Abstract
The tumor microenvironment is created by the tumor and dominated by tumor-induced interactions. Although various immune effector cells are recruited to the tumor site, their anti-tumor functions are downregulated, largely in response to tumor-derived signals. Infiltrates of inflammatory cells present in human tumors are chronic in nature and are enriched in regulatory T cells (Treg) as well as myeloid suppressor cells (MSC). Immune cells in the tumor microenvironment not only fail to exercise antitumor effector functions, but they are co-opted to promote tumor growth. Sustained activation of the NF-κB pathway in the tumor milieu represents one mechanism that appears to favor tumor survival and drive abortive activation of immune cells. The result is tumor escape from the host immune system. Tumor escape is accomplished through the activation of one or several molecular mechanisms that lead to inhibition of immune cell functions or to apoptosis of anti-tumor effector cells. The ability to block tumor escape depends on a better understanding of cellular and molecular pathways operating in the tumor microenvironment. Novel therapeutic strategies that emerge are designed to change the pro-tumor microenvironment to one favoring acute responses and potent anti-tumor activity.
Similar content being viewed by others


Introduction
It has been well documented that human tumors are generally infiltrated by inflammatory cells (Whiteside, 1993; Mihm et al., 1996; Balkwill and Mantovani, 2001). Although these infiltrates of inflammatory cells can vary in size and composition from tumor to tumor, their presence has been taken as evidence that the host is not ignorant of the developing tumor, but rather attempts to interfere with tumor progression, a process referred to as immune surveillance (Zitvogel et al., 2006). In this context, inflammatory infiltrates in tumors are considered to be a host attempt at the detection of emerging tumor cells and their elimination (Zitvogel et al., 2006). Indeed, numerous reports in the literature have linked the presence of inflammatory infiltrates in human tumors with an improved prognosis or better patient survival (Kornstein et al., 1983; Baxevanis et al., 1994; Naito et al., 1998; Pages et al., 2005). More recent data based on analyses of multiple immune markers, including heat maps and microarrays, suggest that type, density and location of immune cells in the tumor have a prognostic value (Pages et al., 2005; Galon et al., 2006; Galon et al., 2007). At the same time, equally numerous reports have indicated a lack of significant correlations between lymphocytic infiltrate intensity and improved prognosis or have linked immune cell infiltration to a poor prognosis (Stewart and Tsai, 1993; Sheu et al., 1999; Nakano et al., 2001). These contradictory results emerging from reputable laboratories have remained unexplained for many years, until it became possible to explore functional properties of tumor-infiltrating lymphocytes (TIL), which often represented the major component of immune infiltrates in tumors (Whiteside, 1993; Mihm et al., 1996). In nearly all cases, TIL obtained from human tumor tissues showed inhibited proliferation in response to mitogens or antigens, compromised signaling through T cell receptor, decreased ability to mediate cytotoxicity of tumor targets or to produce Th1-type cytokines upon stimulation with tumor antigens (Kiessling et al., 1996; Reichert et al., 1998b, 2002; Kuss et al., 1999; Uzzo et al., 1999). Functional impairments in TIL were more pronounced in patients with advanced cancer than in early disease, and they seemed to differ in the frequency as well as magnitude, depending on the tumor type or its tissue of origin (reviewed in Whiteside, 1993). Importantly, the functional status of TIL was now shown to be an independent and significant correlate of improved prognosis and longer overall survival in patients with malignancy (Reichert et al., 1998a). In time, as the methods used to study functions and attributes of tissue-infiltrating immune cells improved, and the understanding of their local interactions with other cells enlarged, the role of a microenvironment in shaping cellular events in health and disease came to be appreciated. A recent comprehensive multivariate analysis of cellular interactions in the tumor microenvironment based on the nature, function, density and localization of immune cells within human colorectal cancers demonstrated that immune reactions within the tumor influence clinical outcome (Pages et al., 2005; Galon et al., 2006, 2007). The current view of the tumor microenvironment is that it exerts a key influence on tumor progression and that re-shaping of its character might offer unexpected therapeutic benefits.
Cells found in the tumor microenvironment
A tissue microenvironment of developing tumor is comprised of proliferating tumor cells, the tumor stroma, blood vessels, infiltrating inflammatory cells and a variety of associated tissue cells. It is a unique environment that emerges in the course of tumor progression as a result of its interactions with the host. It is created by and at all times shaped and dominated by the tumor, which orchestrates molecular and cellular events taking place in surrounding tissues (Figure 1).

A diagram depicting the tumor microenvironment. Interactions of various cells with each other, fibroblasts (F) in the tumor stroma and blood vessels (V) are indicated by arrows. Although the tumor (TU) generates signals inducing dysfunction and death of immune cells, the latter are a source of signals promoting tumor growth. TAM, tumor-associated macrophages; DC, dendritic cells; EL, effector lymphocytes; Treg, regulatory T cells; TA, tumor-derived antigens; ROS, reactive oxygen species; PGE2, prostaglandin E2.
