
Abstract
Pediatric neurodegenerative diseases are a heterogeneous group of diseases that result from specific genetic and biochemical defects. In recent years, studies have revealed a wide spectrum of abnormal cellular functions that include impaired proteolysis, abnormal lipid trafficking, accumulation of lysosomal content, and mitochondrial dysfunction. Within neurons, elaborated degradation pathways such as the ubiquitin–proteasome system and the autophagy–lysosomal pathway are critical for maintaining homeostasis and normal cell function. Recent evidence suggests a pivotal role for autophagy in major adult and pediatric neurodegenerative diseases. We herein review genetic, pathological, and molecular evidence for the emerging link between autophagy dysfunction and lysosomal storage disorders such as Niemann–Pick type C, progressive myoclonic epilepsies such as Lafora disease, and leukodystrophies such as Alexander disease. We also discuss the recent discovery of genetically deranged autophagy in Vici syndrome, a multisystem disorder, and the implications for the role of autophagy in development and disease. Deciphering the exact mechanism by which autophagy contributes to disease pathology may open novel therapeutic avenues to treat neurodegeneration. To this end, an outlook on novel therapeutic approaches targeting autophagy concludes this review.
Similar content being viewed by others

Main
Pediatric neurodegenerative diseases are a heterogeneous group of diseases that result from specific genetic and biochemical defects. Although the age of onset and the clinical course are variable, neurodegenerative disorders of childhood are characterized by progressive neurologic and cognitive dysfunction with loss of speech, vision, hearing, and motor skills, and a frequent association with seizures, feeding difficulties, and failure to thrive. The degenerative process can affect white and gray matter and spreads in a disease-specific manner. Current genetic and biochemical testing and neuroimaging can establish a diagnosis. The outcome for most diseases, however, is still poor, as therapeutic approaches are limited.
Accumulation of proteins, polysaccharides, and lipids are common mechanisms shared by major adult-onset neurodegenerative diseases1,2,3 and many neurodegenerative diseases of childhood.4 These conditions are, therefore, often referred to as “storage diseases” or “proteinopathies.” Cells and postmitotic cells such as neurons, in particular, rely on effective cargo targeting, tagging, transportation, and degradation to maintain intracellular homeostasis under normal conditions and metabolic stress. Within this network, autophagy plays an indispensible role by eliminating misfolded and aggregated proteins, lipids, saccharides, and damaged organelles. Recent evidence suggests that autophagy is dysregulated in a number of pediatric neurodegenerative and metabolic diseases.
Autophagy: An Overview
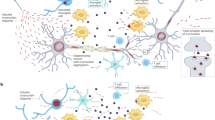
Autophagy describes intracellular pathways that converge into degradation of target material in lysosomes.5 The route of cargo delivery to the lysosome distinguishes the respective subtype of autophagy, i.e., macroautophagy, chaperone-mediated autophagy, and microautophagy ( Figure 1 ). The final step of these processes is the degradation of the substrate, yielding basic building blocks that can be further broken down and recycled.

Neuronal pathways of protein and cargo degradation. A substantial portion of newly synthesized proteins is folded incorrectly, a process that is facilitated under conditions of metabolic stress. Chaperones can mediate refolding and can thereby prevent misfolding and oligomer or even aggregate formation. If refolding, however, is not possible or the capacity of the chaperone system is exceeded, protein degradation pathways are necessary to maintain protein homeostasis. Pathways that remove proteins and other cargo under normal and pathologic conditions are the ubiquitin–proteasome system and autophagy. The latter consists of three distinct subtypes: macroautophagy, chaperone-mediated autophagy, and microautophagy.
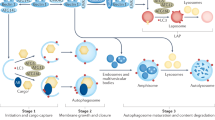
Most studies on autophagy in pediatric neurodegenerative diseases have focused on macroautophagy (hereafter referred to as “autophagy”), a “bulk” degradation process that involves the de novo formation of double-membrane–bound vesicles that elongate and, as they grow, sequester complete regions of the cytosol ( Figures 1 and 2 ). The resulting mature double-membrane vesicles are referred to as autophagosomes. While this compartment lacks proteolytic enzymes, the cargo can only be degraded by fusion with late endosomes or lysosomes, which provide the necessary enzymes. The fusion product of autophagosomes and lysosomes has been termed autolysosome.

The autophagy cascade can be divided into the several key steps. Autophagosome formation includes initiation, nucleation, elongation, closure, and maturation. Fusion with the lysosome creates autolysosomes in which cargo degradation and recycling finally take place. The regulation of the autophagic cascade involves both mTORC1-dependent and mTORC1-independent signaling pathways and signaling by the transcription factor EB (TFEB) (also see Figure 7 ).
