
Key Points
-
An imbalance between the production and clearance of amyloid-β (Aβ) is an early, often initiating, factor in Alzheimer disease (AD)
-
Peripheral systems are suggested to be involved in Aβ production and clearance
-
The central and peripheral pathways of Aβ metabolism communicate with each other, and work synergistically to clear Aβ from the brain
-
Increasing experimental, epidemiologic and clinical evidence suggests that AD manifestations extend beyond the brain, and that AD pathogenesis is closely associated with systemic abnormalities
-
The systemic abnormalities in patients with AD might not be secondary to the cerebral degeneration; instead, they might reflect underlying disease processes
-
A systemic view of AD provides a novel perspective for understanding the role of Aβ in AD pathogenesis and offers opportunities for the development of new treatments and diagnostic biomarkers for AD
Abstract
Alzheimer disease (AD) is the most common type of dementia, and is currently incurable; existing treatments for AD produce only a modest amelioration of symptoms. Research into this disease has conventionally focused on the CNS. However, several peripheral and systemic abnormalities are now understood to be linked to AD, and our understanding of how these alterations contribute to AD is becoming more clearly defined. This Review focuses on amyloid-β (Aβ), a major hallmark of AD. We review emerging findings of associations between systemic abnormalities and Aβ metabolism, and describe how these associations might interact with or reflect on the central pathways of Aβ production and clearance. On the basis of these findings, we propose that these abnormal systemic changes might not only develop secondary to brain dysfunction but might also affect AD progression, suggesting that the interactions between the brain and the periphery have a crucial role in the development and progression of AD. Such a systemic view of the molecular pathogenesis of AD could provide a novel perspective for understanding this disease and present new opportunities for its early diagnosis and treatment.
Similar content being viewed by others


Main
The global burden of Alzheimer disease (AD), already the most common type of dementia, is expected to increase still further owing to population ageing. AD not only causes severe distress for patients and caregivers, but also results in a large economic burden on society. Current major challenges in AD include the lack of reliable biomarkers for its early diagnosis, as well as the lack of effective preventive strategies and treatments1,2. Thus, increased understanding of the molecular pathogenesis of AD could lead to the development of improved diagnostic and therapeutic strategies.
AD is conventionally regarded as a CNS disorder. However, increasing experimental, epidemiological and clinical evidence has suggested that manifestations of AD extend beyond the brain. These systemic alterations might not be simply secondary effects of the cerebral degeneration seen in AD, but could reflect underlying processes linked to progression of the disease. AD pathogenesis is complex, involving abnormal amyloid-β (Aβ) metabolism, tau hyperphosphorylation, oxidative stress, reactive glial and microglial changes, and other pathological events. Given that Aβ is a major hallmark of AD and a fertile area of research in this disease, this Review focuses on the systemic role of Aβ in AD. We discuss the communication between peripheral and central pools of Aβ, and describe interactions between systemic abnormalities and AD pathogenesis in the brain. We review emerging findings of associations between systemic abnormalities and Aβ metabolism, and describe how these associations might interact with or reflect on the central pathways of Aβ production and clearance. On the basis of these findings, we suggest that interactions between the brain and the periphery might have a crucial role in the development and progression of AD.
Aβ biogenesis and catabolism
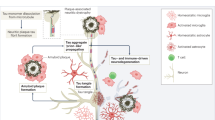
A steady accrual of data from laboratories and clinics is providing increasing support for the concept that an imbalance between the production and clearance of Aβ is a very early (and often initiating) factor in AD3. Normal metabolism of Aβ and maintenance of the homeostatic balance between Aβ production and clearance is, therefore, essential to maintain brain health. In fact, physiological metabolism of Aβ occurs not only in the brain but also in the periphery, and communication between these regions is possible (Fig. 1).

Amyloid-β (Aβ) is generated by neurons, microglia and astrocytes in the brain, and by platelets, skin fibroblasts, osteoblasts, and skeletal muscle cells in the periphery. The CNS and peripheral pools of Aβ can interact; some Aβ peptides in the CNS are cleared via phagocytosis or proteolytic degradation, whereas others are released into the blood via the blood–brain barrier (BBB), interstitial fluid (ISF) bulk flow or cerebrospinal fluid (CSF) egress pathways. Some Aβ peptides in blood are phagocytosed, including by monocytes or neutrophils, some are degraded by Aβ-degrading enzymes, and some are transported by carriers (such as erythrocytes, albumin and lipoproteins) to peripheral organs or tissues, where they are degraded by macrophages or hepatocytes, or excreted via the liver or kidney. BCSFB, blood–CSF barrier; RAGE, receptor for advanced glycation end products; RBC, red blood cell.
Central and peripheral production of Aβ
Aβ is derived from the proteolytic cleavage of amyloid precursor protein (APP), which is expressed not only in brain cells, including neurons, astrocytes and microglia, but also in peripheral organs and tissues, such as the adrenal gland, kidney, heart, liver, spleen, pancreas, muscles, and various blood and endothelial cells4,5. Aβ levels (in both peripheral tissues and the brain) are known to be lower in cognitively normal elderly individuals than in patients with AD (Table 1). The accumulation of Aβ aggregates in elderly patients with and without AD is an important factor that could influence the ratio of Aβ42 to Aβ40 in both brain and periphery. Given that skeletal muscle represents about one-quarter of body weight in humans and is just one of many peripheral sources of Aβ, peripherally derived Aβ is likely to represent a substantial proportion of the total. However, levels and profiles of the dominant Aβ species in brain and periphery still need to be measured in young people without Aβ deposition in future studies, for comparison purposes.
Important differences have been found between the central and peripheral pools of Aβ. First, Aβ42, which is the most aggregation-prone and most neurotoxic form of Aβ, is the dominant molecular species in the brain, whereas Aβ40 is dominant in the periphery, although the mechanism underlying this difference is still unclear. Differential expression of APP isoforms in the brain and periphery is one possible explanation. APP695 is the dominant species produced by neurons, whereas APP751 and APP770 are the dominant species produced by peripheral cells, including platelets and leukocytes6. Another possible explanation is that the tissue microenvironment differs between the CNS and periphery, which could result in differential processing of APP by γ-secretase and generation of different Aβ species4,5. Second, levels of Aβ in the central pool are higher than those in the peripheral pool. Concentrations of Aβ in cerebrospinal fluid (CSF) are at least 5–15 times higher than those in plasma7,8. One possible interpretation is that APP processing in peripheral cells probably occurs via α-cleavage (rather than the β-cleavage used in neurons)9,10, resulting in decreased peripheral production of Aβ. Another explanation for the lower Aβ levels in the periphery is that the periphery contains abundant Aβ-binding proteins (lipoproteins and albumin)11 and Aβ-binding cells, such as erythrocytes12, which all contribute to Aβ transportation and clearance. Additionally, the high volume of the circulatory system and the blood-dilution effect efficiently reduce systemic Aβ concentrations.
These factors might also help to explain why Aβ aggregates are mainly deposited in the brain and cerebral vessel walls, and only rarely in peripheral organs (although detection of Aβ aggregates has been claimed in skin, subcutaneous tissue, intestinal tissues, and heart)13,14,15. The aggregation (oligomerization and fibrillogenesis) of Aβ peptides is determined by the relative proportions of Aβ species, their concentrations, the pH, temperature and ionic strength of solution, and incubation time16. In the periphery, therefore, an increased proportion of Aβ40 might lead to sequestration of Aβ42 in a stable mixed formation, thereby preventing its oligomerization and aggregation17, whereas an increased proportion of Aβ42 in the brain might render Aβ42 susceptible to aggregation.
Central and peripheral clearance of Aβ
The failure to clear Aβ (especially Aβ42) is an important cause of sporadic AD, which accounts for 99% of AD cases. Understanding how Aβ is physiologically cleared from the brain is, therefore, essential. Several potential pathways could clear Aβ from the brain: phagocytosis, endocytosis and macropinocytosis by various cells (such as microglia, perivascular macrophages, astrocytes, oligodendroglia and neurons); proteolytic degradation by various enzymes (including neprilysin, insulin-degrading enzyme (IDE) and matrix metalloproteinases); and efflux of Aβ to the peripheral circulation, via transportation across the blood–brain barrier (BBB) and blood–CSF barrier, interstitial fluid bulk flow and CSF egress pathways, including arachnoid villi and glymphatic–lymphatic pathways18 (Fig. 1). Some endogenous inhibitors of Aβ aggregation, such as the secreted ectodomain of tumour necrosis factor receptor superfamily member 16 (also known as low affinity neurotrophin receptor p75NTR)19 and the N-terminal domain of myelin basic protein20, prevent Aβ deposition in the brain and facilitate its efflux into the circulation.
