
Key Points
-
Traumatic brain injury (TBI) is an important public health issue: the global incidence of TBI is on the rise, and mild, repetitive and blast injuries, in particular, are increasingly recognized in the popular press
-
Neuroinflammation, triggered by release of endogenous danger signals and innate immune activation, is crucial to recovery after TBI; however, a dysregulated immune response can result in secondary injury
-
After TBI, the activity of microglia and infiltrating macrophages and adaptive immune cells is driven by extracellular injury signals and intracellular molecular pathways that might represent novel therapeutic targets
-
Trials assessing immunomodulatory interventions should account for changes in neuroinflammation that occur over time, between injury type and severity, and across patient characteristics such as age, sex and genetic variability
-
Some individuals with TBI develop chronic neuroinflammation, which can last for years after the injury, and is being investigated as a link to accelerated neurodegeneration and chronic traumatic encephalopathy
Abstract
The 'silent epidemic' of traumatic brain injury (TBI) has been placed in the spotlight as a result of clinical investigations and popular press coverage of athletes and veterans with single or repetitive head injuries. Neuroinflammation can cause acute secondary injury after TBI, and has been linked to chronic neurodegenerative diseases; however, anti-inflammatory agents have failed to improve TBI outcomes in clinical trials. In this Review, we therefore propose a new framework of targeted immunomodulation after TBI for future exploration. Our framework incorporates factors such as the time from injury, mechanism of injury, and secondary insults in considering potential treatment options. Structuring our discussion around the dynamics of the immune response to TBI — from initial triggers to chronic neuroinflammation — we consider the ability of soluble and cellular inflammatory mediators to promote repair and regeneration versus secondary injury and neurodegeneration. We summarize both animal model and human studies, with clinical data explicitly defined throughout this Review. Recent advances in neuroimmunology and TBI-responsive neuroinflammation are incorporated, including concepts of inflammasomes, mechanisms of microglial polarization, and glymphatic clearance. Moreover, we highlight findings that could offer novel therapeutic targets for translational and clinical research, assimilate evidence from other brain injury models, and identify outstanding questions in the field.
Similar content being viewed by others
Main
The Centers for Disease Control and Prevention estimates that in the USA, 1.7 million people experience traumatic brain injury (TBI) each year, and 5.3 million are living with TBI-related disability1. These figures may grossly underestimate the scope of the TBI epidemic, particularly for mild TBI (mTBI) which often goes unreported2 and, globally, the incidence of TBI appears to be increasing1. TBI and mTBI are 'signature injuries' of the wars in Iraq and Afghanistan, primarily because many military personnel are exposed to blast injuries from conventional and improvised explosive devices3. In addition, TBI has now been linked to post-traumatic stress disorder, memory deficits, chronic traumatic encephalopathy (CTE), and chronic neuroinflammation.
The inflammatory reaction to TBI was thought to occur solely through peripheral immune mediators entering the CNS via a disturbed blood–brain barrier (BBB); however, inflammation after TBI is now recognized as a robust and complex interaction between central and peripheral cellular and soluble components, influenced by patient age, sex, mechanism of injury (focal, diffuse, blast), degree of injury (mild, repetitive mild, severe), secondary insults (hypoxaemia, hypotension), therapeutic interventions, and genetic variability. TBI leads to early resident microglial activation and peripheral neutrophil recruitment, followed by infiltration of lymphocytes and monocyte-derived macrophages4. Simultaneously, proinflammatory and anti-inflammatory cytokines vie to promote and terminate the post-traumatic neuroinflammatory response, and chemokine signalling results in the activation and recruitment of immune cells towards the lesion5,6,7,8,9.
Post-traumatic inflammation can be beneficial, because it promotes both clearance of debris and regeneration, and/or potentially harmful, by mediating neuronal death and progressive neurodegeneration (Fig. 1). Anti-inflammatory therapies have demonstrated efficacy in preclinical and single-centre trials; unfortunately, however, these therapies failed to show benefit — and several were even deleterious — in multicentre clinical trials10,11.

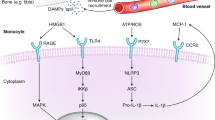
Primary mechanical injury to the CNS may cause cell membrane disruption, vascular rupture and blood–brain barrier (BBB) damage followed by secondary reactions involving ionic imbalance, release of excitatory amino acids, calcium overload, and mitochondrial dysfunction, culminating in cell death pathways. Primary and secondary injury lead to release of damage-associated molecular patterns, cytokines, chemokines, activation of microglia and astrocytes, and recruitment of circulating immune cells. Temporally, these immune responses largely overlap. The inflammatory response is crucial to clearance of debris, repair, and regeneration after traumatic brain injury. However, dysregulated inflammation can lead to additional acute and chronic brain injury. 4-HNE, 4-hydroxynonenol; HMGB1, high mobility group protein B1; mtDNA, mitochondrial DNA; ROS, reactive oxygen species.
In this Review, we propose a new framework to guide future preclinical and clinical trials to optimize the immune response to TBI. The purpose of immune-targeting interventions in TBI is to limit the acute proinflammatory response to the level needed for clearance of debris and danger signals, promote an anti-inflammatory and pro-regenerative immune phenotype, and prevent the development of chronic neuroinflammation. Using this framework, we review the dynamics of the immune response to TBI, progressing from initiation of acute inflammation by danger signals and early inflammatory mediators, to subacute inflammation occurring days to weeks after injury and, finally, to chronically activated elements of the immune system that can remain active for months to years and have been linked to the development of traumatic encephalopathies. Mechanisms that balance proinflammatory and pro-reparative immune activation are discussed, as well as the potential for therapies to promote beneficial aspects of inflammation. We discuss recent discoveries in immunology and our current understanding of the possible roles of processes and systems in the immune response to TBI. Acknowledging the limitations of TBI models12,13, we incorporate in this comprehensive Review what is known from human studies over the past two decades of TBI research, although notably, limited human data are available for mTBI. Lastly, considering the current knowledge of post-traumatic neuroinflammation, we propose new areas for advancing translational and clinical research.
Acute and subacute neuroinflammation
Inflammatory triggers
Damage-associated molecular patterns. Cellular membrane disruption as a result of primary mechanical insult or secondary injury causes release of damage-associated molecular patterns (DAMPs)4,14,15 (Table 1). In response, tumour necrosis factor (TNF), IL-6 and IL-1β are upregulated rapidly by local glial cells and infiltrating immune cells16 and represent early effectors that drive post-traumatic neuroinflammation (Table 1).
The dual nature of inflammation was demonstrated in experimental models investigating the role of TNF and inducible nitric oxide synthase (iNOS) after TBI. TNF is linked to brain oedema, BBB disruption, and recruitment of leukocytes9. However, TNF−/− mice had impaired motor function and larger lesions at 4 weeks after injury, despite showing early neuroprotection17. Similarly, although TBI increased iNOS expression in the brain, with multiple proinflammatory and neurotoxic effects, genetic or chemical iNOS blockade resulted in worsened spatial memory 2–3 weeks after injury18.
Cell death via programmed necrosis, such as necroptosis through TNF-mediated RIP kinase activation19, can lead to a vicious cycle: necrosis increases membrane disruption, which promotes DAMP release, which in turn further exacerbates necrosis and amplifies inflammation. The prototypical DAMP, high mobility group protein B1 (HMGB1), is increased in cerebrospinal fluid (CSF) of patients after severe TBI, and is associated with elevated intracranial pressure in adults and poor outcome in children20,21. HMGB1 is a structural DNA-binding protein that regulates transcription by stabilizing nucleosomes under normal conditions21. This protein can be released from cells by membrane disruption or actively secreted by monocytes or macrophages, and it signals through RAGE (receptor for advanced glycation end products) and Toll-like receptor (TLR) 2 and TLR4 to increase production and release of cytokines22.
One mechanism of cytokine production triggered by DAMPs is the activation of the inflammasome complex. Binding to intracellular pattern recognition receptors such as the NLR (NOD-like receptor) family or absent in melanoma (AIM) leads to autoactivation of caspase-1 and processing of pro-IL-1β and pro-IL-18 to their active forms23. Relatively few inflammasome complexes are expressed in the brain: NLRP1 and AIM2 are present in neurons24,25 and NLRP3 is found in astrocytes25 and microglia in both mice and humans26,27. In patients, NLRP1 and caspase-1 levels are increased in the CSF after severe TBI, and are associated with unfavourable outcomes25. In mice, neutralization of the NLRP1 and NLRP3 inflammasomes attenuated IL-1β processing and reduced lesion volume24,28. Inflammasome-dependent cytokine production also contributes to disease progression in mouse models of multiple sclerosis, Alzheimer disease (AD) and amyotrophic lateral sclerosis26,29,30. However, it remains unclear which inflammasome complexes are the primary producers of IL-1β and IL-18 after TBI, and whether neurons, microglia or astrocytes are the key cellular mediators of inflammasome-mediated tissue damage.
Glutamate excitotoxicity. Concurrent with the release of DAMPs, a massive increase in extracellular glutamate (and other excitatory amino acids)31,32 can occur, leading to excitotoxic neuronal injury via activation of neuronal glutamate receptors, such as N-methyl-D-aspartate (NMDA) and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors, followed by Ca2+-dependent degeneration33. In mice, multiple interactions between inflammatory mediators and glutamate signalling have been demonstrated, including changes in cell surface expression, distribution and function of NMDA and AMPA receptors, mediated by TNF and IL-1β; NMDA receptor-mediated induction of inflammatory gene expression; and a TNF-and-IL-1β-mediated reduction in astrocytic glutamate transporters, resulting in impaired glutamate clearance from the synaptic cleft34. NMDA receptor blockade is an attractive therapeutic strategy to suppress inflammation after TBI; however, NMDA receptor antagonists have failed in clinical TBI trials, partly owing to a limited therapeutic window and off-target neurotoxicity, as well as the effects of inhibiting normal synaptic function and plasticity35.
In response to TBI and glutamate toxicity, high levels of the endogenous neuroprotectant adenosine are produced through breakdown of adenosine triphosphate and mRNA36. Activation of adenosine receptor A1 after TBI has anti-excitotoxic37 and anti-inflammatory effects in mice38; however, systemic administration of adenosine to patients can result in bradycardia and hypotension. A variety of adenosine-associated strategies are being actively investigated to mitigate excitoxicity and various facets of acute and chronic neuroinflammation39.
Mitochondrial dysfunction. Mitochondrial dysfunction and reactive oxygen species (ROS) generation, caused by direct and indirect injury after TBI, have also been identified as triggers of neuroinflammation40. Translocation of the phospholipid cardiolipin from the inner to the outer mitochondrial membrane, shown to occur after experimental TBI, tags damaged mitochondria for mitophagy, and can also be a final pathway for inflammasome activation41,42. Failure of mitophagy and resultant cell death can lead to release of mitochondrial DAMPs: in children with TBI, increased levels of mitochondrial DNA have been reported in the CSF43. These mitochondrial danger signals produce local and systemic responses through interaction with receptors on immune cells: mitochondrial DNA via TLR9 on dendritic cells and N-formyl peptides via formyl peptide receptor 1 on neutrophils44. Membranes with mitochondrial cardiolipins on their surface are engulfed via phagocytosis dependent on the CD36 protein45. There is a paucity of data regarding the CD36-mediated inflammatory response after TBI; however, one study has indicated that CD36 has a beneficial role in neurological outcomes in patients with intracranial haemorrhage, probably by facilitating haematoma resorption46. Although individuals with TBI were excluded from the study, intracranial haemorrhage is present in many patients with moderate to severe TBI, and CD36-mediated haematoma resorption may outweigh the harm of CD36-mediated inflammation.
Vascular injury. In addition to inflammatory triggers released from neurons and glia, trauma to the vasculature can lead to leakage of blood components, including complement and the potent neurotoxin and immune modulator cell-free haemoglobin (fHb), into the cerebral parenchyma47. Complement factors have been detected in brain tissue48 and CSF49 of patients within hours of severe TBI, and they correlate with increased BBB permeability49. Evidence from murine models50 suggests that although complement system activation is required for normal wound healing, TBI can cause acute dysregulation of complement, resulting in secondary injury51. Indeed, in a weight-drop model of TBI in mice, inhibition of the alternative complement pathway52 or the membrane attack complex53 reduced neurodegeneration and axonal loss, and improved neurological outcomes. fHb–haptoglobin complexes are released in the contused areas of the brain, where they are cleared by CD163 receptors on microglia and macrophages which, as a result, differentiate to an anti-inflammatory phenotype54. If the haptoglobin–CD163 pathway is dysfunctional or is outcompeted by the amount of fHb present, fHb and its breakdown products haem and iron can, presumably, induce direct neuronal toxicity by generating ROS and scavenging nitric oxide55, as demonstrated in rodent models of post-traumatic epilepsy56. CSF levels of soluble CD163 and the iron-binding protein ferritin are increased after TBI in children, and they correlate with injury severity and unfavourable outcome57. Thus, inflammation triggered by fHb and its degradation products could be a therapeutic target after TBI.