Autophagy integrates numerous other signaling pathways. The first proteomic analysis of the basal autophagy interactions in human cells has revealed a complex network of 751 interactions between 409 proteins.6 The cascade of events can be divided into six steps: initiation, nucleation, elongation, closure, maturation, and fusion5,7 ( Figure 2 ). Initiation is regulated by several signaling pathways, the best known of which involves mammalian target of rapamycin (mTOR), a serine/threonine kinase that integrates various cell signals such as growth factors and energy status. A second recently described pathway for initiation of autophagy is mediated by the basic helix-loop-helix leucine zipper transcription factor EB, which is involved in the regulation of both lysosomal biogenesis and autophagy.8,9
Autophagy initiation is mediated by a regulatory complex that involves Beclin-1, the class III phosphatidylinositol-3-phosphate kinase Vps34, and other interacting proteins.10 Beclin-1 promotes Vps34-dependent generation of phosphatidylinositol-3-phosphate, which in turn facilitates the nucleation of autophagosomal membrane.10
Elongation of evolving autophagosomes is governed by two unique ubiquitin-like conjugation systems that interact in a hierarchical order: Atg12-Atg5 and microtubule-associated proteins 1A/1B light chain 3 (LC3 or Atg8)-phosphatidylethanolamine conjugation7 ( Figure 2 ). The latter is of particular importance as the resulting lipidated form of LC3, LC3-II, is attached to both sides of the autophagosome membrane, making this protein a useful tool to quantify autophagosome formation and turnover.11 Interestingly, autophagosomes can be formed anywhere within the cell, independent of the close presence of lysosomes. Indeed in neurons, autophagosome formation and cargo sequestration takes place in neuronal processes requiring transport along the microtubule system for subsequent fusion and cargo degradation.12
Accumulating evidence suggests that autophagy is not only a “bulk” degradation process for random cargo in the cytosol, but, by using specific cargo recognition molecules, can also be a very selective means to degrade tagged proteins or organelles. For example, mitophagy, the autophagic removal of dysfunctional mitochondria, is of particular importance for neurons and many other cell types.13
Although the three autophagy subtypes have individual characteristics, they “communicate” as they display compensatory adaptation to preserve homeostasis. Crosstalk is of outmost importance for neurodegenerative diseases where a primary failure of degradation mechanisms has been proposed.1,2,3,14–16 Dysfunction of autophagy can result from a block at every level of the pathway ( Figure 2 ). With regards to neuronal pathology, defects in the autophagy pathway can be divided into those that cause impaired autophagic turnover and those that result in deregulated and overactive autophagy.
Niemann–Pick type C: Linking Autophagy to Lipid Metabolism
Niemann–Pick type C (NP-C, OMIM no. 257220; no. 607625) is a devastating neurodegenerative disease caused by autosomal-recessive mutations in the NPC117 and NPC218 genes, which encode for two proteins that are instrumental to the efflux of cholesterol from late-endosomal and lysosomal compartments. NPC1, a transmembrane protein with a “sterol sensing domain” and a luminal cholesterol binding domain, and NPC2, a soluble intraluminal protein capable of extracting cholesterol from lipid layers, act together to promote the recycling of cholesterol from the late-endosomal system.19,20 Consequently, impairment of these proteins leads to the endosomal and lysosomal accumulation of cholesterol and secondary storage of glycophospholipids in the brain and other organs.19
In recent years, studies have demonstrated accumulation of autophagosomes in degenerating neurons of NPC1 knockout mice,21,22,23 primary fibroblasts derived from NP-C patients,23 and NPC1-deficient cell culture models.24 The activation of basal autophagy was found to be accompanied by impaired degradation of long-lived proteins, abnormal levels of the lysosomal enzyme cathepsin D, and accumulation of undigested polyubiquitinated proteins in the endolysosomal and lysosomal fraction.22 These findings indicate that disruption of lipid trafficking in NP-C results in a global enhancement of autophagy that is, however, not sufficient to handle the quantity of proteins targeted for degradation. Importantly, induction of autophagy in NPC1-deficient cells is associated with an increased expression of Beclin-1, whereas mTOR signaling remains unchanged.23 Consistent with the findings in NPC1- and NPC2-deficient cells, pharmacological induction of unesterified cholesterol leads to enhanced Beclin-1 expression, further demonstrating that Beclin-1 is induced by lipid trafficking defects and may act as an important regulator of autophagy in sphingolipid storage diseases.23
In view of these findings, a critical question remains whether enhanced autophagy is a compensatory response that promotes cell survival or rather has deleterious effects. Interestingly, the acute knockdown of the microtubule-binding protein tau, a protein implicated in many neurodegenerative diseases including NP-C, was found to significantly impair microtubule-dependent autophagy induction and flux in NPC1-deficient fibroblasts.25 NPC1/TAU double-null mice, however, exhibited an exacerbated NP-C phenotype and a decreased life span (but no exacerbated cell loss), providing evidence that the integrity of the described induction of autophagy can affect the severity of the disease phenotype.25 These findings have significant implications as they suggest that hyperphosphorylated tau could lead to a progressive loss of tau’s normal function, which in turn impairs the ability to upregulate autophagic flux in response to NPC deficiency,25 indicating a “protective” role of increased autophagy in NP-C.
In contrast to this notion, however, autophagy activation in NP-C was recently shown to impair both mitochondrial turnover26 and lysosomal protease function.27 Using human embryonic stem cell–derived neurons, it was shown that both induction of basal autophagy and impaired autophagic flux concomitantly occur with NPC1 knockdown and that this significantly compromises mitochondrial clearance.26 Remarkably, these changes were partially reversible through the application of the autophagy inhibitor 3-methyladenine, which reduces autophagosome formation and, by treatment with cyclodextrin, a compound capable of mobilizing cholesterol from the lysosomal compartment.26 Concurrent with the notion that NPC deficiency interferes with the completion of the autophagic cascade, recent findings demonstrate that the autophagic response increases delivery of cholesterol to the lysosome and thereby causes dysfunction of lysosomal proteases, which in turn could impair turnover of autolysosomes, therefore creating a pathological feedback loop that promotes induction of dysfunctional autophagy27 ( Figure 3 ). The cascade of deleterious events was again ameliorated with pharmacological inhibition of autophagy initiation, and vice versa, unesterified cholesterol significantly increased in NP-C fibroblasts following treatment with the mTOR-dependent autophagy inducer rapamycin.27 Cholesterol accumulation in cultured neurons was shown to lead to leakage of the lysosomal protease cathepsin D into the cytosol, which may trigger neurodegeneration, and interestingly, extracellular cathepsin D was found to trigger defective autophagy in exposed cells, a finding that might have implications for disease progression28 ( Figure 3 ).

Deregulated autophagy in Niemann–Pick type C disease. Lipids accumulate in the endosomal pathway, which induces autophagosome formation through the Beclin-1 complex as a potential compensatory action. The accumulating lipids, however, also impair cargo degradation and lysosomal recycling at the autolysosome levels leading to a downstream block in the autophagic cascade. Therefore, autophagosome and their cargo, including lipids, accumulate, and a vicious cycle ensues. Finally, cargo accumulation and leakage of lysosomal enzymes into the cytosol might contribute to neurodegeneration. Pathological changes in the autophagy pathway and lysosomal lipid metabolism are highlighted in red. LDL, low-density lipoprotein.