How Aβ is cleared in the periphery is poorly understood. Previous studies have suggested that ∼60% of brain Aβ is cleared via transportation to the periphery21,22. Our group has demonstrated, in a mouse model of AD, that brain-derived Aβ can be physiologically cleared in the periphery, and that a singular peripheral system can remove ∼40% of the Aβ produced in the brain23. These findings indicate that peripheral clearance has a crucial role in removing brain-derived Aβ, and suggest that effective peripheral Aβ clearance can improve the efficacy of Aβ efflux from the brain. In fact, several peripheral tissues or organs participate in Aβ catabolism and constitute potential Aβ clearance pathways. These include uptake and phagocytosis or endocytosis by monocytes, macrophages, neutrophils, lymphocytes, and hepatocytes24,25; excretion via bile or urine26,27; proteolytic degradation by Aβ-degrading enzymes28; and clearance from blood mediated by Aβ-binding proteins and cells, such as erythrocytes, albumin, antithrombin III and lipoproteins, including apolipoprotein E (ApoE) and apolipoprotein J (ApoJ)11,12. These central and peripheral pathways might interact with each other and work synergistically to clear Aβ from the brain.
Communication between Aβ pools
Brain-derived Aβ can be transported into the peripheral pool via the BBB, blood–CSF barrier, arachnoid villi or glymphatic–lymphatic pathway. Several transporters mediate Aβ flow out of the brain across the BBB, including LDL-related protein 1 (LRP1) and ATP-dependent efflux transporter P-glycoprotein29. The arachnoid villi absorb Aβ in the CSF and mediate its release into the circulation30. The glymphatic–lymphatic pathway, which consists of the glymphatic pathway in the brain and the CNS lymphatic vessels (discovered in 2015)31,32, might also transport Aβ from the brain to the periphery for clearance18,33. However, the glymphatic–lymphatic pathway and arachnoid villi are unidirectional; they only mediate Aβ efflux from the CNS to the periphery18.
Whether peripherally generated Aβ can enter the brain and exert neurotoxic effects there remains poorly understood. In the absence of a relevant transport mechanism, systemic amyloidosis might not necessarily lead to AD. However, peripheral inoculation of Aβ-containing brain extracts induces cerebral Aβ deposition in both mice and humans, suggesting that peripherally generated Aβ is able to enter the brain and participate in the pathogenesis of AD34,35,36,37. Receptor for advanced glycation end products (RAGE) has been suggested to transport Aβ across the BBB, from the blood into the brain38. Expression of the AGER gene (encoding RAGE) is upregulated in the AD brain vasculature39,40, indicating that influx of peripheral Aβ into the brain is increased in AD. The contribution of peripherally derived Aβ to amyloidosis in the AD brain needs to be determined in future studies.
A decline in peripheral Aβ clearance might also impede efflux of Aβ from the brain to the periphery, and thereby attenuate central clearance of Aβ. Moreover, the influx and efflux of Aβ might result in equilibrium between the central and peripheral pools of Aβ. Mechanisms that might regulate this equilibrium need to be understood.
Systemic abnormalities in AD
An increasing number of studies indicate that a series of systemic abnormalities can exacerbate the progression of AD (Fig. 2). In turn, the downstream effects of processes in the AD brain can also drive these systemic disorders, forming feedback loops. Mechanisms that might underlie the effects of systemic abnormalities or alterations on Aβ metabolism are outlined in Table 2. Here, we discuss the interactions between Aβ metabolism in the brain and periphery, and place them in a systemic context.

Various systemic abnormalities have been found in patients with Alzheimer disease (AD). Red boxes highlight AD risk variants in genes related to innate immunity, phagocytosis of amyoid-β (Aβ) by immune cells, and lipid metabolism in periphery and brain, respectively, which were identified in genome-wide association studies and candidate-gene studies of sporadic AD. APP, amyloid precursor protein; BA, basilar artery; COPD, chronic obstructive pulmonary disease; CRP, C-reactive protein; HDL-C, HDL-cholesterol; IGF, insulin-like growth factor; LDL-C, LDL-cholesterol; LPS, lipopolysaccharide; LTICA, left terminal internal carotid artery; OSA, obstructive sleep apnoea; PI, pulsatility index; RBC, red blood cells; RI, resistance index; RTICA, right terminal internal carotid artery; TNF, tumour necrosis factor.
Disorders of systemic immunity
One of the primary pathways of Aβ clearance in the brain is phagocytosis or endocytosis by professional phagocytes and microglia, as well as by astrocytes, oligodendrocytes and neurons. Accumulations of Aβ in the periphery can similarly be phagocytosed by monocytes and neutrophils in the blood, and by macrophages in tissues41. Of note, in transgenic mice with AD, expression of Aβ scavenger receptors and Aβ-degrading enzymes in circulating mononuclear phagocytes decreases substantially as these mice age42, and the phagocytic functions of these cells are impaired in both mice and humans with AD43,44,45. Infusion of monocytes derived from peripheral human umbilical cord blood reduces the Aβ burden and improves cognitive deficits in a mouse model of AD46, implying that peripheral mononuclear phagocytes have an important role in Aβ clearance. Promoting the phagocytic function of peripheral blood monocytes or promoting the recruitment of peripheral macrophages into the brain might, therefore, improve Aβ clearance in the brain47, although the existence of conflicting data48 renders this approach controversial.
In this regard, a cluster of genes associated with the risk of sporadic AD (including CD33, CR1, MS4A6A, MS4A4E, ABCA7 and TREM2)49,50 encode proteins that are involved in innate immunity. Variants in these genes, especially in TREM2 and CD33, are associated with compromised phagocytic function of peripheral monocytes or macrophages and altered Aβ accumulation in AD brains24,51. Interestingly, CR1 (encoding complement receptor-1, also known as CD35) is expressed primarily in peripheral leukocytes and erythrocytes, but not in any brain cells.
In regard to adaptive immunity, much attention has been focused on autoimmunity and autoreactive antibodies related to the pathogenesis of AD, including naturally occurring antibodies and autoantibodies. These autoreactive antibodies are ubiquitous in human blood and CSF, and profiles of these antibodies are altered in patients with AD52,53,54,55,56. Identification of the most antigenic epitopes targeted by human antibodies against Aβ aggregates could lead to development of an effective immunotherapy for AD. Aducanumab, derived from a naturally occurring human autoantibody against Cu2+-modified Aβ aggregates (which are the most neurotoxic Aβ species in the AD brain), showed promise in clearing brain Aβ deposits and improving cognition in a 2016 phase Ib trial57. Lymphocytes (including B cells, T cells and natural killer cells) also participate in Aβ clearance via immunoglobulin-mediated adaptive phagocytosis58,59. Future studies will help to elucidate the crosstalk between innate immunity and adaptive immunity, and to discover how the interaction of these two immune systems might synergistically affect AD pathogenesis.
Blood abnormalities
Besides monocytes and leukocytes, other blood components are also involved in Aβ metabolism. Increased expression of APP, altered APP isoform ratios and processing patterns, and enhanced β-secretase activity are observed in platelets from patients with AD60,61,62, and these changes could result in overproduction of Aβ in the periphery. Blood levels of albumin, an Aβ carrier, are reduced in patients with AD63. The quantity and function of erythrocytes (another Aβ carrier) are also altered in such patients, and their erythrocytes show compromised binding of Aβ64,65. In addition, the activity of Aβ-degrading enzymes in serum is thought to be decreased in AD28. These changes impede Aβ transportation and clearance in the periphery.
Some anti-ageing molecules, such as growth and differentiation factor 11, granulocyte–macrophage colony stimulating factor, and metalloproteinase inhibitor 2, have been identified in blood from young mice and in human umbilical cord plasma66,67. Whether levels of these anti-ageing molecules are reduced in patients with AD, and whether they are pathophysiologically relevant to this disease, remain unknown. However, identification of these protective components could be of importance in understanding the pathogenesis of AD and in developing systemic rejuvenation therapies68,69,70,71.
Metabolic disorders
Diabetes mellitus. How diabetes mellitus affects Aβ catabolism and AD risk is not yet well understood. Patients with diabetes mellitus are estimated to be 1.4–2.0-fold more likely than healthy individuals to develop AD72,73, although these claims need to be verified in patients with biomarker-confirmed AD. In patients with diabetes mellitus, insulin resistance substantially compromises the positive effects of insulin on both cognition and hepatic clearance of circulating Aβ74,75, resulting in AD-like alterations in the brain. Moreover, excess insulin can competitively inhibit IDE-mediated Aβ degradation76. Some other pathological features of diabetes mellitus — including oxidative stress, BBB disruption and reduced cell energy supply — can also affect Aβ generation and clearance77,78. In addition, amylin (a misfolded protein deposited in the pancreas in patients with type 2 diabetes mellitus) can enter the brain, where it accelerates and exacerbates the misfolding and aggregation of Aβ79. However, atherosclerosis and small vessel disease (discussed in more detail below) can be important causes of cognitive dysfunction in patients with diabetes mellitus80, and should be considered in the differential diagnosis of AD in this setting.