Several of the biochemical and molecular mechanisms of secondary injury listed above have been reported in blast-induced mTBI animal models. Characterized by axonal, periventricular and hippocampal neuronal injury, blast-induced mTBI is associated with cytokine and chemokine release, adenosine production (probably from mRNA breakdown), and activation of microglia58,59. Promising neuroprotective effects were demonstrated with the anti-inflammatory drug minocycline following blast injury in rats60.
Cellular regulation of inflammation
Dynamics of the cellular response. Several mouse studies have elucidated the time course of inflammatory response in TBI. The first circulating immune cells to infiltrate the CNS after trauma are neutrophils, numbers of which typically peak within 24–48 h of the initial injury then rapidly decline61. Diapedesis between endothelial cells is dependent on binding of integrins to vascular adhesion molecules. Within 4 h of experimental TBI, expression of the neutrophilic vascular adhesion molecules E-selectin (CD62E) and intracellular adhesion molecule 1 (CD54) increases on the endothelium of the injured hemisphere62. Administration of antibody to the CD11d/CD18 integrin reduced leukocyte infiltration to the CNS, as well as the systemic inflammatory response to TBI63,64 (Box 1). Chemokine gradients attract monocytes from the circulation to injured brain tissue, where they differentiate into macrophage subpopulations distinguished by cell-surface expression of chemokine receptors65. Studies of monocyte infiltration in mice have demonstrated accumulation within the lesion through to 3 days post-injury66,67. Dendritic cells, T lymphocytes and natural killer cells are similarly recruited during this period68, but in lower numbers.
Concurrently within the CNS, astrocytes — vital regulators of CNS inflammation — undergo reactive astrogliosis characterized by morphological and functional adaptations, including upregulation of glial fibrillary acidic protein (GFAP) and production of cytokines and chemokines that further recruit and activate immune cells69,70. YKL-40, a marker of reactive astrocytes, is significantly elevated in the CSF of adults with severe TBI on day 2, and peaks on day 4 post-injury (Table 1). Microglia undergo a similar transformation in morphology and function, with an initial peak approximately 7 days post-injury54,71,72.
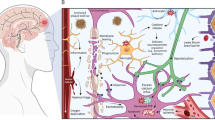
Multiple phenotypes of microglia. The concept of post-traumatic neuroinflammation as a 'double-edged sword' (Ref. 73), with both beneficial and injurious effects, has recently been expanded to include the function of microglia74,75. Like peripheral macrophages, microglia respond to changes in their microenvironment to become polarized along an activation spectrum ranging from the classic M1-like phenotype to an alternative M2-like phenotype76 (Fig. 2). This concept has evolved from the canonical M1 versus M2 classification to reflect mixed phenotypes and the functional plasticity of tissue macrophages and microglia to changes in the microenvironment. Stimulation by DAMPs, free radicals, or proinflammatory cytokines such as IFNγ induces a M1-like phenotype77. Although M1-like 'proinflammatory' cells are often presumed to be harmful, a well-regulated M1-like response could be neuroprotective after TBI. An exaggerated or prolonged M1-like response, however, can lead to secondary brain injury and drive a self-propagating hyperinflammatory state78,79. The M2a-like 'alternative' phenotype, a response to IL-4 and IL-13 stimulation, is associated with production of anti-inflammatory cytokines and increased phagocytic activity77. The M2c-like 'de-activated' phenotype occurs in response to IL-10, glucocorticoids or uptake of apoptotic cells, and regulates tissue repair and remodelling77. The M2b-like 'intermediate' phenotype is stimulated by immune complex exposure or TLR ligands76,77, and has both proinflammatory (mediated by IL-1, IL-6 and TNF) and anti-inflammatory (mediated by IL-10) effects77. The degree to which microglia assume a particular phenotype (or multiple phenotypes) depends on these and other changes in the lesion microenvironment driving complex intracellular signalling pathways, influenced by genetic and epigenetic factors, that might offer additional opportunities for therapeutic intervention77,80.

Molecular signals from injured tissue drive phenotypic and functional responses in microglia or macrophages after traumatic brain injury (TBI). Damage-associated molecular patterns (DAMPs) released by injured neurons, and proinflammatory or oxidative mediators released by infiltrating immune cells polarize cells towards an M1-like phenotype. M1-like populations are characterized by expression of proteins such as IL-1β, TNF, IL-6, NOS2, IL-12p40, and NOX2. Molecular pathways that regulate the M1 phenotype include signal transducer and activator of transcription 1 (STAT1), interferon regulatory factor (IRF)-3/5, nuclear factor-κB (NF-κB), p50/p65 and microRNA (miR)-155. M1-like cells release proinflammatory factors and free radicals that promote neuroinflammation, oxidative stress and neurodegeneration. In response to anti-inflammatory and neurotrophic signals, microglia and macrophages can be polarized towards an M2-like phenotype, characterized by expression of proteins such as CD206, CD163, arginase-1, FCγR, Ym1, IL-10, and TGFβ. Molecular pathways that regulate M2-like phenotypic transitions include STAT6/3, IRF-4/7, NF-κB p50/p50, Nrf2 and miR-124. M2-like microglia and macrophages release anti-inflammatory and trophic factors that resolve inflammation. They have increased phagocytic activity and promote repair by modulating neurogenesis, axonal regeneration, synaptic plasticity, and angiogenesis. Microglia and macrophages demonstrate marked plasticity and can switch between M1-like and M2-like phenotypes. Following TBI, mixed phenotypes are present during the acute phase, transitioning to an M1-like-dominant phenotype in the chronic phase.
Microglial polarization has been shown to vary over time and between different TBI models. In mice, activated microglia demonstrate a bimodal increase after focal contusion with an initial M2-like peak at 7 days followed by an M1-like peak at 21–28 days, although it should be noted that the bulk of activated microglia have mixed M1-like and M2-like activation markers68,71,79. In diffuse brain injury, M1-like versus M2-like polarization dynamics are strikingly different, probably as a result of altered cellular immune responses that include reduced neutrophil infiltration and restricted macrophage or microglial accumulation in white matter regions that incurred the greatest damage. Diffuse brain injury results in transient increases in IL-1β, TNF and CD14 expression in the cortex and hippocampus of mice as early as 4 h after injury; the levels of these molecules return to baseline by 72 h (Ref. 81). In addition, mixed M1-like and M2-like activation markers are present on microglia or macrophages 24 h after injury in mice82. However, the functional role of M1-like versus M2-like phenotypes in axonal injury and repair following diffuse brain injury remains to be elucidated.
Links to adaptive immune response. The adaptive immune response, mediated by T cells and B cells, can strongly influence microglial phenotype and function, but the involvement of this system after TBI remains unclear. Data from rodent studies have shown that T cells infiltrate the injured tissues to promote inflammation after experimental spinal cord injury; fingolimod administration sequestered lymphocytes in lymph nodes, thereby suppressing inflammation and promoting recovery83,84,85. However, fingolimod also acts directly on CNS cells, complicating the interpretation of these findings. Mice genetically engineered to lack T cells have worse outcomes after experimental CNS injury86,87, suggesting that T cells are predominantly neuroprotective.
Somewhat counterintuitively, activation of autoimmune T cells protects mice from secondary neurodegeneration in CNS injury, a phenomenon termed 'protective autoimmunity' (Refs 86,88). One potential mechanism underlying this protection is the production, by T cells, of neurotrophic factors that promote survival and repair of neurons and astrocytes89,90. Mice lacking T cells show cognitive, behavioural and developmental abnormalities, suggesting that T cells contribute to neurodevelopment; these cells might also participate in maintenance of normal brain function91. As well as regulating the M1-like versus M2-like balance, T-cell-produced IL-4 protects neurons by amplification of neurotrophin signalling86. Although T-cell responses typically require antigen presentation to the T-cell receptor, IL-4-mediated protection of injured CNS tissue does not seem to require antigen-specific receptor activation of T cells, and DAMPs from injured neurons directly induce differentiation of IL-4-producing T cells86. IL-33, an interleukin belonging to the IL-1 family that is released from damaged cells, is also neuroprotective after CNS injury in mice92. IL-33 is known to act on type 2 T-helper (TH2) cells that produce IL-4 (Ref. 93); hence, IL-33 could provide a link between CNS injury and activation of IL-4 production. An IL-33-responsive population of tissue-resident regulatory T cells has been identified in the muscle94,95 and gut of mice96, and contributes to resolution of inflammation and wound repair in those tissues. It is intriguing to speculate that a similar IL-33–regulatory T-cell axis could operate in the human brain after TBI.
To harness the neuroprotective benefits of T-cell activity without adverse effects, the specific mechanisms of T-cell-mediated protection versus damage need to be precisely targeted. The methods used to evoke an autoimmune response that protects from injury in mouse models of TBI are also used to induce experimental autoimmune encephalomyelitis (EAE), which is used to model multiple sclerosis in rodents. TH17 cells are thought to drive inflammatory demyelination of the spinal cord in EAE97. TH17 cells and other type 17 T cells have been associated with myriad autoimmune and inflammatory conditions98 but have not yet been investigated in TBI.
Potential involvement of the gut–brain axis. Type 17 responses are promoted by cytokines, particularly IL-1β, that are known to be released after TBI in humans, and induce CXCL8 and neutrophil recruitment. In ischaemic stroke modelled in mice, harmful IL-17 is largely produced by 'type 17' γδT cells that rapidly infiltrate the injured brain99. These cells are strongly influenced by the remote gut environment, as shown in another mouse study in which antibiotic-induced dysbiosis of gut microbial flora resulted in protection from stroke, an effect that could be linked to reduced numbers of IL-17+ γδT cells100. The profound impact of the gut microbiome on peripheral tissue immune responses, including the CNS, is a recurring theme in immunology101,102. Besides CNS injury, the gut–CNS communication has been suggested to influence cognition, mood and anxiety103,104. Thus, it is possible that administration of antibiotics or changes in diet during intensive care unit hospitalization after severe TBI could inadvertently alter this gut microbiome–brain inflammation axis.
Lymphatic and glymphatic systems. Lymphatic drainage of body tissues regulates the flow of interstitial fluid and removal of waste products. The lymphatic system also supports immune surveillance by carrying macromolecules and activated antigen-bearing dendritic cells to local lymph nodes, where the antigens can be presented to activate the adaptive immune response. Antigen presentation may represent a critical step, as naive T cells and B cells typically circulate through lymph nodes via blood and efferent lymphatics but do not enter non-lymphoid tissues until primed.
Until recently, the brain was considered an immune-privileged site: the apparent lack of a lymphatic drainage system in the brain supported the assumption that the peripheral immune system was unresponsive to pathologies in the CNS. However, adaptive immune responses are primed and recruited as a response to CNS injury, and waste products must be rapidly cleared from the brain. Technological advances in imaging that enabled interrogation of brain drainage in closed-skull systems have resolved these paradoxes and, in the past few years, two — most probably intersecting — systems that drain the brain tissue have been delineated in mice.
The glymphatic system105 allows small molecules to rapidly enter the brain and, perhaps more importantly, enables fluid drainage and clearance of metabolites, soluble proteins and waste products, including amyloid-β (Aβ), from the brain interstitial space. Glymphatic flow is greatly increased during sleep, when the brain interstitial space volume is increased; this phenomenon seems to be at least partly attributable to passive mechanisms, as lateral posture in awake mice replicated the increased flow compared with upright posture106,107.
The second CNS clearance system consists of lymphatic vessels that line the dural sinuses and meningeal arteries108,109. These vessels have classic lymphatic architecture and drain to the deep cervical lymph nodes, providing a direct conduit between the brain and the peripheral immune system. These vessels also contain immune cells and macromolecules, mimicking peripheral lymphatic vessels. Brain lymphatic vessels include populations of T cells and B cells109, which have presumably migrated through and surveyed the brain tissue.
