
Abstract
Determining the right dose for drugs used to treat neonates is critically important. Neonates have significant differences in physiology affecting drug absorption, distribution, metabolism, and elimination that make extrapolating dosages from adults and older children inappropriate. In spite of recent legislative efforts requiring drug studies in this population, most drugs given to neonates remain insufficiently studied. Many ethical and logistical concerns make designing studies in this age group difficult. Fortunately, specialized analytical techniques, such as the use of dried blood spots, scavenged sampling, population pharmacokinetics analyses, and sparse sampling, have helped investigators better define doses that maximize efficacy and safety. Through the use of these methods, successful clinical trials have resulted in recent changes to drug dosing in this population.
Similar content being viewed by others


Main
A critical goal of drug development is getting the dose right. Under-dosing can result in a lack of efficacy, and overdosing can result in adverse effects. Most drugs given to neonates have not been sufficiently studied in this population and are often dosed based on information extrapolated from adults or older children (1). This approach to drug dosing is subject to error. The neonatal period is a time of incredible physiological change leading to unpredictable responses to doses of drugs deemed safe and efficacious in adults (2). Rapid developmental changes in neonatal organ systems influence pharmacologic safety and efficacy due to changes in the way drugs are absorbed, distributed, metabolized, and eliminated.
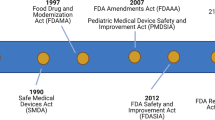
The need for determining the correct drug doses for children is becoming increasingly recognized. In the United States, several legislative efforts have addressed the lack of pediatric drug studies, including the Food and Drug Administration Modernization Act (1997), Best Pharmaceuticals for Children Act (2002), Pediatric Research Equity Act (2003), the Food and Drug Administration Amendments Act (2007), and the Food and Drug Administration Safety and Innovation Act (2012) (3,4). While these efforts have greatly improved labeling of drugs in older children, neonates remain understudied. Between 1997 and 2010, 406 labeling changes resulted from this legislation; however, only 24 (6%) labeling changes included neonates (5). Clinicians continue to lack access to data on neonatal drug safety, efficacy, and pharmacokinetics. Almost all patients in the neonatal intensive care unit are exposed to at least 1 off-label, unapproved, or extemporaneously prepared drug (1).
Contributing to this problem is the fact that clinical trials are difficult to conduct in neonates. Challenges in designing neonatal studies range from the ethical to the logistical (3). Several research and analytical techniques have been developed to address the current barriers to conducting neonatal drug studies. Through use of these techniques, a number of antimicrobials have been successfully studied, resulting in improvements in dosing in this population.
Unique Physiology in Neonates
Compared with older children and adults, neonates have significant differences in physiology affecting drug absorption, distribution, metabolism, and elimination. Disease, critical illness, specialized therapies, and developmental changes in the expression of organ-specific drug transporters may further contribute to these differences (6,7,8). Differences in neonatal physiology can also affect pharmacodynamics, resulting in differences in the expected potency, efficacy, or toxicity of drugs (9).
Drug Absorption
Drug absorption in neonates is largely affected by the maturation process of organ systems. Characteristics of the neonatal gastrointestinal tract that affect absorption of orally administered drugs include increased gastric pH, decreased intestinal motility, delayed gastric emptying time, and a reduction in bile acid synthesis (2,10,11,12).
Characteristics of neonatal skin that lead to increased absorption of drugs administered transdermally include a thinner stratum corneum, increased skin perfusion secondary to immature vasomotor control, increased water content, and higher body surface area-to-weight ratio (2,12,13). These differences are most pronounced at the extreme of prematurity. In premature neonates, pharmacologic predictions based on the condition of the stratum corneum at birth may be inaccurate by 1 wk of life due to rapid postnatal maturation (14).
Characteristics that affect intramuscular absorption in neonates include decreased muscle mass, reduced overall muscular perfusion, and decreased contractility (2,10,12,15). Additionally, intramuscular drug absorption in neonates can vary depending on the physiochemical properties of the drug, such as pH, molecular weight, solubility, ester salt formulation, or dissolution rates (2,12). Reduction in muscle perfusion due to hypotension, sepsis, or decreased cardiac output can lead to reduced absorption and unpredictable pharmacokinetics of drugs administered intramuscularly (11). Decreased muscle contractility in neonates can result in slower rates of intramuscular drug absorption and lower peak serum concentrations (16). Water soluble drugs tend to have greater intramuscular absorption in neonates than children or adults due to higher muscular water content and increased density of skeletal muscle capillaries in neonates (2,10,16).