Immune cells present in the tumor include those mediating adaptive immunity, T lymphocytes, dendritic cells (DC) and occasional B cells, as well as effectors of innate immunity, macrophages, polymorphonuclear leukocytes and rare natural killer (NK) cells (Whiteside, 2007). NK cells, which mediate innate immunity and are rich in perforin- or granzyme-containing granules, are conspicuously absent from most tumor infiltrates or even pre-cancerous lesions (Whiteside et al., 1998). Although NK cells represent ‘the first line’ of defense against pathogens (Lanier, 2003) and mediate potent antitumor cytotoxicity in vitro, in tumor milieu, they are infrequent, despite the fact that tumor cells frequently downregulate expression of HLA antigens and are enriched in MICA and MICB molecules (Chang et al., 2005). These features make the tumor susceptible to NK cell-mediated cytotoxicity (Lee et al., 2004), and their paucity in tumor infiltrates may be an example of the evasion mechanism preventing NK-cell recruitment to the tumor site.
Tumor-infiltrating lymphocytes, containing various proportions of CD3+CD4+ and CD3+CD8+ T cells, are usually a major component of the tumor microenvironment (Whiteside, 2007). Many of these T cells are specific for tumor-associated antigens, as indicated by clonal analyses (Miescher et al., 1987) and tetramer staining of CD8+ T cells isolated from human tumors (Albers et al., 2002). In some tumors, for example, medullary breast carcinomas, infiltrating lymphocytes form lymph node-like structures suggesting that the immune response is operating in situ (Coronella et al., 2002). Also, TIL are a source of tumor-specific lymphocytes used for adoptive transfers after expansion in IL-2-containing cultures (Zhou et al., 2004). TIL clones with the specificity to a broad variety of the tumor-associated antigens can be outgrown from human tumors, confirming that immune responses directed not only at ‘unique’ antigens expressed by the tumor, but also at a range of differentiation or tissue-specific antigens, are generated by the host (Romero et al., 2006). Although accumulations of these effector T cells in the tumor might be considered as evidence of immune surveillance by the host, they are largely ineffective in arresting tumor growth. Among CD4+ T cells present in the tumor, a subset of CD4+CD25high Foxp3+ cells is expanded (5–15% of CD3+CD4+ T cells in TIL) relative to their significantly lower frequency in the peripheral circulation of patients with cancer (Woo et al., 2001; Strauss et al., 2007a). These cells are regulatory T cells (Treg) capable of suppressing proliferation of other T cells in the microenvironment through contact-dependent mechanisms or IL-10 and TGF-β secretion (Figure 2). They come in different flavors (for example, nTreg, Tr1) and are a characteristic feature of the microenvironment in human tumors (Bergmann et al., 2007; Strauss et al., 2007a).

Accumulation and expansion of Treg in the tumor microenvironment may be a result of the cross talk between the tumor and DC. The tumor coopts DC differentiation, and in the presence of tumor-derived factors, immature DC develop abnormalities in APM, have decreased expression levels of MHC molecules, upregulate B7-H1 (PD-L1) and produce excess of IL-10 and TGF-β1. Cross-presentation of TA by these DC to T cells (as in the lower part of figure) lead to emergence of Treg and their expansion. Once generated, Treg interfere with functions of anti-tumor effector cells (CTL). Even if cross-presentation of TA by DC to naive CD8+ T cells is successful (as in the upper part of figure), Treg proceed to block CTL functions. Reproduced with changes from Ferrone and Whiteside, 2007.
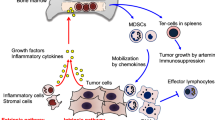
Macrophages present in tumors are known as tumor-associated macrophages or TAMs. They are re-programmed to inhibit lymphocyte functions through release of inhibitory cytokines such as IL-10, prostaglandins or reactive oxygen species (ROS) (Mantovani et al., 2003; Martinez et al., 2008). Myeloid suppressor cells (MSC) accumulating in human tumors are CD34+CD33+CD13+CD15(−) bone marrow-derived immature dendritic cells, an equivalent to CD11b+/Gr1+ cells in mice (Serafini et al., 2006). They promote tumor growth and suppress immune cell functions through copious production of an enzyme involved in L-arginine metabolism, arginase 1, which synergizes with iNOS to increase superoxide and NO production, blunting lymphocyte responses (Ochoa et al., 2007) and by induction of iNOS in surrounding cells (Tsai et al., 2007). Relatively little is known about human MSC. A recent report describes expansion of CD14+HLA-DR−/low myeloid-derived cells exerting immune suppression through TGF-β production in the peripheral circulation of patients with metastatic melanoma treated with GM-CSF-based vaccines (Filipazzi et al., 2007). The recruitment of MSC to the tumor site is orchestrated by the tumor (see Figure 3). Tumors produce many factors, including IL-10, VEGF, GM-CSF, which promote MSC accumulation and block DC maturation as well as lymphocyte functions (Serafini et al., 2006). Current data support the active role of MSC in tumor-induced immune suppression in mice and in man.

Tumors recruit MSC from the bone marrow by means of tumor-derived soluble factors. Immature myeloid cells migrate to lymph nodes, where DC cross-prime T cells, and interfere with this process. They also migrate to the tumor site and become tumor-associated MSC, which are adept in blocking T cell functions through the production of arginase I and activation of iNOs.
Polymorphonuclear leukocytes are infrequently seen in infiltrates of human tumors, with the exception of nests of eosinophils that may be present in association with tumor cells in various squamous cell tumors, for example. In contrast, granulocytes tend to be a major cellular component of many murine tumor models (Loukinova et al., 2000). This disparity may be because of a different nature of infiltrates, which in man are chronic rather than acute. Acute cellular responses may be long gone by the time human tumors are diagnosed, biopsied and examined.
