
Abstract
ADP-ribosylation is an important post-translational protein modification (PTM) that regulates diverse biological processes. ADP-ribosyltransferase diphtheria toxin-like 10 (ARTD10, also known as PARP10) mono-ADP-ribosylates acidic side chains and is one of eighteen ADP-ribosyltransferases that catalyze mono- or poly-ADP-ribosylation of target proteins. Currently, no enzyme is known that reverses ARTD10-catalyzed mono-ADP-ribosylation. Here we report that ARTD10-modified targets are substrates for the macrodomain proteins MacroD1, MacroD2 and C6orf130 from Homo sapiens as well as for the macrodomain protein Af1521 from archaebacteria. Structural modeling and mutagenesis of MacroD1 and MacroD2 revealed a common core structure with Asp102 and His106 of MacroD2 implicated in the hydrolytic reaction. Notably, MacroD2 reversed the ARTD10-catalyzed, mono-ADP-ribose–mediated inhibition of glycogen synthase kinase 3β (GSK3β) in vitro and in cells, thus underlining the physiological and regulatory importance of mono-ADP-ribosylhydrolase activity. Our results establish macrodomain-containing proteins as mono-ADP-ribosylhydrolases and define a class of enzymes that renders mono-ADP-ribosylation a reversible modification.
Similar content being viewed by others

Main
ADP-ribosylation is a PTM involved in many biological processes including the regulation of chromatin structure, transcription and DNA repair1,2,3,4,5,6. ARTD10 is a mono-ADP-ribosyltransferase that transfers ADP-ribose to aspartate or glutamate residues by substrate-assisted catalysis7. Recently, GSK3β was identified as a new substrate of ARTD10. Mono-ADP-ribosylation noncompetitively inhibits GSK3β kinase activity8.
Although many ADP-ribosyltransferases (including ARTDs and ADP-ribosyltransferases cholera toxin-like (ARTCs)) that ADP-ribosylate different amino acid acceptor sites have been identified9, the enzymes able to reverse this modification are largely unknown. Irreversible ADP-ribosylation is highly detrimental and causes embryonic lethality10,11. In humans, the deficiency of an ADP-ribose hydrolase is the cause of fatal glutamyl ribose 5-phosphate storage disease12. Therefore, enzymes that remove ADP-ribose modifications must exist. The mammalian hydrolases characterized so far include the mono-ADP-ribosylarginine hydrolase 1 (ARH1) as well as poly-ADP-ribose glycohydrolases (PARG and ARH3). Whereas ARH1 is the only hydrolase that specifically removes mono-ADP-ribose from arginine residues, ARH3 and PARG hydrolyze the O-glycosidic ribose-ribose 1-2′ bonds within ADP-ribose polymers13,14. In contrast, enzymes catalyzing the removal of specific mono-ADP-ribose marks of modified aspartates or glutamates, such as those synthesized by ARTD10, are currently unknown (Supplementary Fig. 1a).
Macrodomains are a family of evolutionarily conserved proteins that bind mono- or poly-ADP-ribose (PAR), poly(A) or O-acetyl-ADP-ribose (OAADPr)15,16,17,18. Macrodomain proteins are involved in diverse cellular processes15 and have been implicated in transcriptional regulation19,20,21,22, chromatin remodeling23,24 and developmental processes as well as in B-cell lymphomagenesis25,26,27. Macrodomain-containing proteins localize to the nucleus (for example, MacroD1v2) or to mitochondria (MacroD1v1) or are found in the cytoplasm (MacroD1v2, MacroD2 or C6orf130)18. In addition to the binding of ADP-ribose, the human MacroD1, MacroD2 and C6orf130 possess C2- or C3-specific OAADPr deacetylase activity28,29 and weak C1-specific phosphatase activity toward ADP-ribose-1″-phosphate (Appr-1″-p), a product of ARTD18 (TPT1) and cyclic nucleotide phosphodiesterases17,30,31. On the basis of structural and functional analyses, several residues in the active centers of macrodomains were identified that participate in the catalytic mechanism28,29. Notably, the catalytically important residues of MacroD1 are not conserved in C6orf130, which indicates that sequence variation within the macrodomain family allows a different set of catalytic residues to perform OAADPr hydrolysis29.
Here we test the hypothesis that macrodomain-containing proteins possess ADP-ribosylhydrolase activity and thereby to characterize the missing mono-ADP-ribosylhydrolases. We provide evidence that the human proteins MacroD1, MacroD2 and C6orf130 as well as the archaebacterial macrodomain Af1521 are able to hydrolyze ARTD10-catalyzed mono-ADP-ribosylation. Notably, MacroD2 rendered the inhibitory effect of GSK3β ADP-ribosylation reversible. Treatment with MacroD2 removed the ADP-ribose moiety from GSK3β, which was sufficient to restore kinase activity in vitro and in cells. These data highlight the important physiological function of endogenous mono-ADP-ribosylation for intracellular signaling and regulatory processes.