Rectal absorption of drugs is generally increased in the neonate compared with children and adults (10,15). However, variability in the depth of insertion or retention of drug in the rectal vault can lead to variability in absorption (13). Drugs absorbed deep inside the rectum undergo first-pass metabolism by accessing the liver through the superior rectal veins whereas drugs inserted more shallowly will enter the systemic circulation directly through the inferior and middle rectal veins (17).
Drug Distribution
Compared with children and adults, neonates have higher volumes of extracellular fluid and total body water, lower proportions of adipose tissue, and decreased muscle mass (2,18,19). Premature neonates have lower fat and higher water content than term neonates (11,19). Initial resorption of fetal lung fluid can result in expansion of extracellular volume during the first few days of life with a robust diuresis and concomitant natriuresis occurring afterwards (20). The presence of a patent ductus arteriosus, renal injury, or use of extracorporeal membrane oxygenation can result in increased volumes of distribution leading to lower peak serum drug concentrations (8,21).
Neonates have a decreased drug protein-binding affinity relative to children and adults. Only unbound drug travels across membranes, exerts biological effect, and is eliminated from the body. Theophylline exhibits decreased protein binding in premature neonates, so equivalent total plasma concentrations will achieve higher unbound concentrations in neonates compared to adults (22). Consequently, efficacy and toxicity of theophylline can be achieved with lower total plasma concentrations in premature neonates.
Neonates have decreased plasma concentrations of albumin and α1-acid glycoprotein, resulting in increased plasma concentrations of unbound drug (2,10,11,19). At the time of birth, neonates have lower concentrations of α1-acid glycoprotein and albumin, which gradually increase to adult levels by 1 y of age (10,23). Elevated plasma levels of bilirubin can increase the concentration of unbound drug by displacing highly bound drugs from protein-binding sites (2).
Drug penetration into the neonatal central nervous system can also be different. Higher concentrations of drug in the brain are more likely in neonates than in children and adults due to decreased protein binding, a higher relative brain weight, and higher ratio of cerebral to systemic blood flow (24).
Blood is sequestered from the brain interstitial fluid and cerebrospinal fluid by the blood–brain and blood–cerebrospinal fluid barriers, respectively (25). The blood–brain barrier is formed by the cerebral microvasculature endothelium, and the blood–cerebrospinal fluid barrier comprises the choroid plexus endothelium. These barriers are commonly believed to be immature and more permeable to drugs in neonates (2,10,18). However, intercellular tight junctions are fully functional at the age of viability and restrict passage of most compounds except for specific inorganic ions, solutes, and water (25,26).
The ontogeny of drug transporters at these interfaces can affect the distribution of drugs into the neonatal central nervous system (27). In the blood–brain barrier of rats and nonhuman primates, the efflux transporter P-glycoprotein demonstrates increasing expression and activity with age, suggesting that neonates may have higher brain drug concentrations due to reduced outward drug transport (28).
Drug Metabolism and Elimination
Renal clearance of drugs increases with increasing gestational age, postnatal age, and body weight (7,18). Mechanisms of renal excretion affected by these factors are glomerular filtration (GFR), active tubular secretion, and tubular reabsorption.
GFR normalized to body surface area is lower in neonates compared with children and adults, with lowest values seen in the most premature neonates (10). Term neonates experience a rapid increase in GFR during the first 2 wk of life, followed by a steady rise to adult values by 6–12 mo of age (2). Premature infants demonstrate similar trends, with an initial rise in GFR that is less steep due to nephrogenesis not being complete until 34 wk gestation (7,10,29). Reduced renal blood flow or renal damage from nephrotoxic drugs such as indomethacin or diseases such as patent ductus arteriosus and perinatal asphyxia can result in lower GFR (7,8).