Lipid and lipoprotein risk factors. Some evidence suggests that abnormal lipid metabolism is associated with an increased risk of AD81. Several potential AD risk genes (including APOE, BIN1, CLU, SORL1, PICALM and PLD3) encode proteins linked to lipid metabolism82. Among these, ApoE is known to participate in Aβ production, aggregation, and clearance in an isoform-dependent manner83,84.
Cholesterol levels in the brain can affect Aβ synthesis, clearance and neurotoxicity. High serum cholesterol levels are associated with an increased cerebral burden of Aβ85,86. Abnormal cholesterol levels could reflect unmeasured genetic factors or dietary patterns that might affect the pathogenesis of AD85,86. Cell membrane fluidity is strongly affected by its lipid composition, and increased membrane fluidity of platelets and leukocytes has been reported in patients with AD, as well as in individuals with Down syndrome (who have a greatly increased risk of developing dementia and AD)87,88,89,90. Cell membrane fluidity also influences the processing of APP and cellular phagocytosis, both of which might affect Aβ generation and clearance. A high dietary intake of polyunsaturated fatty acids such as docosahexaenoic acid, which maintain membrane fluidity, have a beneficial effect on cognition in patients with AD91,92. The study of membrane fluidity in AD might provide insights into the alteration of membrane-dependent biological functions related to Aβ, such as phagocytosis, endocytosis, macropinocytosis and autophagy.
Cardiovascular disease
Emerging evidence indicates that cardiovascular disease (CVD) is a major comorbidity in patients with sporadic AD. A low cardiac index and heart failure are both associated with dementia, and perhaps also with AD93,94,95. However, as CVD and AD are both complex and multifactorial age-related diseases, the association between them might be attributed partly to shared risk factors, such as diabetes mellitus, hypertension, hypercholesterolemia and stroke96,97.
The presence of compromised myocardial function and intramyocardial deposits of Aβ in patients with AD suggests that peripheral Aβ accumulation could affect heart function in patients with AD15,98. In addition, cardiac systolic dysfunction could affect Aβ generation and clearance in the brain as a result of reduced cerebral blood flow99,100,101,102. Regional cerebral blood flow and glucose uptake or metabolism are consistently decreased in Aβ-positive patients with AD103, and correlate inversely with AD severity104. Indeed, some degree of cerebral small vessel disease almost always accompanies AD. Increased stiffness of small vessel walls might attenuate Aβ clearance via the BBB, interstitial fluid bulk flow and glymphatic pathways, thereby accelerating AD105,106. However, a clear understanding of the interaction between CVD and AD is lacking. Most of the evidence points to CVD being an independent risk factor for cognitive impairment, and having an additive rather than synergistic effect on the AD neurodegenerative process.
Hepatic dysfunction
The liver is the major organ responsible for system-wide metabolic regulation, protein synthesis and metabolic detoxification. Circulating Aβ is predominantly cleared by either degradation in hepatocytes or direct excretion in bile; several peptide clearance experiments have suggested that soluble Aβ has a short half-life of 2.5 min to 2.5 h in the circulation26,107. LRP1 is thought to mediate the uptake of Aβ by hepatocytes25. The liver might also indirectly influence Aβ clearance by regulating albumin levels and Aβ-related lipid metabolism. Plasma Aβ levels inversely correlate with liver function, suggesting that hepatic dysfunction attenuates peripheral Aβ clearance108. Liver tissue from patients with AD contains less Aβ than that from healthy individuals, which implies that the Aβ-clearance function of liver is compromised in patients with AD5.
Whether liver dysfunction also increases the Aβ load in the brain remains unknown; however, treatments that enhance LRP1-mediated Aβ uptake by the liver alleviate both the burden of Aβ in the brain and cognitive impairment74,109. These observations suggest that improving the Aβ clearance capacity of the liver is a potential systemic therapeutic approach for AD.
Renal dysfunction
Soluble Aβ is a normal component of human urine27. In addition, animal experiments have shown that, after intracranial or intravenous infusion of 125I-labelled Aβ, radioactivity is subsequently detected in the kidney and urine23,110. These findings indicate that the kidney might participate in physiological clearance of Aβ by filtering Aβ from blood to urine. Conversely, renal dysfunction probably leads to impaired peripheral Aβ clearance. In support of this notion, serum Aβ levels inversely correlate with measures of renal function (estimated glomerular filtration rate and creatinine levels) in patients with chronic kidney disease111,112. Moreover, human kidney donors have decreased estimated glomerular filtration rates and increased circulating levels of Aβ113, suggesting that the reduction in renal function reserve associated with having a single kidney also attenuates peripheral Aβ clearance.
Whether renal dysfunction increases Aβ burden in the brain or facilitates AD processes remains unknown. Renal dysfunction increases the risk of both cognitive impairment and dementia114, and this association could involve AD pathogenetic pathways. However, kidney transplantation can reduce plasma Aβ levels113, and haemodialysis alleviates Aβ deposition in the brain of patients with chronic kidney disease115. These observations suggest that improvement of renal function is a promising approach to AD prevention and treatment.
Respiratory and sleep disorders
Patients with AD have an increased incidence of respiratory disorders, such as bronchopneumonia, obstructive sleep apnoea (OSA) and sleep-disordered breathing116,117. In addition, sleep-disordered breathing is associated with an increased risk of mild cognitive impairment or dementia and with earlier onset of AD118,119,120.
Compared with healthy control individuals, patients with OSA or chronic obstructive pulmonary disease exhibit higher blood levels of Aβ, which negatively correlate with pulmonary function121,122. OSA is also associated with altered levels of AD biomarkers in CSF, including decreased levels of Aβ42 and elevated levels of phosphorylated tau123. OSA and chronic obstructive pulmonary disease could contribute to AD processes via hypoxia, inflammation, or sleep disruption124. Sleep disruption has been suggested to increase Aβ production and aggregation, suppress glymphatic clearance of AD pathogenic proteins (tau as well as Aβ) and aggravate oxidative stress, inflammation and synaptic damage125,126.
Gut microbiota disturbance and infection
The establishment of the gut–brain axis revealed a clear association between the gastrointestinal microbiota and cognition127. Gram-negative bacterial species (such as Escherichia coli K99) are the predominant sources of bacteria-derived factors in normal human brains, and levels of these molecules are increased in AD brains, along with levels of the bacterial cell wall component lipopolysaccharide128. Lipopolysaccharide colocalizes with Aβ in plaques in AD brains128, suggesting that Gram-negative bacteria are associated with AD pathogenesis. Probiotic supplementation is associated with improved cognition in patients with AD129, which further supports a role for the gut microbiota in AD development.
In addition to its relationship with normal microbial flora, emerging evidence indicates that AD is associated with exposure to an ever-increasing number of pathogens130,131. Moreover, a high infectious burden is associated with increased serum levels of Aβ and proinflammatory cytokines in patients with AD and in healthy controls130. However, the underlying mechanisms through which the microbiota or pathogens influence AD remain to be determined. Whether the microbiota or pathogens contribute to AD development, or whether an increased infectious burden is a consequence of AD, also remains unknown.
Systemic inflammation
Chronic reactive gliosis and microgliosis are neuroinflammatory responses that are important contributors to AD pathology. These processes might participate in a positive feedback loop of Aβ deposition, neurofibrillary tangle formation, and damage to synapses and neurons. Several studies have shown that other conditions involving chronic systemic inflammation, such as rheumatoid arthritis and periodontitis, are associated with an increased risk of AD132,133. These conditions are also associated with elevated levels of C-reactive protein and proinflammatory cytokines, such as tumour necrosis factor, IL-6 and IL-1β. These proinflammatory molecules could participate in AD pathogenesis either directly, by affecting brain Aβ metabolism (via entry to the CNS through the BBB or neural afferent pathways such as the vagus nerve)134,135 or indirectly, by affecting Aβ metabolism in the periphery. In this regard, the results of observational studies show that NSAID use is associated with a reduced risk of AD136. These findings suggest that chronic systemic inflammation promotes the AD process.
By contrast, acute systemic inflammatory responses seem to protect against AD — at least in animal models — by recruiting monocyte-derived macrophages into the brain, where they clear cerebral Aβ47. However, most studies in this area have not been repeated, and their results have not yet been validated in patients with biomarker-confirmed AD.