Results
Macrodomain proteins are mono-ADP-ribosylhydrolases
To investigate whether macrodomain-containing proteins are able to release the mono-ADP-ribose moiety from ARTD10-modified target proteins, we incubated different macrodomains with in vitro–radiolabeled mono-ADP-ribosylated ARTD10 (full length or the catalytic domain, consisting of residues 818–1025) as a substrate (Fig. 1a,b and Supplementary Fig. 1b,c). Human MacroD1, MacroD2 and C6orf130 robustly hydrolyzed the mono-ADP-ribosyl linkage of modified full-length ARTD10 or the ARTD10 catalytic domain. Notably, the structurally related archaebacterial macrodomain protein Af1521 was also active, whereas neither the human histone variant MacroH2A1.1 nor the mouse macrodomains 1 or 2 of ARTD8 (PARP14) were able to remove mono-ADP-ribose under the tested conditions (Fig. 1a,b and Supplementary Fig. 1b,c). Pulldown experiments revealed that mono-ADP-ribosylhydrolase activity of the tested macrodomains correlated with the ability to bind mono-ADP-ribosylated ARTD10 (Supplementary Fig. 1d). In contrast to their activity toward mono-ADP-ribosylated substrates, MacroD1 and MacroD2 were each completely inactive toward PAR synthesized by ARTD1 (PARP1) in the presence of high NAD+ concentrations, whereas the known PAR hydrolase PARG exhibited strong activity toward this substrate (Fig. 1c). However, PARG was not able to completely remove all ADP-ribose modifications from automodified ARTD1, even when tested under low NAD+ concentrations (resulting in mono- and oligo-ADP-ribosylation; Supplementary Fig. 1e and as suggested in ref. 13). The nature of the modified protein did not affect PARG-dependent hydrolysis, because histones modified by ARTD1 under low NAD+ concentrations were also not completely demodified (Supplementary Fig. 1f). Notably, prior PARG treatment rendered ARTD1, histone H1 and core histones at least partially susceptible to hydrolysis by MacroD2 (Supplementary Fig. 1f,g), which suggests that the acceptor residue and/or the linkage (C1 versus C2 or C3) between the ADP-ribose moiety and the acceptor residue are critical for the newly identified enzymatic activity. Consequently, PARG treatment probably generates mono-ADP-ribosylated residues that serve as substrates for hydrolysis by MacroD2. Comparably, PARG was inactive toward ARTD10-catalyzed mono-ADP-ribosylated histone H1, whereas MacroD2 demodified this substrate, irrespective of PARG treatment (Fig. 1d). These results suggested that MacroD2 is able to release mono-ADP-ribose from acceptor proteins but is inactive toward polymers or oligomers of ADP-ribose.

(a,b) Protein mono-ADP-ribosylhydrolase activity of MacroD1, MacroD2, C6orf130, mouse ARTD8 (mMacro1 and mMacro2), macroH2A1.1 and Af1521. Auto-ADP-ribosylated ARTD10 de-ADP-ribosylated by the indicated proteins is shown on SDS-PAGE with Coomassie blue (CB) staining or autoradiography (32P). Shown are representative blots and quantification from two independent experiments, averaged and normalized to the untreated control (n = 2; mean ± range). (c) Hydrolase activity assays showing that MacroD-like macrodomains have no activity toward polymers of ADP-ribose, whereas PARG has strong activity. Auto-poly-ADP-ribosylated ARTD1 (with 160 μM radioactive NAD+ to induce poly-ADP-ribosylation) was used with the indicated hydrolases. (d) Activity assays as in a, showing that PARG cannot hydrolyze ADP-ribose from mono-ADP-ribosylated ARTD10 or from histones. (e) Concentration-dependent removal of the mono-ADP-ribose from ARTD10 (818–1025) by MacroD2 under nonsaturating conditions. (f) Time course of MacroD2 activity toward mono-ADP-ribosylated ARTD10 catalytic domains. (g) Identification of cofactors (1 mM each) for ADP-ribose hydrolysis by MacroD2. (h) Effect of ADP-ribose and nicotinamide (40 μM each) on MacroD2-mediated de-ADP-ribosylation of ARTD10. (i) The ADP-ribose analog ADP-HPD inhibits MacroD2 in a concentration-dependent manner (4, 40 and 400 μM).
To biochemically characterize the enzymatic reaction catalyzed by MacroD2, we performed concentration- and time-dependent experiments. MacroD2 efficiently removed ADP-ribose modifications from ARTD10 (818–1025) in a concentration-dependent manner (Fig. 1e). Furthermore, mono-ADP-ribose hydrolysis by MacroD2 was time dependent and removed >60% of the modifications within 15 min (Fig. 1f). Further characterization of the enzymatic activity revealed that MacroD2 activity was not markedly affected by the addition of magnesium, calcium or manganese as cofactor (Fig. 1g). Notably, MacroD2 activity was inhibited by the addition of the ADP-ribose analog adenosine 5′-diphosphate (hydroxymethyl) pyrrolidinediol (ADP-HPD) and by ADP-ribose itself in a concentration-dependent manner, whereas addition of nicotinamide did not influence the mono-ADP-ribosylhydrolase activity (Fig. 1h,i). These observations indicated that free ADP-ribose is able to inhibit the mono-ADP-ribose hydrolyzing activity by competing with protein-linked mono-ADP-ribose for binding to the active site. Together, these experiments thus defined MacroD1, MacroD2, C6orf130 and Af1521 as new specific mono-ADP-ribosylhydrolases.
MacroD2 is a mono-ADP-ribosylhydrolase for acidic residues
To define the specificity of MacroD2 mono-ADP-ribosylhydrolase activity, we characterized and analyzed different substrates. Hydroxylamine treatment was reported to remove ADP-ribose from glutamate and arginine residues32. Treatment of ARTD10 with hydroxylamine for 60 min at 37 °C released the modification of the enzyme, as already observed earlier (Fig. 2a and ref. 7), which suggests that MacroD2 is able to hydrolyze mono-ADP-ribose from acidic acceptor sites or arginine. To further investigate whether MacroD2 removed ADP-ribose from arginine acceptor amino acids, the arginine-specific ADP-ribosyltransferase CDTa was used33. MacroD2 showed no activity toward actin modified by CDTa (Fig. 2b), which indicates that modified arginine residues cannot be hydrolyzed by MacroD2. In contrast, the arginine-specific hydrolase ARH1 was able to remove the ADP-ribose from arginine-modified β- or γ-actin but was not able to remove ADP-ribose from ARTD10 (Fig. 2c), which confirms that ARTD10 is not modified at arginine residues. Actin modified at threonine residues by the threonine-specific transferase TccC3 (ref. 34) could not be demodified by either ARH1 or MacroD2 (Fig. 2b). To exclude that lysine residues were modified by ARTD10 and consecutively demodified by MacroD2, we mutated all ARTD10 lysine residues to arginines. This mutant (ARTD10-ΔK) showed reduced ADP-ribosylation activity toward both itself and GSK3β, a newly identified ARTD10 target (Fig. 2d and Supplementary Fig. 2a), which indicated that mutation of these lysine residues interferes with the enzymatic activity of ARTD10. However, the inactive mutant ARTD10-G888W-ΔK could be modified in trans by the catalytic domain of ARTD10 to a comparable extent as ARTD10-G888W (Fig. 2e), thus implying that lysines are not the acceptor sites. Of note, ARTD10-ΔK and ARTD10-G888W-ΔK were still demodified by MacroD2 to a comparable extent as were wild-type ARTD10 or ARTD10-G888W, respectively (Fig. 2d,e), which suggests that the same residues (for example, aspartates or glutamates) are automodified in ARTD10-ΔK and wild-type ARTD10 and consecutively demodified by MacroD2. In conclusion, these results suggested that MacroD2 probably releases ADP-ribose from ADP-ribosylated acidic residues.