In a rodent model, TBI has been shown to impair the glymphatic system drainage110, resulting in accumulation of damage and waste products such as tau111, and providing a potential link between injury-induced disruption of glymphatic drainage and development of CTE. Inflammatory astrocyte activation might amplify the effects of mechanical damage on glymphatic flow after TBI, as glymphatic flow is determined in large part by astrocyte function, in particular, aquaporin-4 channels. The effects of TBI on brain lymphatic drainage to deep cervical lymph nodes have not yet been investigated, but one could envision that TBI would alter the associated lymph vessels. Accumulation of waste products as a result of impaired lymphatic drainage might trigger neuroinflammation by activating pattern recognition receptors on microglia. The interaction between altered lymphatic drainage and neuroinflammation and the ensuing long-term consequences warrant further investigation.
Effects of secondary insults
The presence of a concurrent secondary insult such as polytrauma, hypotension and/or hypoxaemia is a critical determinant of outcome after TBI, particularly severe TBI. Secondary insults occur in as many as two-thirds of patients with severe TBI112. These insults are often haemorrhagic, and compromise perfusion and oxygen delivery to the injured brain113. Analysis of >2,000 patients revealed that mortality was 72% in patients with severe TBI and a comorbid secondary injury, compared with 46% in patients with TBI alone114.
Despite the importance of polytrauma and secondary insults in TBI, few preclinical or clinical studies have evaluated their role in the cerebral or systemic inflammatory responses. Although one might anticipate that polytrauma and/or secondary insults would amplify the local inflammatory response in the brain by superimposing tissue hypoxaemia and/or ischaemia onto the traumatic insult, such an effect has, surprisingly, not been observed. Instead, both preclinical and clinical studies have revealed that second insults shift the cytokine response to a more anti-inflammatory phenotype, amplifying the IL-10 response. In a mouse model of polytrauma (controlled cortical-impact TBI followed by a brief period of severe haemorrhagic shock), serum IL-10 levels increased nearly 100-fold compared with TBI alone and 30-fold compared with haemorrhagic shock alone115. Levels of proinflammatory cytokines and chemokines (IP-10, TNF, CXCL1, CCL2, CCL3 and CCL11) increased in serum after TBI alone, but not after TBI plus haemorrhagic shock, and the mice with TBI plus haemorrhagic shock had lower serum IL-6 levels than did animals exposed to TBI alone. Although haemorrhagic shock worsened long-term behavioural outcomes and histologically confirmed pathology after TBI116, the local cytokine and chemokine responses in the brain were not appreciably altered.
In general, clinical data parallel these preclinical findings. Relative to patients with TBI alone, patients with TBI with polytrauma have increased serum concentrations of the anti-inflammatory agents IL-10, IL-1ra, and soluble TNF receptor 1, with no change in the levels of proinflammatory cytokines IL-1β and TNF117. In a longitudinal analysis of cytokines and adhesion molecules in 114 adults with severe TBI, serum IL-10 levels were higher among individuals with TBI plus polytrauma than in patients with TBI alone118.
It remains unclear whether haemorrhagic shock confers a unique effect, or whether all types of secondary insults produce a similar shift toward an anti-inflammatory phenotype. Certain forms of peripheral injury, such as skeletal fracture or hepatic contusion, might increase the circulation of proinflammatory cytokines in patients. Combined-injury animal models that incorporate long-bone fracture with TBI are increasingly used to address the effects of peripheral injuries on TBI outcomes119,120. In a mouse model of tibia fracture combined with diffuse brain injury, mice with combined injury exhibited increased anxiety-related behaviour and brain atrophy, and evidence of increased astrogliosis, neutrophil infiltration and brain tissue IL-1β levels121. Similarly, systemic administration of the proinflammatory mediators IL-1β122 and lipopolysaccharide123 in rodent models of diffuse TBI have been demonstrated to exacerbate the neuroinflammatory response, result in larger contusion volume, and worsen behavioural outcomes. Whether these effects are mediated directly via binding to receptors on microglia and astrocytes is not known, and confounding indirect effects such as hypotension or hyperthermia cannot currently be excluded.
Finally, there has been limited study of the impact of secondary insults in mTBI. In rats, a brief period of imposed hyperthermia to 39 °C, beginning 15 min before mild fluid percussion injury and continued for 4 h, produced cognitive deficits even though the injury level used was so mild that it would not have otherwise resulted in cognitive deficits124. Cooling the rats to normothermia at 15 min after TBI prevented development of the cognitive deficits, a finding that the researchers attributed to amplification of neuroinflammation by hyperthermia. Given the high prevalence of concussions during the summer months, for example, in football and other sports training camps, this observation, if translated to humans, could have important implications.
Neuroinflammation and regeneration
Neurogenesis. The effects of neuronal death after TBI might be mitigated by an increased rate of neuronal progenitor cell (NPC) proliferation, followed by migration to injured brain regions, differentiation to neurons, and integration into neural networks125. Similar to the dual effects of inflammation on secondary brain injury, experimental evidence suggests that inflammatory mediators are a key component of neurogenesis and can either support or hinder the potentially neuroprotective role of NPCs at several stages. For example, microglia stimulated to differentiate to an M1-like phenotype with lipopolysaccharide reduce hippocampal neurogenesis in the adult mouse126, an effect similarly seen with the proinflammatory cytokines TNF, IL-1β and IL-6 (Ref. 127). Reduced neurogenesis was reversed by treatment with minocycline126 or indomethacin128. By contrast, M2-like microglia, induced by IL-4 or low-dose IFNγ, release neurotrophins such as insulin-like growth factor-1 (IGF-1) and induce neurogenesis129. Neurogenesis may be stimulated by addition of a running wheel to the cage after experimentally induced TBI in mice, which was shown to reduce M1-like microglial activation and increase production of IGF-1, IL-10 and brain-derived neurotrophic factor; these changes were associated with improved cognitive outcomes130. NPCs also express chemokine receptors, such as CCR2 and CXCR4, and chemokines may direct the migration of these cells to the area of injury131.
Although brain atrophy increases over time after TBI132,133, most TBI survivors show functional improvements134, which are attributed not only to recovery of function of existing neuronal pathways but also to brain plasticity and reorganization. Although studies in TBI animal models are limited, experiments modelling other forms of brain injury suggest that interactions between the neuronal unit and immune system are critical to formation and strengthening of new synaptic connections135. The degree of activation and the local inflammatory milieu are likely to define whether any particular cytokine or inflammatory cell type benefits or disrupts brain plasticity. For example, in a GFAP–IL-6 transgenic mouse model, overexpression of IL-6 caused a substantial reduction in hippocampal long-term potentiation (LTP)136; this effect was partially reversed by blocking of IL-6 signalling137. Elevated IL-1β also impairs LTP but, surprisingly, promotes neurite outgrowth synergistically with neurotrophin-3, suggesting that IL-1β-blocking therapies would promote neuronal survival at the expense of reinnervation138. In rats subjected to repetitive mTBI, activation of microglia was associated with impaired LTP, attenuated NMDA signalling and impaired memory — effects that were not seen after a single mTBI139. Thus, neuroinflammation is likely to have a critical — though patient-specific — role in neurogenesis and synaptic plasticity after TBI.
Angiogenesis and gliogenesis. Microglial polarization and the local inflammatory milieu might influence repair through activation of angiogenesis and gliogenesis. Although evidence from animal models of TBI is scant, findings from experimental autoimmune and hypoxic–ischaemic brain injury suggest that M1-like microglia impair oligodendrogenesis, and oligodendrocyte maturation and viability, via a TNF-dependent mechanism80,140. By contrast, M2-like microglia or conditioned medium from M2-like microglial culture promote oligodendrogenesis and remyelination in these models. M2-like microglia might also induce angiogenesis and vascular repair via production of pro-angiogenic cytokines (such as TGFβ) and growth factors. Whether these findings translate to recovery from TBI in humans is unknown; however, treatment with pro-angiogenic growth factors released by M2-like microglia has been demonstrated to be neuroprotective in mice, and promotes neurogenesis and angiogenesis in experimental TBI141.
Clinical experience
A key question arising from the data discussed above is whether TBI-responsive neuroinflammation is a clinically relevant therapeutic target (Fig. 3). Several preclinical studies have supported this hypothesis; however, few clinical trials of therapies primarily targeting inflammation TBI have been reported.

Therapies targeting traumatic brain injury (TBI)-responsive inflammation may be effective at different time points depending on the therapeutic target(s). Inflammation triggered by release of damage-associated molecular patterns (DAMPs) and reactive oxygen species (ROS) can be blocked through the use of antioxidants, minocycline and peroxisome proliferator-activated receptor (PPAR) agonists. Inflammasome activation will cause release of IL-1β, the action of which can be inhibited at IL-1 receptors with IL-1ra (anakinra). Over the next few hours to days, invasion of the CNS by circulating immune cells can be inhibited by therapies such as NK1 and chemokine antagonists. Microglial polarization to an M2-like phenotype can be promoted by mesenchymal stem cells (MSCs), PPAR agonists and CCR2 antagonists. T cells must be primed to enter the CNS — this process might be inhibited by therapies such as intravenous immunoglobulin. Additionally, alterations in the gut microbiome may affect the relative numbers of proinflammatory and anti-inflammatory T lymphocytes. Glymphatic clearance may be impaired after TBI, possibly reducing clearance of proinflammatory mediators. Investigations are ongoing to determine ways to improve glymphatic flow, such as increased clearance observed during sleep. Chronic microglial activation might develop and lead to neurodegeneration. Activation of mGluR5 on microglia, such as with (RS)-2-chloro-5-hydroxyphenylglycine (CHPG), attenuates chronic M1-like microglial activation. Rehabilitation and exercise have also been shown to reduce M1-like microglial activation. CSF, cerebrospinal fluid.
Steroids. Surprisingly, corticosteroids have been the least successful anti-inflammatory class of drugs in TBI (Table 2). Despite the potent suppressive effects on inflammation, high-dose methylprednisolone (5 mg/kg daily)142, large doses of dexamethasone (100 mg daily for 4 days, followed by 16 mg daily from days 5–7 and tapering of the dose from days 8–10 (Ref. 143)), ultra-high-dose dexamethasone (2.3 g over 51 h)144, the aminosteroid tirilazad145, and a trial of hydrocortisone and fludrocortisone146 all failed to demonstrate beneficial effects on neurological outcome. Off-target effects with systemic administration of corticosteroids are likely to affect outcomes and, in the case of tirilazad, limited brain exposure might have been a confounder147.
Other anti-inflammatory drugs. Other anti-inflammatory strategies have been evaluated in humans, including the bradykinin B2 receptor antagonist anatibant, which produced a trend toward worse outcome in 228 patients enrolled148. A provocative randomized controlled trial (RCT) used recombinant human granulocyte colony stimulating factor (G-CSF) to enhance the cellular inflammatory response149. The number of patients was low, and no differences were seen between G-CSF-treated and placebo groups in the primary outcome (nosocomial infections), mortality or hospital length of stay. Minocycline has shown promise in a phase II trial in patients with spinal cord injury150; however, its efficacy has not been tested in human TBI, and preclinical data are equivocal151. Of note, the studies targeting inflammation after TBI have all been performed in adult patients. Given the observed association between heightened inflammation and younger age in children with severe TBI, anti-neuroinflammatory strategies might be more impactful in the developing brain57,152.
Non-selective versus targeted therapies. Inferences can be made by extrapolating from clinical trials that evaluated multifaceted therapies with anti-inflammatory consequences. The anti-inflammatory effect of hypothermia was touted as one of its main modes of efficacy153. Disappointingly, although single-centre studies in adults with severe TBI showed a reduction of inflammatory cytokine levels154, multicentre RCTs of therapeutic hypothermia after TBI have failed to show benefit in either adults155,156 or children11,157. Progesterone, which blunted the neuroinflammatory response to trauma in mice158, was evaluated in two large multicentre RCTs159,160 of adults with moderate to severe TBI, and failed to show benefit with regard to 6-month Glasgow Outcome Scale scores or mortality.
The results from these clinical studies suggest that non-selective attenuation of the inflammatory response early after severe TBI is not beneficial, and can potentially be detrimental. The existing literature lacks studies using targeted, single-pathway anti-inflammatory strategies in humans, and more-personalized approaches that individualize treatments according to genotype, inflammatory biomarkers, timing and duration of therapy, and patient age and sex. Identification of specific patient subsets to target enrolment criteria for clinical studies could increase the success of future trials. For example, a variant of a gene encoding IL-1β was associated with increased risk of post-traumatic epilepsy161, making it an attractive strategy to target antiseizure interventions to individuals at high risk.