A systemic approach to understanding AD
The close interaction between the brain and the periphery, in terms of Aβ metabolism, provides novel insights into the pathogenesis of AD, and could lead to new approaches to the diagnosis and treatment of AD, based on systems biology and systems neurophysiology paradigms.
Pathogenesis
Our current understanding of the role of Aβ in AD focuses on its contribution to brain pathology and symptoms. However, as already discussed, this view might not be the whole story. First, although Aβ peptides are generated in the brain, a considerable amount of Aβ is also generated in peripheral systems. Second, Aβ can be cleared — from peripheral organs or tissues as well as from the brain — by professional phagocytes, which can transmigrate through the BBB. Third, Aβ deposits have been detected in the periphery — although this claim has not yet been replicated and its pathophysiological relevance remains unknown. Last, a series of systemic abnormalities are both driven by and contribute to AD progression. On the basis of these findings, we propose that AD might not be solely a brain disorder, in the sense that systemic factors might interact with the brain to modify the AD process.
As discussed, the central and peripheral Aβ pools interact with and influence each other. For example, the rate of peripheral catabolism of Aβ seems to affect the rate of Aβ efflux from the brain, and peripherally derived Aβ can enter the brain and accelerate the progression of cerebral AD pathology34,35. Therefore, we hypothesize that the peripheral pool of Aβ is not simply associated with AD, but is causally linked to this disease. Indeed, interactions between the brain and the periphery might have a crucial role in the natural history of AD, and elucidation of the effects of peripheral processes on AD development could lead to improved understanding of its pathogenesis. The crucial questions to answer would be precisely how the brain and periphery interact with each other to affect AD progression, and whether interventions that target systemic factors can modulate the pathogenesis or development of AD.
Diagnosis
Several PET radiotracers can be used to detect Aβ in the brain, and a few biomarkers for AD have been validated for diagnostic use, including CSF levels of Aβ42, total tau and phosphorylated tau. However, these approaches are either invasive or expensive, and are impractical for the early diagnosis of patients without obvious cognitive complaints. The search for peripheral blood or plasma biomarkers for AD that reflect AD-related processes in the brain has, therefore, received considerable attention. However, owing to the complexity of blood components, the accurate measurement of plasma levels of Aβ or tau is very challenging. A study published in 2017 did not find a statistically significant difference in plasma levels of free Aβ between patients with AD and age-matched controls137, and similarly negative results have been published for plasma tau levels138. However, these results do not indicate the end of the road for AD biomarker studies139. With the development of advanced and highly sensitive techniques that are able to promote efflux of Aβ and tau from the brain, and accurately measure their levels even when bound to other serum proteins, researchers might eventually find a method to monitor cerebral Aβ accumulation. In addition, misfolded oligomeric species of Aβ and tau proteins have been detected in CSF and suggested as potential biomarkers for AD140,141. If these misfolded protein species also prove to be present in blood, they might be useful for AD diagnosis, although no published reports yet exist.
Technical advances by the AD Neuroimaging Initiative have enabled the use of microarrays to detect serum anti-Aβ autoantibodies with 100% accuracy55; however, this promising technique requires validation in large independent cohorts. This approach is particularly interesting in the light of the promising results obtained with aducanumab, the first human anti-Aβ autoantibody to be developed for clinical trials57. In addition to Aβ and tau, β-secretase 1 (encoded by BACE1, an enzyme involved in Aβ production) has been suggested as a potential biomarker for AD142. In general, the identification of novel peripheral biomarkers for AD diagnosis and prognosis represents a promising and rapidly expanding research direction. (Box 1)
Treatment
Currently, effective agents for AD prevention or treatment are lacking. The traditional concept 'one target, one treatment' inevitably ignores the complexity of AD pathogenetic mechanisms143. After the failure of over 100 clinical trials of monotherapies targeting Aβ, multitargeted therapies that address various aspects of AD pathogenesis at different disease stages are needed144. We argue that a comprehensive strategy targeting both brain and peripheral (systemic) abnormalities might be more effective than strategies that target CNS abnormalities alone. As discussed, many comorbidities of (and risk factors for) AD — such as diabetes mellitus, metabolic disorders, cardiovascular diseases, and hypertension — are systemic disorders.
Many attempts have been made to prevent AD via peripheral interventions, and some have been associated with beneficial outcomes. Improvements in overall population health have led to a decreased incidence and prevalence of dementia over the past 10–30 years145,146,147,148, perhaps through improved management of cardiovascular risk factors145,146,147,148. For example, administration of statins (which reduce peripheral blood cholesterol levels) to healthy middle-aged individuals was associated with a reduced dementia risk in one large-scale prospective cohort study149; statins have also decreased the brain burden of Aβ in experimental models of AD150, although the results of most studies of statin treatment in patients with AD have been disappointing151. Furthermore, treatment with continuous positive airway pressure for sleep-disordered breathing or OSA might delay the onset of mild cognitive impairment and slow or even improve cognitive decline in patients with AD120,152,153. These observations support the view that systemic management of an individual's known comorbidities or risk factors, with the aim of maintaining bodily homeostasis, might help to prevent or slow the progression of AD.
Active removal of excess peripheral Aβ seems to be a particularly promising therapeutic strategy for AD23. Plasma albumin exchange both improves cognition and decreases the Aβ burden in patients with AD154. Peritoneal dialysis reduces blood Aβ levels in humans and also attenuates AD pathology in an AD mouse model155; patients who have undergone haemodialysis exhibit a reduction in Aβ deposition in the brain115. Approaches to improve peripheral Aβ clearance via enhancing phagocytosis45, proteolytic degradation and excretion155,156, and identification of rejuvenation factors in blood68, are other promising systemic therapeutic strategies for AD157.
Conclusions
AD might be not only a brain disorder, but also a systemic disease with widespread abnormalities beyond the brain. Thus, systemic factors might interact with brain-related factors to modify the AD process. AD diagnosis and treatment should have a corresponding focus not only on pathological changes in the brain but also on peripheral abnormalities, which vary among individuals. Identifying these peripheral abnormalities might offer new opportunities for diagnosis of early AD and lead to the design of specific treatment strategies for individuals with preclinical, prodromal or frank AD. In conclusion, the systemic view of AD proposed in this Review provides a novel perspective for understanding the pathogenesis of this disease, and fosters new opportunities for its early diagnosis and successful management.
Review criteria
Articles for inclusion in this Review were identified by searches of the PubMed database using the following terms: “Alzheimer disease”, “amyloid beta”, “peripheral clearance”, “innate immunity”, “adaptive immunity”, “macrophage”, “monocyte”, “phagocytosis”, “blood”, “circulation”, “plasma”, “platelet”, “erythrocyte”, “exosome”, “diabetes”, “insulin”, “amylin”, “apolipoprotein E”, “cholesterol”, “lipid”, “lipoprotein”, “cerebrovascular diseases”, “heart failure”, “cerebral blood flow”, “hypoperfusion”, “hypoxia”, “liver”, “hepatic function”, “kidney”, “renal function”, “respiratory”, “sleep”, “microbiome”, “gastrointestinal microbiota”, “gut flora”, “infection”, “inflammation”, “immune responses”, “GWAS”, “risk factor”, “biomarker”, “diagnosis”, “therapy”, “treatment”, “prevent”. Only articles published in English were retrieved. Full-text papers were available for most of the articles that were chosen for review, and the reference lists of these articles were searched for further relevant material.
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Change history
13 October 2017
In the version of this article originally published online, Figure 1 incorrectly labelled the main Aβ molecule in the peripheral pool as Aβ42 instead of Aβ40. This error has been corrected in the HTML and PDF versions of the article.
References
Mangialasche, F., Solomon, A., Winblad, B., Mecocci, P. & Kivipelto, M. Alzheimer's disease: clinical trials and drug development. Lancet Neurol. 9, 702–716 (2010).
Berk, C., Paul, G. & Sabbagh, M. Investigational drugs in Alzheimer's disease: current progress. Expert Opin. Investig. Drugs 23, 837–846 (2014).
Selkoe, D. J. & Hardy, J. The amyloid hypothesis of Alzheimer's disease at 25 years. EMBO Mol. Med. 8, 595–608 (2016). This article reviews new evidence supporting the concept that an imbalance between production and clearance of Aβ is a very early, often initiating factor, in AD — a widely debated issue.
Yankner, B. A. & Mesulam, M. M. Seminars in medicine of the Beth Israel Hospital, Boston. β-Amyloid and the pathogenesis of Alzheimer's disease. N. Engl. J. Med. 325, 1849–1857 (1991).
Roher, A. E. et al. Amyloid β peptides in human plasma and tissues and their significance for Alzheimer's disease. Alzheimers Dement. 5, 18–29 (2009). This study evaluates Aβ levels in brain, peripheral organs and tissues, suggesting that brain as well as plasma Aβ levels are the consequence of intricate relationships between central and peripehral sources.
