
Abstract
The treatment of primary Sjögren's syndrome (SS) is based principally on the management of sicca features and systemic manifestations. Sicca manifestations are treated symptomatically through administration of topical therapies, such as saliva substitutes and artificial tears; in patients with residual salivary gland function, stimulation of salivary flow with a sialogogue is the therapy of choice. The management of extraglandular features must be tailored to the specific organ or organs involved; however, limited data have been obtained from controlled trials in SS to guide the treatment of systemic symptoms using therapies including antimalarials, glucocorticoids, immunosuppressive drugs and biologic agents. Nevertheless, randomised controlled trials of biologic agents that target molecules and receptors involved in the aetiopathogenesis of primary SS have initiated a new era in the therapeutic management of the disease, although the potential risks and benefits of these agents must be carefully considered. In this Review, we analyse the evidence regarding the efficacy of the therapeutic agents currently available to treat the manifestations of SS. On the basis of this evidence, we provide guidance on the use of these agents in different clinical scenarios.
Key Points
-
The primary therapeutic approach to sicca manifestations in Sjögren's syndrome (SS) should be symptomatic relief, using artificial tears and saliva substitutes
-
The treatment of choice for patients with moderate or severe oral dryness and residual salivary gland function is an oral muscarinic agonist
-
Patients with severe or refractory keratoconjunctivitis sicca might require the addition of topical cyclosporine A to suppress the underlying inflammation
-
The management of extraglandular features must be tailored to the specific organ involved, mainly using corticosteroids and immunosuppressive agents
-
Controlled trials of biologic agents in patients with SS have demonstrated the lack of efficacy of TNF blockade, whereas more promising results have been obtained with B-cell-targeted therapies, especially rituximab
-
The overall low level of evidence from therapeutic studies in primary SS underscores the need for considerably larger trials
Similar content being viewed by others


Introduction
Patients with Sjögren's syndrome (SS) primarily present with sicca symptoms involving the major mucosal surfaces. The main sicca features of this systemic autoimmune disease are xerophthalmia (dry eyes) and xerostomia (dry mouth), which are diagnosed using specific ocular (Rose Bengal or Lissamine Green staining, or the Schirmer test) and oral (salivary flow measurement or parotid scintigraphy) assessments, respectively.1 Patients with SS can present with a broad spectrum of disease, extending from sicca syndrome to systemic manifestations (extraglandular involvement), and the disease can be further complicated by lymphomas. A wide range of analytical features are associated with SS, including cytopenia, hypergammaglobulinemia and a high erythrocyte sedimentation rate (ESR), and cryoglobulins and hypocomplementemia are the main prognostic markers.2 SS is also associated with autoantibody production; antinuclear antibodies are the most frequently detected, whereas anti-Ro/SSA are the most specific for SS.2 Focal lymphocytic infiltration of the exocrine glands, usually determined by minor labial salivary gland biopsy, is the histological hallmark of SS.
At present, no cure exists for primary SS, and no therapy prevents disease progression. The classic therapeutic approach is based on symptomatic treatment of glandular manifestations and broad-spectrum immunosuppression directed against organ-specific extraglandular disease. Over the past decade, SS research has centred on the development of more effective targeted therapies, including various completed and ongoing randomised controlled trials (RCTs), a number of which have shown promising results. These new therapies provide hope for better disease management in patients with SS.3 The objective of this Review is to summarize the reported data on targeted therapies for the treatment of SS, with the aim of providing physicians with a rational therapeutic approach tailored to the different clinical scenarios that patients with SS can present. In the following sections, we deem the highest-to-lowest quality of evidence to be derived from study designs in the following order: RCTs, prospective cohort studies, case–control studies, retrospective studies and case series.
Topical medications—level of evidence
Saliva substitutes
Saliva substitutes are frequently used in clinical practice, owing to the near-complete absence of appreciable adverse events and their beneficial effects on nocturnal oral dryness. A number of studies in SS have evaluated different saliva substitutes (Table 1).4,5,6,7,8,9,10 In one RCT,9 no marked differences in efficacy were observed between three different saliva substitutes and placebo. By contrast, three RCTs in patients with SS found that saliva substitutes considerably improved several oral symptoms, but not salivary flow rates.4,5,6 In a comparison of mucin-containing and carboxymethylcellulose-containing substitutes, neither was shown to provide superior therapeutic benefit, although the former preparation was reportedly preferred by patients.10 Data from uncontrolled studies have shown considerable improvement in xerostomia and salivary flow after application of a hydroxymethylcellulose-containing oral spray,8 and a reduction in the dental plaque burden in patients treated with chlorhexidine mouthrinses.7
Eye drops
Glucanes
Marked improvements in xerophthalmia compared with baseline measurements were observed for topical eye drops containing glucanes; five studies using sodium hyaluronate11,12,13,14,15 and one using hydroxypropylmethylcellulose16 as the active ingredient (Table 1). Condon et al.14 found that the use of artificial tear drops containing hyaluronate resulted in a greater improvement in dry eye symptoms and ocular tests than saline eye drops. A comparative study showed greater therapeutic benefit for hypotonic than isotonic hyaluronate preparations.11 No marked differences in efficacy were observed between hyaluronate-containing and carboxymethylcellulose-containing eye drops.13
NSAIDs
Limited evidence is available for the use of artificial tear drops containing topical NSAIDs in patients with primary SS. In an uncontrolled comparative study that enrolled 20 patients with SS,17 ocular symptoms improved after treatment with eye drops containing diclofenac or indomethacin, with greater improvement in corneal sensitivity observed for diclofenac. However, a small RCT (32 patients enrolled) found no substantial difference in the resolution of filamentary keratitis at day 28 after initiation of treatment with diclofenac-containing tears compared with saline eye drops.18 Adverse events are the key point to consider when using topical NSAIDs. Guidera et al.19 retrospectively described severe adverse events associated with the use of topical NSAIDs in 18 eyes of 16 patients with different medical conditions, including SS. The adverse events occurred after 2–4 weeks treatment with topical NSAIDs in most patients, and included corneal–scleral melts, perforation, ulceration and severe keratopathy.
Corticosteroids
At present, the use of topical corticosteroids for treatment of ocular symptoms in patients with dry eyes has been analysed in four studies; considerable improvements were demonstrated after treatment with fluorometholone or methylprednisolone, but not loteprednol, in comparison with placebo.20,21,22,23 One of these studies also compared topical corticosteroids and NSAIDs and found that corticosteroids were superior.20 Adverse events associated with prolonged corticosteroid use should be taken into account; Marsh et al.21 reported serious adverse effects (increased intraocular pressure, worsening or development of cataracts) in 14% of patients with SS treated with topical nonpreserved methylprednisolone sodium succinate.
Cyclosporine A
Cyclosporine A has been used to treat autoimmune diseases for more than three decades, and an ophthalmic formulation was approved by the FDA in December 2002 for the treatment of dry eye disease. Improvements have been observed after administration of twice daily topical cyclosporine A in a number of studies, with respect to baseline subjective and objective measurements of dry eyes (Table 1).24,25,26,27,28,29,30,31,32,33,34 The largest RCT,32 which included 877 patients with dry eye syndrome (31% with SS) who were evaluated after 6 months, tested two different cyclosporine A doses (0.05% and 0.1%). A significant improvement in Schirmer test scores was observed for both groups (P<0.007) compared with placebo, but corneal staining scores only significantly improved after treatment with 0.05% cyclosporine A (P = 0.008, P = 0.06 in the 0.1% group);32 the 0.05% group also showed significantly reduced blurred vision—one of seven symptoms of ocular discomfort—(P <0.01) and artificial tear use (P = 0.006).32 Therefore, the recommended dosing regimen is 0.05% cyclosporine A drops twice daily. In a publication reporting the extension of two RCTs in patients initially treated for 6–12 months, no additional improvement in subjective and objective measurements was observed during the extended study period (mean of 20 months).34 With respect to adverse events, in the largest RCT of cyclosporine A eye drops,32 the only marked difference between the cyclosporine A and placebo groups was in the percentage of patients who experienced burning eye (15% versus 6%, respectively); most events were mild-to-moderate and transient, with only 2% of patients discontinuing treatment because of burning or stinging.32 Only two patients, both in the placebo group, experienced ocular infection during the study period.32 Taken together, these studies have led to ocular cyclosporine A being licensed for use in the USA and 27 other countries worldwide, but not in Europe, Canada or Australia.
