
Abstract
Many mutations have been detected in the SLC12A3 gene of Gitelman syndrome (GS, OMIM 263800) patients. In previous studies, only one mutant allele was detected in ∼20 to 41% of patients with GS; however, the exact reason for the nonidentification has not been established. In this study, we used RT-PCR using mRNA to investigate for the first time transcript abnormalities caused by deep intronic mutation. Direct sequencing analysis of leukocyte DNA identified one base insertion in exon 6 (c.818_819insG), but no mutation was detected in another allele. We analyzed RNA extracted from leukocytes and urine sediments and detected unknown sequence containing 238bp between exons 13 and 14. The genomic DNA analysis of intron 13 revealed a single-base substitution (c.1670–191C>T) that creates a new donor splice site within the intron resulting in the inclusion of a novel cryptic exon in mRNA. This is the first report of creation of a splice site by a deep intronic single-nucleotide change in GS and the first report to detect the onset mechanism in a patient with GS and missing mutation in one allele. This molecular onset mechanism may partly explain the poor success rate of mutation detection in both alleles of patients with GS.
Similar content being viewed by others

Main
Gitelman syndrome (GS, OMIM 263800) is an autosomal recessive renal disorder characterized by hypokalemia, hypomagnesemia, metabolic alkalosis, and hypocalciuria (1). Mild weakness, cramps, and general fatigue are clinically observable, but they are often so slight that patients with GS are not diagnosed until late childhood or even adulthood; however, a reduced health quality of life for patients with GS, compared with a control group, has been referred (2). Simon et al. (3) demonstrated that mutations in the SLC12A3 gene encoding the thiazide-sensitive sodium-chloride cotransporter (NCCT) are responsible for GS. This gene, which is located in chromosome 16 and comprises 26 exons, is a 1021-amino acid protein with 12 predicted transmembrane domains. The lack of a functional NCCT leads to a decrease in sodium and chloride reabsorption in the distal convoluted tubule and an increase in solute delivery to the collecting duct. These changes result in blood volume reduction, activation of the renin-angiotensin-aldosterone system, and increased secretion of potassium and hydrogen ions into the collecting duct.
To date, >100 different mutations throughout the SLC12A3 gene have been identified in patients with GS, but only one mutant allele was detected in ∼20 to 41% of patients with GS (3–7). However, the exact reason for the nonidentification has not been established.
This report concerns a patient with GS and only one mutation in the exons and exon-intron boundaries. However, we succeeded in detecting a deep intronic mutation that creates a new donor splice site resulting in the inclusion of a novel cryptic exon in mRNA. This novel disease onset mechanism may partly explain the poor success rate of mutation detection in both alleles in patients with GS and how single-base changes deep within introns can cause GS.
PATIENTS AND METHODS
Case report.
The subject of this study was a 12-y-old girl who was suffering from paralysis, muscle stiffness, and pain. When she was referred to our hospital, a blood examination disclosed that her serum potassium level was low. No medicine, such as diuretics or laxatives, had been prescribed previously. On admission, she was 145-cm tall (−2.5 SD), weighed 46 kg, her blood pressure was 102/62 mm Hg, and physical examination findings were normal. Laboratory results showed that serum potassium was low (1.9 mEq/L; normal: 3.5–4.7 mEq/L), whereas plasma renin activity (20 pg mL−1 h−1; normal: 0.2–3.9 ng mL−1 h−1) and aldosterone concentration (297 pg/mL; normal: 29.9–159 pg/mL) were elevated. Serum magnesium (1.4–1.6 mg/dL; normal: 1.7–2.5 mg/dL), and urinary calcium (urine calcium/creatinine: 0.02) and chloride excretion (FeCl 0.88%; normal: 1.6–3.2%) were also low. Her parents were nonconsanguineous.
This study was approved by the Institutional Review Board of Kobe University Graduate School of Medicine and consent for this study was obtained from the patient's parents.
Genetic analyses.