(a) Demodification of mono-ADP-ribosylated ARTD10 by hydroxylamine treatment. SDS-PAGE analysis of in vitro–labeled ARTD10 demodified by MacroD2, PARG or hydroxylamine. (b,c) Quantification of ADP-ribosylhydrolase assays with different substrates, analyzed by SDS-PAGE. The mean ± range is shown (n = 2). MacroD2 does not remove ADP-ribose modifications generated by the arginine-specific enzyme CDTa (gray bars) or TccC3 (white bars) (b), and ARH1 does not demodify mono-ADP-ribosylated ARTD10 (c). (d) ADP-ribosylation assay with [32P]-NAD+ and subsequent incubation with MacroD2. Samples are immunoprecipitates of overexpressed hemagglutinin (HA)-tagged HA-ARTD10 or HA-ARTD10-ΔK. (e) Trans-ADP-ribosylation by the indicated overexpressed, immunoprecipitated ARTD10 (818–1025) constructs. The samples treated with MacroD2 (lanes 3 and 4) were washed to terminate the reaction by removing ARTD10 (818–1025) and [32P] NAD+ before incubation with MacroD2. An enhanced contrast is shown to visualize immunoprecipitated HA-ARTD10. (f) Extracted ion chromatograms of mass 558.064, the monoisotopic mass of the ADP-ribose [M-H] adduct. The elution time window of ADP-ribose was between 6 and 7 min. (g) MS-MS spectrum of mass 558.064. Typical mass fragments of ADP-ribose as seen in the ADP-ribose standard (top spectrum) are present in the MS-MS spectrum of ARTD10 + MacroD2 samples (bottom spectrum).
To confirm that MacroD2 removes ADP-ribose from its target protein, the reaction products were analyzed by LC-MS and HPLC. Upon incubation of ARTD10 with MacroD2, a product that eluted at the same time as the ADP-ribose standard and had the expected mass of 558.064 Da was detected, thus showing that MacroD2 indeed removed ADP-ribose from ARTD10 (Fig. 2f,g and Supplementary Fig. 2b–d).
Modeling of MacroD2 and of residues implicated in catalysis
Homology modeling and atomistic simulations were carried out to shed light on the hydrolysis reaction mechanism. First, the three-dimensional structure of MacroD2 was modeled by using the X-ray structure of MacroD1 as a template (Fig. 3a). Except for their flexible loops, the human MacroD1, MacroD2 and C6orf130 structures are highly similar, as emerges from their superposition (Fig. 3b). This structural likeness, along with their similar enzymatic activities, suggested a conserved mode of action.

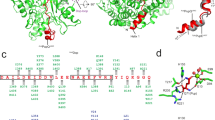
(a) Structure of MacroD2 modeled on the basis of the PDB 2X47 crystal structure of MacroD1. Residues highlighted in yellow represent those having a low r.m.s. fluctuation value as compared to the crystal-structure B factors. The primary macrodomain binding-site loops are marked in violet. (b) Overlay of the MacroD1 (yellow), MacroD2 (green) and C6orf130 (violet) structures with the ADP-ribose product in the binding pocket. (c) Mutational analysis of MacroD2 highlights residues implicated in enzymatic activity toward mono-ADP-ribosylated ARTD10 (818–1025). Quantifications from blots of two independent experiments are shown normalized to the untreated control (n = 2; mean ± range). (d) ADP-ribose binding to wild-type (WT) and mutant macrodomain proteins, assessed with histidine- and glutathione-S-transferase (GST)-tagged, recombinantly expressed macrodomain proteins immobilized on glutathione or nickel Sepharose and incubated with automodified ARTD10. (e) Structural model of MacroD2 with the mutated residues highlighted. (f) Model for MacroD2 protein–catalyzed hydrolysis of mono-ADP-ribosylated glutamate residues.
Second, the ADP-ribose product of the hydrolysis reaction was automatically docked to MacroD2 and followed by multiple explicit solvent molecular dynamics simulations of the complex to validate the binding mode. The mono-ADP-ribosylhydrolase activity was then studied by mutational analysis of MacroD2 and by comparison with existing mutants of MacroD1 (ref. 29) because these two macrodomain-containing proteins are closely related.
To confirm that the hydrolase activity of MacroD2 is dependent specifically on the macrodomain, we mutated the conserved glycine at position 100 of MacroD2 (Supplementary Fig. 3a) to a glutamate, which is predicted to block the ADP-ribose–binding site in macrodomain proteins35, or to an isoleucine. The resulting MacroD2 mutants lacked hydrolase and ADP-ribose–binding activity under the same assay conditions, which provided evidence that the macrodomain is responsible for the catalytic activity toward ARTD10-mediated mono-ADP-ribosylation and that the interaction with ADP-ribose is specific (Fig. 3c,d and data not shown). Notably, MacroH2A1.1 contains a glutamate at the corresponding position 225 instead of a glycine (Supplementary Fig. 3a), which possibly explains its inactivity due to its inability to bind mono-ADP-ribosylated ARTD10. However, although mutation of Glu225 of MacroH2A1.1 to a glycine resulted in a gain of binding, it did not restore its enzymatic activity (Supplementary Fig. 3b), thus indicating that additional residues are important for the activity.
Multiple explicit solvent molecular dynamics simulations of the MacroD2–ADP-ribose complex, together with defining conserved residues between MacroD1, MacroD2 and C6orf130, was used to propose site-specific mutants of MacroD2. Of particular note were Asp102 and its buried neighbor His106, which by modeling were predicted to be located near the 2- and 3-hydroxyl groups of the distal ribose (Fig. 3e). Mutational analysis, guided by the in silico predictions, indicated partial involvement in catalysis of Asp102 and His106 of MacroD2 as well as Asp184 and His188 of MacroD1 (Fig. 3c and Supplementary Fig. 3c). Pulldown experiments revealed that the MacroD2 mutants were still able to bind mono-ADP-ribosylated ARTD10 under the conditions tested for their enzymatic activity but that increasing the salt concentration to 500 mM reduced their affinity to the substrate to some extent (Supplementary Fig. 3d). On the basis of our findings, a model emerges for MacroD2-catalyzed hydrolysis of mono-ADP-ribosylated aspartate or glutamate residues (Fig. 3f). In this model, which is similar to the one previously suggested for the hydrolysis of OAADPr by MacroD1 (ref. 28), Asp102 or Asp184 acts as a general base that deprotonates a water molecule, which then acts as a nucleophile to attack the carbonyl carbon. We note that our mutagenesis data do not exclude participation of additional residues and/or transition-state stabilization due to induced fit.