Active tubular secretion and tubular reabsorption are also immature at birth and are ~20–30% of adult values (10). Maturation of active tubular occurs gradually, reaching adult values by 7–12 mo of life (2,23). Maturation of tubular reabsorption continues slowly into adolescence, with the steepest rise occurring between 1 and 3 y of age (10). Elimination by these processes is dependent on renal blood flow, which increases over time with GFR (10). Reduced protein binding in neonates will increase the clearance of drugs by these renal processes due to higher concentrations of unbound drug available.
The capacity for drug metabolism by the neonatal liver is affected by the ontogeny of many drug-metabolizing enzymes. Rates of hepatic drug metabolism generally correspond with the expression of these enzymes, which is typically low at birth and gradually increases over time (2,13,15,24,30,31,32,33). Neonates are often exposed to drugs affected by enzymes with these changes in expression ( Table 1 ). Despite lower enzyme expression, reduced protein binding in neonates can sometimes lead to unexpectedly higher metabolic clearance of drugs such as micafungin (34). Rates of change in the expression of an enzyme can vary significantly among individuals and do not always correlate with changes in other enzymes (31).
Diet and special therapies can also alter the metabolism of drugs given to the neonate. For example, formula-fed neonates demonstrate quicker maturation and higher expression of CYP1A2 activity compared with breast-fed neonates (35). Neonates receiving therapeutic hypothermia for hypoxic–ischemic encephalopathy had decreased clearance and higher concentrations of morphine than normothermic neonates with hypoxic–ischemic encephalopathy, suggesting that lower body temperatures could impair enzyme activity (38).
Drug Transporters
Drug transporters are responsible for the cellular uptake and efflux of drugs within organ systems. Age-related differences in the expression of transporters have been demonstrated through in vitro and animal studies in the hepatic, intestinal, renal, and central nervous systems (6,28,39). However, data characterizing the impact of transporter ontogeny on human drug disposition are limited (7).
Developmental Pharmacodynamics
When at comparable drug exposures, neonates can respond differently than older populations due to immaturity of drug targets and receptors (9). Increased drug sensitivity and higher risk for toxicity may result. Because calcium stores in the neonatal heart are reduced compared with adults, neonatal cardiac contractility is more sensitive to administration of calcium (40). Calcium channel blocking agents are more likely to result in life-threatening bradycardia and hypotension in the neonate (40). Neonates may also be more sensitive to morphine than adults due to increased expression of the mu opioid receptor (9).
Immaturity of receptors can also result in decreased drug efficacy. Maturational changes in intestinal motilin receptors explain why erythromycin has minimal effect on intestinal motility in neonates <32 wk gestation (41). Organ immaturity can also confer protection against toxicity. Observations in neonatal dogs and rats show decreased renal accumulation of gentamicin and reduced risk for nephrotoxicity than their adult counterparts (42). Tubular secretion of gentamicin is partly mediated by the organic cation transporter in the renal brush border, which does not fully mature in mice until 4 wk postnatal age (43,44).
Challenges with Neonatal Drug Study Design
Clinical trials in neonates, especially premature neonates, are difficult. Lack of expertise in neonatal pharmacology, difficulty in obtaining informed consent, concerns about exposing this vulnerable population to the risks associated with clinical trials, low blood volumes, difficulty accurately measuring drug concentrations in small sample volumes, and lack of validated clinical end points are just a few examples of obstacles responsible for the lack of clinical trials in this population (3,45).
One source of great difficulty in conducting neonatal pharmacokinetic studies involves limitations on blood sampling. The World Health Organization recommends that a maximum limit of 3 ml/kg within 24 h be allowed for blood sampling in children involved in clinical research, with even lower limits advisable for critically ill subjects (46). For a 1,000 g neonate with a total blood volume of 90 ml, this equates to a maximum of 3 ml of blood allowed.
Other limitations include constraints around sampling timing and frequency. Acquisition of sample from central venous catheters used for drug administration is likely to result in inaccurate concentration measurements due to adherence of drug to the catheter, and repeated venipuncture and heel lancing are invasive and painful (47). Umbilical and peripheral arterial catheters can serve as an outstanding source of blood sampling in neonates who have them. However, prolonged use of these catheters places neonates at risk for complications such as infection, thromboembolism, and ischemic injury to distal appendages (48). Several strategies reducing the number of samples and the volume of blood needed per sample are currently being used to aid in the successful completion of pharmacokinetic studies in neonates.