Inflammatory cells present in the tumor microenvironment either contribute to tumor progression or actively interfere with its development. It is clear today that the former takes precedence, largely because the tumor generally proceeds to establish mechanisms responsible for its ‘immune evasion’ or escape from the immune intervention. The tumor not only manages to escape from the host immune system, but it effectively contrives to benefit from infiltrating cells by modifying their functions to create the microenvironment favorable to tumor progression. To this end, immune cells infiltrating the tumor together with fibroblasts and extracellular matrix forming a scaffold supporting its expansion, contribute to establish an inflammatory milieu that nourishes the tumor and promotes its growth. Tumor escape from the host is facilitated by the ability of human tumors to actively subvert anti-tumor immunity by downregulating or completely suppressing local and systemic innate as well as adaptive antitumor immunity by a variety of mechanisms as discussed below.
Inflammation and cancer
Almost 20 years ago, H.F. Dworak coined the phrase describing human tumors as ‘wounds that do not heal’ (Dworak, 1986). Indeed, changes occurring in the microenvironment of the progressing tumor resemble the process of chronic inflammation, which begins with ischemia followed by interstitial and cellular edema, appearance of immune cells and, finally, growth of blood vessels and tissue repair (Aller et al., 2004). Chronic inflammation is clearly involved in shaping the tumor microenvironment and has been referred to as ‘host reaction’ to the tumor, although it might be more appropriate to think of it as ‘tumor promoting’ reaction.
An initial goal of the inflammatory response is to destroy an invader, which in this case is the tumor. Therefore, the ‘immune phase’ of tumor-driven inflammation involves a recruitment and influx of antitumor effector cells to the tissue site. However, compared with vigorous cellular and humoral responses that are generated in tissues upon infections by exogenous pathogens, those mediated by the tumor are weak. This is probably because most tumor-associated antigens are considered ‘self,’ in contrast to infections with bacteria or viruses which are viewed by the host as ‘danger signals’ (Gallucci and Matzinger, 2001). Indeed, it is entirely possible that accumulations of regulatory T cells (Treg) in the tumor microenvironment represent an attempt of the host to downregulate response against ‘self’ with an unfortunate concomitant suppression of antitumor immunity.
Among the factors that determine the nature of inflammatory infiltrates found in the tumor microenvironment is the hypoxic environment. It is created early in the tumor development through activation of hypoxia-responsive genes in tumor cells (Denko et al., 2003). It favors the influx of those inflammatory cells that depend on the glycolytic pathway for survival, namely, phagocytic macrophages and granulocytes (Aller et al., 2004). These cells not only survive in the hypoxic environment but contribute to it by hyperproduction of ROS upon local activation. In the tumor milieu, where apoptosis of rapidly expanding tumor cells is common, infiltrating phagocytes receive ample activation signals and produce an abundance of ROS. Immunoinhibitory activities of ROS are mediated by the NF-κB pathway, which in turn is regulated by hypoxia and/or re-oxygenation (Lluis et al., 2007). It has been proposed that the NF-κB pathway plays a key role in activation of signaling in cancer cells as well as tumor-infiltrating leukocytes (Balkwill and Coussens, 2004; Greten et al., 2004; Pikarsky et al., 2004). NF-κB activation in these cells lead to secretion of TNF-α or other pro-inflammatory cytokines which initiate and drive regulated expression of the cytokine genes responsible for cell proliferation. Tumor cells depend on these cytokines for growth, and infiltrating leukocytes become programmed to continually release these growth factors. Responding to this NF-κB-driven pro-inflammatory cytokine cascade, tumor and stromal cells produce a variety of soluble mediators with wide-ranging biologic effects. Thus, cell proliferation and differentiation, matrix remodeling, blood vessel growth and cell migration/recruitment are all re-programmed to benefit the tumor. The role of TNF-α in driving tumor progression has long been emphasized by Balkwill and coworkers (Malik et al., 1989). It provides an example of how tumors usurp a normal process of inflammation to promote their own progression. It also suggests that blocking of TNF-α, for example, by anti-TNF-α antibodies, might be therapeutically useful (Harrison et al., 2007). Similarly, inhibition of NF-κB activation in the tumor microenvironment represents a potentially effective strategy for arresting tumor growth (Karin and Greten, 2004).
Mechanisms of tumor escape
The tumor microenvironment, once established, represents a consistently effective barrier to immune cell functions. This is because tumors are not passive targets for host immunity; instead, they actively downregulate all phases of anti-tumor immune responses using a spectrum of different strategies and mechanisms (Figure 4). To date, many mechanisms responsible for dysfunction of immune cells in the tumor microenvironment have been identified. Some are directly mediated by factors produced by tumors, whereas others result from alterations of normal tissue homeostasis occurring in the presence of cancer. Until recently, little was known about molecular alterations in tumor cells in situ as they progressed from the pre-malignant to metastatic phenotype. Genetic instability, now recognized as a principal characteristic of all tumors, may result in changes in their epitope profile. Molecular changes, already detectable during early stages of tumorigenesis, become more pronounced as the tumor progresses. The net result of these changes is increased resistance of tumor cells to immune surveillance. In addition, most human tumors appear to be able to interfere with one or more stages of immune cell development, differentiation, migration, cytotoxicity and other effector functions. Thus, all phases of an antitumor immune response are subject to adverse intervention in the tumor microenvironment as indicated in Table 1. These escape mechanisms and their consequences in terms of tumor progression have been recently reviewed (Whiteside, 2006).