MacroD2 regulates GSK3β function in vitro and in cells
GSK3β is a key regulator in processes ranging from cell structure and survival to diseases such as Alzheimer's disease, cancer and diabetes36, and it was recently identified in a screen for ARTD10 target proteins (ref. 8 and Supplementary Fig. 2a). To address whether the ARTD10-catalyzed ADP-ribosylation of GSK3β is reversible (in addition to the observed ribosylation of ARTD10 and histones (Figs. 1a,b,d and 2e)), the ADP-ribosylhydrolase activity of MacroD2 on GSK3β was analyzed. Notably, MacroD2 removed the ADP-ribosylation from both ARTD10 and GSK3β in vitro (Fig. 4a). These findings support the notion that ARTD10-mediated mono-ADP-ribosylation of target proteins in general is a reversible PTM. To test whether removal of the inhibitory mono-ADP-ribose by MacroD2 is sufficient to restore GSK3β kinase activity, mono-ADP-ribosylated GSK3β was demodified by MacroD2 and consecutively used in kinase assays with a primed peptide substrate. Although the ADP-ribosylated protein showed little in vitro kinase activity compared to the control, de-ADP-ribosylation restored the GSK3β activity (Fig. 4b).

MacroD2 but not the catalytically inactive macrodomains of ARTD8 removes mono-ADP-ribose from ARTD10 and GSK3β. (a) Coomassie blue (CB) staining and autoradiography (32P) results of in vitro assays with tandem-affinity purification (TAP)-tagged ARTD10 and GST-GSK3β coupled to beads. (b) Scintillation counting of kinase assays using [γ-32P]ATP and substrate peptide with modified or demodified GST-GSK3β. (c) Scintillation counting results as in b with HA-GSK3β from U2OS cells expressing DsRed-ARTD10, GFP-MacroD2 or both. (d) Input blot showing expression of DsRed-ARTD10, GFP-MacroD2 and HA-GSK3β in U2OS cells and immunoprecipitation efficiency of HA-GSK3β (lower blot). Data are represented as mean ± s.d. of at least triplicate measurements from representative experiments. *P < 0.05; NS, not significant by two-tailed Student's t test.
To address whether MacroD2 also reversed ARTD10-mediated ADP-ribosylation in cells, HA-GSK3β was coexpressed with the fluorescently tagged DsRed-ARTD10 alone or together with GFP-MacroD2, and subsequently immunoprecipitated and included in a kinase assay (Fig. 4c). The presence of MacroD2 enhanced the kinase activity of GSK3β, which suggested that MacroD2 antagonized the ARTD10-mediated inactivation of GSK3β without affecting protein expression (Fig. 4d). These data indicated that MacroD2 hydrolyzes mono-ADP-ribosyl linkages in cells, thus rendering mono-ADP-ribosylation a dynamic modification that can regulate the activation or inactivation of proteins such as GSK3β.
Discussion
The biochemical experiments, mutational analyses and structural predictions presented here suggest that certain macrodomain-containing proteins such as MacroD1, MacroD2, C6orf130 or Af1521, but neither the macrodomains 1 and 2 of ARTD8 nor macroH2A1.1, are bona fide protein mono-ADP-ribosylhydrolases. These results thus fill a major gap in understanding of the ADP-ribosylation cycle and define these proteins as new mono-ADP-ribosylhydrolases that reverse the PTM catalyzed by ARTD10.
The Appr-1′′-p phosphatase activity and the OAADPr-hydrolyzing activity of macrodomain proteins have been previously described and studied28,29,30. OAADPr represents an ADP-ribose that is O-acetylated at the C2 or C3 atom, whereas the PAR chain features C1 linkages37,38,39. The OAADPr hydrolysis by MacroD-like macrodomains suggests that these proteins hydrolyze ADP-riboses with C2 or C3 linkages at the proximal ribose. In contrast, the lack of MacroD1 or MacroD2 and C6orf130 activity toward PAR suggests that the glycosidic (ribose-ribose 1′-2′) linkages at the C1 atom of ADP-ribose polymers are not attacked, whereas these bonds within PAR are efficiently degraded by PARG and ARH3. The inefficient hydrolysis of ADP-ribose from ARTD10 by PARG suggests that the C2 or C3 atoms form the glycosidic bond to a glutamate or aspartate (similar to the linkage in OAADPr) rather than to the C1 atom as in PAR. Alternatively, MacroDs might recognize not only the ADP-ribose but also parts of the modified target protein. Moreover, the inability of PARG and MacroD1 or MacroD2 to completely remove the protein-linked ADP-ribose unit from ARTD1 suggests that at least some of the modifications catalyzed by ARTD1 are linked to yet another acceptor site.
The MacroD2-mediated reaction seems to be very efficient because at a 1:10 ratio (MacroD2/ARTD10) more than 60% of the modification synthesized by ARTD10 was removed within 15 min in vitro. Owing to the low rates of enzymatic mono-ADP-ribosylation, it was not possible to generate sufficient amounts of mono-ADP-ribosylated MacroD2 substrate that would allow substrate-saturated conditions to exactly determine Vmax and Km. In addition, it has been previously shown that ARTD10 is ADP-ribosylated at multiple sites7. Because the MacroD2 affinity for these different acceptor sites probably varies, it is impossible to determine exact kinetics with such a substrate. However, chemical synthesis of defined mono-ADP-ribosylated peptides, which could serve as substrates, is currently not possible.