Furthermore, it is unclear whether acute inhibition of inflammation after single or repetitive mTBI might prevent chronic sequelae such as CTE. The key question is whether a single exposure or multiple exposures to TBI can prime the brain for chronic neuroinflammation and, if so, would modulation of the inflammatory response early after mTBI, or at a later stage, provide clinically meaningful benefit? Finally, from a therapeutic perspective, given the aforementioned beneficial effects of strategies mimicking rehabilitation on neuroinflammation neurogenesis and cognitive outcome, it is possible that enhancement of beneficial aspects of neuroinflammation — rather than inhibition of detrimental effects — could represent a more successful avenue for future clinical investigation.
Chronic neuroinflammation
Bystander or driver of pathology?
Findings from patients. As already discussed, one might expect the acute inflammatory response after TBI to eventually resolve to a normal state. However, a subset of patients develop chronic neuroinflammation that can last for years after injury162,163,164,165. Areas under active investigation include the proportion of patients in whom chronic inflammation will develop, the dominant triggers and intracellular pathways propagating inflammation, and genetic susceptibilities to chronic inflammation.
An examination of autopsy specimens from patients surviving >1 year after TBI, and in some cases as long as 18 years, revealed an increase in amoeboid microglia in subcortical white matter tracts versus control tissue163,166. Activated microglia were observed in 28% of the autopsies, and were associated with thinning of the corpus callosum163,166. These findings are supported by PET studies in which translocator protein (TSPO) ligands, which are thought to bind to activated microglia, were used to examine chronic neuroinflammation in TBI survivors164,167. In one study of adult patients with moderate to severe TBI, diffuse binding of the TSPO ligand 11C-(R)-PK11195 was observed in areas remote to the trauma, including the thalamus, putamen and occipital cortex, up to 17 years after injury164. Inflammation in the thalamus was associated with more-severe cognitive impairments164. In another study, the second-generation TPSO ligand 11C-DPA-713 was used to study retired National Football League (NFL) players with self-reported histories of career concussions. The supramarginal gyrus and right amygdala exhibited ligand binding to levels greater than seen in age-matched controls167.
Serum cytokines might also demonstrate a chronic immune activation state after TBI. For example, a prospective TBI biomarker study reported chronically elevated expression of TNF in the serum after TBI, and an association between increased TNF and unfavourable long-term neuropsychiatric outcomes168.
Findings from animal models. Animal studies substantiate the clinical evidence for a chronic inflammatory state after TBI, and indicate underlying molecular mechanisms and potential therapeutic strategies139,169,170,171,172,173. In mice, chronic microglial activation with upregulation of the cell surface markers MHC class II, CD68 and NADPH oxidase (NOX2) is seen 1 year after moderate to severe contusion78. These markers would indicate an M1-like phenotype, with proinflammatory cytokine production and reduced phagocytic activity that would be less effective at protective functions such as Aβ clearance. Over the course of the year, the mice demonstrated progressive neurodegeneration with enlarging lesion volume, persistent oxidative stress, demyelination, and cognitive impairments. Chronic neuroinflammation is also observed in several murine models of repetitive mTBI. As in the acute inflammatory response to mild repetitive injury, microglia are characteristically localized to white matter tracts and may be seen in regions bordering degenerating axons, with associated neurobehavioural changes, 12–18 months after injury139,170,174,175,176,177.
Prospects for therapy. These findings have raised interest in the development of pharmacological and nonpharmacological approaches to reduce chronic neuroinflammation after TBI, thereby greatly expanding the window for targeted interventions. The concept of targeting chronic neuroinflammation has been examined in preclinical studies. One of the compounds tested was the selective metabotropic glutamate receptor 5 agonist (RS)-2-chloro-5-hydroxyphenylglycine (CHPG), which was previously shown to reduce microglial activation178 and improve functional recovery179 when given acutely after trauma. Mice given CHPG at 1 month after focal brain injury displayed improved neurological recovery, decreased neuroinflammation, arrested lesion expansion, sparing of white matter, and reduced neurodegeneration at 4 months180. Similarly, though with a more generalized anti-inflammatory approach, administration of the phosphodiesterase inhibitor ibudilast on days 30–34 after fluid percussion injury in rats reduced anxiety-like behaviour and gliosis at 6 months181.
As previously stated, exercise regimens that simulate physical rehabilitation might alleviate neuroinflammation and promote release of neurotrophic factors after TBI. In mice, a 4-week treatment regime involving voluntary exercise attenuated IL-1β gene expression and chronic microglial activation, increased production of IL-10 and neurotrophic factors, improved behavioural outcomes, and reduced lesion volume130. Importantly, the authors compared two start dates for the exercise regimen: exercise was only effective if started 5 weeks after injury, and exercise initiated at 1 week after injury was potentially proinflammatory. Thus, accumulating evidence from preclinical research indicates that chronic neuroinflammation and related neurodegeneration can be treated weeks after TBI, which suggests exciting potential for clinical translation of delayed anti-inflammatory therapies.
Progressive neurodegeneration
Accelerated neurodegeneration and CTE have been reported after single and repetitive TBI in athletes182 and military personnel3, who have a high incidence of head trauma and concussion183. Besides its association with CTE, TBI increases the risk of developing non-AD dementia years after the initial injury184.
Recently, the role of chronic inflammation in the pathophysiology of neurodegenerative disorders has attracted considerable attention185,186, leading investigators to speculate about the role of post-traumatic neuroinflammation in mediating neurodegeneration, non-AD dementias, and CTE. Autopsies from patients with CTE associated with repetitive mTBI have shown activated microglia in perivascular regions of subcortical white matter and, in advanced disease, throughout the brain3,187,188. Two recent neuroimaging studies of retired NFL players support a role for microglial neuroinflammation in the development of CTE following repetitive head injury189,190.
Early intervention with progesterone to prevent neurodegeneration after repetitive mTBI has been evaluated in one rat study191. The animals were administered three mild diffuse injuries, each separated by 5 days, and randomly assigned to vehicle or progesterone for 15 days after the first injury. At 12 weeks after the end of the treatment, a time point considered chronic in rodent TBI models, progesterone-treated rats exhibited improved neurocognitive outcomes, reduced brain atrophy, and attenuated neuroinflammation compared with vehicle-treated rats. These promising data warrant further preclinical studies.
It remains unclear whether persistent inflammation initiates the characteristic neuropathology in CTE and should be targeted by anti-inflammatory treatments. It is also possible that the accumulation of these abnormal proteins triggers the inflammatory response, which may be aberrant and ill-suited to restore normal function. Further research is required to understand the mechanisms that underlie the chronic pathologies of TBI, including chronic neuroinflammation, and their relationship to development of neurodegenerative disease (Fig. 4). Advances in clinical neuroimaging of TBI, including the use of selective PET ligands to reflect amyloid deposition192, tau deposits193 and neuroinflammation164,167, could clarify the mechanisms driving chronic neurodegeneration after TBI, and provide opportunities to develop targeted therapies for the long-term sequelae.

Neuroinflammation and microglial activation are key mediators of repair and recovery after traumatic brain injury (TBI). However, recent clinical and laboratory data have shown that TBI can cause persistent neuroinflammation and microglial activation, in some cases lasting many years, and can lead to chronic neurodegeneration, dementia and encephalopathy. Prospective studies of TBI biomarkers in adults with severe TBI have shown that serum levels of IL-1β, IL-6, CXCL8, IL-10, and tumour necrosis factor (TNF) are chronically increased. Experiments in animal models have demonstrated persistently increased numbers of microglia expressing MHC class II (MHC II), CD68 and NOX2 at the margins of the lesion and in the thalamus at 1 year post-injury associated with oxidative stress and white matter disruption. These inflammatory findings correlate with chronic neurodegeneration, the development of dementia, and encephalopathies (which may subsequently cause additional inflammation in a self-perpetuating deleterious feedback mechanism).
Conclusions
Advances in understanding TBI-responsive neuroinflammation have raised exciting new questions (Box 2), identified new therapeutic targets, and expanded the time window for treatment. Clinical trials of therapies that modulate inflammation after TBI — even in severe TBI — are still in their infancy, and therapies targeting neuroinflammation after mTBI in patients remain completely unexplored. The approach of treating all patients with TBI using a broad-acting anti-inflammatory agent has so far not shown benefit in RCTs. Clearly, there is a need to define the inflammatory phenotypes of our patients on the basis of injury characteristics such as patient age, sex, genetic predisposition, presence or absence of secondary insults, and serum, CSF and/or imaging biomarkers. An initial approach towards a more comprehensive inflammatory phenotyping of patients by principle component analysis — incorporating classic inflammatory markers as well as steroid hormones, neurotrophic factors, and brain-derived TBI biomarkers in serum and CSF — was recently reported by Kumar et al.194. Such an approach will allow us to answer questions posited in our initial framework, including how to target inflammation for clearance of debris, which patients will benefit from therapies to promote reparative aspects of inflammation, and when therapies targeting chronic inflammation should be initiated. This approach should be combined with improved preclinical trials, which incorporate different injury types and severities, and include secondary insults. In addition, preclinical trials should be expanded to define clinically relevant therapeutic window(s) and treatment durations, and include outcomes to examine both harmful and protective aspects of inflammation at acute and chronic end points. Coupled with important new clinical trial design strategies, such as adaptive design, this approach will enable trials to be conducted that target individual patients with personalized immunomodulatory treatments. We hope that this patient-tailored approach to harness inflammation after TBI will reduce secondary injury, enhance repair, and improve patient outcomes.
Review criteria
To identify human data on neuroinflammation associated with traumatic brain injury (TBI), we searched PubMed for articles published in English from January 1950 to March 2016 using the following query: “traumatic brain injury” or “closed head injury” or “closed-head injury” or “head trauma” AND ((Case Reports[ptyp] OR Clinical Study[ptyp] OR Clinical Trial[ptyp] OR Clinical Trial, phase I[ptyp] OR Clinical Trial, phase II[ptyp] OR Clinical Trial, phase III[ptyp] OR Clinical Trial, phase IV[ptyp] OR Controlled Clinical Trial[ptyp] OR Comparative Study[ptyp] OR Meta-Analysis[ptyp] OR Multicenter Study[ptyp] OR Randomized Controlled Trial[ptyp]) AND Humans[Mesh]). We selected articles reporting clinical findings of neuroinflammation in human TBI. Reference lists and the authors' expertise were used for inclusion of additional relevant studies.
Change history
04 August 2017
In the initially published version of this article, reference 194 was incorrectly cited as reference 193 in the Conclusions section. This error has been corrected in the HTML and PDF versions of the article.
References
Roozenbeek, B., Maas, A. I. & Menon, D. K. Changing patterns in the epidemiology of traumatic brain injury. Nat. Rev. Neurol. 9, 231–236 (2013).
Feigin, V. L. et al. Incidence of traumatic brain injury in New Zealand: a population-based study. Lancet Neurol. 12, 53–64 (2013).
Goldstein, L. E. et al. Chronic traumatic encephalopathy in blast-exposed military veterans and a blast neurotrauma mouse model. Sci. Transl Med. 4, 134ra60 (2012).
Corps, K. N., Roth, T. L. & McGavern, D. B. Inflammation and neuroprotection in traumatic brain injury. JAMA Neurol. 72, 355–362 (2015).
Csuka, E. et al. IL-10 levels in cerebrospinal fluid and serum of patients with severe traumatic brain injury: relationship to IL-6, TNF-α, TGF-β1 and blood–brain barrier function. J. Neuroimmunol. 101, 211–221 (1999).
Frugier, T., Morganti-Kossmann, M. C., O'Reilly, D. & McLean, C. A. In situ detection of inflammatory mediators in post mortem human brain tissue after traumatic injury. J. Neurotrauma 27, 497–507 (2010).
Kossmann, T. et al. Interleukin-8 released into the cerebrospinal fluid after brain injury is associated with blood–brain barrier dysfunction and nerve growth factor production. J. Cereb. Blood Flow Metab. 17, 280–289 (1997).
Semple, B. D., Kossmann, T. & Morganti-Kossmann, M. C. Role of chemokines in CNS health and pathology: a focus on the CCL2/CCR2 and CXCL8/CXCR2 networks. J. Cereb. Blood Flow Metab. 30, 459–473 (2010). Excellent review on the several crucial roles of chemokines and their receptors in response to traumatic brain injury, with a focus on two of the best-studied chemokines, CCL2 and CXCL8.
Ziebell, J. M. & Morganti-Kossmann, M. C. Involvement of pro- and anti-inflammatory cytokines and chemokines in the pathophysiology of traumatic brain injury. Neurotherapeutics 7, 22–30 (2010).
Roberts, I. et al. Effect of intravenous corticosteroids on death within 14 days in 10008 adults with clinically significant head injury (MRC CRASH trial): randomised placebo-controlled trial. Lancet 364, 1321–1328 (2004).