Mechanisms responsible for ‘immunoediting’ of tumor cells in the tumor microenvironment. The various mechanisms listed collaborate in immunoediting of the tumor cells. The tumor, stromal cells and infiltrating leukocytes all contribute to pro-inflammatory milieu. Infiltrating immune cells and stromal elements are re-programmed by the tumor to the pro-inflammatory mode favoring its survival. In this milieu, the tumor evolves a phenotype allowing it to escape and to counterattack immune cells. Tumor stem cells are resistant to anti-tumor therapies and thus represent yet another means of escape.
Of the various escape mechanisms listed in Table 1, two have received special attention in recent years, possibly because they appear to be ubiquitous and are clearly associated with disease progression. Accumulations in tumors of Treg (CD4+CD25bright Foxp3+ T cells) and myeloid-derived cells (CD34+CD33+CD13+CD11b+CD15−) are common features of human tumors, and the frequency as well as suppressor activity subsets of these cells mediate locally and systemically have been linked to poor prognosis in patients with cancer (Almand et al., 2000).
Under normal physiologic conditions, Treg have a beneficial role in preventing autoimmunity (Shevach, 2000). However, in cancer, they expand, migrate to tumor sites, downregulate autologous effector T-cell proliferation and suppress anti-tumor responses of both CD4+CD25− and CD8+CD25− T cells using distinct molecular pathways (Roncarolo et al., 2006; Bacchetta et al., 2007). They are a heterogenous population of regulatory CD3+CD4+ T cells, comprising natural Treg, antigen-specific Tr1 cells and other less well defined subsets of suppressor cells (Roncarolo et al., 2006). Tr1 cells are induced in the tumor microenvironment, which is rich in IL-10, TGF-β, and prostaglandin E2 (PGE2), all of which have been shown to promote Tr1 generation (Bergmann et al., 2007). Today, the nature of human Treg is only partially defined. The phenotype, functions (antigenic specificity, stability, trafficking or survival), lineage, differentiation and the relationship between the various Treg subsets are under intense investigation. No single specific marker is sufficient for distinguishing Treg subpopulations. Given the expansion of these populations in the circulation and tumor tissues of cancer patients (Woo et al., 2001; Liyanage et al., 2002; Curiel et al., 2004; Shevach, 2004; Strauss et al., 2007b), it is important to perform Treg phenotypic and functional evaluations to be able to define their role in the regulation of tumor-specific responses. From a practical point of view, it is important to distinguish Treg from activated CD4+CD25+ T cells which mediate helper functions and are sensitive to activation induced cell death (AICD). In contrast, Treg appear to be resistant to apoptosis (Strauss et al., 2007c). Recent reports suggest that oncologic therapies, surgery, radiation, chemotherapy, expand Treg and enhance their suppressor functions (Banerjee et al., 2006; Zhou et al., 2006; Strauss et al., 2007b). These intriguing data support the need for serial follow-up studies of Treg in cancer patients treated with oncologic therapies.
MSC also suppress T-cell responses in the tumor microenvironment. Most human tumors secrete TGF-β or induce TGF-β secretion from MSC that accumulate in the tumor microenvironment (Gallina et al., 2006; Serafini et al., 2006). Pak et al. first reported accumulations of CD34+ cell-derived myeloid cells with immunosuppressive ability the peripheral blood of HNC patients (Pak et al., 1995). These cells correspond to CD11b+/Gr-1+ myeloid progenitor cells in mice (Serafini et al., 2006). In tumor-bearing mice, MSC accumulate in the spleen and peripheral circulation, reaching very high proportions and exerting potent immunosuppression, thus favoring tumor growth. MSC also control the availability of essential amino acids such as L-arginine and produce high levels of ROS. MSC present in tumors constitutively express iNOS and arginase 1, an enzyme involved in metabolism of L-arginine, which also synergizes with iNOS to increase superoxide and NO production, blunting lymphocyte responses (Bronte et al., 2003; Ochoa et al., 2007; Tsai et al., 2007). Another enzyme that might be produced by MSC is indoleamine-2,3-dioxygenase (IDO) involved in the catabolism of tryptophan, an essential amino acid for T-cell proliferation and differentiation (Munn and Mellor, 2007). The frequency of MSC with high levels of suppressive functions were found to be increased in the peripheral blood of patients with various cancers (Almand et al., 2001). Further, maturation defects in DC of patients with cancer have been described (Almand et al., 2000) and are attributable, in part, to vascular endothelial growth factor (VEGF) production by human tumors (Gabrilovich et al., 1999; Fricke and Gabrilovich, 2006). GM-CSF, which is also a frequently secreted product of tumor cells, recruits MSC and induces dose-dependent in vivo immune suppression and tumor promotion (Serafini et al., 2006). At the same time, GM-CSF is widely used as immune adjuvant in antitumor vaccines (Dranoff et al., 1993). In fact, GM-CSF was observed to significantly enlarge a subset of TGF-β-producing MSC phenotypically defined as CD14+HLA-DR−/low in the circulation of patients with metastatic melanoma (Filipazzi et al., 2007). This dual role of GM-CSF (stimulatory and suppressive) suggests that GM-CSF and MSC are involved in maintaining immune homeostasis under normal physiologic conditions but in the tumor presence are subverted to promote its escape.