Diquafosol
Tauber et al.35 performed a RCT using an ophthalmic formulation of 2% diquafosol tetrasodium (INS365), an agonist of the P2Y purinoceptor 2 protein that is involved in ocular surface hydration. The study, which enrolled 527 patients with keratoconjunctivitis sicca (76 secondary to SS), revealed no marked difference in therapeutic effect between topical diquafosol and placebo, except for a greater improvement in corneal staining score with the former.
Systemic medications—level of evidence
Secretagogues
For patients with SS who have residual salivary gland function, stimulation of saliva flow with a secretagogue is the treatment of choice and is, at present, the most efficacious means of preventing long-term oral complications (Table 2). Nonpharmacological secretagogues might be useful in patients with reasonable levels of salivary flow, such as mechanical and/or gustatory stimulation with sugar-free gums and candies. With respect to pharmacological salivary stimulation, some choleretic (anetholtrithione) and mucolytic (bromhexine, N-acetylcysteine) agents have been used as secretagogues in primary SS since the 1980s, although without solid scientific evidence of efficacy. Uncontrolled studies found only marginal therapeutic benefits for anetholtrithione and bromhexine,36,37 whereas a small trial, in which 26 patients were treated with 200 mg N-acetylcysteine three times daily, demonstrated substantial improvements in sicca symptoms and ocular test results.38
Two muscarinic acetylcholine receptor agonists (pilocarpine and cevimeline) are licensed for the treatment of sicca symptoms in SS. These agents stimulate the muscarinic acetylcholine receptors M1 and M3 present on salivary glands, leading to increased secretory function.
Pilocarpine
11 studies have analysed the use of the muscarinic acetylcholine receptor agonist pilocarpine in patients with SS.39,40,41,42,43,44,45,46,47,48,49 In the two largest RCTs,42,48 pilocarpine doses of 5 mg and 7.5 mg every 6 h resulted in markedly improved oral, ocular, nasal, vaginal and skin dryness, and salivary flow rates. Uncontrolled studies have also reported a substantial therapeutic benefit of this drug, based on the findings of ocular tests and reduction of candidiasis.39,40,41,43,44,45,46 Data from RCTs have revealed a high frequency of adverse events associated with pilocarpine use, including sweating, increased urinary frequency and flushing (observed in 43%, 10% and 10% of patients, respectively).42,47,48,49 In a dose-adjustment RCT,42 23% of patients were switched from the 7.5 mg to the 5 mg regimen after 6 weeks of treatment, suggesting the lower dose is better tolerated.
Cevimeline
The efficacy of another muscarinic acetylcholine receptor agonist, cevimeline, has been assessed in eight studies that enrolled patients with SS.50,51,52,53,54,55,56,57 Three RCTs have compared dosages ranging between 15 mg and 60 mg taken three times daily, and the best results—including considerable improvements in dry mouth and dry eyes, salivary flow rates and ocular test results—were achieved with a dose of 30 mg.50,52,54 A crossover trial also found a marked reduction in candidiasis, dental plaque burden and gingival bleeding.52 In the two largest trials of cevimeline in SS,50,54 both the 30 mg and 60 mg regimens were associated with higher frequencies of nausea (relative risk [RR] = 1.68 and 2.77, respectively) and sweating (RR = 2.16 and 3.00, respectively) in comparison with placebo, and rigors were more common with the 60 mg dose than with placebo (RR = 1.92).50
Glucocorticoids
The frequent use of glucocorticoids in clinical practice for the treatment of primary SS is not supported by reliable scientific evidence, as no study has specifically evaluated these agents as therapies for the extraglandular features of this disease. Five studies—all but one uncontrolled—have analysed the use of corticosteroids in SS (Table 3),58,59,60,61,62 but have focused principally on their effects on sicca symptoms. In 1993, Fox et al.60 performed a RCT in a small series of patients with SS (eight patients in each arm) comparing placebo to 30 mg daily prednisone combined with 20 mg daily piroxicam, and found appreciable differences in subjective symptoms but not in objective tests. Increased salivary flow with prednisolone, administered either orally59 or by parotid irrigation,58 together with reduced levels of IgG, rheumatoid factor (RF) and anti-Ro/SSA and anti-La/SSB antibodies has been demonstrated in uncontrolled studies in SS; however, limited information can be drawn from these studies owing to substantial methodological flaws in their design. By contrast, a prospective study in 60 patients with primary SS, followed for a mean of nearly 4 years,61 found that the use of corticosteroids did not influence the progressive disease-associated decrease in salivary flow rates. The use of systemic corticosteroids in patients with SS has been associated with adverse events, including increased appetite and weight gain in comparison with placebo,60 and a twofold higher frequency of diabetes mellitus compared with age-matched and gender-matched individuals without systemic autoimmune diseases.62
Antimalarial agents
The use of antimalarial agents in primary SS is based on their effectiveness in similar diseases, such as systemic lupus erythematosus. Only five studies have analysed the use of hydroxychloroquine (HCQ) in patients with SS (Table 3).63,64,65,66,67,68,69 A retrospective study of 50 patients described considerable improvements in sicca features, parotid gland enlargement, oral infection, myalgia, arthralgia, fatigue and joint swelling after 12 months of HCQ therapy.67 All studies of HCQ use in patients with SS found substantial improvements in baseline analytical and immunological parameters, including ESR and levels of gamma globulins, IgG, IgM, C-reactive protein (CRP), RF, anti-La/SSB antibodies and haemoglobin.63,64,65,66,67,68,69 No cases of retinal toxicity or severe adverse events were reported in any of these studies.63,64,65,66,67,68,69 Furthermore, a retrospective case–control study noted a lower prevalence of diabetes mellitus in patients with SS treated with antimalarial agents than in age-matched and gender-matched individuals without systemic autoimmune diseases.62
Immunosuppressive agents
The use of immunosuppressive agents in primary SS is based on the same level of evidence as that of glucocorticoids (Table 3). The use of cyclosporine A, azathioprine, methotrexate, leflunomide or mycophenolic acid has been studied in five small series of patients with SS.70,71,72,73,74 One of these studies, a RCT using azathioprine, observed no improvements in disease.74 Although improvements in sicca symptoms were found in the studies using cyclosporine A,70 methotrexate71 and mycophenolic acid,73 no study found statistically significant improvements in objective tests. Treatment with leflunomide72 or mycophenolic acid73 was associated with substantial improvements in some SF-36® (36-item Short Form Health Survey) and Multidimensional Fatigue Inventory domains, and with a reduction in analytical abnormalities, including levels of gamma globulin, IgM, IgG, RF and C3 and/or C4. The results of these studies provided three key messages regarding the use of immunosuppressive agents: these agents offer limited benefits for sicca features; specific analysis of their effects on extraglandular features is inadequate; and they are associated with unacceptable rates of adverse events (frequency of 41–100%).70,71,72,73,74
Immunomodulatory agents
Several immunomodulatory agents have been tested in SS (Table 3), with marginal benefits (D-penicillamine)75 or with an unacceptable rate of adverse events (thalidomide).76 Mizoribine, an imidazole nucleoside, was evaluated in two uncontrolled studies performed by the same group,77,78 with improvements in sicca visual analogue scale scores and salivary flow being reported. Somewhat surprisingly, however, 29 adverse drug reactions were reported in 18 of 59 patients included in the first study,77 compared with none in the second study.78
Other therapies
A wide variety of other drugs have been tested in patients with primary SS, including steroid hormones, antimicrobial agents, antiulcer drugs and fatty acids. Some of the drugs tested showed no change in objective and subjective sicca features in comparison with placebo, including rebamipide79 and doxycycline.80 Other agents were found to be associated with an unacceptable rate of adverse events (zidovudine),81 or marginal therapeutic benefits (dehydroepiandrosterone, linolenic and γ-linolenic acids and nizatidine).82,83,84,85,86,87,88
Biologic agents—level of evidence
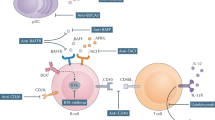
The emergence of biologic therapies has increased the therapeutic armamentarium available for the treatment of SS, but use of these agents is limited by the lack of licensing. 17 studies have analysed the therapeutic potential of five biologic agents in primary SS (Table 4): IFN-α;89,90,91,92 two anti-TNF agents—infliximab93,94 and etanercept95,96 (a monoclonal antibody against TNF and a recombinant soluble TNF receptor, respectively); and two B-cell-targeted therapies—rituximab97,98,99,100,101,102,103,104,105 and epratuzumab106 (monoclonal antibodies against CD20 and CD22, respectively).