Hutchison, J. S. et al. Hypothermia therapy after traumatic brain injury in children. N. Engl. J. Med. 358, 2447–2456 (2008).
Xiong, Y., Mahmood, A. & Chopp, M. Animal models of traumatic brain injury. Nat. Rev. Neurosci. 14, 128–142 (2013).
Marklund, N. & Hillered, L. Animal modelling of traumatic brain injury in preclinical drug development: where do we go from here? Br. J. Pharmacol. 164, 1207–1229 (2011).
Sheikh, A. M. et al. Lysophosphatidylcholine induces glial cell activation: role of rho kinase. Glia 57, 898–907 (2009).
Uchida, K. Redox-derived damage-associated molecular patterns: ligand function of lipid peroxidation adducts. Redox Biol. 1, 94–96 (2013).
Ransohoff, R. M. & Brown, M. A. Innate immunity in the central nervous system. J. Clin. Invest. 122, 1164–1171 (2012).
Scherbel, U. et al. Differential acute and chronic responses of tumor necrosis factor-deficient mice to experimental brain injury. Proc. Natl Acad. Sci. USA 96, 8721–8726 (1999).
Sinz, E. H. et al. Inducible nitric oxide synthase is an endogenous neuroprotectant after traumatic brain injury in rats and mice. J. Clin. Invest. 104, 647–656 (1999).
You, Z. et al. Necrostatin-1 reduces histopathology and improves functional outcome after controlled cortical impact in mice. J. Cereb. Blood Flow Metab. 28, 1564–1573 (2008).
Laird, M. D. et al. High mobility group box protein-1 promotes cerebral edema after traumatic brain injury via activation of toll-like receptor 4. Glia 62, 26–38 (2014).
Au, A. K. et al. Cerebrospinal fluid levels of high-mobility group box 1 and cytochrome C predict outcome after pediatric traumatic brain injury. J. Neurotrauma 29, 2013–2021 (2012).
Frank, M. G., Weber, M. D., Watkins, L. R. & Maier, S. F. Stress sounds the alarmin: the role of the danger-associated molecular pattern HMGB1 in stress-induced neuroinflammatory priming. Brain Behav. Immun. 48, 1–7 (2015).
Martinon, F., Burns, K. & Tschopp, J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-β. Mol. Cell 10, 417–426 (2002).
de Rivero Vaccari, J. P. et al. Therapeutic neutralization of the NLRP1 inflammasome reduces the innate immune response and improves histopathology after traumatic brain injury. J. Cereb. Blood Flow Metab. 29, 1251–1261 (2009).
Adamczak, S. E. et al. Pyroptotic neuronal cell death mediated by the AIM2 inflammasome. J. Cereb. Blood Flow Metab. 34, 621–629 (2014).
Halle, A. et al. The NALP3 inflammasome is involved in the innate immune response to amyloid-β. Nat. Immunol. 9, 857–865 (2008).
Liu, H. D. et al. Expression of the NLRP3 inflammasome in cerebral cortex after traumatic brain injury in a rat model. Neurochem. Res. 38, 2072–2083 (2013).
de Rivero Vaccari, J. P., Lotocki, G., Marcillo, A. E., Dietrich, W. D. & Keane, R. W. A molecular platform in neurons regulates inflammation after spinal cord injury. J. Neurosci. 28, 3404–3414 (2008).
Lukens, J. R., Barr, M. J., Chaplin, D. D., Chi, H. & Kanneganti, T. D. Inflammasome-derived IL-1β regulates the production of GM-CSF by CD4+ T cells and γδ T cells. J. Immunol. 188, 3107–3115 (2012).
Meissner, F., Molawi, K. & Zychlinsky, A. Mutant superoxide dismutase 1-induced IL-1β accelerates ALS pathogenesis. Proc. Natl Acad. Sci. USA 107, 13046–13050 (2010).
Palmer, A. M., Marion, D. W., Botscheller, M. L., Bowen, D. M. & DeKosky, S. T. Increased transmitter amino acid concentration in human ventricular CSF after brain trauma. Neuroreport 6, 153–156 (1994).
Ruppel, R. A. et al. Excitatory amino acid concentrations in ventricular cerebrospinal fluid after severe traumatic brain injury in infants and children: the role of child abuse. J. Pediatr. 138, 18–25 (2001).
Kochanek, P. M. et al. Biochemical, cellular, and molecular mechanisms in the evolution of secondary damage after severe traumatic brain injury in infants and children: lessons learned from the bedside. Pediatr. Crit. Care Med. 1, 4–19 (2000).
Viviani, B., Boraso, M., Marchetti, N. & Marinovich, M. Perspectives on neuroinflammation and excitotoxicity: a neurotoxic conspiracy? Neurotoxicology 43, 10–20 (2014).
Ikonomidou, C. & Turski, L. Why did NMDA receptor antagonists fail clinical trials for stroke and traumatic brain injury? Lancet Neurol. 1, 383–386 (2002).
Verrier, J. D. et al. Expression of the 2′,3′-cAMP-adenosine pathway in astrocytes and microglia. J. Neurochem. 118, 979–987 (2011).
Kochanek, P. M. et al. Adenosine A1 receptor knockout mice develop lethal status epilepticus after experimental traumatic brain injury. J. Cereb. Blood Flow Metab. 26, 565–575 (2006).
Haselkorn, M. L. et al. Adenosine A1 receptor activation as a brake on the microglial response after experimental traumatic brain injury in mice. J. Neurotrauma 27, 901–910 (2010).
Jackson, E. K., Boison, D., Schwarzschild, M. A. & Kochanek, P. M. Purines: forgotten mediators in traumatic brain injury. J. Neurochem. 137, 142–153 (2016).
Suliman, H. B. & Piantadosi, C. A. Mitochondrial quality control as a therapeutic target. Pharmacol. Rev. 68, 20–48 (2016).
Chu, C. T., Ji, J. & Dagda, R. K. Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat. Cell Biol. 15, 1197–1205 (2013).
Iyer, S. S. et al. Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity 39, 311–323 (2013).
Walko, T. D. III et al. Cerebrospinal fluid mitochondrial DNA: a novel DAMP in pediatric traumatic brain injury. Shock 41, 499–503 (2014).
Galluzzi, L., Kepp, O. & Kroemer, G. Mitochondria: master regulators of danger signalling. Nat. Rev. Mol. Cell Biol. 13, 780–788 (2012).
Balasubramanian, K. et al. Dichotomous roles for externalized cardiolipin in extracellular signaling: promotion of phagocytosis and attenuation of innate immunity. Sci. Signal. 8, ra95 (2015).
Fang, H. et al. CD36-mediated hematoma absorption following intracerebral hemorrhage: negative regulation by TLR4 signaling. J. Immunol. 192, 5984–5992 (2014).
Wagner, K. R., Sharp, F. R., Ardizzone, T. D., Lu, A. & Clark, J. F. Heme and iron metabolism: role in cerebral hemorrhage. J. Cereb. Blood Flow Metab. 23, 629–652 (2003).
Bellander, B. M., Singhrao, S. K., Ohlsson, M., Mattsson, P. & Svensson, M. Complement activation in the human brain after traumatic head injury. J. Neurotrauma 18, 1295–1311 (2001).
Stahel, P. F. et al. Intrathecal levels of complement-derived soluble membrane attack complex (sC5b-9) correlate with blood–brain barrier dysfunction in patients with traumatic brain injury. J. Neurotrauma 18, 773–781 (2001).
Stahel, P. F. et al. Absence of the complement regulatory molecule CD59a leads to exacerbated neuropathology after traumatic brain injury in mice. J. Neuroinflammation 6, 2 (2009).
Brennan, F. H., Anderson, A. J., Taylor, S. M., Woodruff, T. M. & Ruitenberg, M. J. Complement activation in the injured central nervous system: another dual-edged sword? J. Neuroinflammation 9, 137 (2012). Review of complement activation and dysregulation after brain and spinal cord injury, including several complement-targeted therapeutics.
Rich, M. C. et al. Site-targeted complement inhibition by a complement receptor 2-conjugated inhibitor (mTT30) ameliorates post-injury neuropathology in mouse brains. Neurosci. Lett. 617, 188–194 (2016).
Ruseva, M. M., Ramaglia, V., Morgan, B. P. & Harris, C. L. An anticomplement agent that homes to the damaged brain and promotes recovery after traumatic brain injury in mice. Proc. Natl Acad. Sci. USA 112, 14319–14324 (2015).
Zhang, Z., Zhang, Z. Y., Wu, Y. & Schluesener, H. J. Lesional accumulation of CD163+ macrophages/microglia in rat traumatic brain injury. Brain Res. 1461, 102–110 (2012).
Wang, X., Mori, T., Sumii, T. & Lo, E. H. Hemoglobin-induced cytotoxicity in rat cerebral cortical neurons: caspase activation and oxidative stress. Stroke 33, 1882–1888 (2002).
Willmore, L. J. & Ueda, Y. Posttraumatic epilepsy: hemorrhage, free radicals and the molecular regulation of glutamate. Neurochem. Res. 34, 688–697 (2009).
Newell, E. et al. Cerebrospinal fluid markers of macrophage and lymphocyte activation after traumatic brain injury in children. Pediatr. Crit. Care Med. 16, 549–557 (2015).
Kochanek, P. M. et al. Screening of biochemical and molecular mechanisms of secondary injury and repair in the brain after experimental blast-induced traumatic brain injury in rats. J. Neurotrauma 30, 920–937 (2013).
Bandak, F. A., Ling, G., Bandak, A. & De Lanerolle, N. C. Injury biomechanics, neuropathology, and simplified physics of explosive blast and impact mild traumatic brain injury. Handb. Clin. Neurol. 127, 89–104 (2015).
Kovesdi, E. et al. Acute minocycline treatment mitigates the symptoms of mild blast-induced traumatic brain injury. Front. Neurol. 3, 111 (2012).
Clark, R. S., Schiding, J. K., Kaczorowski, S. L., Marion, D. W. & Kochanek, P. M. Neutrophil accumulation after traumatic brain injury in rats: comparison of weight drop and controlled cortical impact models. J. Neurotrauma 11, 499–506 (1994).
Carlos, T. M., Clark, R. S., Franicola-Higgins, D., Schiding, J. K. & Kochanek, P. M. Expression of endothelial adhesion molecules and recruitment of neutrophils after traumatic brain injury in rats. J. Leukoc. Biol. 61, 279–285 (1997).
Bao, F. et al. A CD11d monoclonal antibody treatment reduces tissue injury and improves neurological outcome after fluid percussion brain injury in rats. J. Neurotrauma 29, 2375–2392 (2012).
Weaver, L. C. et al. CD11d integrin blockade reduces the systemic inflammatory response syndrome after traumatic brain injury in rats. Exp. Neurol. 271, 409–422 (2015).
Auffray, C. et al. Monitoring of blood vessels and tissues by a population of monocytes with patrolling behavior. Science 317, 666–670 (2007).
Hsieh, C. L. et al. Traumatic brain injury induces macrophage subsets in the brain. Eur. J. Immunol. 43, 2010–2022 (2013).
Morganti, J. M. et al. CCR2 antagonism alters brain macrophage polarization and ameliorates cognitive dysfunction induced by traumatic brain injury. J. Neurosci. 35, 748–760 (2015).
Jin, X., Ishii, H., Bai, Z., Itokazu, T. & Yamashita, T. Temporal changes in cell marker expression and cellular infiltration in a controlled cortical impact model in adult male C57BL/6 mice. PLoS ONE 7, e41892 (2012).
de Lanerolle, N. C., Lee, T. S. & Spencer, D. D. Astrocytes and epilepsy. Neurotherapeutics 7, 424–438 (2010).
Burda, J. E., Bernstein, A. M. & Sofroniew, M. V. Astrocyte roles in traumatic brain injury. Exp. Neurol. 275, 305–315 (2016). Excellent review of astrocyte function in health and after traumatic brain injury, including proinflammatory and anti-inflammatory roles and importance in repair and regeneration.
Turtzo, L. C. et al. Macrophagic and microglial responses after focal traumatic brain injury in the female rat. J. Neuroinflammation 11, 82 (2014).
Loane, D. J. & Byrnes, K. R. Role of microglia in neurotrauma. Neurotherapeutics 7, 366–377 (2010).
Morganti-Kossmann, M. C., Rancan, M., Stahel, P. F. & Kossmann, T. Inflammatory response in acute traumatic brain injury: a double-edged sword. Curr. Opin. Crit. Care 8, 101–105 (2002).