Li, Q. X., Fuller, S. J., Beyreuther, K. & Masters, C. L. The amyloid precursor protein of Alzheimer disease in human brain and blood. J. Leukoc. Biol. 66, 567–574 (1999).
Toledo, J. et al. Factors affecting Aβ plasma levels and their utility as biomarkers in ADNI. Acta Neuropathol. 122, 401–413 (2011).
Mehta, P. D., Pirttila, T., Patrick, B. A., Barshatzky, M. & Mehta, S. P. Amyloid β protein 1–40 and 1–42 levels in matched cerebrospinal fluid and plasma from patients with Alzheimer disease. Neurosci. Lett. 304, 102–106 (2001).
Delvaux, E., Bentley, K., Stubbs, V., Sabbagh, M. & Coleman, P. Differential processing of amyloid precursor protein in brain and in peripheral blood leukocytes. Neurobiol. Aging 34, 1680–1686 (2013).
Evin, G., Zhu, A., Holsinger, R. M., Masters, C. & Li, Q.-X. Proteolytic processing of the Alzheimer's disease amyloid precursor protein in brain and platelets. J. Neurosci. Res. 74, 386–392 (2003).
Biere, A. L. et al. Amyloid β-peptide is transported on lipoproteins and albumin in human plasma. J. Biol. Chem. 271, 32916–32922 (1996).
Kuo, Y. M. et al. Amyloid-β peptides interact with plasma proteins and erythrocytes: implications for their quantitation in plasma. Biochem. Biophys. Res. Commun. 268, 750–756 (2000).
Joachim, C. L., Mori, H. & Selkoe, D. J. Amyloid β-protein deposition in tissues other than brain in Alzheimer's disease. Nature 341, 226–230 (1989).
Koronyo, Y., Salumbides, B., Black, K. & Koronyo Hamaoui, M. Alzheimer's disease in the retina: imaging retinal Aβ plaques for early diagnosis and therapy assessment. Neurodegener. Dis. 10, 285–293 (2012).
Troncone, L. et al. Aβ amyloid pathology affects the hearts of patients with Alzheimer's disease: mind the heart. J. Am. Coll. Cardiol. 68, 2395–2407 (2016). This article was the first to describe the presence of compromised myocardial function and intramyocardial deposits of Aβ in patients with AD.
Stine, W. B., Dahlgren, K., Krafft, G. & LaDu, M. In vitro characterization of conditions for amyloid-β peptide oligomerization and fibrillogenesis. J. Biol. Chem. 278, 11612–11622 (2003).
Murray, M. et al. Amyloid β protein: Aβ40 inhibits Aβ42 oligomerization. J. Am. Chem. Soc. 131, 6316–6317 (2009).
Tarasoff-Conway, J. M. et al. Clearance systems in the brain — implications for Alzheimer disease. Nat. Rev. Neurol. 11, 457–470 (2015). This review summarizes the clearance systems of Aβ and tau in the brain.
Yao, X. Q. et al. p75NTR ectodomain is a physiological neuroprotective molecule against amyloid-β toxicity in the brain of Alzheimer's disease. Mol. Psychiatry 20, 1301–1310 (2015).
Liao, M. C. et al. N-Terminal domain of myelin basic protein inhibits amyloid β-protein fibril assembly. J. Biol. Chem. 285, 35590–35598 (2010).
Qosa, H. et al. Differences in amyloid-β clearance across mouse and human blood–brain barrier models: kinetic analysis and mechanistic modeling. Neuropharmacology 79, 668–678 (2014).
Yuede, C. M. et al. Rapid in vivo measurement of β-amyloid reveals biphasic clearance kinetics in an Alzheimer's mouse model. J. Exp. Med. 213, 677–685 (2016).
Xiang, Y. et al. Physiological amyloid-β clearance in the periphery and its therapeutic potential for Alzheimer's disease. Acta Neuropathol. 130, 487–499 (2015). This article demonstrates that peripheral clearance systems are potent in clearing brain Aβ and preventing AD.
Bradshaw, E. M. et al. CD33 Alzheimer's disease locus: altered monocyte function and amyloid biology. Nat. Neurosci. 16, 848–850 (2013).
Kanekiyo, T. & Bu, G. The low-density lipoprotein receptor-related protein 1 and amyloid-β clearance in Alzheimer's disease. Front. Aging Neurosci. 6, 93 (2014).
Ghiso, J. et al. Systemic catabolism of Alzheimer's Aβ40 and Aβ42 . J. Biol. Chem. 279, 45897–45908 (2004). This article demonstrates that the liver is the major organ responsible for uptake and degradation of circulating Aβ 42 and Aβ 40 , followed by the kidney.
Ghiso, J. et al. Alzheimer's soluble amyloid β is a normal component of human urine. FEBS Lett. 408, 105–108 (1997).
Liu, Z. et al. Characterization of insulin degrading enzyme and other amyloid-β degrading proteases in human serum: a role in Alzheimer's disease? J. Alzheimers Dis. 29, 329–340 (2012).
Mackic, J. B. et al. Human blood–brain barrier receptors for Alzheimer's amyloid-β1–40. Asymmetrical binding, endocytosis, and transcytosis at the apical side of brain microvascular endothelial cell monolayer. J. Clin. Invest. 102, 734–743 (1998).
Silverberg, G. D., Mayo, M., Saul, T., Rubenstein, E. & McGuire, D. Alzheimer's disease, normal-pressure hydrocephalus, and senescent changes in CSF circulatory physiology: a hypothesis. Lancet Neurol. 2, 506–511 (2003).
Aspelund, A. et al. A dural lymphatic vascular system that drains brain interstitial fluid and macromolecules. J. Exp. Med. 212, 991–999 (2015).
Louveau, A. et al. Structural and functional features of central nervous system lymphatic vessels. Nature 523, 337–341 (2015).
Iliff, J. J., Goldman, S. A. & Nedergaard, M. Implications of the discovery of brain lymphatic pathways. Lancet Neurol. 14, 977–979 (2015).
Eisele, Y. S. et al. Peripherally applied Aβ-containing inoculates induce cerebral β-amyloidosis. Science 330, 980–982 (2010). This study suggests that peripherally derived Aβ might enter the brain and participate in AD pathogenesis.
Eisele, Y. S. et al. Multiple factors contribute to the peripheral induction of cerebral β-amyloidosis. J. Neurosci. 34, 10264–10273 (2014).
Ritchie, D. L. et al. Amyloid-β accumulation in the CNS in human growth hormone recipients in the UK. Acta Neuropathol. 134, 221–240 (2017).
Jaunmuktane, Z. et al. Evidence for human transmission of amyloid-β pathology and cerebral amyloid angiopathy. Nature 525, 247–250 (2015).
Deane, R. et al. RAGE mediates amyloid-β peptide transport across the blood–brain barrier and accumulation in brain. Nat. Med. 9, 907–913 (2003).
Donahue, J. E. et al. RAGE, LRP-1, and amyloid-β protein in Alzheimer's disease. Acta Neuropathol. 112, 405–415 (2006).
Yan, S. D. et al. RAGE and amyloid-β peptide neurotoxicity in Alzheimer's disease. Nature 382, 685–691 (1996).
Zenaro, E. et al. Neutrophils promote Alzheimer's disease-like pathology and cognitive decline via LFA-1 integrin. Nat. Med. 21, 880–886 (2015).
Frenkel, D. et al. Scara1 deficiency impairs clearance of soluble amyloid-β by mononuclear phagocytes and accelerates Alzheimer's-like disease progression. Nat. Commun. 4, 2030 (2013).
Krabbe, G. et al. Functional impairment of microglia coincides with β-amyloid deposition in mice with Alzheimer-like pathology. PLoS ONE 8, e60921 (2013).
Zaghi, J. et al. Alzheimer disease macrophages shuttle amyloid-β from neurons to vessels, contributing to amyloid angiopathy. Acta Neuropathol. 117, 111–124 (2009).
Gu, B. J. et al. Innate phagocytosis by peripheral blood monocytes is altered in Alzheimer's disease. Acta Neuropathol. 132, 377–389 (2016). This human study demonstrates that innate immunity is compromised in patients with AD.
Darlington, D. et al. Human umbilical cord blood-derived monocytes improve cognitive deficits and reduce amyloid-β pathology in PSAPP mice. Cell Transplant. 24, 2237–22350 (2015).
Baruch, K. et al. PD-1 immune checkpoint blockade reduces pathology and improves memory in mouse models of Alzheimer's disease. Nat. Med. 22, 135–137 (2016).
Prokop, S. et al. Impact of peripheral myeloid cells on amyloid-β pathology in Alzheimer's disease-like mice. J. Exp. Med. 212, 1811–1818 (2015).