The activity of MacroD-like proteins toward other acceptor sites remains to be tested, but the different chemical nature of these linkages probably requires specific enzymes for the different acceptor sites (for example, ARH1). Furthermore, the specificity may additionally be determined by protein-protein interactions. However, as MacroD2 is expressed in the cytoplasm and MacroD1 exhibits nuclear and mitochondrial localization18, these two proteins are likely to encounter different protein substrates but catalyze the same mono-ADP-ribosylhydrolysis reaction. ARTD10 and most of the other mono-ARTDs are mainly localized in the cytoplasm40. ARTD10-modified target proteins thus require cytoplasmic MacroD proteins for demodification.
On the basis of crystal structures, several residues in MacroD1 and C6orf130 were mutated in other studies to identify side chains involved in OAADPr hydrolysis28,29. Notably, we find that corresponding residues are implicated in MacroD1- and MacroD2-mediated hydrolysis of residues ADP-ribosylated by ARTD10. Mutating any of the putative catalytic residues (Asp102 and His106 of MacroD2) individually or together resulted in only a partial loss of activity, which indicates that other residues might compensate for the single mutations, to a certain extent. This observation is further supported by the fact that C6orf130 is active even though the residues identified in MacroD1 and MacroD2 are not conserved in C6orf130 (Supplementary Fig. 2a), which again points at sequence variation allowing different sets of residues to confer hydrolase activity. The residues Ser35 and Asp125, crucial for the hydrolysis of OAADPr by C6orf130, would indeed be strong candidates28.
The removal of ADP-ribose from GSK3β is sufficient to restore kinase activity, which indicates that mono-ADP-ribosylation is a dynamic PTM that directly influences the catalytic activity of its substrates in a reversible manner. The hydrolases identified here represent the missing link in the regulatory network formed by mono-ADP-ribosylation, which may prove highly important for diverse signaling networks as implied by the diversity of ARTD10 substrates identified8.
In summary, the findings presented here define the macrodomain-containing proteins MacroD1, MacroD2 and C6orf130 as protein mono-ADP-ribosylhydrolases and thus establish mono-ADP-ribosylation of acidic residues by ARTD10 as a reversible PTM. The MacroD-like proteins unite specific ADP-ribose binding with ADP-ribose degradation and thereby define new players in ADP-ribose metabolism and function. MacroD-like hydrolases form the functional antagonists of intracellular mono-ADP-ribosyltransferases. Notably, substrates of the mono-ADP-ribosyltransferase ARTD10 include many kinases, which may thus be activated and inactivated by the opposite activities of MacroD2 and ARTD10.
Methods
Purification of recombinant proteins.
Macrodomain cDNAs were amplified by PCR, cloned into a pET28-GST vector according to standard protocols, recombinantly expressed in E. coli BL21 and purified by using Ni Sepharose High Performance beads (Amersham Biosciences) according to the manufacturer's protocol. GSK3β was purified from SF9 cells8. Bound MacroD2 was cleaved from the beads with PreScission protease 3C (GE Healthcare) or eluted with 200 mM imidazole. Protein concentration was determined with a spectrophotometer ND-1000 (Nanodrop), and cleavage was verified by SDS-PAGE. ARTD10 was purified by using the TAP-tagging method as described before7. Cloning and purification of mMacro1 and mMacro2 of ARTD8 has been described41.
De-ADP-ribosylation assay with recombinant proteins.
Unless otherwise stated, 50 pmol recombinant purified GST-ARTD10 or His-ARTD1 were incubated with 100 nM [32P]nicotinamide adenine dinucleotide ([32P]NAD+, PerkinElmer) for 15 min at 30 °C in reaction buffer (10 mM potassium phosphate, pH 7.2, 10 mM MgCl2, 1.25 mM DTT, 1 μg/ml pepstatin, 1 μg/ml bestatin, 1 μg/ml leupeptin) and in the case of His-ARTD1 supplemented with 5 pmol annealed double-stranded oligomer (5′-GGAATTCC-3′). The reaction was stopped by filtration through a G50 column (GE Healthcare). De-ADP-ribosylation reactions were performed with 10 pmol MacroD2 protein at 30 °C for 15 min and stopped by the addition of SDS-PAGE loading buffer and boiling (5 min, 95 °C). De-ADP-ribosylation of automodified proteins was visualized by SDS-PAGE and autoradiography. Bands were quantified by using GelEval (http://www.frogdance.dundee.ac.uk).
ADP-ribosylation assays with immunoprecipitated proteins.
ADP-ribosylation assays were carried out at 30 °C for 30 min. The reaction mixture (50 mM Tris-HCl, pH 8.0, 0.2 mM DTT, 5 mM MgCl2 and 50 μM β-NAD+ (Sigma) and 1 μCi [32P]β-NAD+ (Amersham Biosciences)) was added to IgG beads with immunoprecipitated HA-ARTD10 or HA-ARTD10-ΔK and optionally 0.5 μg substrate protein in a total reaction volume of 30 μl. Reactions were stopped by adding SDS sample buffer and were subsequently boiled and run on SDS-PAGE. Incorporated radioactivity was analyzed by autoradiography.
ADP-ribosylation of actin by bacterial toxins.
In vitro ADP-ribosylation of actin was performed as reported42. Briefly, 2 μg β/γ actin was incubated with either 100 ng recombinant TccC3hvr or 50 ng CDTa in the presence of 100 nM [32P]NAD+, 150 μM cold NAD+ and reaction buffer (5 mM HEPES, pH 7.5, 0.1 mM CaCl2, 0.5 mM NaAc, 0.1 mM ATP). TccC3hvr and CDT1 reactions were incubated for 30 min at room temperature or 37 °C, respectively.
De-ADP-ribosylation assays with immunoprecipitated proteins.
ADP-ribosylation assays were terminated by placing on ice and washing with high-salt buffer (50 mM Tris-HCl, pH 8.0, 0.2 mM DTT, 5 mM MgCl2, 200 mM NaCl). MacroD2 (500 ng) was added to the beads in 30 μl high-salt buffer. After incubation (30 °C, 20 min), the reaction was stopped by addition of SDS sample buffer and boiling for analysis by SDS-PAGE and autoradiography. For subsequent kinase assays, beads with coupled GST-GSK3β were cooled and washed after incubation with MacroD2.
Chemical de-ADP-ribosylation.