Collectively, these findings demonstrate that autophagy is induced in the setting of NPC deficiency and lysosomal cholesterol storage but remains dysfunctional and thereby promotes lysosomal cholesterol storage, lysosomal dysfunction, and disease pathology, a conclusion that points inhibition of autophagy as an interesting novel target in NP-C.
Vici Syndrome: Linking Autophagy to Embryonic (Brain) Development
Vici syndrome (OMIM no. 242840) is a rare recessively inherited multisystem disorder first described in 1988 based on its clinical phenotype consisting of callosal agenesis, bilateral cataracts, cardiomyopathy, immunodeficiency, and skin and hair hypopigmentation.29 Further neurological manifestations include microcephaly, nonlissencephalic cortical dysplasia, cerebellar vermis hypoplasia, severe cognitive and motor impairment, seizures, muscular hypotonia, and failure to thrive.29,30,31 The disease is usually fatal in early childhood. Reports of available muscle biopsies record myopathic features with type 1 atrophy and glycogen accumulation.30,32 Electron microscopy studies further revealed altered mitochondrial morphology and distribution besides abundant vacuoles and dense bodies, possibly of lysosomal origin.30,31,32
Recently, mutations in the EPG5 gene, the human homolog of the metazoan-specific autophagy gene Epg5 (ectopic P-granules autophagy protein 5), which encodes the key autophagy regulatory protein EPG5, were identified as the genetic cause of Vici syndrome.31 Epg5 in Caenorhabditis elegans is one of the four eukaryote-specific autophagy genes that regulate different steps of the autophagic pathway in multicellular organisms.31,33 It is ubiquitously expressed during embryonic development and appears to play an essential role in starvation-induced autophagy.33 In silico analysis predicts the expression of Epg5 in different mammalian organs, including the central nervous system, skeletal and heart muscle, immune cells, thymus, and others,34 which are all involved in Vici syndrome. Knockdown of the EPG5 gene in mammalian cells results in the accumulation of nondegenerative autolysosomes, possibly indicating that EPG5 is involved in the rate-limiting late steps of the autophagic pathway, such as the proteolysis within autolysosomes.33
Characterization of autophagy in Vici syndrome revealed a wide spectrum of abnormalities, making it a paradigm for a human multisystem disorder associated with defective autophagy31 ( Figure 4 ). In skeletal muscle tissue and fibroblasts derived from Vici syndrome patients, accumulation of autophagosomes and the autophagy linker proteins NBR1 and p62 was observed, potentially reflecting a block in autophagic flux.31 This was further supported by reduced colocalization of autophagosomes and lysosomes and by increased levels of K63-polyubiquitinated proteins,31 which are usually targeted for degradation by autophagy.35 On the regulatory level, altered phosphorylation in the mTOR pathway was noted, potentially indicating deranged transcriptional regulation via this pathway.31 Taken together, these data point to defective autophagosome clearance due to mutated EPG5, which leads to the accumulation of autophagic cargo and potentially impaired fusion of autophagosomes and lysosomes or downstream defects thereof.

Vici syndrome—a paradigm for a multisystem disorder associated with impaired autophagy. Deficiency of EPG5, a key autophagy regulator, leads to impaired autophagy in multiple organ systems, creating a complex clinical picture. The role of autophagy in brain development is highlighted by impaired neuronal migration and brain development in Vici syndrome. Neurodegeneration might also occur. Cargo accumulation, such as glycogen storage in skeletal muscle, and impaired turnover of mitochondria might also contribute to disease progression.
The pivotal findings in Vici syndrome have great implications for our understanding of autophagy in multiple organ systems and their embryonic development ( Figure 4 ). With regards to the central nervous system, the clinical findings of callosal agenesis, cortical hypoplasia, and cerebellar malformation connect EPG5-dependent autophagy to central nervous system development. Regulators of neuronal autophagy, such as Ambra1 or Atg5, have been found to play a crucial role during early neuronal differentiation36,37 and postnatal adaptation38 by assuring that basal autophagy disposes cellular components and provides energy to enable the dynamic changes necessary in the developing brain.
It remains to be elucidated whether patients with Vici syndrome develop a neurodegenerative phenotype in addition to brain malformation. In this case, a neurodegenerative phenotype might include the accumulation of classic polyubiquitinated inclusion bodies as observed with genetic ablation of autophagosome formation.39,40 Supporting this idea, a recently published study in Epg5-deficient mice demonstrated selective neurodegeneration of cortical and spinal cord motor neurons leading to a phenotype that resembles key features of amyotrophic lateral sclerosis.41 Accumulation of p62 and ubiquitin-positive inclusions were found in neurons and glia cells, underscoring impairment of autophagosome maturation and autophagic flux.41
Further characterizing findings in Vici syndrome and Epg5-deficient mice might indeed enhance our understanding of the detrimental consequences of deficits in late stages of the autophagic pathway for neuronal maturation, integrity, and neurodegeneration. Likewise, exploring the mitochondrial abnormalities observed in Vici syndrome could further facilitate our insights into the role of autophagy in maintaining mitochondrial quality and function.