Hollingworth, P. et al. Common variants at ABCA7. MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer's disease. Nat. Genet. 43, 429–435 (2011).
Jonsson, T. et al. Variant of TREM2 associated with the risk of Alzheimer's disease. N. Engl. J. Med. 368, 107–116 (2013).
Jay, T. R. et al. TREM2 deficiency eliminates TREM2+ inflammatory macrophages and ameliorates pathology in Alzheimer's disease mouse models. J. Exp. Med. 212, 287–295 (2015).
Bartos, A., Fialova, L., Svarcova, J. & Ripova, D. Patients with Alzheimer disease have elevated intrathecal synthesis of antibodies against tau protein and heavy neurofilament. J. Neuroimmunol. 252, 100–105 (2012).
Liu, Y. H. et al. Immunity and Alzheimer's disease: immunological perspectives on the development of novel therapies. Drug Discov. Today 18, 1212–1220 (2013).
Wang, T. et al. Naturally occurring autoantibodies against Aβ oligomers exhibited more beneficial effects in the treatment of mouse model of Alzheimer's disease than intravenous immunoglobulin. Neuropharmacology 105, 561–576 (2016).
DeMarshall, C. A. et al. Detection of Alzheimer's disease at mild cognitive impairment and disease progression using autoantibodies as blood-based biomarkers. Alzheimers Dement. (Amst.) 3, 51–62 (2016).
Monning, U. E. A. in Alzheimer's Disease: Basic Mechanisms, Diagnosis and Therapeutic Strategies (eds Iqbal, K. et al.) 557–563 (Wiley–Blackwell, 1991).
Sevigny, J. et al. The antibody aducanumab reduces Aβ plaques in Alzheimer's disease. Nature 537, 50–56 (2016).
Marsh, S. E. et al. The adaptive immune system restrains Alzheimer's disease pathogenesis by modulating microglial function. Proc. Natl Acad. Sci. USA 113, E1316–E1325 (2016).
Baruch, K. et al. Breaking immune tolerance by targeting Foxp3+ regulatory T cells mitigates Alzheimer's disease pathology. Nat. Commun. 6, 7967 (2015).
Rosenberg, R. N. et al. Altered amyloid protein processing in platelets of patients with Alzheimer disease. Arch. Neurol. 54, 139–144 (1997).
Di Luca, M. et al. Abnormal pattern of platelet APP isoforms in Alzheimer disease and Down syndrome. Arch. Neurol. 53, 1162–1166 (1996).
Srisawat, C. et al. The platelet amyloid precursor protein ratio as a diagnostic marker for Alzheimer's disease in Thai patients. J. Clin. Neurosci. 20, 644–648 (2013).
Doecke, J. D. et al. Blood-based protein biomarkers for diagnosis of Alzheimer disease. Arch. Neurol. 69, 1318–1325 (2012).
Rogers, J. et al. Peripheral clearance of amyloid β peptide by complement C3-dependent adherence to erythrocytes. Neurobiol. Aging 27, 1733–1739 (2006).
Chen, S. H. et al. Altered peripheral profile of blood cells in Alzheimer disease: a hospital-based case–control study. Medicine (Baltimore) 96, e6843 (2017).
Loffredo, F. S. et al. Growth differentiation factor 11 is a circulating factor that reverses age-related cardiac hypertrophy. Cell 153, 828–839 (2013).
Castellano, J. M. et al. Human umbilical cord plasma proteins revitalize hippocampal function in aged mice. Nature 544, 488–492 (2017).
Villeda, S. A. et al. Young blood reverses age-related impairments in cognitive function and synaptic plasticity in mice. Nat. Med. 20, 659–663 (2014). This article suggests that anti-ageing molecules exist in young blood. Identification of these protective components could be important in understanding the pathogenesis of AD and in developing systemic rejuvenation therapeutics.
Villeda, S. A. et al. The ageing systemic milieu negatively regulates neurogenesis and cognitive function. Nature 477, 90–94 (2011).
Katsimpardi, L. et al. Vascular and neurogenic rejuvenation of the aging mouse brain by young systemic factors. Science 344, 630–634 (2014).
Middeldorp, J. et al. Preclinical assessment of young blood plasma for Alzheimer disease. JAMA Neurol. 73, 1325–1333 (2016).
Xu, W. et al. Meta-analysis of modifiable risk factors for Alzheimer's disease. J. Neurol. Neurosurg. Psychiatry 86, 1299–1306 (2015).
Velayudhan, L. et al. Risk of developing dementia in people with diabetes and mild cognitive impairment. Br. J. Psychiatry 196, 36–40 (2010).
Tamaki, C., Ohtsuki, S. & Terasaki, T. Insulin facilitates the hepatic clearance of plasma amyloid β-peptide (1–40) by intracellular translocation of low-density lipoprotein receptor-related protein 1 (LRP-1) to the plasma membrane in hepatocytes. Mol. Pharmacol. 72, 850–855 (2007).
Kang, S., Lee, Y. H. & Lee, J. E. Metabolism-centric overview of the pathogenesis of Alzheimer's disease. Yonsei Med. J. 58, 479–488 (2017).
Gasparini, L. et al. Stimulation of β-amyloid precursor protein trafficking by insulin reduces intraneuronal β-amyloid and requires mitogen-activated protein kinase signaling. J. Neurosci. 21, 2561–2570 (2001).
Salameh, T. S., Shah, G. N., Price, T. O., Hayden, M. R. & Banks, W. A. Blood–brain barrier disruption and neurovascular unit dysfunction in diabetic mice: protection with the mitochondrial carbonic anhydrase inhibitor topiramate. J. Pharmacol. Exp. Ther. 359, 452–459 (2016).
Leuner, K. et al. Mitochondrion-derived reactive oxygen species lead to enhanced amyloid β formation. Antioxid. Redox Signal. 16, 1421–1433 (2012).
Moreno-Gonzalez, I. et al. Molecular interaction between type 2 diabetes and Alzheimer's disease through cross-seeding of protein misfolding. Mol. Psychiatry 22, 1327–1334 (2017). This study proposes a new molecular interaction between AD and diabetes mellitus: misfolded amylin (generated in the pancreas in type 2 diabetes mellitus) and Aβ accelerate or exacerbate the misfolding and aggregation of each other by cross-seeding.
Biessels, G. J. & Reijmer, Y. D. Brain changes underlying cognitive dysfunction in diabetes: what can we learn from MRI? Diabetes 63, 2244–2252 (2014).
Lesser, G. T. Association of Alzheimer disease pathology with abnormal lipid metabolism: the Hisayama study. Neurology 78, 1280 (2012).
Sato, N. & Morishita, R. The roles of lipid and glucose metabolism in modulation of β-amyloid, tau, and neurodegeneration in the pathogenesis of Alzheimer disease. Front. Aging Neurosci. 7, 199 (2015).
Yu, J. T., Tan, L. & Hardy, J. Apolipoprotein E in Alzheimer's disease: an update. Annu. Rev. Neurosci. 37, 79–100 (2014).
Lee, C. Y., Tse, W., Smith, J. D. & Landreth, G. E. Apolipoprotein E promotes β-amyloid trafficking and degradation by modulating microglial cholesterol levels. J. Biol. Chem. 287, 2032–2044 (2012).
Zissimopoulos, J. M., Barthold, D., Brinton, R. D. & Joyce, G. Sex and race differences in the association between statin use and the incidence of Alzheimer disease. JAMA Neurol. 74, 225–232 (2016).
Reed, B. et al. Associations between serum cholesterol levels and cerebral amyloidosis. JAMA Neurol. 71, 195–200 (2014).
Zubenko, G. S. et al. Platelet membrane fluidity in Alzheimer's disease and major depression. Am. J. Psychiatry 144, 860–868 (1987).
Collins, J. M., Scott, R. B., McClish, D. K., Taylor, J. R. & Grogan, W. M. Altered membrane anisotropy gradients of plasma membranes of living peripheral blood leukocytes in aging and Alzheimer's disease. Mech. Ageing Dev. 59, 153–162 (1991).
Zubenko, G. S. & Howland, R. Markedly increased platelet membrane fluidity in Down syndrome with a (14q, 21q) translocation. J. Geriatr. Psychiatry Neurol. 1, 218–219 (1988).
Scott, R. B., Collins, J. M. & Hunt, P. A. Alzheimer's disease and Down syndrome: leukocyte membrane fluidity alterations. Mech. Ageing Dev. 75, 1–10 (1994).
Yassine, H. N. et al. Association of serum docosahexaenoic acid with cerebral amyloidosis. JAMA Neurol. 73, 1208–1216 (2016).