For chemical de-ADP-ribosylation with hydroxylamine, automodified ARTD10 was supplemented with 0.8 M hydroxylamine in a 1:1 ratio (v/v). The reactions were incubated at 37 °C for 1 h and subsequently stopped by the addition of SDS loading buffer.
GST-macro pulldown assays with ARTD10 proteins.
GST- or His-tagged macrodomains were immobilized on glutathione or Ni Sepharose (Amersham Biosciences) at 4 °C (wild type and mutants) and incubated with automodified ARTD10 (full length; 100 ng) or GST-ARTD10 (818–1025; 50 ng) proteins for 2 h at 4 °C in 1,200 μl of binding buffer (100 mM Tris, pH 7.6, 250 mM NaCl, 125 mM KCl, 50 mM KAc, 1.5% NP-40 (high salt) or 75 mM NaCl, 25 mM KCl, 15 mM KAc, 1% NP-40 (low salt), 10% glycerol and protease inhibitors) and washed 5× with binding buffer (1,200 μl) for 25 min at 4 °C. Bound proteins were dissolved by boiling and were loaded on an SDS-PAGE gel for subsequent autoradiography.
Kinase assays.
[γ32P]ATP was diluted to 0.16 μCi/μl in 250 μM ATP in 3× kinase assay buffer (5 mM MOPS, pH 7.2, 2.5 mM β-glycerophosphate, 1 mM EGTA, 0.4 mM EDTA, 4 mM MgCl2, 50 μM DTT and 40 ng/μl BSA). GST-GSK3β (25 ng) or precipitate was incubated in a reaction volume of 25 μl (5 μl 0.16 μCi/μl [32P]ATP solution, 5 μg substrate peptide RRRPASVPPSPSLSRHS(pS)HQRR (Millipore)). After incubating (30 °C, 15 min), the reaction was stopped by placing on ice. Aliquots of 10 μl were spotted on P81 paper in duplicate, washed with 0.5% phosphoric acid and air dried before scintillation counting. Data are presented as mean ± s.d. of at least triplicate measurements from representative experiments. Statistical significance was determined by employing two-tailed Student's t test.
LC-MS and HPLC analysis of ADP-ribose.
For HPLC analysis, released ADP-ribose or ADP-ribose standards were purified over Microcon Ultracel YM-3 columns and subjected to reversed-phase liquid chromatography on an Accucore C18 2.7 μm, 150 × 2.1 mm ID Column. A water-methanol gradient from 0% to 20% MeOH at a flow rate of 200 μl/min was applied. Free ADP-ribose was monitored by UV absorbance at 260 nm. For LC-MS analysis, 10 μM ADP-ribose standard and samples were analyzed by using hydrophilic interaction chromatography (HILIC) coupled to accurate MS. The chromatographic separation of ADP-ribose was performed on a 0.2 μm × 150 mm BEH amide column, using a linear gradient of acetonitrile to water, 0.5 mM ammonium acetate, pH 9. For MS, we used negative mode with a capillary voltage of 1.2 kV. Data were acquired in MS and MS-MS mode. Extracted ion chromatograms from the MS data were generated by using the monoisotopic mass of ADP-ribose adduct [M-H] 558.064 and a mass window of 10 mDa. For relative quantification of ADP-ribose, MS scans in the elution time range of ADP-ribose were combined, and the ion abundance of mass 558.064 was calculated.
Homology modeling of MacroD2.
The MacroD2 homology model (obtained by Modeller43,44,45,46; Supplementary Note) with the lowest discrete optimized protein energy assessment score47 was selected for minimization and molecular-dynamics refinement. Following 0.5 ns of NVT and subsequent 0.5 ns of NPT equilibration during which the protein heavy atoms and protein Cα atoms were, respectively, positionally restrained, two 100-ns trajectories were generated by using different random seeds (MD runs I and II). Trajectory analyses (Supplementary Fig. 4a–c) were performed with the MD-analysis tool WORDOM48,49.
Docking and molecular-dynamics simulations of ADP-ribose in MacroD2.
From the MD simulations of the apo-MacroD2 homology model, a trajectory frame was selected that maximized the solvent-accessible surface area of putative binding-site residues within the mixed α/β macrodomain fold35, and which maintained similar Asp78 χ1 and χ2 angles relative to the corresponding residue of Af1521 (PDB 2BFQ).
Water molecules and ions were removed from this frame. AutoDock Vina50 was employed to dock the ADP-ribose ligand to the mixed α/β fold. The 20 top-ranking poses were minimized with CHARMM by using the CHARMM27 force field for the protein atoms and the CHARMM general force field51,52 for ADP-ribose. Upon structural superposition of MacroD2 and Af1521, the minimized pose of ADP-ribose having the lowest r.m.s. deviation value relative to the one in Af1521 (PDB 2BFQ) was immersed in a box of TIP3P water molecules and subjected to ten explicit water MD runs of 10 ns each at 300 K.
For each run, the distance between the center of mass of the putative binding site of MacroD2 and that of ADP-ribose was calculated by using WORDOM48,49. Residues forming the putative binding site were those having at least one atom within 5 Å of any ligand atom following equilibration.
A total of 65% of the obtained MD trajectory frames presented a binding-site ligand center-of-mass distance of <6 Å; above this distance, the distal ribose of the ligand rarely re-entered the binding site. Unbinding of ADP-ribose within 10 ns in almost half of the runs is consistent with its high μM inhibition of human MacroD1 activity28.
Residues participating in hydrogen-bonding to ADP-ribose among trajectory frames in which the ligand remained bound were identified by using WORDOM with a distance cutoff of 4.0 Å between donor (D) and acceptor (A) atoms and a D-H··A angle larger than 130°. Predicted interactions of the distal ribose of ADP-ribose are predominantly with the carboxylate of Asp102 and with protein backbone atoms (Supplementary Fig. 4d,e).
MacroD2 residues forming stable hydrogen bonds to structural water molecules were identified by using the GROMACS g_hbond function. The most stable water molecules in the vicinity of the distal ribose are shown (Supplementary Fig. 4f) along with their interaction partners in MacroD2.
Figures were created with Pymol (http://pymol.sourceforge.net/).
References
Erener, S. et al. ARTD1 deletion causes increased hepatic lipid accumulation in mice fed a high-fat diet and impairs adipocyte function and differentiation. FASEB J. 26, 2631–2638 (2012).