Lafora Disease: Linking Autophagy to Carbohydrate Metabolism
Lafora disease (LD) (OMIM no. 254780) is a fatal autosomal-recessive neurodegenerative disease that belongs to the group of progressive myoclonic epilepsies. The disease usually manifests in early adolescence with different types of seizures, myoclonus, and rapidly progressive deterioration of neurologic function, leading patients to a vegetative state and death within ~10 y after diagnosis. The histological hallmarks of LD are insoluble intracellular inclusion bodies, termed as Lafora bodies (LBs). LBs accumulate in cells of most tissues and mainly contain insoluble poorly branched and phosphorylated glycogen molecules, called polyglucosans, besides a minor fraction of partly ubiquitinated proteins.42,43,44
Mutations causing LD have been identified in two genes, EPM2A45 and EPM2B,46 which encode the proteins laforin and malin, respectively. Laforin is a dual-specificity phosphatase with a multitude of functions,47 and malin acts as an E3-ubiquitin ligase that regulates laforin levels by tagging it for degradation.48 A subtype of LD, early-onset LB disease, has been recently identified, and the mutated protein PRDM8 was shown to interact with both laforin and malin by sequestering the two proteins in the nucleus, therefore potentially leading to an effective shortage in the cytosol.49
Laforin is involved in the formation of LBs by at least two mechanisms. It contains a carbohydrate-binding domain by which it can bind glycogen for subsequent dephosphorylation, thus it may help to prevent the formation of insoluble polyglucosans.50,51,52 Second, it aids protein degradation by recruiting substrates to malin for subsequent ubiquitination and degradation.48 Importantly, substrates of the functional laforin–malin complex include a number of enzymes involved in glycogen synthesis, further linking its function to glycogen metabolism.47
The laforin–malin complex also appears to have a key role in the endoplasmic reticulum–unfolded protein response pathway as laforin depletion in cells leads to increased endoplasmic reticulum stress, which was found to correlate with impaired degradation of proteins through the ubiquitin–proteasome system and increased apoptosis.53 In addition, proteasome inhibition induced accumulation of laforin and malin leads to polyubiquitinated protein aggregates that contain chaperone proteins and proteasomal subunits and locate to perinuclear aggresomes.54 Similarly, LB in laforin-deficient mice recruit and potentially sequester proteasomal subunits, endoplasmic reticulum–associated chaperone proteins and the autophagy substrate and polyubiquitin-binding protein p62.55 Important to protein metabolism in neurodegenerative diseases, the laforin–malin complex can suppress cytotoxicity and accumulation of aggregate-prone proteins by interaction with the chaperone proteins, heat-shock protein 70 and C-terminus of Hsc70-interacting protein, to effectively shuttle target proteins to degradation via the proteasome.56,57,58
Linking LD to autophagy, a recent study provided evidence for a role of laforin in the regulation of autophagy, potentially through the mTOR pathway.59 In laforin-deficient fibroblasts obtained from patients and in mouse embryonic fibroblasts and liver tissue derived from laforin knockout mice, the levels of the autophagosome marker LC3-II were significantly reduced, indicating compromised autophagosome formation.59 Accumulating proteins in these models were found to include the autophagy substrate p62. Furthermore, protein aggregates could be reduced by the overexpression of functional laforin, thus providing evidence for a positive regulation of autophagy by laforin.59 Consistent with these findings, impaired autophagy was observed in brain tissue of malin knockout mice, and experiments in embryonic fibroblasts of these mice revealed a block in autophagosome formation similar to that found in laforin-deficient cells.60
In contrast, another recent study revealed no changes in LC3-II or other autophagy proteins in brain lysates of another laforin-deficient mouse model, arguing that autophagosome formation and function in the brain of these mice are not critically abnormal.55 Nevertheless, the recruitment of crucial proteins of the proteasomal, endosomal, and autophagy pathway to LBs, as observed in laforin-deficient mice55 and brain biopsy tissue of a LD patient,61 seems to indicate that secondary changes in protein degradation pathways are important contributors to neuronal pathology.
In summary, current data provide evidence for a crucial involvement of protein degradation pathways in LD. On the one hand, abnormal autophagosome formation as a consequence of a genetic defect in the laforin–malin complex appears to contribute to neuronal pathology and LB formation; on the other hand, defects in late stages of the autophagic pathway secondary to LB formation could be critical contributors to disease progression ( Figure 5 ). Both scenarios open exciting opportunities to explore modulating autophagy to prevent or ameliorate disease evolution and progression in LD.

Impaired protein degradation in Lafora disease. Protein degradation pathways including the ubiquitin–proteasome system (UPS) and autophagy are impaired what may promote Lafora body (LB) formation and disease progression. Secondary sequestration of crucial components of protein degradation pathways into LBs may enhance existing deficits. Pathological changes in protein degradation pathways are highlighted in red. CHIP, C-terminus of Hsc70-interacting protein; ER, endoplasmic reticulum; HSP70, heat-shock protein 70.
Alexander Disease: A link to Adaptive Autophagy in Astrocytes
Infantile-onset Alexander disease (AxD) (OMIM no. 203450) is a rare neurological disorder of childhood that is characterized by leukodystrophy, megalencephaly, delayed psychomotor development, seizures, and spasticity, with progression to death within commonly 3–4 y.62,63 AxD is an important example of a degenerative brain disorder in which the pathological cascade is primarily driven by astrocyte pathology. The disease is caused by autosomal-dominant gain-of-function mutations in the gene encoding the glial fibrillary acidic protein (GFAP), an intermediate filament of the astrocyte cytoskeleton.64 Histologically, AxD is characterized by the accumulation of Rosenthal fibers (RFs) in astrocytes throughout the white matter, most prominently in the subpial, perivascular, and subependymal regions.65,66 Major components of RFs are GFAP and small heat shock proteins such as α-B-crystallin and Hsp27 along other ubiquitinated proteins.66 Using different mouse models, the accumulation of RFs was shown to be caused by mutant GFAP protein and also by overexpression of wild-type human GFAP, suggesting that elevation of total levels of GFAP is a critical element.67,68 All GFAP mutations discovered in AxD patients allow production of functional full-length mutant protein,69 but mutant GFAP tends to form abnormally large, soluble oligomers,70 and hence, progressive accumulation of mutant GFAP impacts protein metabolism in several ways.
