization skills (executive function) and visuospatial perceptual abilities.
- Verbal memory
-
Short-term memory for words and verbal information that is partially mediated by language function (for example, memory for words tested through a list-learning task).
- Transmission
-
The spread of a pathological protein in an altered conformation (for example, PrPSc) between neurons within an individual; transmission does not necessarily imply that the disease protein is infectious (that is, it can be spread between individuals).
- Amyloid fibrils
-
Insoluble filamentous structures composed of polymerized protein monomers with notable β-sheet conformation, which can be detected with amyloid-binding dyes (for example, thioflavin S).
- Cognitive reserve
-
This refers to the notion of relative resistance to clinical symptoms of neurodegeneration and other CNS insults that is thought to be mediated by neuroplasticity or an ability to recruit additional brain networks to compensate for the disease state; such plasticity may be influenced by education or other environmental or genetic factors.
- Cerebrovascular disease
-
(CVD). Damage to intracerebral blood vessels from atherosclerosis and lipohyalinosis, which are caused by systemic cardiovascular risk factors (for example, hypertension, diabetes and hyperlipidaemia) and result in ischaemic damage to the brain parenchyma (for example, lacunar infarcts).
- Amyloid angiopathy
-
A form of cerebral vasculopathy that is caused by fibrillar amyloid-β deposition in blood vessel walls.
- Tauopathies
-
A family of neurodegenerative disease proteinopathies that are characterized by inclusions composed primarily of the microtubule-associated protein tau.
Rights and permissions
About this article
Cite this article
Irwin, D., Lee, VY. & Trojanowski, J. Parkinson's disease dementia: convergence of α-synuclein, tau and amyloid-β pathologies. Nat Rev Neurosci 14, 626–636 (2013). https://doi.org/10.1038/nrn3549
Published:
Issue Date:
DOI: https://doi.org/10.1038/nrn3549
This article is cited by
-
Glypican-4 serum levels are associated with cognitive dysfunction and vascular risk factors in Parkinson’s disease
Scientific Reports (2024)
-
Liquid–liquid phase separation in Alzheimer’s disease
Journal of Molecular Medicine (2024)
-
The cervical lymph node contributes to peripheral inflammation related to Parkinson’s disease
Journal of Neuroinflammation (2023)
-
Role of dopamine in the pathophysiology of Parkinson’s disease
Translational Neurodegeneration (2023)
-
Translational molecular imaging and drug development in Parkinson’s disease
Molecular Neurodegeneration (2023)