Targeting of the tumor microenvironment for therapy
The failure of immune surveillance in tumor-bearing hosts has been one of the major incentives for the development of cancer immunotherapy, including anti-tumor vaccines, adoptive transfers of T cells and exogenous cytokine delivery. Numerous animal tumor models have provided strong evidence that in the presence of effective anti-tumor immunity, tumors fail to progress and established tumors regress (Ostrand-Rosenberg, 2004). Hence, recovery of immune surveillance and protection of immune cells from tumor-induced suppression are well-rationalized objectives of current anti-tumor therapies. As the molecular mechanisms responsible for tumor escape or involved in tumor-induced immune suppression are defined, therapeutic options for blocking tumor-induced suppression are becoming more realistic. With an improved understanding of mechanisms underlying tumor-induced immune suppression, future therapeutic strategies will likely focus on combined approaches designed to restore antitumor immune responses, eliminate tumor escape and correct tumor-induced immune deviation to enable the host immune system to more effectively control tumor growth. Table 2 lists some of the therapeutic approaches aimed at the modification of the tumor microenvironment and targeting Treg. On the basis of the currently available data, it seems reasonable to hypothesize that in situ interactions of the tumor with the host tissues, including infiltrating leukocytes, are a critical factor for tumor promotion. Therefore, disrupting or otherwise altering these interactions in favor of the host might result in therapeutic benefits. For example, the elimination of Treg before delivering antitumor vaccines might be considered to enhance their anti-tumor potential in patients with advanced malignancies. Table 2 lists some of the potential therapeutic strategies targeting Treg that are already in the clinic or are being experimented with animal models of cancer. Similar strategies for elimination or blocking of MSC activities might also be considered.
Conclusions
Current evidence suggests that chronic inflammation is associated with tumor development and progression. The NF-κB pathway may form a link between inflammation and cancer. Its activation in cells present in the tumor results in sustained production of pro-inflammatory cytokines, which promote tumor survival. The tumor co-opts functions of leukocytes in the microenvironment to support its growth using a variety of molecular mechanisms, which are beginning to be elucidated. At the same time, the tumor manages to hide from the immune attack, and either mounts a ‘counterattack’ or develops resistance to immune cells. The nature and intensity of inflammatory infiltrates may vary as the tumor progresses, depending on the local milieu that is created and shaped by the tumor. Consequently, mechanisms evolved by tumors for disarming host defenses and escape from the immune control vary in different cancers, and the unique signature of each tumor is reflected by its microenvironment. Therefore, understanding of cellular and molecular interactions operative in the tumor microenvironment is of crucial importance. Changing of chronic to acute inflammation at the tumor site might be therapeutically beneficial. Molecular tools are now available for devising novel and more effective anticancer therapies targeting not only the tumor but also its microenvironment.
References
Albers AE, Kim G, Ferris RL, Chikamatsu K, DeLeo AB, Whiteside TL et al. (2002). Immune responses to p53 in patients with cancer: enrichment in tetramer+p53 peptide-specific T cells and regulatory CD4+CD25+ cells at tumor sites. Cancer Immunol Immunother 62: 670–679.
Aller MA, Arias JL, Nava MP, Arias J . (2004). Posttraumatic inflammation is a complex response based on the pathological expression of the nervous, immune and endocrine function systems. Exp Biol Med 229: 170–181.
Almand B, Clark JI, Nikitina E, van Beynen J, English NR, Knight SC et al. (2001). Increased production of immature myeloid cells in cancer patients: a mechanism of immunosuppression in cancer. J Immunol 166: 678–689.
Almand B, Resser JR, Lindman B, Nadaf S, Clark JI, Kwon ED et al. (2000). Clinical significance of defective dendritic cell differentiation in cancer. Clin Cancer Res 6: 1755–1766.
Bacchetta R, Gambineri E, Roncarolo MG . (2007). Role of regulatory T cells and FOXP3 in human diseases. J Allergy Clin Immunol 120: 227–235.
Balkwill F, Coussens LM . (2004). Cancer: an inflammatory link. Nature 431: 405–406.
Balkwill F, Mantovani A . (2001). Inflammation and cancer: back to Virchow? Lancet 357: 539–545.
Banerjee DK, Dhodapkar MV, Matayeva E, Steinman RM, Dhodapkar KM . (2006). Expansion of FOXP3 high regulatory T cells by human dendritic cells (DCs) in vitro and after injection of cytokine-matured DCs in myeloma patients. Blood 108: 2655–2661.
Baxevanis CN, Dedoussis GV, Papadopoulos NG, Missitzis I, Stathopoulos GP, Papamichail M . (1994). Tumor specific cytolysis by tumor infiltrating lymphocytes in breast cancer. Cancer 74: 1275–1282.
Bergmann C, Strauss L, Zeidler R, Lang S, Whiteside TL . (2007). Expansion of human T regulatory type 1 cells in the microenvironment of COX-2-overexpressing head and neck squamous cell carcinoma. Cancer Res 67: 8865–8873.
Bronte V, Serafini P, Mazzoni A, Segal DM, Zanovello P . (2003). L-arginine metabolism in myeloid cells controls T-lymphocyte functions. Trends Immunol 24: 302–306.
Chang CC, Campoli M, Ferrone S . (2005). Classical and nonclassical HLA class I antigen and NK cell-activating ligand changes in malignant cells: current challenges and future direction. Adv Cancer Res 93: 189–234.