IFN-α
Controlled studies (two double-blind, placebo-controlled and one single-blind, controlled with sucralfate) comprising a total of 569 patients with SS have evaluated the use of 150 IU (international units) of oral IFN-α three times daily.89,90,91 Two of these studies reported that sicca symptoms, salivary flow and lymphocytic infiltration all improved with IFN-α treatment.90,91 However, the largest of the three controlled trials—a RCT that included 497 of the 569 patients treated with IFN-α in controlled trials to date—observed that IFN-α use only improved unstimulated salivary flow and was associated with a higher percentage of gastrointestinal adverse events in comparison with placebo.89 A small uncontrolled comparative study found increased tear and saliva production in patients treated with 3.1 MU (million units) of oral IFN-α2 three times per week in comparison with patients receiving 6 mg per kg of HCQ daily.92
TNF blockade
Infliximab
A prospective open-label study in 16 patients with SS demonstrated efficacy of infliximab in this disease, based on subjective and objective measures.93 By contrast, the subsequent TRIPSS study,94 a RCT that enrolled 103 patients with SS, found no statistically significant changes in symptoms, salivary flow rates, quality of life or in the results of ocular tests or salivary biopsy in comparison with placebo. The only therapeutic benefits of infliximab treatment reported in the TRIPSS study were improvements in some analytical parameters.94 With respect to adverse events, infusion-related reactions were reported in 10% of patients and infections in 4% (one patient developed pneumococcal septicaemia).
Etanercept
Two studies using etanercept (one RCT, one prospective) revealed no difference in the main sicca characteristics and symptoms associated with SS after treatment.95,96 Nevertheless, substantial improvements in some analytical parameters (ESR and CRP levels) were observed. Three of 29 patients treated with etanercept in these trials experienced adverse events.
B-cell-targeted therapies
The largest number of clinical trials investigating the efficacy of biologic agents in patients with SS used the B-cell-depleting antibody rituximab;97,98,99,100,101,102,103,104,105 Table 5 summarizes data reported for the 125 patients with SS treated with this agent. Two placebo-controlled RCTs comprising 47 patients demonstrated considerable improvements in sicca features, salivary flow, ocular tests results, fatigue and quality of life scores after treatment with rituximab.97,98
With respect to systemic involvement, one of these trials also reported a reduction in extraglandular features in patients treated with rituximab, compared with placebo.97 Similar results of rituximab therapy were also observed in uncontrolled studies, especially for articular, vasculitic, pulmonary and neurological involvement. Seror et al.101 reported a decrease in the daily dose of corticosteroids used by patients with systemic involvement of primary SS after treatment with rituximab, which might have implications for reduction of the risk of steroid-associated adverse events. All studies that have investigated the effects of rituximab in patients with SS97,98,99,100,101,102,103,104,105 have reported statistically significant reductions in analytical parameters, such as ESR, CRP levels, cryoglobulinemia and/or RF titres.
In one study,100 antichimeric-antibodies were found in five of 23 (22%) patients with SS, four of whom had early disease. An overall analysis of rituximab-related adverse events in patients with SS revealed early and late infusion-related reactions in 19% of patients—particularly in individuals with early disease who were not pretreated with corticosteroids—and infections in 13% (Table 5).
The anti-CD22 antibody epratuzumab has only been tested in one study in patients with SS. This open-label prospective study, which included 16 patients treated with epratuzumab, demonstrated marked improvements in fatigue and subjective patient-reported and physician-assessed outcomes.106
Therapy tailored to clinical scenarios
Management of sicca features
The primary therapeutic approach for sicca manifestations should be centred on symptomatic treatment, using artificial tears and saliva substitutes (Figure 1). This recommendation is based on evidence from a number of studies demonstrating that daily use of these treatments offers relief from sicca symptoms and can improve quality of life, without the risk of adverse events.

Level of evidence (1–4; recommendations based on evidence from studies that predominantly included patients with sicca syndrome or patients with SS are denoted by + and ++, respectively) and strength of recommendation (A–D) are shown in parentheses for each therapy according to the grading recommendations of Harbour and Miller.123 The first step in the management of sicca symptoms always consists of topical therapies; systemic therapies represent the second step, if topical interventions are not effective. Patients with acceptable salivary flow outputs might have poor tolerance of saliva substitutes owing to the 'sticky' feeling they can cause; in these patients, mechanical and/or gustatory stimulation (sugar-free candies and chewing gum) might be a useful first-line therapy before intervention with muscarinic receptor agonists (pilocarpine or cevimeline) is considered. Although a high level of evidence supports the use of certain topical ocular therapies, these agents have a grade B recommendation because studies were overwhelmingly performed in patients with keratoconjunctivitis sicca, with only a variable proportion of patients with SS included. Limited evidence is available for the treatment of sicca features other than those affecting the eyes and mouth in primary SS.1 Lacrimal punctal occlusion using plugs or thermal cautery can be useful in refractory and/or severe cases. Abbreviations: ENT, ear, nose and throat; SS, Sjögren's syndrome.
No controlled studies have directly compared saliva substitutes and secretagogues in individuals with moderate-to-severe sicca symptoms; therefore, it seems reasonable that, in clinical practice, patients with mild symptoms should first be treated with agents not associated with adverse events (such as saliva substitutes) before drugs with a higher rate of adverse effects (including muscarinic acetylcholine receptor agonists) are introduced.107 The available data on management of oral dryness do not show conclusively that one specific saliva substitute is superior to another in patients with SS; thus, more than one substitute should always be tried before turning to alternative approaches, especially when the first agent prescribed is poorly tolerated. In addition, patients with acceptable salivary flow outputs might show poor tolerance of saliva substitutes owing to the 'sticky feeling' caused by these agents, which can provoke a sensation of oral dryness. In these patients, mechanical and/or gustatory stimulation of saliva production using sugar-free chewing gums and sweets might be useful. The use of anticholinergic medications, intake of alcohol (including mouthwashes and fluoride rinses that contain alcohol) and smoking should be discouraged, whereas nonpharmacological approaches such as water intake, mechanical and gustatory stimulation, and fluoride toothpaste can be useful and should be promoted.
With respect to eye drops for relief of ocular symptoms, evidence supports the use of preservative-free formulations of artificial tears (containing hyaluronate or methylcellulose) 3–4 times daily, reducing the interval between dosing to as little as 1 h when necessary.1,2,3 Alternatives include topical 0.05% retinyl palmitate and artificial tears containing polyethylene or propylene glycols.
Individuals with severe sicca features can require more intensive therapy than those with mild or moderate sicca symptoms, owing to the substantial implications for the patient's general health status. In patients with moderate or severe oral dryness and residual salivary gland function, oral muscarinic receptor agonists (pilocarpine or cevimeline) are the treatment of choice, as long as contraindications are taken into account (Figure 1). Evidence suggests that the doses with the best ratios of efficacy to adverse events are 5 mg every 6 h for pilocarpine and 30 mg every 8 h for cevimeline, although no studies have directly compared the efficacy of the two drugs. Tolerance must be considered, as the frequency of reported adverse events for these agents was as high as 40%, with some events being more commonly associated with pilocarpine (increased urinary frequency and flushing) and others with cevimeline (gastrointestinal effects). In patients intolerant to muscarinic receptor agonists, N-acetylcysteine might be an alternative. Although RCTs have demonstrated a considerable improvement in sicca features after rituximab therapy,97,100 we consider that its off-label use to treat only sicca involvement—even when severe—is not warranted.