Astrocytes and RFs in AxD contain abundant amounts of chaperone proteins suggesting cell stress potentially through the accumulation of misfolded GFAP. α-B-crystallin is a chaperone protein normally found in a pool of soluble cytosolic proteins; however, in cells expressing mutant GFAP, α-B-crystallin shifts its localization to a cytoskeletal fraction, likely because of increased binding to abundant GFAP.66,71 By genetically manipulating the levels of α-B-crystallin in a mouse model of AxD, deficiency of α-B-crystallin exacerbates the phenotype, causing a dose-dependent increase in mortality.72 Vice versa, by increasing α-B-crystallin levels beyond naturally occurring levels, AxD mice could be rescued from the otherwise lethal effects of GFAP mutation and excess.72
Interestingly, proteasome activity was found to be significantly impaired by mutant GFAP, leading to a disruption of normal protein turnover, a finding that could be partly reversed by expressing α-B-crystallin.70,73 Importantly, proteasome inhibition in AxD has been linked to the induction of autophagy via mTOR.74 This is in agreement with the general notion that autophagic degradation is enhanced when the proteasome is dysfunctional or overwhelmed.16,75,76,77 Autophagy can degrade GFAP, and increased autophagosome formation indicative of enhanced autophagic flux was found in astrocytes of AxD patients and mutant GFAP mice.74 Surprisingly though, the increase in autophagosome generation was not accompanied by an overall increase in the degradation of long-lived proteins, suggesting a rather specific modulation of GFAP levels as opposed to simply enhancing bulk protein degradation.74 Blocking autophagy at different stages of the autophagic cascade using specific inhibitors resulted in further GFAP accumulation, whereas upregulating autophagic flux by starvation or rapamycin treatment suppresses GFAP accumulation. Similarly in Atg5-deficient mouse fibroblasts, the lack of autophagy was found to result in enhanced GFAP aggregation and inclusion formation.74 Collectively, these findings suggest that rapid mobilization of autophagy in AxD may reflect an attempt, although unsuccessful, to remove accumulating GFAP and relief protein burden.66,74
Based on current evidence, AxD can be viewed as a classic “proteinopathy” in which accumulating GFAP overwhelms the capacity of chaperone-mediated refolding and proteasomal degradation with accumulating protein burden leading to further impairment of these pathways, therefore eventually creating a vicious cycle. Crosstalk between protein degradation pathways drives the induction of autophagy, which however is not capable of rescuing protein homeostasis, leading to cellular dysfunction and spreading degeneration ( Figure 6 ). This hypothetical cascade has striking similarities to other proteinopathies such as the synucleinopathies (Parkinson’s disease and others),1 what may allow researchers to share common concepts and findings between these seemingly unrelated diseases.

Pathological cascade of events in Alexander disease with a focus on altered protein metabolism. Accumulation of mutant glial fibrillary acidic protein (GFAP) oligomers may overwhelm the capacity of the chaperone network and proteasomal degradation with accumulating protein burden leading to further impairment of these pathways, therefore eventually creating a vicious cycle. Autophagy is induced as a compensatory action but is not sufficient to preserve protein homeostasis, which may promote cellular dysfunction and degeneration. Pathological changes in protein degradation pathways are highlighted in red, whereas compensatory changes are marked in green.
Autophagy as a Novel Therapeutic Target and Concluding Remarks
Protein degradation pathways have a key role in the development and progression of neurodegenerative diseases. In recent years, autophagy has emerged as a key pathway in many pediatric neurodegenerative diseases including the examples discussed in this review. Despite the obvious differences among the individual diseases, a common theme can be recognized: primary or secondary impairment of autophagy may lead to the accumulation of substrates such as proteins, lipids, or even organelles like mitochondria. On the other hand, the compensatory upregulation of autophagic flux as observed in many diseases may become uncontrolled leading to detrimental consequences besides initial cytoprotective effects.
Increased understanding of the role of autophagy in many diseases, including common neurodegenerative diseases such as Alzheimer’s or Parkinson’s disease, has led to an interest in pharmacological- or gene therapy–based approaches to modulate autophagy1,2,3,78,79 ( Figure 7 ). Because of similar changes in the autophagic pathway in pediatric brain diseases, related approaches could possibly be applied, for example, inhibiting autophagy in NP-C to ameliorate the induction of defective autophagy or enhancing autophagic flux in LD where a block in autophagosome has been described. However, a challenge for modulating autophagy remains to be the target selectivity as most if not all-available small-molecule modulators of autophagy are rather nonspecific and have collateral targets outside the autophagy pathway. In addition, reliable biomarkers for measuring autophagy are clearly needed in order to determine drug effectiveness and kinetics in patients.

Signaling pathways involved in the regulation of autophagy and their pharmacological modulators. The main signaling pathways are as follows: (i) the mTORC1 pathway, (ii) mTORC1-independent pathways that converge into signaling via IP3 and intracellular calcium release, and (iii) a pathway that involves the basic helix-loop-helix leucine zipper transcription factor EB (TFEB)–mediated expression of genes involved in different stages of autophagy. Examples of the main pharmacological modulators of autophagy are shown with new compounds being constantly discovered. Inducers of autophagy include the following: rapamycin and rapalogs (via mTORC1 inhibition); 10-NC (via AKT1 inhibition); metformin, nilotinib, and resveratrol (via AMPK activation); lithium, valproate, and carbamazepine (via inhibition of intracellular inositol synthesis and recycling); calpain inhibitors (via inhibition of calpains); and trehalose (unknown mechanism). Inhibitors of autophagy include the following: 3-MA and wortmannin (via class III phosphatidylinositol 3-kinase inhibition); bafilomycin A1 and concanamycin A (via inhibition of the lysosomal V-ATPase); vinblastine (via depolymerization of acetylated microtubules); CQ and HCQ (via elevation of the vacuolar/lysosomal pH); leupeptin, E64-D, and pepstatin A (via inhibition of lysosomal proteases). 3-MA, 3-methyladenine; 10-NC, 10-[4′-(N-diethylamino)butyl]-2-chlorophenoxazine; AMPK, 5′ AMP-activated protein kinase; CQ, chloroquine; HCQ, hydroxychloroquine; IP3, inositol 1,4,5-trisphosphate; mTORC1, mammalian target of rapamycin complex 1; PI3-kinases, phosphatidylinositide 3-kinases; PKB, protein kinase B; RHEB, Ras homolog enriched in brain; TFEB, transcription factor EB; TSC1, tuberous sclerosis protein 1; TSC2, tuberous sclerosis protein 2; ULK1 complex, UNC51-like-kinase 1 complex.