Population Pharmacokinetics and Sparse Sampling
In traditional pharmacokinetic data analysis, individual pharmacokinetic parameters are first estimated using concentration-time data obtained from each subject. These individual estimates are then used to calculate an average parameter estimate for the entire population. Because this method depends on parameter estimates calculated for every subject, missing or limited data for each subject can lead to inaccurate overall pharmacokinetic estimates.
With population pharmacokinetic data analysis techniques, concentration–time data collected from every subject are combined and used to calculate a pharmacokinetic parameter estimate for the entire population in a single step. In neonatal studies, selecting a physiologically and developmentally homogenous population is important to avoid confounding and to reduce variability. Because this method treats the entire population as a single entity, this allows for the use of sparse sampling techniques. With sparse sampling, two to three samples are collected per subject, often with different collection times for each subject. Because all data points are combined and analyzed as a single unit, population pharmacokinetic data analysis avoids inaccurate pharmacokinetic characterizations associated with the use of limited data (3,49).
By reducing the number of samples collected per patient, sparse sampling schemes improve the feasibility of neonatal pharmacokinetic trials. Population pharmacokinetic data analysis offers several other advantages over traditional pharmacokinetic methods. Subjects in population pharmacokinetics studies often represent patients in the drug’s target population, whereas subjects in traditional pharmacokinetics studies are typically healthy volunteers. Population pharmacokinetics allow investigators to compare differences in drug responses among different subgroups, particularly among patients for whom the drug is intended (49).
Scavenged Sampling Techniques
Scavenged sampling is a novel strategy that uses surplus blood collected for laboratory tests done as part of standard of care that would otherwise be discarded. This strategy minimizes risk to the neonate by avoiding venous punctures and removal of blood volume solely for study purposes; further benefits include higher rates of parental consent and an increased number of samples per subject available for analysis (3). Scavenged sampling has been successfully used in population pharmacokinetic studies of antimicrobials involving neonates (50,51,52). Potential problems with scavenged sampling include drug instability with improper sample storage, sample collection times that are not optimal for pharmacokinetic analyses, and inaccurate documentation of time of blood draw (3,50). With proper study planning, many of these disadvantages can be avoided.
Dried Blood Spot Sampling
Dried blood spot sampling is another recently developed technique that uses ultra-low volumes to evaluate drug levels. The obvious advantage is the reduced blood volumes needed. For each sample, 15–30 μl of whole blood is collected onto blotting paper. Dried blood spot sampling techniques offer other benefits, as they require less training of research personnel, no additional sample processing, storage at room temperature, and simple bioanalytical analysis methods (3,53). This technique has been successfully used in pharmacokinetic studies of metronidazole and caffeine in premature neonates (53,54,55).
Neonatal Drug Trials
A number of recent studies describing the pharmacokinetics ( Table 2 ) of antibiotics in neonates have incorporated several of the techniques described above and have highlighted the differences in dosing between neonates and older children and adults. These studies, while not an exhaustive list, highlight the importance of conducting neonatal drug trials through the following observations: (i) antimicrobials exhibit a wide range of differences in pharmacokinetics that cannot be predicted through extrapolation of similar studies in older populations; (ii) age-related changes in pharmacokinetics occur at different rates and extents for different drugs; and (iii) pharmacokinetics of drugs not only differ between neonates and older children and adults, but also among neonates of different ranges of maturity. Changes in dosing recommendations that resulted from some of these trials illustrate the possibilities that efficacious doses in neonates can be less, similar, or more than the adult recommended dose ( Table 3 ). Additionally, recent studies of two antifungal drugs described below have demonstrated the importance of drug trials in getting the dose right in neonates.
Micafungin
Micafungin is a semisynthetic echinocandin antifungal agent that inhibits the synthesis of 1,3-β-D-glucan, an essential component of fungal cell walls. It exhibits concentration-dependent fungicidal activity against most relevant species of Candida (70). Micafungin is currently labeled for use in adults and children ages 4 mo and older. It is highly protein-bound; extensively metabolized by CYP1A2, CYP2D6, CYP2C, and CYP3A4; and primarily cleared by biliary excretion (34,71). Micafungin is dosed without adjustment in patients with renal impairment, suggesting only a minor contribution from renal clearance (34,72).