Genomic DNA was isolated from peripheral blood leukocytes of the patient as well as from normal control subjects with the Qiagen kit (Qiagen Inc., Chatsworth, CA), according to the manufacturer's instructions. Primer pairs for the SLC12A3 gene and the CLCNKB gene were generated following previous reports (8,9). Polymerase chain reaction (PCR) was performed and the resultant products were analyzed, including every intron-exon boundary, by direct sequencing with a DNA sequencer (Perkin-Elmer-ABI, Foster City, CA). Multiplex ligation-dependent probe amplification (MLPA) was also conducted by means of the SALSA P136-Gitelman MLPA assay (MRC-Holland, Amsterdam, The Netherlands) and following the manufacturer's instructions.
Total RNA was extracted from blood leukocytes and urine sediments. The urine sediments were obtained by centrifugation at 1500 × g for 10 min from 100 mL of early morning urine. Microscopic examination of these sediments confirmed that they contained sufficient renal tubular epithelial cells having large round nucleus surrounded by a large granular cytoplasm which is larger than granulocytes (10). RNA was isolated with the aid of Isogen Kit (Nippon Gene Co., Toyama, Japan) and was then reverse-transcribed onto cDNA by using random hexamers and the Superscript III kit (Invitrogen). cDNA was amplified by means of nested PCR using primer pairs. The primer sequences of exons 13 to 15 were as follows: exon13, first forward: GTACCCACTGATCGGCTTCT; second forward: TTCCTCTGCTCCTATGCC; exon 15, first reverse: TCCTCGGCAATGACATCC; second reverse: GGGCGGTAGTTCTTGATGT. After 40 cycles of amplification, PCR products were separated on 2% agarose and sequenced with a DNA sequencer (Perkin-Elmer-ABI, Foster City, CA). For sequencing, amplified products were separated by electrophoresis, and the RT-PCR products were purified. The purified products were subcloned into pT7 vector (Novagen, Inc., Madison, WI), and the inserted products were sequenced. Normal control kidney cDNA was obtained from the Human Kidney cDNA Library (Invitrogen). Normal control blood leukocytes and urinary sediments samples were obtained from a healthy control to extract total RNA.
RESULTS
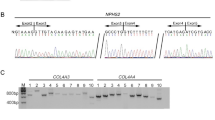
Direct sequencing of the PCR-amplified products disclosed a novel heterozygous single-base insertion of G between nucleotides 818 and 819 in exon 6. This mutation was also seen in the father's genomic DNA (Fig. 1A). Because the second abnormality was not detectable, we conducted an MLPA analysis using the MLPA kit for GS. However, no large heterozygous deletion was detected with this approach, either. Next, we performed a complete transcript sequence analysis using mRNA extracted from leukocytes. In addition to the normal band in the fragment of exons 13 to 15, an abnormal band was detected in the RT-PCR products from the patient's sample, which showed two bands, one the same size as the control and the other larger (Fig. 2). After subcloning of the two bands, sequencing was conducted, and the larger product disclosed a normal sequence containing a 238-bp insertion between exons 13 and 14, which are part of intron 13 (Figs. 1B and 3). This insertion was not detected in cDNA from a normal blood leukocytes, kidney, and urinary sediments (Fig. 2). To shed light on the mechanism involved in this intronic sequence exonization, we sequenced this part of intron 13 and identified the c.1670–191C>T heterozygous base substitution at the position immediately after the insertion sequence and created a novel splicing donor site for cryptic exon resulting in the 238-bp insertion in the mRNA (Fig. 4). This mutation was found in the genomic DNA of both the patient and the mother (Fig. 1C), whereas not detected in 100 normal control samples (200 chromosomes). Moreover, no mutations were detected in the CLCNKB gene. It was thus demonstrated that this patient possessed a compound heterozygous mutation of c.818_819insG and c.1670–191C>T and the GS phenotype. Both mutations will lead to create premature stop codons in exon 7 and in cryptic exon, respectively.