Deciphering mechanisms by which autophagy contributes to and can prevent neurodegeneration may lead to the identification of novel targets for future diagnostic tests and treatment. Although the diseases discussed in this review are rare, the extent to which they allow us to understand the contribution of autophagy in health and disease is significant. Translating these mechanistic insights into therapeutic strategies amenable to clinical testing may eventually provide novel effective therapies for treating pediatric neurodegenerative diseases and common adult neurologic disorders.
Statement of Financial Support
D.E.-F. and L.W. received support from the German National Academic Foundation (Studienstiftung des deutschen Volkes e.V.) and the Young Investigator Award Program at Ruprecht-Karls-University Heidelberg Faculty of Medicine.
Disclosure:
The authors declare no conflict of interest.
Change history
26 August 2014
Darius Ebrahimi-Fakhari, Lara Wahlster, Georg F. Hoffmann and Stefan Kölker Pediatr Res 75:217–226 (2014); doi:10.1038/pr.2013.185 In the published Figure 7, the labels for two signaling molecules—RHEB and TSC1-TSC2—were transposed. The corrected figure is shown below.
References
Ebrahimi-Fakhari D, Wahlster L, McLean PJ . Protein degradation pathways in Parkinson’s disease: curse or blessing. Acta Neuropathol 2012;124:153–72.
Ihara Y, Morishima-Kawashima M, Nixon R . The ubiquitin-proteasome system and the autophagic-lysosomal system in Alzheimer disease. Cold Spring Harb Perspect Med 2012;2.
Ebrahimi-Fakhari D, Wahlster L, McLean PJ . Molecular chaperones in Parkinson’s disease–present and future. J Parkinsons Dis 2011;1:299–320.
Lieberman AP, Puertollano R, Raben N, Slaugenhaupt S, Walkley SU, Ballabio A . Autophagy in lysosomal storage disorders. Autophagy 2012;8:719–30.
Mizushima N, Komatsu M . Autophagy: renovation of cells and tissues. Cell 2011;147:728–41.
Behrends C, Sowa ME, Gygi SP, Harper JW . Network organization of the human autophagy system. Nature 2010;466:68–76.
Mizushima N, Yoshimori T, Ohsumi Y . The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol 2011;27:107–32.
Settembre C, Di Malta C, Polito VA, et al. TFEB links autophagy to lysosomal biogenesis. Science 2011;332:1429–33.
Sardiello M, Palmieri M, di Ronza A, et al. A gene network regulating lysosomal biogenesis and function. Science 2009;325:473–7.
Funderburk SF, Wang QJ, Yue Z . The Beclin 1-VPS34 complex–at the crossroads of autophagy and beyond. Trends Cell Biol 2010;20:355–62.
Klionsky DJ, Abdalla FC, Abeliovich H, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012;8:445–544.
Jahreiss L, Menzies FM, Rubinsztein DC . The itinerary of autophagosomes: from peripheral formation to kiss-and-run fusion with lysosomes. Traffic 2008;9:574–87.
Ashrafi G, Schwarz TL . The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ 2013;20:31–42.
Ebrahimi-Fakhari D, McLean PJ, Unni VK . Alpha-synuclein’s degradation in vivo: opening a new (cranial) window on the roles of degradation pathways in Parkinson disease. Autophagy 2012;8:281–3.
Cook C, Stetler C, Petrucelli L . Disruption of protein quality control in Parkinson’s disease. Cold Spring Harb Perspect Med 2012;2:a009423.
Ebrahimi-Fakhari D, Cantuti-Castelvetri I, Fan Z, et al. Distinct roles in vivo for the ubiquitin-proteasome system and the autophagy-lysosomal pathway in the degradation of a-synuclein. J Neurosci 2011;31:14508–20.
Carstea ED, Morris JA, Coleman KG, et al. Niemann-Pick C1 disease gene: homology to mediators of cholesterol homeostasis. Science 1997;277:228–31.
Naureckiene S, Sleat DE, Lackland H, et al. Identification of HE1 as the second gene of Niemann-Pick C disease. Science 2000;290:2298–301.
Schulze H, Sandhoff K . Lysosomal lipid storage diseases. Cold Spring Harb Perspect Biol 2011;3.
Vance JE, Peake KB . Function of the Niemann-Pick type C proteins and their bypass by cyclodextrin. Curr Opin Lipidol 2011;22:204–9.
Ko DC, Milenkovic L, Beier SM, Manuel H, Buchanan J, Scott MP . Cell-autonomous death of cerebellar purkinje neurons with autophagy in Niemann-Pick type C disease. PLoS Genet 2005;1:81–95.
Liao G, Yao Y, Liu J, et al. Cholesterol accumulation is associated with lysosomal dysfunction and autophagic stress in Npc1 -/- mouse brain. Am J Pathol 2007;171:962–75.
Pacheco CD, Kunkel R, Lieberman AP . Autophagy in Niemann-Pick C disease is dependent upon Beclin-1 and responsive to lipid trafficking defects. Hum Mol Genet 2007;16:1495–503.
Ishibashi S, Yamazaki T, Okamoto K . Association of autophagy with cholesterol-accumulated compartments in Niemann-Pick disease type C cells. J Clin Neurosci 2009;16:954–9.