An initial single-dose pharmacokinetic study of intravenous micafungin in 18 premature neonates weighing <1,000 g demonstrated total drug clearances that were 1.7-fold greater than those in children aged 2–8 y and 2.6-fold greater than those in children aged 9–17 y (71). Overall volumes of distribution were also greater in these premature neonates. This study was followed by a multidose, open-label, pharmacokinetic and safety trial of 12 premature neonates with suspected systemic infections given micafungin at 15 mg/kg per dose (73). This study confirmed the previous findings that neonates demonstrated higher clearances and volumes of distribution compared with older children and adults. Due to these differences in pharmacokinetic parameters, neonates needed a threefold higher dose compared with adults (15 vs. 5 mg/kg) to achieve similar drug exposures (73). A subsequent, open-label study in 13 preterm infants found that doses of 7 and 10 mg/kg/day were well tolerated and provided exposure levels adequate for coverage of the central nervous system (70). Simulations based on population pharmacokinetic data from 47 infants demonstrated that a dose of 10 mg/kg/day resulted in a target attainment rate of 83% for the area under the concentration–time curve associated with adequate central nervous system coverage (72). Currently, the dose recommended for neonates is 10 mg/kg/day compared with the adult dose of 150 mg (~2 mg/kg/day for a 70 kg adult) (74).
The finding of increased micafungin clearance was surprising considering that the drug-metabolizing enzymes involved exhibit decreased expression in the neonatal period. A neonate who failed to achieve target plasma concentrations of micafungin was noted to have lower levels of serum albumin at baseline and during treatment (70). Comparison among serum samples from six neonates and six adults demonstrated an increased fraction of unbound drug in the neonates (96.7% bound drug in neonates vs. 99.6% in adults) (34). There was no difference in the expression of hepatic transporter proteins between neonatal and adult liver tissue samples, suggesting that there was no difference in intrinsic hepatic clearance and that age-dependent serum protein-binding had a significant role in the faster clearance of micafungin in neonates (34).
Fluconazole
Fluconazole is a triazole antifungal that inhibits lanosterol 14-α-demethylase, an enzyme that is responsible for the formation of compounds essential for fungal cell membrane integrity (75). It exhibits time-dependent fungicidal activity and is used in the prophylaxis and treatment of systemic neonatal candidiasis (75,76). Fluconazole exhibits low plasma protein-binding, demonstrates excellent cerebrospinal fluid penetration, and is predominantly eliminated through the renal system in unchanged form (75,76). Pediatric studies involving subjects aged 3 mo and older showed that children and adolescents had higher fluconazole clearance, with a drug half-life of 22 h compared with 30 h in adults (75,76). To ensure 70% efficacy against fungal infections, a 24-h area under the curve (AUC)-to-minimum inhibitory concentration (MIC) ratio of >50 is needed (76). This equates to a minimum 24-h AUC of 400 mg*h/l for an MIC of <8 µg/ml.
Fifty-five neonates between 25 and 42 wk gestational age, <120 d postnatal age, and receiving fluconazole intravenously provided 357 samples used in a population pharmacokinetics analysis (51). The final pharmacokinetic model developed found that drug clearance increased with increasing weight, birth gestational age, and postnatal age, and decreased with increasing serum creatinine levels. Bayesian estimates of pharmacokinetic parameters showed that fluconazole clearance was much lower at the time of birth and nearly doubled over the first month of life. Neonates with serum creatinine levels >1.3 mg/dl had clearances 70% lower than neonates with preserved renal function. Monte Carlo simulations performed using the final model predicted half-lives of 30 and 50 h for neonates 23–29 and 30–40 wk birth gestational age, respectively. These simulations also showed that achievement of therapeutic steady-state concentrations would take 5–7 d, demonstrating the potential need for a loading dose in this population.
Using this model, the investigators performed Monte Carlo simulations to evaluate the exposure–dose responses of fluconazole in neonates (77). Doses of 12 mg/kg/day during the first 90 d of life were required to achieve a goal AUC >400 mg*h/l and AUC/MIC >50 in 90% of neonates <30 wk gestational age and 80% of neonates 30–40 wk gestational age. This dose achieved similar exposures provided by the recommended adult dose of 400 mg (~6 mg/kg/day for a 70 kg adult) (77). Furthermore, a loading dose of 25 mg/kg was necessary to achieve the target AUC by day 2 of treatment.