Nishihira, J. et al. Associations between serum omega-3 fatty acid levels and cognitive functions among community-dwelling octogenarians in Okinawa, Japan: the KOCOA study. J. Alzheimers Dis. 51, 857–866 (2016).
Rusanen, M. et al. Heart diseases and long-term risk of dementia and Alzheimer's disease: a population-based CAIDE study. J. Alzheimers Dis. 42, 183–191 (2014).
Qiu, C. et al. Heart failure and risk of dementia and Alzheimer disease: a population-based cohort study. Arch. Intern. Med. 166, 1003–1008 (2006).
Jefferson, A. L. et al. Low cardiac index is associated with incident dementia and Alzheimer disease: the Framingham Heart Study. Circulation 131, 1333–1339 (2015).
Luchsinger, J. A. et al. Aggregation of vascular risk factors and risk of incident Alzheimer disease. Neurology 65, 545–551 (2005).
Li, J. et al. Vascular risk factors promote conversion from mild cognitive impairment to Alzheimer disease. Neurology 76, 1485–1491 (2011).
Jin, W. S. et al. Reduced cardiovascular functions in patients with Alzheimer's disease. J. Alzheimers Dis. 58, 919–925 (2017).
Okamoto, Y. et al. Cerebral hypoperfusion accelerates cerebral amyloid angiopathy and promotes cortical microinfarcts. Acta Neuropathol. 123, 381–394 (2012).
Zetterberg, H. et al. Hypoxia due to cardiac arrest induces a time-dependent increase in serum amyloid β levels in humans. PLoS ONE 6, e28263 (2011).
Wang, L. et al. Chronic cerebral hypoperfusion induces memory deficits and facilitates Aβ generation in C57BL/6J mice. Exp. Neurol. 283, 353–364 (2016).
Cermakova, P. et al. Heart failure and Alzheimer's disease. J. Intern. Med. 277, 406–425 (2015).
Mattsson, N. et al. Association of brain amyloid-β with cerebral perfusion and structure in Alzheimer's disease and mild cognitive impairment. Brain 137, 1550–1561 (2014).
Leeuwis, A. E. et al. Lower cerebral blood flow is associated with impairment in multiple cognitive domains in Alzheimer's disease. Alzheimers Dement. 13, 531–540 (2017).
Marnane, M. & Hsiung, G. Y. Could better phenotyping small vessel disease provide new insights into Alzheimer disease and improve clinical trial outcomes? Curr. Alzheimer Res. 13, 750–763 (2016).
Kester, M. I. et al. Associations between cerebral small-vessel disease and Alzheimer disease pathology as measured by cerebrospinal fluid biomarkers. JAMA Neurol. 71, 855–862 (2014).
Mackic, J. B. et al. Cerebrovascular accumulation and increased blood-brain barrier permeability to circulating Alzheimer's amyloid-β peptide in aged squirrel monkey with cerebral amyloid angiopathy. J. Neurochem. 70, 210–215 (1998).
Wang, Y. R. et al. Associations between hepatic functions and plasma amyloid-β levels.— implications for the capacity of liver in peripheral amyloid-β clearance. Mol. Neurobiol. 54, 2338–2344 (2017).
Sehgal, N. et al. Withania somnifera reverses Alzheimer's disease pathology by enhancing low-density lipoprotein receptor-related protein in liver. Proc. Natl Acad. Sci. USA 109, 3510–3515 (2012).
Ghersi-Egea, J. F. et al. Fate of cerebrospinal fluid-borne amyloid β-peptide: rapid clearance into blood and appreciable accumulation by cerebral arteries. J. Neurochem. 67, 880–883 (1996).
Arvanitakis, Z., Lucas, J. A., Younkin, L. H., Younkin, S. G. & Graff-Radford, N. R. Serum creatinine levels correlate with plasma amyloid β protein. Alzheimer Dis. Assoc. Disord. 16, 187–190 (2002).
Liu, Y. H. et al. Association between serum amyloid-β and renal functions: implications for roles of kidney in amyloid-β clearance. Mol. Neurobiol. 52, 115–119 (2015).
Gronewold, J. et al. Factors responsible for plasma β-amyloid accumulation in chronic kidney disease. Mol. Neurobiol. 53, 3136–3145 (2016).
Deckers, K. et al. Dementia risk in renal dysfunction: a systematic review and meta-analysis of prospective studies. Neurology 88, 198–208 (2017).
Sakai, K. et al. Patients that have undergone hemodialysis exhibit lower amyloid deposition in the brain: evidence supporting a therapeutic strategy for Alzheimer's disease by removal of blood amyloid. J. Alzheimers Dis. 51, 997–1002 (2016).
Emamian, F. et al. The association between obstructive sleep apnea and Alzheimer's disease: a meta-analysis perspective. Front. Aging Neurosci. 8, 78 (2016).
Brunnstrom, H. R. & Englund, E. M. Cause of death in patients with dementia disorders. Eur. J. Neurol. 16, 488–492 (2009).
Pan, W. & Kastin, A. J. Can sleep apnea cause Alzheimer's disease? Neurosci. Biobehav. Rev. 47, 656–669 (2014).
Yaffe, K. et al. Sleep-disordered breathing, hypoxia, and risk of mild cognitive impairment and dementia in older women. JAMA 306, 613–619 (2011).
Osorio, R. S. et al. Sleep-disordered breathing advances cognitive decline in the elderly. Neurology 84, 1964–1971 (2015).
Bu, X. L. et al. Serum amyloid-β levels are increased in patients with obstructive sleep apnea syndrome. Sci. Rep. 5, 13917 (2015).
Bu, X. L. et al. Serum amyloid-β levels are increased in patients with chronic obstructive pulmonary disease. Neurotox. Res. 28, 346–351 (2015).
Osorio, R. S. et al. Interaction between sleep-disordered breathing and apolipoprotein E genotype on cerebrospinal fluid biomarkers for Alzheimer's disease in cognitively normal elderly individuals. Neurobiol. Aging 35, 1318–1324 (2014).
Rosenzweig, I. et al. Sleep apnoea and the brain: a complex relationship. Lancet Respir. Med. 3, 404–414 (2015).
Musiek, E. S. & Holtzman, D. M. Mechanisms linking circadian clocks, sleep, and neurodegeneration. Science 354, 1004–1008 (2016).
Cedernaes, J. et al. Candidate mechanisms underlying the association between sleep–wake disruptions and Alzheimer's disease. Sleep Med. Rev. 31, 102–111 (2017).
Gareau, M. Microbiota–gut–brain axis and cognitive function. Adv. Exp. Med. Biol. 817, 357–371 (2014).
Zhan, X. et al. Gram-negative bacterial molecules associate with Alzheimer disease pathology. Neurology 87, 2324–2332 (2016).
Akbari, E. et al. Effect of probiotic supplementation on cognitive function and metabolic status in Alzheimer's disease: a randomized, double-blind and controlled trial. Front. Aging Neurosci. 8, 256 (2016).
Bu, X. L. et al. A study on the association between infectious burden and Alzheimer's disease. Eur. J. Neurol. 22, 1519–1525 (2015). This study offers the first evidence that an increased infectious burden is associated with AD, supporting the role of systemic infection and/or inflammation in the aetiopathogenesis of AD.
Harris, S. A. & Harris, E. A. Herpes simplex virus type 1 and other pathogens are key causative factors in sporadic Alzheimer's disease. J. Alzheimers Dis. 48, 319–353 (2015).
Abbayya, K., Puthanakar, N. Y., Naduwinmani, S. & Chidambar, Y. S. Association between periodontitis and Alzheimer's disease. N. Am. J. Med. Sci. 7, 241–246 (2015).
Wallin, K. et al. Midlife rheumatoid arthritis increases the risk of cognitive impairment two decades later: a population-based study. J. Alzheimers Dis. 31, 669–676 (2012).
Rivest, S. Regulation of innate immune responses in the brain. Nat. Rev. Immunol. 9, 429–439 (2009).
Gao, H. M. & Hong, J. S. Why neurodegenerative diseases are progressive: uncontrolled inflammation drives disease progression. Trends Immunol. 29, 357–365 (2008).
Wang, J. et al. Anti-inflammatory drugs and risk of Alzheimer's disease: an updated systematic review and meta-analysis. J. Alzheimers Dis. 44, 385–396 (2015).
Lövheim, H. et al. Plasma concentrations of free amyloid-β cannot predict the development of Alzheimer's disease. Alzheimers Dement. 13, 778–782 (2017).
Mattsson, N. et al. Plasma tau in Alzheimer disease. Neurology 87, 1827–1835 (2016).
Wood, H. Alzheimer disease: biomarkers of AD risk — the end of the road for plasma amyloid-β? Nat. Rev. Neurol. 12, 613 (2016).