Patients with SS-associated severe or refractory keratoconjunctivitis sicca might require the addition of topical anti-inflammatory agents. Ocular NSAIDs or corticosteroids should only be prescribed by ophthalmologists and for the minimum time necessary, as adverse events seem to be more frequently reported after use of these agents for >2 weeks. By contrast, controlled trials support the use of topical 0.05% cyclosporine A twice daily, as this therapy has an acceptable safety profile, although further benefits are not seen beyond 6 months of treatment.
The role of autologous serum has only been tested in two small studies at present,108,109 and its efficacy in the treatment of sicca symptoms should be confirmed in larger studies. For the most refractory cases of occular dryness, temporary occlusion of the puncta through the insertion of plugs is recommended.1,2,3
Management of general symptoms
Patients with primary SS often present with nonspecific general symptoms, including noninflammatory muscle and joint involvement, fatigue and weakness, which can have a greater impact on the quality of life than sicca features. In patients with these manifestations, the first step should be a differential diagnosis with associated conditions such as hypothyroidism, neoplasia, depression and especially fibromyalgia, which is reported in 22–33% of patients with primary SS and can heavily influence both patient and physician evaluations of health status.110,111 After excluding these disorders, HCQ should be the cornerstone of therapy, with clinical benefits of this agent being reported beyond fatigue and musculoskeletal pain; uncontrolled studies found additional improvements in subjective and objective sicca features as well as analytical and immunological parameters, and reductions in parotid enlargement and oral infections.63,64,65,66,67,68,69
The four biologic agents investigated in trials that enrolled patients with primary SS have been associated with improvements in fatigue (including one small RCT using rituximab).93,95,97,98,106 However, we consider that the off-label use of these new drugs to treat only general symptoms of SS (even when severe) is not justified.
Management of systemic involvement
As a general rule, the management of extraglandular features in primary SS should be organ-specific, using mainly corticosteroids and immunosuppressive agents (Figure 2).1,2,3 Unfortunately, studies analysing the effects of immunosuppressive agents in patients with primary SS, which are overwhelmingly uncontrolled, are designed to evaluate sicca rather than systemic outcomes. These trials have shown poor results of immunosuppressive therapies, with an excess of adverse events.

Available data for treatment of extraglandular SS symptoms come from nonanalytical studies, such as retrospective series or case reports (evidence level 3, on a scale of 1–4) representing the lowest strength of recommendation (grade D, rated from A–D) according to the grading recommendations of Harbour and Miller.123 The sole exception is a RCT using rituximab, which showed a reduction of the number of reported extraglandular manifestations compared with placebo (though the number of patients was too small to provide a specific recommendation). Corticosteroids should be used at the minimum dose and length of time necessary. Classification of drugs as first-line, second-line and third-line therapies is based on the number of case reports that have shown them to be effective and the authors' experience. No data are available on immunosuppressive maintenance therapy; thus, regimens similar to those used in other autoimmune diseases (systemic lupus erythematosus, vasculitis) are recommended. Abbreviations: CNS, central nervous system; IVIg, intravenous immunoglobulin; SS, Sjögren's syndrome.
Similarly, studies of biologic agents in SS have primarily focused on evaluating their effects on sicca involvement; nevertheless, several studies have reported promising results regarding the influence of these therapies on the extraglandular features of SS. These studies include a small RCT97 and uncontrolled trials of rituximab,98,99,100,101 which have reported a clinical response in >80% of patients with systemic involvement, improvements in several immunological parameters and a reduction in the mean daily corticosteroid dose. Therefore, current scientific evidence suggests that rituximab can be considered for the treatment of patients with systemic involvement refractory to standard therapies (limited response or intolerance to corticosteroids and immunosuppressive agents).112 The amount and quality of evidence on the off-label use of rituximab in the management of SS-related extraglandular features is higher than that available for corticosteroids and immunosuppressive drugs; however, a reasonable assessment of costs and the risk of serious adverse events versus the benefits of treatment should always be made on an individual basis.
Some retrospective studies have specifically analysed the use of corticosteroids and immunosuppressive agents for the treatment of organ-specific manifestations in patients with primary SS. These studies support the use of corticosteroids and cyclophosphamide in myelitis, azathioprine in interstitial lung disease and methotrexate in joint involvement (Figure 2).113 Evidence for the therapeutic management of other extraglandular SS features comes from isolated case reports or small case series.1,2,3 The most-frequently used immunosuppressive agent for the treatment of glomerulonephritis, vasculitis, multiple neuritis and central nervous system (CNS) involvement is cyclophosphamide.1,2,3 By contrast, some extraglandular features, such as interstitial nephritis or ataxic neuronopathy, seem to have a poor or no response to corticosteroids and immunosuppressive agents.113 Given the low level of evidence from studies in patients with SS, the choice of drugs for organ-by-organ management is, therefore, usually heavily influenced by therapeutic strategies accepted in clinically similar, but aetiopathogenically different, diseases such as systemic lupus erythematosus or systemic vasculitis.
Management of life-threatening situations
Severe, life-threatening disease manifestations have rarely been reported in patients with primary SS. In nine studies comprising 2,241 patients with primary SS, in which mortality rates and causes of death were detailed, only 17 deaths were attributable to SS-related systemic involvement, representing less than 8% of the 221 deaths reported.114 Vasculitis—overwhelmingly cryoglobulinemic and involving vital organs such as the kidneys, lungs and gastrointestinal tract—was the major life-threatening presentation of primary SS.114 Other severe complications of SS include CNS involvement, progressive ataxic neuronopathy, pulmonary arterial hypertension and severe cytopenia.115
No controlled studies have evaluated the therapeutic management of patients with SS-associated life-threatening conditions; at present, data is only available from small (<10 patients) retrospective studies and isolated case reports. However, this small body of evidence, together with expert opinion, suggests that methylprednisolone and cyclophosphamide pulses should be used in patients with severe systemic vasculitis or CNS involvement, combined with plasma exchange in the most severe cases.115 Rituximab is increasingly reported as a promising therapy, not only in patients with life-threatening SS but also in individuals with associated B-cell lymphoma (Figure 2).115,116
Future directions
Advances in our understanding of the molecular mechanisms underlying the aetiopathogenesis of primary SS could lead to the development of novel therapies. After the demonstration of the effectiveness of TNF inhibitors in rheumatoid arthritis, these agents were studied in patients with SS, but RCTs revealed a lack of efficacy. At present, B-cell-targeted biologic agents seem to be the most promising therapies in patients with SS, especially rituximab, which has been used in more than 100 reported cases. Other B-cell-targeting agents with therapeutic potential include those against CD22+ cells (epratuzumab) and, in particular, inhibitors of B lymphocyte stimulator (also known as TNF ligand superfamily member 13B or B-cell-activating factor)-signalling (belimumab). On the basis of data from preliminary studies,117,118,119 the suggestion of a role for B-cell-depleting therapies in the modification of the aetiopathogenesis of primary SS—a disease characterized by B-cell hyperactivity—seems reasonable. Ongoing trials in patients with SS are concentrated on the evaluation of rituximab and belimumab, but the efficacy of HCQ, anakinra and oromucosal surfactants is also being investigated. In the future, the therapeutic management of sicca features will probably be based on muscarinic receptor agents with increased selectivity and, possibly, on salivary gland electrostimulation.120 New immunosuppressive agents, especially B-cell-targeted therapies, will probably have key roles in the treatment of complicated cases of primary SS, especially those with extraglandular involvement.