Chen A, Liu S, Park D, Kang Y, Zheng G . (2007). Depleting intratumoral CD4+CD45+ regulatory T cells via Fasl protein transfer enhances the therapeutic efficacy of adoptive T cell transfer. Cancer Res 67: 1291–1298.
Coronella JA, Spier C, Welch M, Trevor KT, Stopeck AT, Villar H et al. (2002). Antigen-driven oligoclonal expansion of tumor-infiltrating B cells in infiltrating ductal carcinoma of the breast. J Immunol 169: 1829–1836.
Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P et al. (2004). Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nature Med 10: 942–949.
Denko NC, Fontana LA, Hudson KM, Sutphin PD, Raychaudhuri S, Altman R et al. (2003). Investigating hypoxic tumor physiology through gene expression patterns. Oncogene 22: 5907–5914.
Dranoff G, Jaffee E, Lazenby A, Golumbek P, Levitsky H, Brose K et al. (1993). Vaccination with irradiated tumor cells engineered to secrete murine granulocyte-macrophage colony-stimulating factor stimulates potent, specific, and long-lasting anti-tumor immunity. Proc Natl Acad Sci USA 90: 3539–3543.
Dworak HF . (1986). Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N Engl J Med 315: 1650–1659.
Ferrone S, Whiteside TL . (2007). Tumor microenvironment and immune escape. Surg Oncol Clinics N Amer 16: 755–774.
Filipazzi P, Valenti R, Huber V, Pilla L, Canese P, Iero M et al. (2007). Identification of a new subset of myeloid suppressor cells in peripheral blood of melanoma patients with modulation by a ganulocyte-macrophage colony-stimulation factor-based antitumor vaccine. J Clin Oncol 25: 2546–2553.
Fricke I, Gabrilovich DI . (2006). Dendritic cells and tumor microenvironnment: a dangerous liaison. Immunol Invest 35: 459–483.
Gabrilovich DI, Ishida T, Nadaf S, Ohm JE, Carbone DP . (1999). Antibodies to vascular endothelial growth factor enhance the efficacy of cancer immunotherapy by improving endogenous dendritic cell function. Clin Cancer Res 5: 2963–2970.
Gallina G, Dolcetti L, Serafini P, DeSanto C, Marigo I, Colombo MP et al. (2006). Tumors induce a subset of inflammatory monocytes with immunosuppressive activity on CD8+ T cells. J Clin Invest 116: 2777–2790.
Gallucci S, Matzinger P . (2001). Danger signals: SOS to the immune system. Curr Opin Immunol 13: 114–119.
Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pagès C et al. (2006). Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 313: 1960–1964.
Galon J, Fridman W-H, Pages F . (2007). The adaptive immunologic microenvironment in colorectal cancer: A novel perspective. Cancer Res 67: 1883–1886.
Gondek DC, Lu LF, Quezada SA, Sakaguchi S, Noelle RJ . (2005). Cutting edge: contact-mediated suppression by CD4+cd25+ regulatory cells involves a granzyme B-dependent, perforin-independent mechanism. J Immunol 174: 1783–1786.
Greten FR, Eckmann L, Greten TF, Park JM, Li ZW, Egan LJ et al. (2004). IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell 118: 285–296.
Harrison ML, Obermuller E, Maisey NR, Hoare S, Edmonds K, Li NF et al. (2007). Tumor necrosis factor alpha as a new target for renal cell carcinoma: two sequential phase II trials of infliximab at standard and high dose. J Clin Oncol 25: 4542–4549.
Janssens W, Carlier V, Wu B, VanderElst L, Jacquemin MG, Saint-Remy JM . (2003). CD4+CD25+ T cells lyse antigen-presenting B cells by Fas-Fas ligand interaction in an epitope-specific manner. J Immunol 171: 4604–4612.
Johnson BD, Jing W, Orentas RJ . (1997). CD25+ regulatory T cell inhibition enhances vaccine-induced immunity to neuroblastoma. J Immunother 30: 203–214.
Karin M, Greten FR . (2004). NF-κB: linking inflammation and immunity to cancer development and progression. Nature 431: 461–466.
Kiessling R, Kono K, Petersson M, Wasserman K . (1996). Immunosuppression in human tumor-host interaction: role of cytokines and alterations in signal-transducing molecules. Springer Sem Immunopathol 18: 227–242.
Kornstein MJ, Brooks JS, Elder DE . (1983). Immunoperoxidase localization of lymphocyte subsets in the host response to melanoma and nevi. Cancer Res 43: 2749–2753.
Kuss I, Saito T, Johnson JT, Whiteside TL . (1999). Clinical significance of decreased ζ chain expression in peripheral blood lymphocytes of patients with head and neck cancer. Clin Cancer Res 5: 329–334.
Lanier LL . (2003). Natural killer cell receptor signaling. Curr Opin Immunol 15: 308–314.
Lee J-C, Lee K-M, Kim D-W, Heo DS . (2004). Elevated TGF-β1 secretion and down-modulation of NKG2D underlies impaired NK cytotoxicity in cancer patients. J Immunol 172: 7335–7340.
Liyanage UK, Moore TT, Joo HG, Tanaka Y, Herrmann V, Doherty G et al. (2002). Prevalence of regulatory T cells is increased in peripheral blood and tumor microenvironment of patients with pancreas or breast adenocarcinoma. J Immunol 169: 2756–2761.