David, S. & Kroner, A. Repertoire of microglial and macrophage responses after spinal cord injury. Nat. Rev. Neurosci. 12, 388–399 (2011).
Kumar, A. & Loane, D. J. Neuroinflammation after traumatic brain injury: opportunities for therapeutic intervention. Brain Behav. Immun. 26, 1191–1201 (2012).
Colton, C. A. Heterogeneity of microglial activation in the innate immune response in the brain. J. Neuroimmune Pharmacol. 4, 399–418 (2009).
Sica, A. & Mantovani, A. Macrophage plasticity and polarization: in vivo veritas. J. Clin. Invest. 122, 787–795 (2012).
Loane, D. J., Kumar, A., Stoica, B. A., Cabatbat, R. & Faden, A. I. Progressive neurodegeneration after experimental brain trauma: association with chronic microglial activation. J. Neuropathol. Exp. Neurol. 73, 14–29 (2014).
Wang, G. et al. Microglia/macrophage polarization dynamics in white matter after traumatic brain injury. J. Cereb. Blood Flow Metab. 33, 1864–1874 (2013).
Loane, D. J. & Kumar, A. Microglia in the TBI brain: the good, the bad, and the dysregulated. Exp. Neurol. 275, 316–327 (2016).
Fenn, A. M. et al. Immune activation promotes depression 1 month after diffuse brain injury: a role for primed microglia. Biol. Psychiatry 76, 575–584 (2014).
Fenn, A. M. et al. Methylene blue attenuates traumatic brain injury-associated neuroinflammation and acute depressive-like behavior in mice. J. Neurotrauma 32, 127–138 (2015).
Lee, K. D. et al. FTY720 reduces inflammation and promotes functional recovery after spinal cord injury. J. Neurotrauma 26, 2335–2344 (2009).
Zhang, J., Zhang, A., Sun, Y., Cao, X. & Zhang, N. Treatment with immunosuppressants FTY720 and tacrolimus promotes functional recovery after spinal cord injury in rats. Tohoku J. Exp. Med. 219, 295–302 (2009).
Norimatsu, Y. et al. FTY720 improves functional recovery after spinal cord injury by primarily nonimmunomodulatory mechanisms. Am. J. Pathol. 180, 1625–1635 (2012).
Walsh, J. T. et al. MHCII-independent CD4+ T cells protect injured CNS neurons via IL-4. J. Clin. Invest. 125, 699–714 (2015).
Kipnis, J. et al. Neuroprotective autoimmunity: naturally occurring CD4+CD25+ regulatory T cells suppress the ability to withstand injury to the central nervous system. Proc. Natl Acad. Sci. USA 99, 15620–15625 (2002).
Yoles, E. et al. Protective autoimmunity is a physiological response to CNS trauma. J. Neurosci. 21, 3740–3748 (2001).
Hammarberg, H. et al. Neuroprotection by encephalomyelitis: rescue of mechanically injured neurons and neurotrophin production by CNS-infiltrating T and natural killer cells. J. Neurosci. 20, 5283–5291 (2000).
Linker, R., Gold, R. & Luhder, F. Function of neurotrophic factors beyond the nervous system: inflammation and autoimmune demyelination. Crit. Rev. Immunol. 29, 43–68 (2009).
Filiano, A. J., Gadani, S. P. & Kipnis, J. Interactions of innate and adaptive immunity in brain development and function. Brain Res. 1617, 18–27 (2015).
Gadani, S. P., Walsh, J. T., Smirnov, I., Zheng, J. & Kipnis, J. The glia-derived alarmin IL-33 orchestrates the immune response and promotes recovery following CNS injury. Neuron 85, 703–709 (2015).
Lohning, M. et al. T1/ST2 is preferentially expressed on murine Th2 cells, independent of interleukin 4, interleukin 5, and interleukin 10, and important for Th2 effector function. Proc. Natl Acad. Sci. USA 95, 6930–6935 (1998).
Burzyn, D. et al. A special population of regulatory T cells potentiates muscle repair. Cell 155, 1282–1295 (2013).
Kuswanto, W. et al. Poor repair of skeletal muscle in aging mice reflects a defect in local, interleukin-33-dependent accumulation of regulatory T cells. Immunity 44, 355–367 (2016).
Schiering, C. et al. The alarmin IL-33 promotes regulatory T-cell function in the intestine. Nature 513, 564–568 (2014).
Fletcher, J. M., Lalor, S. J., Sweeney, C. M., Tubridy, N. & Mills, K. H. T cells in multiple sclerosis and experimental autoimmune encephalomyelitis. Clin. Exp. Immunol. 162, 1–11 (2010).
Patel, D. D. & Kuchroo, V. K. Th17 cell pathway in human immunity: lessons from genetics and therapeutic interventions. Immunity 43, 1040–1051 (2015).
Shichita, T. et al. Pivotal role of cerebral interleukin-17-producing γδT cells in the delayed phase of ischemic brain injury. Nat. Med. 15, 946–950 (2009).
Benakis, C. et al. Commensal microbiota affects ischemic stroke outcome by regulating intestinal γδ T cells. Nat. Med. 22, 516–523 (2016).
Belkaid, Y. & Hand, T. W. Role of the microbiota in immunity and inflammation. Cell 157, 121–141 (2014).
Kasper, L. H. The evolving role of the gut microbiome in human disease. FEBS Lett. 588, 4101 (2014).
Cryan, J. F. & Dinan, T. G. Mind-altering microorganisms: the impact of the gut microbiota on brain and behaviour. Nat. Rev. Neurosci. 13, 701–712 (2012).
Kelly, J. R. et al. Breaking down the barriers: the gut microbiome, intestinal permeability and stress-related psychiatric disorders. Front. Cell. Neurosci. 9, 392 (2015).
Iliff, J. J. et al. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid β. Sci. Transl Med. 4, 147ra111 (2012).
Lee, H. et al. The effect of body posture on brain glymphatic transport. J. Neurosci. 35, 11034–11044 (2015).
Xie, L. et al. Sleep drives metabolite clearance from the adult brain. Science 342, 373–377 (2013).
Aspelund, A. et al. A dural lymphatic vascular system that drains brain interstitial fluid and macromolecules. J. Exp. Med. 212, 991–999 (2015).
Louveau, A. et al. Structural and functional features of central nervous system lymphatic vessels. Nature 523, 337–341 (2015).
Plog, B. A. et al. Biomarkers of traumatic injury are transported from brain to blood via the glymphatic system. J. Neurosci. 35, 518–526 (2015).
Iliff, J. J., Chen, M. J., Plog, B. A., Zeppenfeld, D. M. & Soltero, M. Impairment of glymphatic pathway function promotes tau pathology after traumatic brain injury. J. Neurosci. 34, 16180–16193 (2014).
Tortella, F. C. & Leung, L. Y. Traumatic brain injury and polytrauma in theaters of combat: the case for neurotrauma resuscitation? Shock 44 (Suppl. 1), 17–26 (2015).
Chesnut, R. M. et al. The role of secondary brain injury in determining outcome from severe head injury. J. Trauma 34, 216–222 (1993).
Tisherman, S. A. et al. Detailed description of all deaths in both the shock and traumatic brain injury hypertonic saline trials of the Resuscitation Outcomes Consortium. Ann. Surg. 261, 586–590 (2015).
Shein, S. et al. Hemorrhagic shock shifts the serum cytokine profile from pro-to anti-inflammatory after experimental traumatic brain injury in mice. J. Neurotrauma 31, 1386–1395 (2014).
Hemerka, J. N. et al. Severe brief pressure-controlled hemorrhagic shock after traumatic brain injury exacerbates functional deficits and long-term neuropathological damage in mice. J. Neurotrauma 29, 2192–2208 (2012).
Shiozaki, T. et al. Cerebrospinal fluid concentrations of anti-inflammatory mediators in early-phase severe traumatic brain injury. Shock 23, 406–410 (2005).
Kumar, R. G. et al. Acute CSF interleukin-6 trajectories after TBI: associations with neuroinflammation, polytrauma, and outcome. Brain Behav. Immun. 45, 253–262 (2015).
Simon, D. W., Vagni, V. M., Kochanek, P. M. & Clark, R. S. Combined neurotrauma models: experimental models combining traumatic brain injury and secondary insults. Methods Mol. Biol. 1462, 393–411 (2016).
McDonald, S. J., Sun, M., Agoston, D. V. & Shultz, S. R. The effect of concomitant peripheral injury on traumatic brain injury pathobiology and outcome. J. Neuroinflammation 13, 90 (2016).
Shultz, S. R. et al. Tibial fracture exacerbates traumatic brain injury outcomes and neuroinflammation in a novel mouse model of multitrauma. J. Cereb. Blood Flow Metab. 35, 1339–1347 (2015).
Utagawa, A., Truettner, J. S., Dietrich, W. D. & Bramlett, H. M. Systemic inflammation exacerbates behavioral and histopathological consequences of isolated traumatic brain injury in rats. Exp. Neurol. 211, 283–291 (2008).
Hang, C. H. et al. Effect of systemic LPS injection on cortical NF-κB activity and inflammatory response following traumatic brain injury in rats. Brain Res. 1026, 23–32 (2004).
Titus, D. J., Furones, C., Atkins, C. M. & Dietrich, W. D. Emergence of cognitive deficits after mild traumatic brain injury due to hyperthermia. Exp. Neurol. 263, 254–262 (2015).
Richardson, R. M., Sun, D. & Bullock, M. R. Neurogenesis after traumatic brain injury. Neurosurg. Clin. N. Am. 18, 169–181 (2007).
Ekdahl, C. T., Claasen, J. H., Bonde, S., Kokaia, Z. & Lindvall, O. Inflammation is detrimental for neurogenesis in adult brain. Proc. Natl Acad. Sci. USA 100, 13632–13637 (2003).
Whitney, N. P., Eidem, T. M., Peng, H., Huang, Y. & Zheng, J. C. Inflammation mediates varying effects in neurogenesis: relevance to the pathogenesis of brain injury and neurodegenerative disorders. J. Neurochem. 108, 1343–1359 (2009).
Monje, M. L., Toda, H. & Palmer, T. D. Inflammatory blockade restores adult hippocampal neurogenesis. Science 302, 1760–1765 (2003).
Butovsky, O. et al. Microglia activated by IL-4 or IFN-γ differentially induce neurogenesis and oligodendrogenesis from adult stem/progenitor cells. Mol. Cell. Neurosci. 31, 149–160 (2006).
Piao, C. S. et al. Late exercise reduces neuroinflammation and cognitive dysfunction after traumatic brain injury. Neurobiol. Dis. 54, 252–263 (2013).
Wang, B. & Jin, K. Current perspectives on the link between neuroinflammation and neurogenesis. Metab. Brain Dis. 30, 355–365 (2015).
Bendlin, B. B. et al. Longitudinal changes in patients with traumatic brain injury assessed with diffusion-tensor and volumetric imaging. Neuroimage 42, 503–514 (2008).
Trivedi, M. A. et al. Longitudinal changes in global brain volume between 79 and 409 days after traumatic brain injury: relationship with duration of coma. J. Neurotrauma 24, 766–771 (2007).
Shaklai, S., Peretz, R., Spasser, R., Simantov, M. & Groswasser, Z. Long-term functional outcome after moderate-to-severe paediatric traumatic brain injury. Brain Inj. 28, 915–921 (2014).
Ziebell, J. M., Adelson, P. D. & Lifshitz, J. Microglia: dismantling and rebuilding circuits after acute neurological injury. Metab. Brain Dis. 30, 393–400 (2015).
Bellinger, F. P., Madamba, S. G., Campbell, I. L. & Siggins, G. R. Reduced long-term potentiation in the dentate gyrus of transgenic mice with cerebral overexpression of interleukin-6. Neurosci. Lett. 198, 95–98 (1995).
Balschun, D. et al. Interleukin-6: a cytokine to forget. FASEB J. 18, 1788–1790 (2004).
Boato, F. et al. Interleukin-1 beta and neurotrophin-3 synergistically promote neurite growth in vitro. J. Neuroinflammation 8, 183 (2011).
Aungst, S. L., Kabadi, S. V., Thompson, S. M., Stoica, B. A. & Faden, A. I. Repeated mild traumatic brain injury causes chronic neuroinflammation, changes in hippocampal synaptic plasticity, and associated cognitive deficits. J. Cereb. Blood Flow Metab. 34, 1223–1232 (2014).
Miron, V. E. et al. M2 microglia and macrophages drive oligodendrocyte differentiation during CNS remyelination. Nat. Neurosci. 16, 1211–1218 (2013).