Conclusions
During the past three decades, the use of substitute agents for sicca features and corticosteroids and immunosuppressive agents for systemic involvement has been the foundation of SS therapy. Unfortunately, a major problem in reviewing therapies and offering solid therapeutic recommendations in primary SS is the limited level of reported evidence (Box 1).3 The overall low level of evidence from therapeutic studies in primary SS highlights the requirement for substantially larger trials of the most promising therapies. Large RCTs, which are considered the gold standard in clinical research for the assessment of treatment efficacy and safety, are scarce in primary SS. This limitation might be explained by the heterogeneous clinical presentation (both sicca-related and systemic), the lack of consensus end points for evaluating outcomes and the often poor correlation between the intensity of self-reported symptoms and the results of objective tests. Consequently, therapeutic decisions are based on a combination of personal experience and reported scientific evidence, principally from uncontrolled studies. The complexity of the therapeutic approach in primary SS is increased by the fact that not all patients respond to first-line therapies, and even less scientific evidence is available to guide treatment interventions in this situation; thus, allocation of second-line drugs is frequently based on the opinion of the treating clinician. Objective, standardized evaluation of disease burden has been facilitated by two proposed international indexes.121,122 However, international efforts are also required to enrol and characterize large, multicentre cohorts of patients with SS, and to develop consensus end points for clinical trials.
Management of patients with SS requires a multidisciplinary approach; ophthalmologists, odontologists and gynaecologists should all be routinely involved in the follow-up monitoring of sicca manifestations. Likewise, extraglandular involvement requires organ-specific specialist care, and patients with lymphoproliferative neoplasia should be referred to a haematologist.
The emergence of new immunosuppressive and biologic agents has increased the therapeutic armamentarium available for the treatment of severe disease, but their use in primary SS is limited by the absence of specific regulatory approval. Nevertheless, considerable progress has been made in the targeting of specific aetiopathogenic pathways in primary SS. Ultimately, this approach could result in the development of new effective and highly selective therapies, without the adverse effects often associated with the less selective immunosuppressive agents that represent the current standard-of-care therapies.
Review criteria
We searched MEDLINE for English-language articles published between 1st January 1986 and 30th November 2011 that reported studies in adult humans using the MeSH term “Sjögren's syndrome” and the MeSH subheading “therapy”. We read the titles and abstracts (if available) looking for articles on currently available drugs for Sjögren's syndrome therapy (inclusion criterion). Duplicate publications, case reports, reviews, experimental studies and uncontrolled series with fewer than 10 patients were excluded. We also manually searched the reference list of relevant articles retrieved for further publications.
References
Fox, R. I. Sjögren's syndrome. Lancet 366, 321–331 (2005).
Kassan, S. S. & Moutsopoulos, H. M. Clinical manifestations and early diagnosis of Sjögren syndrome. Arch. Intern. Med. 164, 1275–1284 (2004).
Ramos-Casals, M., Tzioufas, A. G., Stone, J. H., Sisó, A. & Bosch, X. Treatment of primary Sjögren syndrome: a systematic review. JAMA 304, 452–460 (2010).
Alpöz, E. et al. The efficacy of Xialine in patients with Sjögren's syndrome: a single-blind, cross-over study. Clin. Oral Investig. 12, 165–172 (2008).
Alves, M. B., Motta, A. C., Messina, W. C. & Migliari, D. A. Saliva substitute in xerostomic patients with primary Sjögren's syndrome: a single-blind trial. Quintessence Int. 35, 392–396 (2004).
Aagaard, A., Godiksen, S., Teglers, P. T., Schiødt, M. & Glenert, U. Comparison between new saliva stimulants in patients with dry mouth: a placebo-controlled double-blind crossover study. J. Oral Pathol. Med. 21, 376–380 (1992).
Johansson G. et al. Effects of mouthrinses with linseed extract Salinum without/with chlorhexidine on oral conditions in patients with Sjögren's syndrome. A double-blind crossover investigation. Gerodontology 18, 87–94 (2001).
Rhodus, N. L. & Bereuter, J. Clinical evaluation of a commercially available oral moisturizer in relieving signs and symptoms of xerostomia in postirradiation head and neck cancer patients and patients with Sjögren's syndrome. J. Otolaryngol. 29, 28–34 (2000).
van der Reijden, W. A., van der Kwaak, H., Vissink, A., Veerman, E. C. & Amerongen, A. V. Treatment of xerostomia with polymer-based saliva substitutes in patients with Sjögren's syndrome. Arthritis Rheum. 39, 57–63 (1996).
Visch, L. L., Gravenmade, E. J., Schaub, R. M., Van Putten, W. L. & Vissink, A. A double-blind crossover trial of CMC- and mucin-containing saliva substitutes. Int. J. Oral Maxillofac. Surg. 15, 395–400 (1986).
Aragona, P., Di Stefano, G., Ferreri, F., Spinella, R. & Stilo, A. Sodium hyaluronate eye drops of different osmolarity for the treatment of dry eye in Sjögren's syndrome patients. Br. J. Ophthalmol. 86, 879–884 (2002).
Aragona, P., Papa, V., Micali, A., Santocono, M. & Milazzo, G. Long term treatment with sodium hyaluronate-containing artificial tears reduces ocular surface damage in patients with dry eye. Br. J. Ophthalmol. 86, 181–184 (2002).
Brignole, F., Pisella, P. J., Dupas, B., Baeyens, V. & Baudouin, C. Efficacy and safety of 0.18% sodium hyaluronate in patients with moderate dry eye syndrome and superficial keratitis. Graefes Arch. Clin. Exp. Ophthalmol. 243, 531–538 (2005).
Condon, P. I. et al. Double blind, randomised, placebo controlled, crossover, multicentre study to determine the efficacy of a 0.1% (w/v) sodium hyaluronate solution (Fermavisc) in the treatment of dry eye syndrome. Br. J. Ophthalmol. 83, 1121–1124 (1999).
McDonald, C. C., Kaye, S. B., Figueiredo, F. C., Macintosh, G. & Lockett, C. A randomised, crossover, multicentre study to compare the performance of 0.1% (w/v) sodium hyaluronate with 1.4% (w/v) polyvinyl alcohol in the alleviation of symptoms associated with dry eye syndrome. Eye (Lond.) 16, 601–607 (2002).
Toda, I., Shinozaki, N. & Tsubota, K. Hydroxypropyl methylcellulose for the treatment of severe dry eye associated with Sjögren's syndrome. Cornea 15, 120–128 (1996).
Aragona, P., Stilo, A., Ferreri, F. & Mobrici, M. Effects of the topical treatment with NSAIDs on corneal sensitivity and ocular surface of Sjögren's syndrome patients. Eye (Lond.) 19, 535–539 (2005).
Avisar, R., Robinson, A., Appel, I., Yassur, Y. & Weinberger, D. Diclofenac sodium, 0.1% (Voltaren Ophtha), versus sodium chloride, 5%, in the treatment of filamentary keratitis. Cornea 19, 145–147 (2000).
Guidera, A. C., Luchs, J. I. & Udell, I. J. Keratitis, ulceration, and perforation associated with topical nonsteroidal anti-inflammatory drugs. Ophthalmology 108, 936–944 (2001).
Avunduk, A. M., Avunduk, M. C., Varnell, E. D. & Kaufman, H. E. The comparison of efficacies of topical corticosteroids and nonsteroidal anti-inflammatory drops on dry eye patients: a clinical and immunocytochemical study. Am. J. Ophthalmol. 136, 593–602 (2003).
Marsh, P. & Pflugfelder, S. C. Topical nonpreserved methylprednisolone therapy for keratoconjunctivitis sicca in Sjögren syndrome. Ophthalmology 106, 811–816 (1999).
Pflugfelder, S. C. et al. A randomized, double-masked, placebo-controlled, multicenter comparison of loteprednol etabonate ophthalmic suspension, 0.5%, and placebo for treatment of keratoconjunctivitis sicca in patients with delayed tear clearance. Am. J. Ophthalmol. 138, 444–457 (2004).