Nucleotide changes in the SLC12A3 gene. A, A heterozygous 1-bp insertion between nucleotides 818 and 819 located in exon 6 (c.818_819insG) was detected in the paternal allele. B, Fragments of the sequence after subcloning of the sense strand of cDNA from the patient. The larger product (upper figure) containing 238-bp cryptic exon sequence and the smaller product (lower figure) showed the normal sequence. C, A heterozygous single-base substitution of C to T in intron 13 (c.1670-191C>T) was detected in the maternal allele.

A transcript abnormality in the SLC12A3 gene. Electrophoresis of cDNA after PCR. A, Normal control leukocytes. B, Normal control kidney. C, Normal control urinary sediments. D, Patient's leukocytes. E, Patient's urinary sediments. F, The mother's leukocytes. For PCR, the forward primer located in exon 13 and the reverse primer located in exon 15 was used. Control samples clearly show a single band, whereas the samples extracted from leukocytes of the patient and the mother and urinary sediments of the patient show two bands, one the same size as the control sample and the other a larger one containing the cryptic exon sequence.

Normal sequence surrounding intron 13. The cryptic exon sequence was derived from intron 13, which is positioned between AG and GC in normal genomic DNA.

The schema of the intronic mutation resulting in the new splicing donor site for the cryptic exon.
DISCUSSION
We identified two heterozygous mutations, c.818_819insG in the paternal allele and c.1670–191C>T in the maternal allele, in the genomic DNA from the leukocytes of a typical patient with GS, and we hypothesized that these compound heterozygous mutations of SLC12A3 had caused GS in our patient. The latter mutation was confirmed to result in a splicing abnormality containing a cryptic exon in the transcripts. Amplification of SLC12A3 mRNA extracted from the patient's leukocytes and urinary sediments, and the mother's leukocytes showed the same result for both extracts.
To date, >100 different mutations of the SLC12A3 gene have been identified in the entire gene in patients with GS. These mutations are located all through the coding sequence of the SLC12A3 gene, but most of them are found in the intracellular domains of the protein and missense mutations are the most frequently reported abnormalities. In previous studies, only one mutant allele was detected in 20 to 41% of patients with GS (3–7). There are many possible explanations for the nonidentification of the mutation in the second allele, for example, human error, direct sequencing missing major heterozygous mutations including rearrangements such as duplications, inversions, etc., and the possible presence of mutations in gene-regulating fragments such as promoter or enhancer segments. It has also been hypothesized that there may be a concurrent heterozygous mutation in a gene other than the SLC12A3 gene, particularly in the CLCNKB gene for type III Bartter syndrome (6). However, the exact reason for the nonidentification has not been established.
Because neither MLPA that was a recently established technique for detection of copy number variations nor CLCNKB gene analysis for detection of the modifier gene existence succeeded, we conducted RT-PCR analysis for detection of the deep intronic mutations, which were previously identified in other genes (11–13). As expected, we succeeded in detecting a deep intronic mutation that creates a new donor splice site resulting in the inclusion of a novel cryptic exon in mRNA. This molecular mechanism of intronic mutation may partly explain the poor success rate for detection of mutations in both alleles in patients with GS and demonstrates that single-base changes deep within introns can cause GS. Although, in GS, an intronic single-base substitution that leads to create cryptic splicing site has been reported, this patient showed intron 3 splicing acceptor consensus site mutation and resulted in the activation of a nearby cryptic splice site in intron 3 (14). This mutation can be easily detected by the analysis of exon-intron boundaries and our patient possessed a deep intronic mutation that is impossible to detect by genomic DNA analysis for detecting mutations in exons or exon-intron boundaries.