Thau-Zuchman, O., Shohami, E., Alexandrovich, A. G. & Leker, R. R. Combination of vascular endothelial and fibroblast growth factor 2 for induction of neurogenesis and angiogenesis after traumatic brain injury. J. Mol. Neurosci. 47, 166–172 (2012).
Saul, T. G., Ducker, T. B., Salcman, M. & Carro, E. Steroids in severe head injury: a prospective randomized clinical trial. J. Neurosurg. 54, 596–600 (1981).
Braakman, R., Schouten, H. J., Blaauw-van Dishoeck, M. & Minderhoud, J. M. Megadose steroids in severe head injury. Results of a prospective double-blind clinical trial. J. Neurosurg. 58, 326–330 (1983).
Gaab, M. R. et al. “Ultrahigh” dexamethasone in acute brain injury. Results from a prospective randomized double-blind multicenter trial (GUDHIS). German Ultrahigh Dexamethasone Head Injury Study Group. Zentralbl. Neurochir. 55, 135–143 (1994).
Marshall, L. F. et al. A multicenter trial on the efficacy of using tirilazad mesylate in cases of head injury. J. Neurosurg. 89, 519–525 (1998).
Asehnoune, K. et al. Hydrocortisone and fludrocortisone for prevention of hospital-acquired pneumonia in patients with severe traumatic brain injury (Corti-TC): a double-blind, multicentre phase 3, randomised placebo-controlled trial. Lancet Respir. Med. 2, 706–716 (2014).
Raub, T. J. et al. Use of a biophysical-kinetic model to understand the roles of protein binding and membrane partitioning on passive diffusion of highly lipophilic molecules across cellular barriers. J. Drug Target. 1, 269–286 (1993).
Shakur, H. et al. The BRAIN TRIAL: a randomised, placebo controlled trial of a bradykinin B2 receptor antagonist (Anatibant) in patients with traumatic brain injury. Trials 10, 109 (2009).
Heard, S. O. et al. Effect of prophylactic administration of recombinant human granulocyte colony-stimulating factor (filgrastim) on the frequency of nosocomial infections in patients with acute traumatic brain injury or cerebral hemorrhage. The Filgrastim Study Group. Crit. Care Med. 26, 748–754 (1998).
Casha, S. et al. Results of a phase II placebo-controlled randomized trial of minocycline in acute spinal cord injury. Brain 135, 1224–1236 (2012).
Hanlon, L. A., Huh, J. W. & Raghupathi, R. Minocycline transiently reduces microglia/macrophage activation but exacerbates cognitive deficits following repetitive traumatic brain injury in the neonatal rat. J. Neuropathol. Exp. Neurol. 75, 214–226 (2016).
Satchell, M. A. et al. Cytochrome c, a biomarker of apoptosis, is increased in cerebrospinal fluid from infants with inflicted brain injury from child abuse. J. Cereb. Blood Flow Metab. 25, 919–927 (2005).
Rosomoff, H. L. Protective effects of hypothermia against pathological processes of the nervous system. Ann. NY Acad. Sci. 80, 475–486 (1959).
Marion, D. W. et al. Treatment of traumatic brain injury with moderate hypothermia. N. Engl. J. Med. 336, 540–546 (1997).
Andrews, P. J. et al. Hypothermia for intracranial hypertension after traumatic brain injury. N. Engl. J. Med. 373, 2403–2412 (2015).
Clifton, G. L. et al. Very early hypothermia induction in patients with severe brain injury (the National Acute Brain Injury Study: Hypothermia II): a randomised trial. Lancet Neurol. 10, 131–139 (2011).
Adelson, P. D. et al. Comparison of hypothermia and normothermia after severe traumatic brain injury in children (Cool Kids): a phase 3, randomised controlled trial. Lancet Neurol. 12, 546–553 (2013).
Deutsch, E. R. et al. Progesterone's role in neuroprotection, a review of the evidence. Brain Res. 1530, 82–105 (2013).
Wright, D. W. et al. Very early administration of progesterone for acute traumatic brain injury. N. Engl. J. Med. 371, 2457–2466 (2014).
Skolnick, B. E. et al. A clinical trial of progesterone for severe traumatic brain injury. N. Engl. J. Med. 371, 2467–2476 (2014).
Diamond, M. L. et al. IL-1β associations with posttraumatic epilepsy development: a genetics and biomarker cohort study. Epilepsia 56, 991–1001 (2015).
Gentleman, S. M. et al. Long-term intracerebral inflammatory response after traumatic brain injury. Forensic Sci. Int. 146, 97–104 (2004).
Johnson, V. E. et al. Inflammation and white matter degeneration persist for years after a single traumatic brain injury. Brain 136, 28–42 (2013).
Ramlackhansingh, A. F. et al. Inflammation after trauma: microglial activation and traumatic brain injury. Ann. Neurol. 70, 374–383 (2011).
Smith, D. H., Johnson, V. E. & Stewart, W. Chronic neuropathologies of single and repetitive TBI: substrates of dementia? Nat. Rev. Neurol. 9, 211–221 (2013).
Smith, C. et al. The neuroinflammatory response in humans after traumatic brain injury. Neuropathol. Appl. Neurobiol. 39, 654–666 (2013).
Coughlin, J. M. et al. Neuroinflammation and brain atrophy in former NFL players: an in vivo multimodal imaging pilot study. Neurobiol. Dis. 74, 58–65 (2015).
Juengst, S. B., Kumar, R. G., Arenth, P. M. & Wagner, A. K. Exploratory associations with tumor necrosis factor-alpha, disinhibition and suicidal endorsement after traumatic brain injury. Brain Behav. Immun. 41, 134–143 (2014).
Nagamoto-Combs, K., McNeal, D. W., Morecraft, R. J. & Combs, C. K. Prolonged microgliosis in the rhesus monkey central nervous system after traumatic brain injury. J. Neurotrauma 24, 1719–1742 (2007).
Mouzon, B. C. et al. Chronic neuropathological and neurobehavioral changes in a repetitive mild traumatic brain injury model. Ann. Neurol. 75, 241–254 (2014).
Loane, D. J. et al. Novel mGluR5 positive allosteric modulator improves functional recovery, attenuates neurodegeneration, and alters microglial polarization after experimental traumatic brain injury. Neurotherapeutics 11, 857–869 (2014).
Holmin, S. & Mathiesen, T. Long-term intracerebral inflammatory response after experimental focal brain injury in rat. Neuroreport 10, 1889–1891 (1999).
Acosta, S. A. et al. Long-term upregulation of inflammation and suppression of cell proliferation in the brain of adult rats exposed to traumatic brain injury using the controlled cortical impact model. PLoS ONE 8, e53376 (2013).
Shitaka, Y. et al. Repetitive closed-skull traumatic brain injury in mice causes persistent multifocal axonal injury and microglial reactivity. J. Neuropathol. Exp. Neurol. 70, 551–567 (2011).
Petraglia, A. L. et al. The spectrum of neurobehavioral sequelae after repetitive mild traumatic brain injury: a novel mouse model of chronic traumatic encephalopathy. J. Neurotrauma 31, 1211–1224 (2014).
Petraglia, A. L. et al. The pathophysiology underlying repetitive mild traumatic brain injury in a novel mouse model of chronic traumatic encephalopathy. Surg. Neurol. Int. 5, 184 (2014).
Winston, C. N. et al. Dendritic spine loss and chronic white matter inflammation in a mouse model of highly repetitive head trauma. Am. J. Pathol. 186, 552–567 (2016).
Byrnes, K. R. et al. Metabotropic glutamate receptor 5 activation inhibits microglial associated inflammation and neurotoxicity. Glia 57, 550–560 (2009).
Loane, D. J., Stoica, B. A., Byrnes, K. R., Jeong, W. & Faden, A. I. Activation of mGluR5 and inhibition of NADPH oxidase improves functional recovery after traumatic brain injury. J. Neurotrauma 30, 403–412 (2013).
Byrnes, K. R., Loane, D. J., Stoica, B. A., Zhang, J. & Faden, A. I. Delayed mGluR5 activation limits neuroinflammation and neurodegeneration after traumatic brain injury. J. Neuroinflammation 9, 43 (2012).
Rodgers, K. M. et al. Reversal of established traumatic brain injury-induced, anxiety-like behavior in rats after delayed, post-injury neuroimmune suppression. J. Neurotrauma 31, 487–497 (2014).
McKee, A. C. et al. Chronic traumatic encephalopathy in athletes: progressive tauopathy after repetitive head injury. J. Neuropathol. Exp. Neurol. 68, 709–735 (2009).
McKee, A. C. et al. The spectrum of disease in chronic traumatic encephalopathy. Brain 136, 43–64 (2013).
Gardner, R. C. et al. Dementia risk after traumatic brain injury versus nonbrain trauma: the role of age and severity. JAMA Neurol. 71, 1490–1497 (2014).
Heneka, M. T. et al. Neuroinflammation in Alzheimer's disease. Lancet Neurol. 14, 388–405 (2015).
Perry, V. H., Nicoll, J. A. & Holmes, C. Microglia in neurodegenerative disease. Nat. Rev. Neurol. 6, 193–201 (2010).
McKee, A. C. et al. TDP-43 proteinopathy and motor neuron disease in chronic traumatic encephalopathy. J. Neuropathol. Exp. Neurol. 69, 918–929 (2010).
Saing, T. et al. Frontal cortex neuropathology in dementia pugilistica. J. Neurotrauma 29, 1054–1070 (2012).
Cherry, J. D. et al. Microglial neuroinflammation contributes to tau accumulation in chronic traumatic encephalopathy. Acta Neuropathol. Commun. 4, 112 (2016).
Coughlin, J. M. et al. Imaging of glial cell activation and white matter integrity in brains of active and recently retired National Football League players. JAMA Neurol. 74, 67–74 (2016).
Webster, K. M. et al. Progesterone treatment reduces neuroinflammation, oxidative stress and brain damage and improves long-term outcomes in a rat model of repeated mild traumatic brain injury. J. Neuroinflammation 12, 238 (2015).
Hong, Y. T. et al. Amyloid imaging with carbon 11-labeled Pittsburgh compound B for traumatic brain injury. JAMA Neurol. 71, 23–31 (2014).
Barrio, J. R. et al. In vivo characterization of chronic traumatic encephalopathy using [F-18]FDDNP PET brain imaging. Proc. Natl Acad. Sci. USA 112, E2039–E2047 (2015).
Kumar, R. G., Rubin, J. E., Berger, R. P., Kochanek, P. M. & Wagner, A. K. Principal components derived from CSF inflammatory profiles predict outcome in survivors after severe traumatic brain injury. Brain Behav. Immun. 53, 183–193 (2013).
Junger, W. G. et al. Prehospital hypertonic saline resuscitation attenuates the activation and promotes apoptosis of neutrophils in patients with severe traumatic brain injury. Shock 40, 366–374 (2013).
Pagowska-Klimek, I., Lewkowicz, P., Banasik, M., Krajewski, W. & Tchorzewski, H. Isolated head injury in children affects the neutrophil function and lymphocyte count. J. Trauma 63, 179–186 (2007).
Hazeldine, J., Lord, J. M. & Belli, A. Traumatic brain injury and peripheral immune suppression: primer and prospectus. Front. Neurol. 6, 235 (2015). Excellent review of systemic immune suppression after traumatic brain injury, including future directions for research.
Hayakata, T. et al. Changes in CSF S100B and cytokine concentrations in early-phase severe traumatic brain injury. Shock 22, 102–107 (2004).
Maier, B. et al. Delayed elevation of soluble tumor necrosis factor receptors p75 and p55 in cerebrospinal fluid and plasma after traumatic brain injury. Shock 26, 122–127 (2006).
Ross, S. A., Halliday, M. I., Campbell, G. C., Byrnes, D. P. & Rowlands, B. J. The presence of tumour necrosis factor in CSF and plasma after severe head injury. Br. J. Neurosurg. 8, 419–425 (1994).
Yan, E. B. et al. Post-traumatic hypoxia is associated with prolonged cerebral cytokine production, higher serum biomarker levels, and poor outcome in patients with severe traumatic brain injury. J. Neurotrauma 31, 618–629 (2014).
Waters, R. J. et al. Cytokine gene polymorphisms and outcome after traumatic brain injury. J. Neurotrauma 30, 1710–1716 (2013).
Helmy, A., Carpenter, K. L., Menon, D. K., Pickard, J. D. & Hutchinson, P. J. The cytokine response to human traumatic brain injury: temporal profiles and evidence for cerebral parenchymal production. J. Cereb. Blood Flow Metab. 31, 658–670 (2011).