Hong, S., Kim, T., Chung, S. H., Kim, E. K. & Seo, K. Y. Recurrence after topical nonpreserved methylprednisolone therapy for keratoconjunctivitis sicca in Sjögren's syndrome. J. Ocul. Pharmacol. Ther. 23, 78–82 (2007).
Dastjerdi, M. H., Hamrah, P. & Dana, R. High-frequency topical cyclosporine 0.05% in the treatment of severe dry eye refractory to twice-daily regimen. Cornea 28, 1091–1096 (2009).
Gündüz, K. & Ozdemir, O. Topical cyclosporin treatment of keratoconjunctivitis sicca in secondary Sjögren's syndrome. Acta. Ophthalmol. (Copenh.) 72, 438–442 (1994).
Kunert, K. S., Tisdale, A. S., Stern, M. E., Smith, J. A. & Gipson, I. K. Analysis of topical cyclosporine treatment of patients with dry eye syndrome: effect on conjunctival lymphocytes. Arch. Ophthalmol. 118, 1489–1496 (2000).
Roberts, C. W., Carniglia, P. E. & Brazzo, B. G. Comparison of topical cyclosporine, punctal occlusion, and a combination for the treatment of dry eye. Cornea 26, 805–809 (2007).
Sall, K. N., Cohen, S. M., Christensen, M. T. & Stein, J. M. An evaluation of the efficacy of a cyclosporine-based dry eye therapy when used with marketed artificial tears as supportive therapy in dry eye. Eye Contact Lens 32, 21–26 (2006).
Stevenson, D., Tauber, J. & Reis, B. L. Efficacy and safety of cyclosporin A ophthalmic emulsion in the treatment of moderate-to-severe dry eye disease: a dose-ranging, randomized trial. The Cyclosporin A Phase 2 Study Group. Ophthalmology 107, 967–974 (2000).
Toker, E. & Asfuroğlu, E. Corneal and conjunctival sensitivity in patients with dry eye: the effect of topical cyclosporine therapy. Cornea 29, 133–140 (2010).
Perry, H. D. et al. Evaluation of topical cyclosporine for the treatment of dry eye disease. Arch. Ophthalmol. 126, 1046–1050 (2008).
Sall, K., Stevenson, O. D., Mundorf, T. K. & Reis, B. L. Two multicenter, randomized studies of the efficacy and safety of cyclosporine ophthalmic emulsion in moderate to severe dry eye disease. CsA Phase 3 Study Group. Ophthalmology 107, 631–639 (2000).
Kim, E. C., Choi, J. S. & Joo, C. K. A comparison of vitamin A and cyclosporine A 0.05% eye drops for treatment of dry eye syndrome. Am. J. Ophthalmol. 147, 206–213 (2009).
Barber, L. D., Pflugfelder, S. C., Tauber, J. & Foulks, G. N. Phase III safety evaluation of cyclosporine 0.1% ophthalmic emulsion administered twice daily to dry eye disease patients for up to 3 years. Ophthalmology 112, 1790–1794 (2005).
Tauber, J. et al. Double-masked, placebo-controlled safety and efficacy trial of diquafosol tetrasodium (INS365) ophthalmic solution for the treatment of dry eye. Cornea 23, 784–792 (2004).
Schiødt, M., Oxholm, P. & Jacobsen, A. Treatment of xerostomia in patients with primary Sjögren's syndrome with sulfarlem. Scand. J. Rheumatol. Suppl. 61, 250–252 (1986).
Kriegbaum, N. J., von Linstow, M., Oxholm, P. & Prause, J. U. A follow-up study of the progress of keratoconjunctivitis sicca and response to treatment in primary Sjögren's syndrome. Scand. J. Rheumatol. 18, 193–196 (1989).
Walters, M. T., Rubin, C. E., Keightley, S. J., Ward, C. D. & Cawley, M. I. A double-blind, cross-over, study of oral N-acetylcysteine in Sjögren's syndrome. Scand. J. Rheumatol. Suppl. 61, 253–258 (1986).
Aframian, D. J. et al. Pilocarpine treatment in a mixed cohort of xerostomic patients. Oral Dis. 13, 88–92 (2007).
Aragona, P., Di Pietro, R., Spinella, R. & Mobrici, M. Conjunctival epithelium improvement after systemic pilocarpine in patients with Sjögren's syndrome. Br. J. Ophthalmol. 90, 166–170 (2006).
Gibson, J., Halliday, J. A., Ewert, K. & Robertson, S. A controlled release pilocarpine buccal insert in the treatment of Sjögren's syndrome. Br. Dent. J. 202, E17 (2007).
Papas, A. S. et al. Successful treatment of dry mouth and dry eye symptoms in Sjögren's syndrome patients with oral pilocarpine: a randomized, placebo-controlled, dose-adjustment study. J. Clin. Rheumatol. 10, 169–177 (2004).
Rhodus, N. L., Liljemark, W., Bloomquist, C. & Bereuter, J. Candida albicans levels in patients with Sjögren's syndrome before and after long-term use of pilocarpine hydrochloride: a pilot study. Quintessence Int. 29, 705–710 (1998).
Peluso, G. et al. Proteomic study of salivary peptides and proteins in patients with Sjögren's syndrome before and after pilocarpine treatment. Arthritis Rheum. 56, 2216–2222 (2007).
Rhodus, N. L. Oral pilocarpine HCl stimulates labial (minor) salivary gland flow in patients with Sjögren's syndrome. Oral Dis. 3, 93–98 (1997).
Rosas, J. et al. Usefulness of basal and pilocarpine-stimulated salivary flow in primary Sjögren's syndrome. Correlation with clinical, immunological and histological features. Rheumatology (Oxford) 41, 670–675 (2002).
Tsifetaki, N. et al. Oral pilocarpine for the treatment of ocular symptoms in patients with Sjögren's syndrome: a randomised 12 week controlled study. Ann. Rheum. Dis. 62, 1204–1207 (2003).
Vivino, F. B. et al. Pilocarpine tablets for the treatment of dry mouth and dry eye symptoms in patients with Sjögren syndrome: a randomized, placebo-controlled, fixed-dose, multicenter trial. P92–01 Study Group. Arch. Intern. Med. 159, 174–181 (1999).
Wu, C. H. et al. Pilocarpine hydrochloride for the treatment of xerostomia in patients with Sjögren's syndrome in Taiwan—a double-blind, placebo-controlled trial. J. Formos. Med. Assoc. 105, 796–803 (2006).
Fife, R. S. et al. Cevimeline for the treatment of xerostomia in patients with Sjögren syndrome: a randomized trial. Arch. Intern. Med. 162, 1293–1300 (2002).
Komai, K. et al. Sjögren's syndrome patients presenting with hypergammaglobulinemia are relatively unresponsive to cevimeline treatment. Mod. Rheumatol. 19, 416–419 (2009).
Leung, K. C. et al. The efficacy of cevimeline hydrochloride in the treatment of xerostomia in Sjögren's syndrome in southern Chinese patients: a randomised double-blind, placebo-controlled crossover study. Clin. Rheumatol. 27, 429–436 (2008).
Ono, M. et al. Therapeutic effect of cevimeline on dry eye in patients with Sjögren's syndrome: a randomized, double-blind clinical study. Am. J. Ophthalmol. 138, 6–17 (2004).
Petrone, D. et al. A double-blind, randomized, placebo-controlled study of cevimeline in Sjögren's syndrome patients with xerostomia and keratoconjunctivitis sicca. Arthritis Rheum. 46, 748–754 (2002).
Suzuki, K. et al. Effect of cevimeline on salivary components in patients with Sjögren syndrome. Pharmacology 74, 100–105 (2005).
Takagi, Y., Katayama, I., Tashiro, S. & Nakamura, T. Parotid irrigation and cevimeline gargle for treatment of xerostomia in Sjögren's syndrome. J. Rheumatol. 35, 2289–2291 (2008).
Yamada, H. et al. Efficacy prediction of cevimeline in patients with Sjögren's syndrome. Clin. Rheumatol. 26, 1320–1327 (2007).