Altmeyer, M. & Hottiger, M.O. Poly(ADP-ribose) polymerase 1 at the crossroad of metabolic stress and inflammation in aging. Aging 1, 458–469 (2009).
Hassa, P.O., Haenni, S., Elser, M. & Hottiger, M.O. Nuclear ADP-ribosylation reactions in mammalian cells: where are we today and where are we going? Microbiol. Mol. Biol. Rev. 70, 789–829 (2006).
Telli, M.L. PARP inhibitors in cancer: moving beyond BRCA. Lancet Oncol. 12, 827–828 (2011).
Curtin, N.J. PARP inhibitors for cancer therapy. Expert Rev. Mol. Med. 7, 1–20 (2005).
Schreiber, V., Dantzer, F., Ame, J.-C. & De Murcia, G. Poly(ADP-ribose): novel functions for an old molecule. Nat. Rev. Mol. Cell Biol. 7, 517–528 (2006).
Kleine, H. et al. Substrate-assisted catalysis by PARP10 limits its activity to mono-ADP-ribosylation. Mol. Cell 32, 57–69 (2008).
Feijs, K.L.H. et al. ARTD10 substrate identification on protein microarrays: regulation of GSK3β by mono-ADP-ribosylation. Cell Commun. Signal published online, doi:10.1186/1478-811X-11-5 (19 January 2013).
Hottiger, M.O., Hassa, P.O., Lüscher, B., Schüler, H. & Koch-Nolte, F. Toward a unified nomenclature for mammalian ADP-ribosyltransferases. Trends Biochem. Sci. 35, 208–219 (2010).
Hanai, S. et al. Loss of poly(ADP-ribose) glycohydrolase causes progressive neurodegeneration in Drosophila melanogaster. Proc. Natl. Acad. Sci. USA 101, 82–86 (2004).
Koh, D.W. et al. Failure to degrade poly(ADP-ribose) causes increased sensitivity to cytotoxicity and early embryonic lethality. Proc. Natl. Acad. Sci. USA 101, 17699–17704 (2004).
Williams, J.C., Chambers, J.P. & Liehr, J.G. Glutamyl ribose 5-phosphate storage disease. A hereditary defect in the degradation of poly(ADP-ribosylated) proteins. J. Biol. Chem. 259, 1037–1042 (1984).
Slade, D. et al. The structure and catalytic mechanism of a poly(ADP-ribose) glycohydrolase. Nature 477, 616–620 (2011).
Dunstan, M.S. et al. Structure and mechanism of a canonical poly(ADP-ribose) glycohydrolase. Nat. Commun. 3, 878 (2012).
Han, W., Li, X. & Fu, X. The macro domain protein family: structure, functions, and their potential therapeutic implications. Mutat. Res. 727, 86–103 (2011).
Kim, I.K. et al. Structure of mammalian poly(ADP-ribose) glycohydrolase reveals a flexible tyrosine clasp as a substrate-binding element. Nat. Struct. Mol. Biol. 19, 653–656 (2012).
Karras, G.I. et al. The macro domain is an ADP-ribose binding module. EMBO J. 24, 1911–1920 (2005).
Neuvonen, M. & Ahola, T. Differential activities of cellular and viral macro domain proteins in binding of ADP-ribose metabolites. J. Mol. Biol. 385, 212–225 (2009).
Angelov, D. et al. The histone variant macroH2A interferes with transcription factor binding and SWI/SNF nucleosome remodeling. Mol. Cell 11, 1033–1041 (2003).
Buschbeck, M. et al. The histone variant macroH2A is an epigenetic regulator of key developmental genes. Nat. Struct. Mol. Biol. 16, 1074–1079 (2009).
Changolkar, L.N. et al. Developmental changes in histone macroH2A1-mediated gene regulation. Mol. Cell Biol. 27, 2758–2764 (2007).
Goenka, S., Cho, S.H. & Boothby, M. Collaborator of Stat6 (CoaSt6)-associated poly(ADP-ribose) polymerase activity modulates Stat6-dependent gene transcription. J. Biol. Chem. 282, 18732–18739 (2007).
Ahel, D. et al. Poly(ADP-ribose)-dependent regulation of DNA repair by the chromatin remodeling enzyme ALC1. Science 325, 1240–1243 (2009).
Gottschalk, A.J. et al. Poly(ADP-ribosyl)ation directs recruitment and activation of an ATP-dependent chromatin remodeler. Proc. Natl. Acad. Sci. USA 106, 13770–13774 (2009).
Aguiar, R.C., Takeyama, K., He, C., Kreinbrink, K. & Shipp, M.A. B-aggressive lymphoma family proteins have unique domains that modulate transcription and exhibit poly(ADP-ribose) polymerase activity. J. Biol. Chem. 280, 33756–33765 (2005).
Aguiar, R.C. et al. BAL is a novel risk-related gene in diffuse large B-cell lymphomas that enhances cellular migration. Blood 96, 4328–4334 (2000).
Cho, S.H. et al. Glycolytic rate and lymphomagenesis depend on PARP14, an ADP ribosyltransferase of the B aggressive lymphoma (BAL) family. Proc. Natl. Acad. Sci. USA 108, 15972–15977 (2011).
Chen, D. et al. Identification of macrodomain proteins as novel O-acetyl-ADP-ribose deacetylases. J. Biol. Chem. 286, 13261–13271 (2011).
Peterson, F.C. et al. Orphan macrodomain protein (human C6orf130) is an O-acyl-ADP-ribose deacylase: solution structure and catalytic properties. J. Biol. Chem. 286, 35955–35965 (2011).
Hofmann, A. et al. Structure and mechanism of activity of the cyclic phosphodiesterase of Appr>p, a product of the tRNA splicing reaction. EMBO J. 19, 6207–6217 (2000).
Sawaya, R., Schwer, B. & Shuman, S. Structure-function analysis of the yeast NAD+-dependent tRNA 2′-phosphotransferase Tpt1. RNA 11, 107–113 (2005).
Stone, P.R. & Hilz, H. Quantitation of hydroxylamine sensitive mono(adenosine diphosphate ribose) residues in different hepatic tissues. FEBS Lett. 57, 209–212 (1975).