Lluis JM, Buricchi F, Chiarugi P, Morales A, Fernandez-Checa JC . (2007). Dual role of mitochondrial reactive oxygen species in hypoxia signaling: activation of nuclear factor-kappa B via cSRC and oxidant dependent cell death. Cancer Res 67: 7368–7377.
Loukinova E, Dong G, Enamorado-Ayalya I, Thomas GR, Chen Z, Schreiber H et al. (2000). Growth regulated oncogene-alpha expression by murine aquamous cell carcinoma promotes tumor growth, metastasis, leukocyte infiltration and angiogenesis by a host CXC receptor-2 dependent mechanism. Oncogene 19: 3477–3486.
Mahnke K, Schonfeld K, Fondel S, Ring S, Karakhanova S, Wiedemeyer K et al. (2007). Depletion of CD4+CD25+ human regulatory T cells in vivo: kinetics of Treg depletion and alterations in immune functions in vivo and in vitro. Int J Cancer 120: 2723–2733.
Malik STA, Griffin DB, Fiers W, Balkwill FR . (1989). Paradoxical effects of tumor necrosis factor in experimental ovarian cancer. Int J Cancer 44: 918–925.
Mantovani A, Sozzani A, Locati M, Allavena P, Sica A . (2003). Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol 24: 232–233.
Martinez FO, Sica A, Mantovani A, Locati M . (2008). Macrophage activation and polarization. Front Biosci 13: 453–461.
Miescher S, Whiteside TL, Moretta L, Von Fliedner V . (1987). Clonal and frequency analyses of tumor-infiltrating T lymphocytes from human solid tumors. J Immunol 138: 4004–4011.
Mihm MC, Clemente C, Cascinelli N . (1996). Tumor infiltrating lymphocytes in lymph node melanoma metastases—a histopathologic prognostic indicator and an expression of local immune response. Lab Invest 74: 43–47.
Munn DH, Mellor AL . (2007). Indoleamine 2, 3-dioxygenase and tumor-induced tolerance. J Clin Invest 117: 1147–1154.
Nair S, Boczkowski D, Fassnacht M, Pisetsky D, Gilboa E . (2007). Vaccination against the forkhead family transcription factor Foxp3 enhances tumor immunity. Cancer Res 67: 371–380.
Naito Y, Saito K, Shiiba K, Ohuchi A, Saigenji K, Nagura H et al. (1998). CD8+ T cells infiltrated within cancer cell nests as a prognostic factor in human colorectal cancer. Cancer Res 58: 3491–3494.
Nakano O, Sato M, Naito Y, Suzuki K, Orikasa S, Aizawa M et al. (2001). Proliferative activity of intratumoral CD8+ T lymphocytes as a prognostic factor in human renal cell carcinoma: Clinicopathologic demonstration of antitumor immunity. Cancer Res 61: 5132–5136.
O’Day SJ, Hamid O, Urba WJ . (2007). Targeting cytotoxic T-lymphocyte antigen-4 (CTLA-4_: a novel strategy for the treatment of melanoma and other malignancies. Cancer 110: 2614–2627.
Ochoa AC, Zea AH, Hernandez C, Rodriguez PC . (2007). Arginase, prostaglandins, and myeloid suppressor cells in renal cell carcinoma. Clin Cancer Res 13: 721s–726s.
Ostrand-Rosenberg S . (2004). Animal models of tumor immunity, immunotherapy and cancer vaccines. Curr Opin Immunol 16: 143–150.
Pages F, Berger A, Camus M, Sanchez-Cabo F, Costes A, Molidor R et al. (2005). Effector memory T cells, early metastasis, and survival in colorectal cancer. N Engl J Med 353: 2654–2666.
Pak AS, Wright MA, Matthews JP, Collins SL, Petruzzelli GJ, Young MR . (1995). Mechanisms of immune suppression in patients with head and neck cancer: presence of CD34(+) cells which suppress immune functions within cancers that secrete granulocyte-macrophage colony-stimulating factor. Clin Cancer Res 1: 95–103.
Pikarsky E, Porat RM, Stein I, Abramovitch R, Amit S, Kasem S et al. (2004). NF-kappa B functions as a tumor promoter in inflammation-associated cancer. Nature 431: 461–466.
Reichert TE, Day E, Wagner EM, Whiteside TL . (1998a). Absent of low expression of the ζ chain in T cells at the tumor site correlates with poor survival in patients with oral carcinoma. Cancer Res 58: 5344–5347.
Reichert TE, Rabinowich H, Johnson JT, Whiteside TL . (1998b). Human immune cells in the tumor microenvironment: mechanisms responsible for signaling and functional defects. J Immunother 21: 295–306.
Reichert TE, Strauss L, Wagner EM, Gooding W, Whiteside TL . (2002). Signaling abnormalities and reduced proliferation of circulating and tumor-infiltrating lymphocytes in patients with oral carcinoma. Clin Cancer Res 8: 3137–3145.
Romero P, Cerottini JC, Speiser DE . (2006). The human T cell response to melanoma antigens. Adv Immunol 92: 187–224.
Roncarolo MG, Gregori S, Battaglia M, Bacchetta R, Fleischhauer K, Levings MK . (2006). Interleukin-10 secreting type 1 regulatory T cells in rodents and humans. Immunol Rev 212: 28–50.