Izumi, M. et al. Corticosteroid irrigation of parotid gland for treatment of xerostomia in patients with Sjögren's syndrome. Ann. Rheum. Dis. 57, 464–469 (1998).
Miyawaki, S., Nishiyama, S. & Matoba, K. Efficacy of low-dose prednisolone maintenance for saliva production and serological abnormalities in patients with primary Sjögren's syndrome. Intern. Med. 38, 938–943 (1999).
Fox, P. C. et al. Prednisone and piroxicam for treatment of primary Sjögren's syndrome. Clin. Exp. Rheumatol. 11, 149–156 (1993).
Pijpe, J. et al. Progression of salivary gland dysfunction in patients with Sjogren's syndrome. Ann. Rheum. Dis. 66, 107–112 (2007).
Ramos-Casals, M. et al. High prevalence of serum metabolic alterations in primary Sjögren's syndrome: influence on clinical and immunological expression. J. Rheumatol. 34, 754–761 (2007).
Kruize, A. A. et al. Hydroxychloroquine treatment for primary Sjögren's syndrome: a two year double blind crossover trial. Ann. Rheum. Dis. 52, 360–364 (1993).
Tishler, M., Yaron, I., Shirazi, I. & Yaron, M. Hydroxychloroquine treatment for primary Sjögren's syndrome: its effect on salivary and serum inflammatory markers. Ann. Rheum. Dis. 58, 253–256 (1999).
Rihl, M., Ulbricht, K., Schmidt, R. E. & Witte, T. Treatment of sicca symptoms with hydroxychloroquine in patients with Sjögren's syndrome. Rheumatology (Oxford) 48, 796–799 (2009).
Fox, R. I. et al. Treatment of primary Sjögren's syndrome with hydroxychloroquine. Am. J. Med. 85, 62–67 (1988).
Fox, R. I., Dixon, R., Guarrasi, V. & Krubel, S. Treatment of primary Sjögren's syndrome with hydroxychloroquine: a retrospective, open-label study. Lupus 5 (Suppl. 1), S31–S36 (1996).
Cankaya, H. et al. Effects of hydroxychloroquine on salivary flow rates and oral complaints of Sjögren patients: a prospective sample study. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endod. 110, 62–67 (2010).
Yavuz, S., Asfuroğlu, E., Bicakcigil, M. & Toker, E. Hydroxychloroquine improves dry eye symptoms of patients with primary Sjögren's syndrome. Rheumatol. Int. 31, 1045–1049 (2011).
Drosos, A. A., Skopouli, F. N., Galanopoulou, V. K., Kitridou, R. C. & Moutsopoulos, H. M. Cyclosporin A therapy in patients with primary Sjögren's syndrome: results at one year. Scand. J. Rheumatol Suppl. 61, 246–249 (1986).
Skopouli, F. N., Jagiello, P., Tsifetaki, N. & Moutsopoulos, H. M. Methotrexate in primary Sjögren's syndrome. Clin. Exp. Rheumatol. 14, 555–558 (1996).
van Woerkom, J. M. et al. Safety and efficacy of leflunomide in primary Sjögren's syndrome: a phase II pilot study. Ann. Rheum. Dis. 66, 1026–1032 (2007).
Willeke, P. et al. Mycophenolate sodium treatment in patients with primary Sjögren syndrome: a pilot trial. Arthritis Res. Ther. 9, R115 (2007).
Price, E. J., Rigby, S. P., Clancy, U. & Venables, P. J. A double blind placebo controlled trial of azathioprine in the treatment of primary Sjögren's syndrome. J. Rheumatol. 25, 896–899 (1998).
ter Borg, E. J. et al. Treatment of primary Sjögren's syndrome with D-penicillamine: a pilot study. Neth. J. Med. 60, 402–406 (2002).
Pillemer, S. R. et al. Prominent adverse effects of thalidomide in primary Sjögren's syndrome. Arthritis Rheum. 51, 505–506 (2004).
Nakayamada, S. et al. Efficacy and safety of mizoribine for the treatment of Sjögren's syndrome: a multicenter open-label clinical trial. Mod. Rheumatol. 17, 464–469 (2007).
Nakayamada, S. et al. Usefulness of initial histological features for stratifying Sjögren's syndrome responders to mizoribine therapy. Rheumatology (Oxford) 48, 1279–1282 (2009).
Sugai, S. et al. Efficacy and safety of rebamipide for the treatment of dry mouth symptoms in patients with Sjögren's syndrome: a double-blind placebo-controlled multicenter trial. Mod. Rheumatol. 19, 114–124 (2009).
Seitsalo, H. et al. Effectiveness of low-dose doxycycline (LDD) on clinical symptoms of Sjögren's syndrome: a randomized, double-blind, placebo controlled cross-over study. J. Negat. Results Biomed. 6, 11 (2007).
Steinfeld, S. D., Demols, P., Van Vooren, J. P., Cogan, E. & Appelboom, T. Zidovudine in primary Sjögren's syndrome. Rheumatology (Oxford) 38, 814–817 (1999).
Pillemer, S. R. et al. Pilot clinical trial of dehydroepiandrosterone (DHEA) versus placebo for Sjögren's syndrome. Arthritis Rheum. 51, 601–604 (2004).
Hartkamp, A. et al. Effect of dehydroepiandrosterone administration on fatigue, well-being, and functioning in women with primary Sjögren syndrome: a randomised controlled trial. Ann. Rheum. Dis. 67, 91–97 (2008).
Forsbladd'Elia, H., Carlsten, H., Labrie, F., Konttinen, Y. T. & Ohlsson, C. Low serum levels of sex steroids are associated with disease characteristics in primary Sjogren's syndrome; supplementation with dehydroepiandrosterone restores the concentrations. J. Clin. Endocrinol. Metab. 94, 2044–2051 (2009).
Aragona, P., Bucolo, C., Spinella, R., Giuffrida, S. & Ferreri, G. Systemic omega-6 essential fatty acid treatment and PGE1 tear content in Sjögren's syndrome patients. Invest. Ophthalmol. Vis. Sci. 46, 4474–4479 (2005).
Theander, E., Horrobin, D. F., Jacobsson, L. T. & Manthorpe, R. Gammalinolenic acid treatment of fatigue associated with primary Sjögren's syndrome. Scand. J. Rheumatol. 31, 72–79 (2002).
Oxholm, P., Manthorpe, R., Prause, J. U. & Horrobin, D. Patients with primary Sjögren's syndrome treated for two months with evening primrose oil. Scand. J. Rheumatol. 15, 103–108 (1986).
Kasama, T. et al. Effect of the H2 receptor antagonist nizatidine on xerostomia in patients with primary Sjögren's syndrome. Mod. Rheumatol. 18, 455–459 (2008).
Cummins, M. J., Papas, A., Kammer, G. M. & Fox, P. C. Treatment of primary Sjögren's syndrome with low-dose human interferon α administered by the oromucosal route: combined phase III results. Arthritis Rheum. 49, 585–593 (2003).
Shiozawa, S., Tanaka, Y. & Shiozawa, K. Single-blinded controlled trial of low-dose oral IFN-α for the treatment of xerostomia in patients with Sjögren's syndrome. J. Interferon Cytokine Res. 18, 255–262 (1998).
Khurshudian, A. V. A pilot study to test the efficacy of oral administration of interferon-α lozenges to patients with Sjögren's syndrome. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endod. 95, 38–44 (2003).
Ferraccioli, G. F. et al. Interferon α2 (IFN α2) increases lacrimal and salivary function in Sjögren's syndrome patients. Preliminary results of an open pilot trial versus OH-chloroquine. Clin. Exp. Rheumatol. 14, 367–371 (1996).
Steinfeld, S. D., Demols, P., Salmon, I., Kiss, R. & Appelboom, T. Infliximab in patients with primary Sjögren's syndrome: a pilot study. Arthritis Rheum. 44, 2371–2375 (2001).