Pacheco CD, Elrick MJ, Lieberman AP . Tau deletion exacerbates the phenotype of Niemann-Pick type C mice and implicates autophagy in pathogenesis. Hum Mol Genet 2009;18:956–65.
Ordonez MP, Roberts EA, Kidwell CU, Yuan SH, Plaisted WC, Goldstein LS . Disruption and therapeutic rescue of autophagy in a human neuronal model of Niemann Pick type C1. Hum Mol Genet 2012;21:2651–62.
Elrick MJ, Yu T, Chung C, Lieberman AP . Impaired proteolysis underlies autophagic dysfunction in Niemann-Pick type C disease. Hum Mol Genet 2012;21:4876–87.
Amritraj A, Wang Y, Revett TJ, Vergote D, Westaway D, Kar S . Role of cathepsin D in U18666A-induced neuronal cell death: potential implication in Niemann-Pick type C disease pathogenesis. J Biol Chem 2013;288:3136–52.
Dionisi Vici C, Sabetta G, Gambarara M, et al. Agenesis of the corpus callosum, combined immunodeficiency, bilateral cataract, and hypopigmentation in two brothers. Am J Med Genet 1988;29:1–8.
McClelland V, Cullup T, Bodi I, et al. Vici syndrome associated with sensorineural hearing loss and evidence of neuromuscular involvement on muscle biopsy. Am J Med Genet A 2010;152A:741–7.
Cullup T, Kho AL, Dionisi-Vici C, et al. Recessive mutations in EPG5 cause Vici syndrome, a multisystem disorder with defective autophagy. Nat Genet 2013;45:83–7.
Al-Owain M, Al-Hashem A, Al-Muhaizea M, et al. Vici syndrome associated with unilateral lung hypoplasia and myopathy. Am J Med Genet A 2010;152A:1849–53.
Tian Y, Li Z, Hu W, et al. C. elegans screen identifies autophagy genes specific to multicellular organisms. Cell 2010;141:1042–55.
Halama N, Grauling-Halama SA, Beder A, Jäger D . Comparative integromics on the breast cancer-associated gene KIAA1632: clues to a cancer antigen domain. Int J Oncol 2007;31:205–10.
Tan JM, Wong ES, Kirkpatrick DS, et al. Lysine 63-linked ubiquitination promotes the formation and autophagic clearance of protein inclusions associated with neurodegenerative diseases. Hum Mol Genet 2008;17:431–9.
Vázquez P, Arroba AI, Cecconi F, de la Rosa EJ, Boya P, de Pablo F . Atg5 and Ambra1 differentially modulate neurogenesis in neural stem cells. Autophagy 2012;8:187–99.
Fimia GM, Stoykova A, Romagnoli A, et al. Ambra1 regulates autophagy and development of the nervous system. Nature 2007;447:1121–5.
Kuma A, Hatano M, Matsui M, et al. The role of autophagy during the early neonatal starvation period. Nature 2004;432:1032–6.
Hara T, Nakamura K, Matsui M, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006;441:885–9.
Komatsu M, Waguri S, Chiba T, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006;441:880–4.
Zhao H, Zhao YG, Wang X, et al. Mice deficient in Epg5 exhibit selective neuronal vulnerability to degeneration. J Cell Biol 2013;200:731–41.
Yokoi S, Nakayama H, Negishi T . Biochemical studies on tissues from a patient with Lafora disease. Clin Chim Acta 1975;62:415–23.
Manetto V, Abdul-Karim FW, Perry G, Tabaton M, Autilio-Gambetti L, Gambetti P . Selective presence of ubiquitin in intracellular inclusions. Am J Pathol 1989;134:505–13.
Ganesh S, Delgado-Escueta AV, Sakamoto T, et al. Targeted disruption of the Epm2a gene causes formation of Lafora inclusion bodies, neurodegeneration, ataxia, myoclonus epilepsy and impaired behavioral response in mice. Hum Mol Genet 2002;11:1251–62.
Minassian BA, Lee JR, Herbrick JA, et al. Mutations in a gene encoding a novel protein tyrosine phosphatase cause progressive myoclonus epilepsy. Nat Genet 1998;20:171–4.
Chan EM, Young EJ, Ianzano L, et al. Mutations in NHLRC1 cause progressive myoclonus epilepsy. Nat Genet 2003;35:125–7.
Gentry MS, Romá-Mateo C, Sanz P . Laforin, a protein with many faces: glucan phosphatase, adapter protein, et alii. FEBS J 2013;280:525–37.
Gentry MS, Worby CA, Dixon JE . Insights into Lafora disease: malin is an E3 ubiquitin ligase that ubiquitinates and promotes the degradation of laforin. Proc Natl Acad Sci USA 2005;102:8501–6.
Turnbull J, Girard JM, Lohi H, et al. Early-onset Lafora body disease. Brain 2012;135(Pt 9):2684–98.
Tagliabracci VS, Turnbull J, Wang W, et al. Laforin is a glycogen phosphatase, deficiency of which leads to elevated phosphorylation of glycogen in vivo. Proc Natl Acad Sci USA 2007;104:19262–6.
Tagliabracci VS, Heiss C, Karthik C, et al. Phosphate incorporation during glycogen synthesis and Lafora disease. Cell Metab 2011;13:274–82.
Worby CA, Gentry MS, Dixon JE . Laforin, a dual specificity phosphatase that dephosphorylates complex carbohydrates. J Biol Chem 2006;281:30412–8.
Vernia S, Rubio T, Heredia M, Rodríguez de Córdoba S, Sanz P . Increased endoplasmic reticulum stress and decreased proteasomal function in lafora disease models lacking the phosphatase laforin. PLoS ONE 2009;4:e5907.