Salem ML, Kadima AN, El-Naggar SA, Rubinstein MP, Chen Y, Gillanders WE et al. (1997). Defining the ability of cyclophosphamide preconditioning to enhance the antigen specific CD8+ T-cell response to peptide vaccination: creation of a beneficial host microenvironment involving type I IFNs and myeloid cells. J Immunother 30: 40–53.
Serafini P, Borrelo I, Bronte V . (2006). Myeloid suppressor cells in cancer: recruitment, phenotype, properties, and mechanisms of immune suppression. Semin Cancer Biol 16: 53–65.
Sheu BC, Hsu HN, Ho RH, Lin RH, Torng PL, Huang SC et al. (1999). Reversed CD4/CD8 percentages of tumor-infiltrating lymphocytes correlate with disease progression in human cervical cancer. Cancer 86: 1537–1543.
Shevach EM . (2000). Regulatory T cells in autoimmunity. Annu Rev Immunol 18: 423–449.
Shevach EM . (2004). Fatal attraction: tumors becon regulatory T cells. Nature Med 10: 900–901.
Stewart THM, Tsai S-CJ . (1993). The possible role of stromal cell stimulation in worsening the prognosis of a subset of patients with breast cancer. Clin Expt Metastasis 11: 295–305.
Strauss L, Bergmann C, Gooding W, Johnson JT, Whiteside TL . (2007b). The frequency and suppressor function of CD4+CD25highFoxP3+ T cells in the circulation of patients with squamous cell carcinoma of the head and neck. Clin Cancer Res 13: 6301–6311.
Strauss L, Bergmann C, Szczepanski M, Gooding W, Johnson TJ, Whiteside TL . (2007a). A unique subset of CD4+CD25highFOXP3+ T cells secreting IL-10 and TGF-β1 mediates suppression in the tumor microenvironment. Clin Cancer Res 13: 4345–4354.
Strauss L, Whiteside TL, Knights A, Bergmann C, Knuth A, Zippelius A . (2007c). Selective survival of naturally occurring human CD4+CD25+Foxp3+ regulatory T cells cultured with rapamycin. J Immunol 178: 320–329.
Tsai CS, Chen FH, Wang CC, Huang HL, Jung SM, Wu CJ et al. (2007). Macrophages from irradiated tumors express higher levels of iNOS, arginase I and COX-2, and promote tumor growth. Int J Radiat Oncol Biol Phys 68: 499–507.
Uzzo RG, Clark PE, Rayman P, Bloom T, Rybicki L, Novick AC et al. (1999). Alterations in NFκB activation in T lymphocytes of patients with renal cell carcinoma. J Nat Cancer Inst 91: 718–721.
Whiteside TL, Vujanovic NL, Herberman RB . (1998). Natural killer cells and tumor therapy. Curr Topics Microbiol Immunol 230: 221–244.
Whiteside TL . (1993). Tumor Infiltrating Lymphocytes in Human Malignancies. Medical Intelligence Unit, R.G. Landes Co.: Austin, TX.
Whiteside TL . (2006). Immune suppression in cancer: effects on immune cells, mechanisms and future therapeutic intervention. Sem Cancer Biol 16: 3–15.
Whiteside TL . (2007). The Local Tumor Microenvironment. In: Kaufmann H, Wolchok JD (eds). General Principles of Tumor Immunotherapy: Basic and Clinical Applications of Tumor Immunology. Springer, pp 145–167.
Woo EY, Chu CS, Goletz TJ, Schlienger K, Yeh H, Coukos G et al. (2001). Regulatory CD4+CD25+ T cells in tumors from patients with early-stage non-small cell lung cancer and late-stage ovarian cancer. Cancer Res 61: 4766–4772.
Zhou G, Drake CG, Levitsky HI . (2006). Amplification of tumor-specific regulatory T cells following therapeutic cancer vaccines. Blood 107: 628–636.
Zhou J, Dudley ME, Rosenberg SA, Robbins PF . (2004). Selective growth, in vitro and in vivo, of individual T cell clones from tumor-infiltrating lymphocytes obtained from patients with melanoma. J Immunol 173: 7622–7629.
Zitvogel L, Tesniere A, Kroemer G . (2006). Cancer despite immunosurveillance: immunoselection and immunosubversion. Nat Rev Immunol 6: 715–727.
Acknowledgements
This article was supported in part by the NIH Grant PO-1 CA109688.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Whiteside, T. The tumor microenvironment and its role in promoting tumor growth. Oncogene 27, 5904–5912 (2008). https://doi.org/10.1038/onc.2008.271
Published:
Issue Date:
DOI: https://doi.org/10.1038/onc.2008.271
Keywords
This article is cited by
-
RUNX1 promotes angiogenesis in colorectal cancer by regulating the crosstalk between tumor cells and tumor associated macrophages
Biomarker Research (2024)
-
Unveiling the mechanistic link between extracellular amyloid fibrils, mechano-signaling and YAP activation in cancer
Cell Death & Disease (2024)
-
SMAD7 expression in CAR-T cells improves persistence and safety for solid tumors
Cellular & Molecular Immunology (2024)
-
Prognostic implications of tumor histology and microenvironment in surgically resected intrahepatic cholangiocarcinoma: a single institutional experience
Virchows Archiv (2024)
-
Reprogramming the tumor microenvironment with biotechnology
Biomaterials Research (2023)