Mariette, X. et al. Inefficacy of infliximab in primary Sjögren's syndrome: results of the randomized, controlled Trial of Remicade in Primary Sjögren's Syndrome (TRIPSS). Arthritis Rheum. 50, 1270–1276 (2004).
Zandbelt, M. M. et al. Etanercept in the treatment of patients with primary Sjögren's syndrome: a pilot study. J. Rheumatol. 31, 96–101 (2004).
Sankar, V. et al. Etanercept in Sjögren's syndrome: a twelve-week randomized, double-blind, placebo-controlled pilot clinical trial. Arthritis Rheum. 50, 2240–2245 (2004).
Meijer, J. et al. Effective rituximab treatment in primary Sjögren's syndrome: a randomised, double-blind, placebo-controlled trial. Arthritis Rheum. 62, 960–968 (2010).
Dass, S. et al. Reduction of fatigue in Sjögren syndrome with rituximab: results of a randomised, double-blind, placebo-controlled pilot study. Ann. Rheum. Dis. 67, 1541–1544 (2008).
Devauchelle-Pensec, V. et al. Improvement of Sjögren's syndrome after two infusions of rituximab (anti-CD20). Arthritis Rheum. 57, 310–317 (2007).
Pijpe, J. et al. Rituximab treatment in patients with primary Sjögren's syndrome: an open-label phase II study. Arthritis Rheum. 52, 2740–2750 (2005).
Seror, R. et al. Tolerance and efficacy of rituximab and changes in serum B cell biomarkers in patients with systemic complications of primary Sjögren's syndrome. Ann. Rheum. Dis. 66, 351–357 (2007).
Gottenberg, J. E. et al. Tolerance and short term efficacy of rituximab in 43 patients with systemic autoimmune diseases. Ann. Rheum. Dis. 64, 913–920 (2005).
Voulgarelis, M., Giannouli, S., Tzioufas, A. G. & Moutsopoulos, H. M. Long term remission of Sjögren's syndrome associated aggressive B cell non-Hodgkin's lymphomas following combined B cell depletion therapy and CHOP (cyclophosphamide, doxorubicin, vincristine, prednisone). Ann. Rheum. Dis. 65, 1033–1037 (2006).
Mekinian, A. et al. Efficacy of rituximab in primary Sjögren's syndrome with peripheral nervous system involvement: results from the AIR registry. Ann. Rheum. Dis. 71, 84–87 (2012).
Ramos-Casals, M. et al. Off-label use of rituximab in 196 patients with severe, refractory systemic autoimmune diseases. Clin. Exp. Rheumatol. 28, 468–476 (2010).
Steinfeld, S. D. et al. Epratuzumab (humanised anti-CD22 antibody) in primary Sjögren's syndrome: an open-label phase I/II study. Arthritis Res. Ther. 8, R129 (2006).
Vissink, A., Kallenberg, C. G. & Bootsma, H. Treatment approaches in primary Sjogren syndrome. JAMA 304, 2015–2016 (2010).
Yoon, K. C. et al. Comparison of autologous serum and umbilical cord serum eye drops for dry eye syndrome. Am. J. Ophthalmol. 144, 86–92 (2007).
Noble, B. A. et al. Comparison of autologous serum eye drops with conventional therapy in a randomised controlled crossover trial for ocular surface disease. Br. J. Ophthalmol. 88, 647–652 (2004).
Belenguer, R. et al. Influence of clinical and immunological parameters on the health-related quality of life of patients with primary Sjögren's syndrome. Clin. Exp. Rheumatol. 23, 351–356 (2005).
Ostuni, P. et al. Fibromyalgia in Italian patients with primary Sjögren's syndrome. Joint Bone Spine. 69, 51–57 (2002).
Engel, P., Gómez-Puerta, J. A., Ramos-Casals, M., Lozano, F. & Bosch, X. Therapeutic targeting of B cells for rheumatic autoimmune diseases. Pharmacol. Rev. 63, 127–156 (2011).
Mavragani, C. P. & Kassan, S. S. in Sjögren's Syndrome. Diagnosis and Therapeutics (eds Ramos-Casals, M., Stone, J. & Moutsopoulos, H. M.) 565–570 (Springer-Verlag, Berlin, 2012).
Brito-Zerón, P. & Ramos-Casals, M. Prognosis of patients with primary Sjögren's syndrome [Spanish]. Med. Clin. (Barc.) 130, 109–115 (2008).
Ramos-Casals, M., Brito-Zerón, P., Bové, A. & Sisó, A. in Autoimmune Diseases. Acute and Complex Situations (eds Khamashta, M. A. & Ramos-Casals, M.) 45–66 (Springer-Verlag, London, 2011).
Pollard, R. P. et al. Treatment of mucosa-associated lymphoid tissue lymphoma in Sjögren's syndrome: a retrospective clinical study. J. Rheumatol. 38, 2198–2208 (2011).
Pijpe, J. et al. Clinical and histologic evidence of salivary gland restoration supports the efficacy of rituximab treatment in Sjögren's syndrome. Arthritis Rheum. 60, 3251–3256 (2009).
Jousse-Joulin, S. et al. Ultrasound assessment of salivary glands in patients with primary Sjögren's syndrome treated with rituximab: quantitative and Doppler waveform analysis. Biologics 1, 311–319 (2007).
Lavie, F. et al. Increase of B cell-activating factor of the TNF family (BAFF) after rituximab treatment: insights into a new regulating system of BAFF production. Ann. Rheum. Dis. 66, 700–703 (2007).
Strietzel, F. P. et al. Efficacy and safety of an intraoral electrostimulation device for xerostomia relief: a multicenter, randomized trial. Arthritis Rheum. 63, 180–190 (2011).
Seror, R. et al. EULAR Sjögren's syndrome disease activity index: development of a consensus systemic disease activity index in primary Sjögren's syndrome. Ann. Rheum. Dis. 69, 1103–1109 (2010).
Seror, R. et al. EULAR Sjögren's Syndrome Patient Reported Index (ESSPRI): development of a consensus patient index for primary Sjögren's syndrome. Ann. Rheum. Dis. 70, 968–972 (2011).
Harbour, R. & Miller, J. A new system for grading recommendations in evidence based guidelines. BMJ 323, 334–336 (2001).
Acknowledgements
M. Ramos-Casals, P. Brito-Zerón and A. Sisó-Almirall are supported by funding from Grants La Marató de TV3 (071810) and Fondo de Investigaciones Sanitarias (080103). The authors wish to thank David Buss for his editorial assistance.
Author information
Authors and Affiliations
Contributions
M. Ramos-Casals, P. Brito-Zerón and A. Sisó-Almirall contributed equally to researching the data for the article. M. Ramos-Casals, P. Brito-Zerón and X. Bosch contributed equally to the writing of the article. All authors provided substantial contributions to discussion of the content and reviewing and/or editing the manuscript before submission.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
About this article
Cite this article
Ramos-Casals, M., Brito-Zerón, P., Sisó-Almirall, A. et al. Topical and systemic medications for the treatment of primary Sjögren's syndrome. Nat Rev Rheumatol 8, 399–411 (2012). https://doi.org/10.1038/nrrheum.2012.53
Published:
Issue Date:
DOI: https://doi.org/10.1038/nrrheum.2012.53
This article is cited by
-
Gut microbiota combined with fecal metabolomics reveals the effects of FuFang Runzaoling on the microbial and metabolic profiles in NOD mouse model of Sjögren’s syndrome
BMC Complementary Medicine and Therapies (2023)
-
Treatment with a Lactococcus lactis that chromosomally express E. coli cfaI mitigates salivary flow loss in a Sjögren’s syndrome-like disease
Scientific Reports (2023)
-
Using machine learning model explanations to identify proteins related to severity of meibomian gland dysfunction
Scientific Reports (2023)
-
A tale of two syndromes: nontraumatic Frey’s syndrome in a woman with Sjögren’s syndrome
Clinical Autonomic Research (2023)
-
Antimalarial drugs—are they beneficial in rheumatic and viral diseases?—considerations in COVID-19 pandemic
Clinical Rheumatology (2022)