Igarashi et al. (15) were the first to identify transcript abnormalities by extracting mRNA from urinary sediment cells of patients with Dent disease. This method proved to be very useful for analyzing mRNA expression in renal tubular or glomerular cells in various kidney diseases. Renal biopsy specimens constitute one source of material for analyzing mRNA in inherited kidney diseases, but renal biopsy is invasive and not needed for certain kidney diseases. Urinary sediments, on the other hand, contain cells derived from the kidney, and genetic analysis using those cells constitutes an entirely noninvasive, simple method for the diagnosis of inherited kidney diseases (10,16,17). Therefore, we conducted RT-PCR using urinary sediment cells to determine the presence of transcript abnormalities in the target organ cells.
This patient showed relatively severe symptoms including short stature, paralysis, muscle stiffness, and pain from her younger age. Recent report clearly showed that patients with GS and truncated mutations including out of frame splicing variants in at least one allele tend to show severe symptoms (18). Our patient also possessed truncated mutations caused by one base insertion and deep intronic mutation. These mutations may explain her severe symptoms.
To summarize, we investigated transcript abnormalities in a patient with GS caused by deep intronic mutation. This is the first report of GS associated with creation of a splice acceptor site by a deep intronic single-nucleotide change leading to the inclusion of extra exon structures, and this mutation could only have been detected via mRNA analysis. We identified a novel intronic mutation that may partly explain the poor success rate of mutation detection in both alleles in patients with GS and demonstrated that single-base changes deep within introns can cause GS.
Abbreviations
- GS:
-
Gitelman syndrome
- MLPA:
-
multiplex ligation-dependent probe amplification
References
Gitelman HJ, Graham JB, Welt LG 1966 A new familial disorder characterized by hypokalemia and hypomagnesemia. Trans Assoc Am Physicians 79: 221–235
Cruz DN, Shaer AJ, Bia MJ, Lifton RP, Simon DB 2001 Gitelman's syndrome revisited: an evaluation of symptoms and health-related quality of life. Kidney Int 59: 710–717
Simon DB, Nelson-Williams C, Bia MJ, Ellison D, Karet FE, Molina AM, Vaara I, Iwata F, Cushner HM, Koolen M, Gainza FJ, Gitleman HJ, Lifton RP 1996 Gitelman's variant of Bartter's syndrome, inherited hypokalemic alkalosis, is caused by mutations in the thiazide-sensitive NaCl cotransporter. Nat Genet 12: 24–30
Colussi G, Bettinelli A, Tedeschi S, De Ferrari ME, Syren ML, Borsa N, Mattiello C, Casari G, Bianchetti MG 2007 A thiazide test for the diagnosis of renal tubular hypokalemic disorders. Clin J Am Soc Nephrol 2: 454–460
Lemmink HH, Knoers NV, Karolyi L, van Dijk H, Niaudet P, Antignac C, Guay-Woodford LM, Goodyer PR, Carel JC, Hermes A, Seyberth HW, Monnens LA, van den Heuvel LP 1998 Novel mutations in the thiazide-sensitive NaCl cotransporter gene in patients with Gitelman syndrome with predominant localization to the C-terminal domain. Kidney Int 54: 720–730
Lin SH, Shiang JC, Huang CC, Yang SS, Hsu YJ, Cheng CJ 2005 Phenotype and genotype analysis in Chinese patients with Gitelman's syndrome. J Clin Endocrinol Metab 90: 2500–2507
Monkawa T, Kurihara I, Kobayashi K, Hayashi M, Saruta T 2000 Novel mutations in thiazide-sensitive Na-Cl cotransporter gene of patients with Gitelman's syndrome. J Am Soc Nephrol 11: 65–70