Herskovits, A. Z., Locascio, J. J., Peskind, E. R., Li, G. & Hyman, B. T. A. Luminex assay detects amyloid β oligomers in Alzheimer's disease cerebrospinal fluid. PLoS ONE 8, e67898 (2013).
Sengupta, U. et al. Tau oligomers in cerebrospinal fluid in Alzheimer's disease. Ann. Clin. Transl Neurol. 4, 226–235 (2017).
Shen, Y. et al. Increased plasma β-secretase 1 may predict conversion to Alzheimer's disease dementia in individuals with mild cognitive impairment. Biol. Psychiatry http://dx.doi.org/10.1016/j.biopsych.2017.02.007 (2017).
Brooks, M. One target, one treatment? Not for Alzheimer's disease. Medscape http://www.medscape.com/viewarticle/848322 (2015).
Wang, Y. J. Alzheimer disease: lessons from immunotherapy for Alzheimer disease. Nat. Rev. Neurol. 10, 188–189 (2014).
Larson, E. B., Yaffe, K. & Langa, K. M. New insights into the dementia epidemic. N. Engl. J. Med. 369, 2275–2277 (2013).
Wu, Y. T. et al. Dementia in western Europe: epidemiological evidence and implications for policy making. Lancet Neurol. 15, 116–124 (2016).
Satizabal, C. L. et al. Incidence of dementia over three decades in the Framingham heart study. N. Engl. J. Med. 374, 523–532 (2016). This study finds that the incidence of dementia has decreased over the past three decades, suggesting that control of systemic comorbidities and risk factors, as well as maintenance of body homeostasis, could bring improved results for AD prevention.
Langa, K. M. et al. A comparison of the prevalence of dementia in the United States in 2000 and 2012. JAMA Intern. Med. 177, 51–58 (2017).
Haag, M. D., Hofman, A., Koudstaal, P. J., Stricker, B. H. & Breteler, M. M. Statins are associated with a reduced risk of Alzheimer disease regardless of lipophilicity. The Rotterdam Study. J. Neurol. Neurosurg. Psychiatry 80, 13–17 (2009).
Papadopoulos, P., Tong, X. K. & Hamel, E. Selective benefits of simvastatin in bitransgenic APPSwe, Ind/TGF-β1 mice. Neurobiol. Aging 35, 203–212 (2014).
Richardson, K. et al. Statins and cognitive function: a systematic review. Ann. Intern. Med. 159, 688–697 (2013).
Ancoli-Israel, S. et al. Cognitive effects of treating obstructive sleep apnea in Alzheimer's disease: a randomized controlled study. J. Am. Geriatr. Soc. 56, 2076–2081 (2008).
Cooke, J. R. et al. Sustained use of CPAP slows deterioration of cognition, sleep, and mood in patients with Alzheimer's disease and obstructive sleep apnea: a preliminary study. J. Clin. Sleep Med. 5, 305–309 (2009).
Boada, M. et al. Amyloid-targeted therapeutics in Alzheimer's disease: use of human albumin in plasma exchange as a novel approach for Aβ mobilization. Drug News Perspect. 22, 325–339 (2009).
Jin, W. S. et al. Peritoneal dialysis reduces amyloid-β plasma levels in humans and attenuates Alzheimer-associated phenotypes in an APP/PS1 mouse model. Acta Neuropathol. 134, 207–220 (2017).
Liu, Y. et al. Expression of neprilysin in skeletal muscle reduces amyloid burden in a transgenic mouse model of Alzheimer disease. Mol. Ther. 17, 1381–1386 (2009).
Liu, Y. H. et al. Clearance of amyloid-β in Alzheimer's disease: shifting the action site from center to periphery. Mol. Neurobiol. 51, 1–7 (2015).
Winston, C. N. et al. Prediction of conversion from mild cognitive impairment to dementia with neuronally derived blood exosome protein profile. Alzheimers Dement. (Amst.) 3, 63–72 (2016).
Goetzl, E. J. et al. Cargo proteins of plasma astrocyte-derived exosomes in Alzheimer's disease. FASEB J. 30, 3853–3859 (2016).
Veitinger, M. et al. A platelet protein biochip rapidly detects an Alzheimer's disease-specific phenotype. Acta Neuropathol. 128, 665–677 (2014). This article demonstrates platelet changes in AD, providing potential biomarkers for early diagnosis of AD.
Burnham, S. C. et al. A blood-based predictor for neocortical Aβ burden in Alzheimer's disease: results from the AIBL study. Mol. Psychiatry 19, 519–526 (2014).
Mattsson, N., Andreasson, U., Zetterberg, H., Blennow, K. & Alzheimer's Disease Neuroimaging Initiative. Association of plasma neurofilament light with neurodegeneration in patients with Alzheimer disease. JAMA Neurol. 74, 557–566 (2017).
Chaves, M. L. et al. Serum levels of S100B and NSE proteins in Alzheimer's disease patients. J. Neuroinflammation 7, 6 (2010).
Teunissen, C. E. et al. Brain-specific fatty acid-binding protein is elevated in serum of patients with dementia-related diseases. Eur. J. Neurol. 18, 865–871 (2011).
Zhang, R. et al. Systemic immune system alterations in early stages of Alzheimer's disease. J. Neuroimmunol. 256, 38–42 (2013).
Kayano, M. et al. Plasma microRNA biomarker detection for mild cognitive impairment using differential correlation analysis. Biomark. Res. 4, 22 (2016).
Lu, R. et al. Reduced TRPC6 mRNA levels in the blood cells of patients with Alzheimer's disease and mild cognitive impairment. Mol. Psychiatry http://dx.doi.org/10.1038/mp.2017.136 (2017).
Roberts, B. R. et al. Biochemically-defined pools of amyloid-β in sporadic Alzheimer's disease: correlation with amyloid PET. Brain 140, 1486–1498 (2017).
Bush, A. I. et al. The amyloid precursor protein of Alzheimer's disease is released by human platelets. J. Biol. Chem. 265, 15977–15983 (1990).
Li, Q. X. et al. Secretion of Alzheimer's disease Aβ amyloid peptide by activated human platelets. Lab. Invest. 78, 461–469 (1998).
Citron, M. et al. Excessive production of amyloid β-protein by peripheral cells of symptomatic and presymptomatic patients carrying the Swedish familial Alzheimer disease mutation. Proc. Natl Acad. Sci. USA 91, 11993–11997 (1994).
Li, S., Liu, B., Zhang, L. & Rong, L. Amyloid β peptide is elevated in osteoporotic bone tissues and enhances osteoclast function. Bone 61, 164–175 (2014).
Kuo, Y. M. et al. Elevated Aβ42 in skeletal muscle of Alzheimer disease patients suggests peripheral alterations of AβPP metabolism. Am. J. Pathol. 156, 797–805 (2000).
Zhang, X. et al. Hypoxia-inducible factor 1α (HIF-1α)-mediated hypoxia increases BACE1 expression and β-amyloid generation. J. Biol. Chem. 282, 10873–10880 (2007).
Sun, X. et al. Hypoxia facilitates Alzheimer's disease pathogenesis by up-regulating BACE1 gene expression. Proc. Natl Acad. Sci. USA 103, 18727–18732 (2006).
Acknowledgements
The authors' research work is supported by the National Natural Science Foundation of China (grants 81471296 and 81625007 to Y.-J.W., and grant 81600949 to J.W.), and the Chinese Ministry of Science and Technology (grant 2016YFC1306401 to Y.-J.W.). The authors thank Dr J. Piña-Crespo and Dr H. Xu at Sanford Burnham Prebys Medical Discovery Institute, USA, for critical reading of the paper.
Author information
Authors and Affiliations
Contributions
All authors contributed to researching data for the article, discussion of the content, writing the article, and to review and/or editing of the manuscript before submission.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
About this article

Cite this article
Wang, J., Gu, B., Masters, C. et al. A systemic view of Alzheimer disease — insights from amyloid-β metabolism beyond the brain. Nat Rev Neurol 13, 612–623 (2017). https://doi.org/10.1038/nrneurol.2017.111
Published:
Issue Date:
DOI: https://doi.org/10.1038/nrneurol.2017.111
This article is cited by
-
Therapeutics for neurodegenerative diseases by targeting the gut microbiome: from bench to bedside
Translational Neurodegeneration (2024)
-
Identification of immunogenic cell death-related genes involved in Alzheimer’s disease
Scientific Reports (2024)
-
Flavonoids and Alzheimer’s disease: reviewing the evidence for neuroprotective potential
Molecular and Cellular Biochemistry (2024)
-
Kamikihito reduces β-amyloid25–35-induced axon damage via neurotrophic factors
Journal of Natural Medicines (2024)
-
Myelin in Alzheimer’s disease: culprit or bystander?
Acta Neuropathologica Communications (2023)