Buttram, S. D. et al. Multiplex assessment of cytokine and chemokine levels in cerebrospinal fluid following severe pediatric traumatic brain injury: effects of moderate hypothermia. J. Neurotrauma 24, 1707–1717 (2007).
Chiaretti, A. et al. Interleukin 1β and interleukin 6 relationship with paediatric head trauma severity and outcome. Childs Nerv. Syst. 21, 185–193 (2005).
Hadjigeorgiou, G. M. et al. IL-1RN and IL-1B gene polymorphisms and cerebral hemorrhagic events after traumatic brain injury. Neurology 65, 1077–1082 (2005).
Helmy, A., Antoniades, C. A., Guilfoyle, M. R., Carpenter, K. L. & Hutchinson, P. J. Principal component analysis of the cytokine and chemokine response to human traumatic brain injury. PLoS ONE 7, e39677 (2012).
Mellergard, P., Aneman, O., Sjogren, F., Saberg, C. & Hillman, J. Differences in cerebral extracellular response of interleukin-1β, interleukin-6, and interleukin-10 after subarachnoid hemorrhage or severe head trauma in humans. Neurosurgery 68, 12–19 (2011).
Perez-Barcena, J. et al. Lack of correlation among intracerebral cytokines, intracranial pressure, and brain tissue oxygenation in patients with traumatic brain injury and diffuse lesions. Crit. Care Med. 39, 533–540 (2011).
Hutchinson, P. J. et al. Inflammation in human brain injury: intracerebral concentrations of IL-1α, IL-1β, and their endogenous inhibitor IL-1ra. J. Neurotrauma 24, 1545–1557 (2007).
Bell, M. J. et al. Interleukin-6 and interleukin-10 in cerebrospinal fluid after severe traumatic brain injury in children. J. Neurotrauma 14, 451–457 (1997).
Kossmann, T., Hans, V., Imhof, H. G., Trentz, O. & Morganti-Kossmann, M. C. Interleukin-6 released in human cerebrospinal fluid following traumatic brain injury may trigger nerve growth factor production in astrocytes. Brain Res. 713, 143–152 (1996).
Kossmann, T. et al. Intrathecal and serum interleukin-6 and the acute-phase response in patients with severe traumatic brain injuries. Shock 4, 311–317 (1995).
Maier, B. et al. Differential release of interleukines 6, 8, and 10 in cerebrospinal fluid and plasma after traumatic brain injury. Shock 15, 421–426 (2001).
Shore, P. M. et al. Continuous versus intermittent cerebrospinal fluid drainage after severe traumatic brain injury in children: effect on biochemical markers. J. Neurotrauma 21, 1113–1122 (2004).
Singhal, A. et al. Association between cerebrospinal fluid interleukin-6 concentrations and outcome after severe human traumatic brain injury. J. Neurotrauma 19, 929–937 (2002).
Winter, C. D., Pringle, A. K., Clough, G. F. & Church, M. K. Raised parenchymal interleukin-6 levels correlate with improved outcome after traumatic brain injury. Brain 127, 315–320 (2004).
Kirchhoff, C. et al. Cerebrospinal IL-10 concentration is elevated in non-survivors as compared to survivors after severe traumatic brain injury. Eur. J. Med. Res. 13, 464–468 (2008).
Yan, E. B., Hellewell, S. C., Bellander, B. M., Agyapomaa, D. A. & Morganti-Kossmann, M. C. Post-traumatic hypoxia exacerbates neurological deficit, neuroinflammation and cerebral metabolism in rats with diffuse traumatic brain injury. J. Neuroinflammation 8, 147 (2011).
Morganti-Kossmann, M. C. et al. TGF-β is elevated in the CSF of patients with severe traumatic brain injuries and parallels blood–brain barrier function. J. Neurotrauma 16, 617–628 (1999).
Semple, B. D., Bye, N., Rancan, M., Ziebell, J. M. & Morganti-Kossmann, M. C. Role of CCL2 (MCP-1) in traumatic brain injury (TBI): evidence from severe TBI patients and CCL2−/− mice. J. Cereb. Blood Flow Metab. 30, 769–782 (2010).
Stefini, R. et al. Chemokine detection in the cerebral tissue of patients with posttraumatic brain contusions. J. Neurosurgery 108, 958–962 (2008).
Whalen, M. J. et al. Interleukin-8 is increased in cerebrospinal fluid of children with severe head injury. Crit. Care Med. 28, 929–934 (2000).
Engel, S. et al. Dynamics of microglial activation after human traumatic brain injury are revealed by delayed expression of macrophage-related proteins MRP8 and MRP14. Acta Neuropathol. 100, 313–322 (2000).
Bonneh-Barkay, D. et al. YKL-40 expression in traumatic brain injury: an initial analysis. J. Neurotrauma 27, 1215–1223 (2010).
Zhang, Z. et al. Human traumatic brain injury induces autoantibody response against glial fibrillary acidic protein and its breakdown products. PLoS ONE 9, e92698 (2014).
Wagner, A. K. et al. Adenosine A1 receptor gene variants associated with post-traumatic seizures after severe TBI. Epilepsy Res. 90, 259–272 (2010).
Clark, R. S. et al. Cerebrospinal fluid adenosine concentration and uncoupling of cerebral blood flow and oxidative metabolism after severe head injury in humans. Neurosurgery 41, 1284–1292 (1997).
Bell, M. J., Kochanek, P. M. & Jackson, E. K. Interstitial adenosine, inosine, and hypoxanthine are increased after experimental traumatic brain injury in the rat. J. Neurotrauma 15, 163–170 (1998).
Kossmann, T., Stahel, P. F., Morganti-Kossmann, M. C., Jones, J. L. & Barnum, S. R. Elevated levels of the complement components C3 and factor B in ventricular cerebrospinal fluid of patients with traumatic brain injury. J. Neuroimmunol. 73, 63–69 (1997).
Baker, A. J., Moulton, R. J., MacMillan, V. H. & Shedden, P. M. Excitatory amino acids in cerebrospinal fluid following traumatic brain injury in humans. J. Neurosurg. 79, 369–372 (1993).
Bullock, R. et al. Factors affecting excitatory amino acid release following severe human head injury. J. Neurosurg. 89, 507–518 (1998).
Chamoun, R., Suki, D., Gopinath, S. P., Goodman, J. C. & Robertson, C. Role of extracellular glutamate measured by cerebral microdialysis in severe traumatic brain injury. J. Neurosurg. 113, 564–570 (2010).
Gao, T. L. et al. Expression of HMGB1 and RAGE in rat and human brains after traumatic brain injury. J. Trauma Acute Care Surg. 72, 643–649 (2012).
Adamczak, S. et al. Inflammasome proteins in cerebrospinal fluid of brain-injured patients as biomarkers of functional outcome: clinical article. J. Neurosurg. 117, 1119–1125 (2012).
Clark, R. S. et al. Increases in Bcl-2 and cleavage of caspase-1 and caspase-3 in human brain after head injury. FASEB J. 13, 813–821 (1999).
Mazzeo, A. T. et al. Severe human traumatic brain injury, but not cyclosporin a treatment, depresses activated T lymphocytes early after injury. J. Neurotrauma 23, 962–975 (2006).
Maas, A. I. et al. Efficacy and safety of dexanabinol in severe traumatic brain injury: results of a phase III randomised, placebo-controlled, clinical trial. Lancet Neurol. 5, 38–45 (2006).
Nichol, A. et al. Erythropoietin in traumatic brain injury (EPO-TBI): a double-blind randomised controlled trial. Lancet 386, 2499–2506 (2015).
Robertson, C. S. et al. Effect of erythropoietin and transfusion threshold on neurological recovery after traumatic brain injury: a randomized clinical trial. JAMA 312, 36–47 (2014).
Bulger, E. M. et al. Out-of-hospital hypertonic resuscitation following severe traumatic brain injury: a randomized controlled trial. JAMA 304, 1455–1464 (2010).
Adelson, P. D. et al. Guidelines for the acute medical management of severe traumatic brain injury in infants, children, and adolescents. Chapter 11. Use of hyperosmolar therapy in the management of severe pediatric traumatic brain injury. Pediatr. Crit. Care Med. 4, S40–S44 (2003).
Helmy, A. et al. Recombinant human interleukin-1 receptor antagonist in severe traumatic brain injury: a phase II randomized control trial. J. Cereb. Blood Flow Metab. 34, 845–851 (2014).
Helmy, A. et al. Recombinant human interleukin-1 receptor antagonist promotes M1 microglia biased cytokines and chemokines following human traumatic brain injury. J. Cereb. Blood Flow Metab. 36, 1434–1448 (2016).
Tan, M., Zhu, J. C., Du, J., Zhang, L. M. & Yin, H. H. Effects of probiotics on serum levels of Th1/Th2 cytokine and clinical outcomes in severe traumatic brain-injured patients: a prospective randomized pilot study. Crit. Care 15, R290 (2011).
Tapia-Perez, J. et al. Effect of rosuvastatin on amnesia and disorientation after traumatic brain injury (NCT003229758). J. Neurotrauma 25, 1011–1017 (2008).
Sanchez-Aguilar, M. et al. Effect of rosuvastatin on cytokines after traumatic head injury. J. Neurosurg. 118, 669–675 (2013).
Acknowledgements
The authors acknowledge the following funding sources: NIH grants T32 HD40686 (D.W.S.) and R01 NS082308 (D.J.L.); National Institute of Child Health and Human Development grants R01 NS087978 (P.M.K.), NS061817 and NS076511 (H.B.); National Institute of Allergy and Infectious Diseases grant R01 AI110822-01 (M.J.M.); Department of Defense Grants W81XWH-10-1-0623 and W81XWH-14-2-0018 (P.M.K.); NIA Claude D. Pepper Older Americans Independence Center grant P30-AG028747 (D.J.L.); and Children's Hospital of Pittsburgh — Children's Trust (D.W.S.).
Author information
Authors and Affiliations
Contributions
The authors contributed equally to all aspects of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Glossary
- Chronic traumatic encephalopathy
-
A progressive neurodegenerative disease associated with head trauma and characterized histologically by formation of neurofibrillary tangles, accumulation of phosphorylated TAR DNA-binding protein 43 (TDP-43) accumulation, and deposition of amyloid-β.
- Damage-associated molecular patterns
-
Host-derived molecules that trigger and/or exacarbate the inflammatory response. Prominent examples include DNA and RNA, high mobility group protein B1 (HMGB1), S100 proteins, ATP, uric acid, lysophospholipids, and lipid peroxidation-derived carbonyl adducts of proteins.
- CD11d/CD18 integrin
-
A pattern recognition receptor that is located on the surface of neutrophils and monocytes and is functionally important in recognition of complement, as well as cell–cell interactions and cellular adhesion.
- Chemokine gradients
-
Concentration gradients of chemotactic cytokines with the ability to influence inflammatory cell migration and function. For example, C–C motif chemokine 2 (CCL2), a chemokine for monocytes, macrophages and microglia, and its receptor CCR2 interact to recruit these immune cells to injured tissue after traumatic brain injury.
- Autoimmune T cells
-
Also called autoreactive T cells, these T lymphocytes react to self antigens and may cause autoimmune disease, but are also critical for normal brain function and repair.
- TH17 cells
-
A subset of effector T-helper cells that produce IL-17 and other proinflammatory cytokines.
- Glymphatic system
-
Astrocyte-regulated convective bulk flow of the cerebrospinal fluid from the paravascular space through interstitial fluid in an arterial–venous direction.
Rights and permissions
About this article

Cite this article
Simon, D., McGeachy, M., Bayır, H. et al. The far-reaching scope of neuroinflammation after traumatic brain injury. Nat Rev Neurol 13, 171–191 (2017). https://doi.org/10.1038/nrneurol.2017.13
Published:
Issue Date:
DOI: https://doi.org/10.1038/nrneurol.2017.13
This article is cited by
-
Interaction of high-fat diet and brain trauma alters adipose tissue macrophages and brain microglia associated with exacerbated cognitive dysfunction
Journal of Neuroinflammation (2024)
-
Mastering the brain in critical conditions: an update
Intensive Care Medicine Experimental (2024)
-
Whole blood stimulation provides preliminary evidence of altered immune function following SRC
BMC Immunology (2024)
-
Fibrin promotes oxidative stress and neuronal loss in traumatic brain injury via innate immune activation
Journal of Neuroinflammation (2024)
-
Current state of neuroprotective therapy using antibiotics in human traumatic brain injury and animal models
BMC Neuroscience (2024)