Fukuyama S, Okudaira S, Yamazato S, Yamazato M, Ohta T 2003 Analysis of renal tubular electrolyte transporter genes in seven patients with hypokalemic metabolic alkalosis. Kidney Int 64: 808–816
Nozu K, Fu XJ, Nakanishi K, Yoshikawa N, Kaito H, Kanda K, Krol RP, Miyashita R, Kamitsuji H, Kanda S, Hayashi Y, Satomura K, Shimizu N, Iijima K, Matsuo M 2007 Molecular analysis of patients with type III Bartter syndrome: picking up large heterozygous deletions with semiquantitative PCR. Pediatr Res 62: 364–369
Kaito H, Nozu K, Fu XJ, Kamioka I, Fujita T, Kanda K, Krol RP, Suminaga R, Ishida A, Iijima K, Matsuo M 2007 Detection of a transcript abnormality in mRNA of the SLC12A3 gene extracted from urinary sediment cells of a patient with Gitelman's syndrome. Pediatr Res 61: 502–505
King K, Flinter FA, Nihalani V, Green PM 2002 Unusual deep intronic mutations in the COL4A5 gene cause X linked Alport syndrome. Hum Genet 111: 548–554
Ogino W, Takeshima Y, Nishiyama A, Okizuka Y, Yagi M, Tsuneishi S, Saiki K, Kugo M, Matsuo M 2007 Mutation analysis of the ornithine transcarbamylase (OTC) gene in five Japanese OTC deficiency patients revealed two known and three novel mutations including a deep intronic mutation. Kobe J Med Sci 53: 229–240
Yagi M, Takeshima Y, Wada H, Nakamura H, Matsuo M 2003 Two alternative exons can result from activation of the cryptic splice acceptor site deep within intron 2 of the dystrophin gene in a patient with as yet asymptomatic dystrophinopathy. Hum Genet 112: 164–170
Abuladze N, Yanagawa N, Lee I, Jo OD, Newman D, Hwang J, Uyemura K, Pushkin A, Modlin RL, Kurtz I 1998 Peripheral blood mononuclear cells express mutated NCCT mRNA in Gitelman's syndrome: evidence for abnormal thiazide-sensitive NaCl cotransport. J Am Soc Nephrol 9: 819–826
Igarashi T, Inatomi J, Ohara T, Kuwahara T, Shimadzu M, Thakker RV 2000 Clinical and genetic studies of CLCN5 mutations in Japanese families with Dent's disease. Kidney Int 58: 520–527
Iida K, Nozu K, Takahashi Y, Okimura Y, Kaji H, Matsuo M, Chihara K 2008 Characterization of a splicing abnormality in Gitelman syndrome. Am J Kidney Dis 51: 1077–1078
Krol RP, Nozu K, Nakanishi K, Iijima K, Takeshima Y, Fu XJ, Nozu Y, Kaito H, Kanda K, Matsuo M, Yoshikawa N 2008 Somatic mosaicism for a mutation of the COL4A5 gene is a cause of mild phenotype male Alport syndrome. Nephrol Dial Transplant 23: 2525–2530
Riveira-Munoz E, Chang Q, Godefroid N, Hoenderop JG, Bindels RJ, Dahan K, Devuyst O 2007 Transcriptional and functional analyses of SLC12A3 mutations: new clues for the pathogenesis of Gitelman syndrome. J Am Soc Nephrol 18: 1271–1283
Author information
Authors and Affiliations
Additional information
Supported by Grant in Aid (B-19790720) (to K.N.) from the Japan Society for the Promotion of Science.
Rights and permissions
About this article
Cite this article
Nozu, K., Iijima, K., Nozu, Y. et al. A Deep Intronic Mutation in the SLC12A3 Gene Leads to Gitelman Syndrome. Pediatr Res 66, 590–593 (2009). https://doi.org/10.1203/PDR.0b013e3181b9b4d3
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/PDR.0b013e3181b9b4d3
This article is cited by
-
Allele-specific RT-PCR for the rapid detection of recurrent SLC12A3 mutations for Gitelman syndrome
npj Genomic Medicine (2021)
-
Molecular assay for an intronic variant in NUP93 that causes steroid resistant nephrotic syndrome
Journal of Human Genetics (2019)
-
Cryptic exon activation in SLC12A3 in Gitelman syndrome
Journal of Human Genetics (2017)
-
Deep intronic mutations and human disease
Human Genetics (2017)
-
Natural history of genetically proven autosomal recessive Alport syndrome
Pediatric Nephrology (2014)