Mittal S, Dubey D, Yamakawa K, Ganesh S . Lafora disease proteins malin and laforin are recruited to aggresomes in response to proteasomal impairment. Hum Mol Genet 2007;16:753–62.
Puri R, Suzuki T, Yamakawa K, Ganesh S . Dysfunctions in endosomal-lysosomal and autophagy pathways underlie neuropathology in a mouse model for Lafora disease. Hum Mol Genet 2012;21:175–84.
Garyali P, Siwach P, Singh PK, et al. The malin-laforin complex suppresses the cellular toxicity of misfolded proteins by promoting their degradation through the ubiquitin-proteasome system. Hum Mol Genet 2009;18:688–700.
Rao SN, Sharma J, Maity R, Jana NR . Co-chaperone CHIP stabilizes aggregate-prone malin, a ubiquitin ligase mutated in Lafora disease. J Biol Chem 2010;285:1404–13.
Sengupta S, Badhwar I, Upadhyay M, Singh S, Ganesh S . Malin and laforin are essential components of a protein complex that protects cells from thermal stress. J Cell Sci 2011;124(Pt 13):2277–86.
Aguado C, Sarkar S, Korolchuk VI, et al. Laforin, the most common protein mutated in Lafora disease, regulates autophagy. Hum Mol Genet 2010;19:2867–76.
Criado O, Aguado C, Gayarre J, et al. Lafora bodies and neurological defects in malin-deficient mice correlate with impaired autophagy. Hum Mol Genet 2012;21:1521–33.
Rao SN, Maity R, Sharma J, et al. Sequestration of chaperones and proteasome into Lafora bodies and proteasomal dysfunction induced by Lafora disease-associated mutations of malin. Hum Mol Genet 2010;19:4726–34.
Alexander WS . Progressive fibrinoid degeneration of fibrillary astrocytes associated with mental retardation in a hydrocephalic infant. Brain 1949;72:373–81, 3 pl.
Prust M, Wang J, Morizono H, et al. GFAP mutations, age at onset, and clinical subtypes in Alexander disease. Neurology 2011;77:1287–94.
Brenner M, Johnson AB, Boespflug-Tanguy O, Rodriguez D, Goldman JE, Messing A . Mutations in GFAP, encoding glial fibrillary acidic protein, are associated with Alexander disease. Nat Genet 2001;27:117–20.
Rosenthal W . [On a peculiar swelling of the spinal cord complicated by syringomyelia.]. Beitr Pathol Anat 1898;23:111–143.
Messing A, Brenner M, Feany MB, Nedergaard M, Goldman JE . Alexander disease. J Neurosci 2012;32:5017–23.
Messing A, Head MW, Galles K, Galbreath EJ, Goldman JE, Brenner M . Fatal encephalopathy with astrocyte inclusions in GFAP transgenic mice. Am J Pathol 1998;152:391–8.
Hagemann TL, Connor JX, Messing A . Alexander disease-associated glial fibrillary acidic protein mutations in mice induce Rosenthal fiber formation and a white matter stress response. J Neurosci 2006;26:11162–73.
Liem RK, Messing A . Dysfunctions of neuronal and glial intermediate filaments in disease. J Clin Invest 2009;119:1814–24.
Tang G, Perng MD, Wilk S, Quinlan R, Goldman JE . Oligomers of mutant glial fibrillary acidic protein (GFAP) Inhibit the proteasome system in alexander disease astrocytes, and the small heat shock protein alphaB-crystallin reverses the inhibition. J Biol Chem 2010;285:10527–37.
Perng MD, Wen SF, Gibbon T, et al. Glial fibrillary acidic protein filaments can tolerate the incorporation of assembly-compromised GFAP-delta, but with consequences for filament organization and alphaB-crystallin association. Mol Biol Cell 2008;19:4521–33.
Hagemann TL, Boelens WC, Wawrousek EF, Messing A . Suppression of GFAP toxicity by alphaB-crystallin in mouse models of Alexander disease. Hum Mol Genet 2009;18:1190–9.
Tang G, Xu Z, Goldman JE . Synergistic effects of the SAPK/JNK and the proteasome pathway on glial fibrillary acidic protein (GFAP) accumulation in Alexander disease. J Biol Chem 2006;281:38634–43.
Tang G, Yue Z, Talloczy Z, et al. Autophagy induced by Alexander disease-mutant GFAP accumulation is regulated by p38/MAPK and mTOR signaling pathways. Hum Mol Genet 2008;17:1540–55.
Pandey UB, Nie Z, Batlevi Y, et al. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature 2007;447:859–63.
Iwata A, Riley BE, Johnston JA, Kopito RR . HDAC6 and microtubules are required for autophagic degradation of aggregated huntingtin. J Biol Chem 2005;280:40282–92.
Ding WX, Ni HM, Gao W, et al. Linking of autophagy to ubiquitin-proteasome system is important for the regulation of endoplasmic reticulum stress and cell viability. Am J Pathol 2007;171:513–24.
Rubinsztein DC, Codogno P, Levine B . Autophagy modulation as a potential therapeutic target for diverse diseases. Nat Rev Drug Discov 2012;11:709–30.
Harris H, Rubinsztein DC . Control of autophagy as a therapy for neurodegenerative disease. Nat Rev Neurol 2012;8:108–17.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ebrahimi-Fakhari, D., Wahlster, L., Hoffmann, G. et al. Emerging role of autophagy in pediatric neurodegenerative and neurometabolic diseases. Pediatr Res 75, 217–226 (2014). https://doi.org/10.1038/pr.2013.185
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/pr.2013.185
This article is cited by
-
Aggregation-prone GFAP mutation in Alexander disease validated using a zebrafish model
BMC Neurology (2017)
-
Activation of the AMPK-ULK1 pathway plays an important role in autophagy during prion infection
Scientific Reports (2015)
-
The Parkinsonian mimetic, 6-OHDA, impairs axonal transport in dopaminergic axons
Molecular Neurodegeneration (2014)