Barth, H., Aktories, K., Popoff, M.R. & Stiles, B.G. Binary bacterial toxins: biochemistry, biology, and applications of common Clostridium and Bacillus proteins. Microbiol. Mol. Biol. Rev. 68, 373–402 (2004).
Lang, A.E. et al. Photorhabdus luminescens toxins ADP-ribosylate actin and RhoA to force actin clustering. Science 327, 1139–1142 (2010).
Timinszky, G. et al. A macrodomain-containing histone rearranges chromatin upon sensing PARP1 activation. Nat. Struct. Mol. Biol. 16, 923–929 (2009).
Jope, R.S. & Johnson, G.V. The glamour and gloom of glycogen synthase kinase-3. Trends Biochem. Sci. 29, 95–102 (2004).
Jackson, M.D. & Denu, J.M. Structural identification of 2′- and 3′-O-acetyl-ADP-ribose as novel metabolites derived from the Sir2 family of β-NAD+-dependent histone/protein deacetylases. J. Biol. Chem. 277, 18535–18544 (2002).
Sauve, A.A. et al. Chemistry of gene silencing: the mechanism of NAD+-dependent deacetylation reactions. Biochemistry 40, 15456–15463 (2001).
Jackson, M.D., Schmidt, M.T., Oppenheimer, N.J. & Denu, J.M. Mechanism of nicotinamide inhibition and transglycosidation by Sir2 histone/protein deacetylases. J. Biol. Chem. 278, 50985–50998 (2003).
Kleine, H. et al. Dynamic subcellular localization of the mono-ADP-ribosyltransferase ARTD10 and interaction with the ubiquitin receptor p62. Cell Commun. Signal. 10, 28 (2012).
Forst, A.H. et al. Recognition of mono-ADP-ribosylated ARTD10 substrates by ART8 macrodomains. Structure (in the press).
Gülke, I. et al. Characterization of the enzymatic component of the ADP-ribosyltransferase toxin CDTa from Clostridium difficile. Infect. Immun. 69, 6004–6011 (2001).
Eswar, N. et al. Comparative protein structure modeling using Modeller. in Curr. Protoc. Bioinformatics 15, 5.6 (2006).
Martí-Renom, M.A. et al. Comparative protein structure modeling of genes and genomes. Annu. Rev. Biophys. Biomol. Struct. 29, 291–325 (2000).
Sali, A. & Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234, 779–815 (1993).
Fiser, A., Do, R.K. & Sali, A. Modeling of loops in protein structures. Protein Sci. 9, 1753–1773 (2000).
Shen, M.Y. & Sali, A. Statistical potential for assessment and prediction of protein structures. Protein Sci. 15, 2507–2524 (2006).
Seeber, M., Cecchini, M., Rao, F., Settanni, G. & Caflisch, A. Wordom: a program for efficient analysis of molecular dynamics simulations. Bioinformatics 23, 2625–2627 (2007).
Seeber, M. et al. Wordom: a user-friendly program for the analysis of molecular structures, trajectories, and free energy surfaces. J. Comput. Chem. 32, 1183–1194 (2011).
Trott, O. & Olson, A.J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 31, 455–461 (2010).
Vanommeslaeghe, K. et al. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 31, 671–690 (2010).
Brooks, B.R. et al. CHARMM: the biomolecular simulation program. J. Comput. Chem. 30, 1545–1614 (2009).
Acknowledgements
We are grateful to I. Ahel (Paterson Institute for Cancer Research, Manchester, UK) and M. Neuvonen (Institute of Biotechnology, University of Helsinki, Helsinki, Finland) for MacroD1 and MacroD2 constructs and J. Moss (National Institutes of Health, Bethesda, Maryland, USA) for the ARH1 construct. F. Althaus (University of Zurich, Zurich, Switzerland) and K. Aktories (University of Freiburg, Freiburg, Germany) are acknowledged for generously providing a baculovirus expressing PARG and the CDTa and TccC3 enzymes together with purified actin, respectively. F. Freimoser (University of Zurich, Zurich, Switzerland) provided editorial assistance and critical input during the writing and M. Fey technical assistance. This work was supported by the Deutsche Forschungsgemeinschaft DFG (LU 466/15-1) to B.L. and in part by the University of Zurich (Forschungskredit 54041205 to E.F.), the Swiss National Science Foundation (SNF-31003A_125190 (to P.O.H.) and 31-122421 (to M.O.H.)) and the Kanton of Zurich (to M.O.H.).
Author information
Authors and Affiliations
Contributions
F.R. and P.O.H. performed experiments with MacroD1, MacroD2 and other macrodomains, and MS analysis was done together with D.F.; K.L.H.F. analyzed the influence of MacroD2 on GSK3β; E.F. performed the computer modeling; E.F. and A.C. analyzed the modeling and simulation results; M.B. identified MacroD2 as an ADP-ribosylhydrolase; A.H.F. cloned and purified mMacro1 and mMacro2; R.I. performed the HPLC analysis; H.C.W. compared the macro sequences; A.C., P.O.H., B.L. and M.O.H. supervised the work. B.L. and M.O.H. wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Text and Figures
Supplementary Figures 1–4 and Supplementary Note (PDF 3959 kb)
Rights and permissions
About this article
Cite this article
Rosenthal, F., Feijs, K., Frugier, E. et al. Macrodomain-containing proteins are new mono-ADP-ribosylhydrolases. Nat Struct Mol Biol 20, 502–507 (2013). https://doi.org/10.1038/nsmb.2521
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nsmb.2521
This article is cited by
-
Macrod1 suppresses diabetic cardiomyopathy via regulating PARP1-NAD+-SIRT3 pathway
Acta Pharmacologica Sinica (2024)
-
Mono-ADP-ribosylation by PARP10 inhibits Chikungunya virus nsP2 proteolytic activity and viral replication
Cellular and Molecular Life Sciences (2023)
-
Genetic association study identifies genetic variants for non-alcoholic fatty liver without comorbidities in the Korean population
Genes & Genomics (2023)
-
Poly(ADP-ribosylation) of P-TEFb by PARP1 disrupts phase separation to inhibit global transcription after DNA damage
Nature Cell Biology (2022)
-
Intracellular mono-ADP-ribosyltransferases at the host–virus interphase
Cellular and Molecular Life Sciences (2022)