Following these simulations, an open-label, pharmacokinetic study was performed to evaluate the use of a loading dose in neonates and to confirm the results obtained from the prior modeling and simulation work (78). This study included 57 plasma samples from eight neonates who were 35–38 wk birth gestational age with a median postnatal age of 16 d. All neonates were given a loading dose of 25 mg/kg followed by maintenance doses of 12 mg/kg/day. Under this regimen, five out of eight neonates reached the target 24-h AUC of >400 mg*h/l within the first day of dosing. All neonates achieved the 24-h trough concentration goal of >8 µg/ml during the first 24 h of treatment. Results of this study agreed with the simulation data produced from the population pharmacokinetics model developed earlier. No drug-related adverse events occurred during this study.
Application of Novel Techniques for Future Studies
Dried blood spot and scavenged sampling techniques have been used to guide dosing for only a handful of antimicrobials (50,51,52,53,54,60). These novel techniques will improve feasibility of neonatal studies where the relationship between pharmacokinetics and pharmacodynamics is less clearly defined. When used to prevent bronchopulmonary dysplasia in infants, high-dose regimens of dexamethasone have been associated with increased mortality and long-term neurodevelopmental impairment (79). There remains insufficient data evaluating the use of lower doses of dexamethasone to prevent bronchopulmonary dysplasia (79). A dried blood spot assay has been validated for the quantification of dexamethasone and could be used to facilitate studies evaluating the pharmacokinetics, safety, and efficacy of a low-dose regimen (80).
Conclusion
Determining the right dose for drugs used to treat neonates still remains an immense challenge. Unique and rapidly changing physiological characteristics contribute to unpredictable dose-exposure responses in this population. For this reason, it is not always appropriate to make decisions on dosing through extrapolation from children and adult studies. Many ethical and logistical concerns make designing proper drug studies in this age group difficult. Fortunately, innovative analytical techniques such as the use of dried blood spots, scavenged sampling, population pharmacokinetics analyses, and sparse sampling have helped investigators better define doses that maximize efficacy and safety. Through the use of these methods, successful clinical trials have resulted in changes in standards of care. With many more neonatal drug trials underway, we continue to work toward our goal of improving care and outcomes in these vulnerable patients.
Statement of Financial Support
L.C.K. receives research support from the National Institute of Child Health and Human Development (5T32GM086330-03 (principle investigators (PIs): Brouwer, Benjamin, Watkins)). P.B.S. receives salary support for research from the National Institutes of Health (NIH, Bethesda, MD) and the National Center for Advancing Translational Sciences of the NIH (HHSN267200700051C, HHSN275201000003I, 1K24HD058735-05 (PI: Benjamin), and UL1TR001117); he also receives research support from Astellas Pharma US, GlaxoSmithKline (USA), and Pfizer (USA) for neonatal and pediatric drug development.
References
Kimland E, Odlind V . Off-label drug use in pediatric patients. Clin Pharmacol Ther 2012;91:796–801.
Kearns GL, Abdel-Rahman SM, Alander SW, Blowey DL, Leeder JS, Kauffman RE . Developmental pharmacology–drug disposition, action, and therapy in infants and children. N Engl J Med 2003;349:1157–67.
Laughon MM, Benjamin DK Jr, Capparelli EV, et al. Innovative clinical trial design for pediatric therapeutics. Expert Rev Clin Pharmacol 2011;4:643–52.
Zajicek A . The National Institutes of Health and the Best Pharmaceuticals for Children Act. Paediatr Drugs 2009;11:45–7.
Laughon MM, Avant D, Tripathi N, et al. Drug labeling and exposure in neonates. JAMA Pediatr 2014;168:130–6.
Mooij MG, Schwarz UI, de Koning BA, et al. Ontogeny of human hepatic and intestinal transporter gene expression during childhood: age matters. Drug Metab Dispos 2014;42:1268–74.
Smits A, Annaert P, Allegaert K . Drug disposition and clinical practice in neonates: cross talk between developmental physiology and pharmacology. Int J Pharm 2013;452:8–13.
van den Anker JN, Schwab M, Kearns GL . Developmental pharmacokinetics. Handb Exp Pharmacol 2011;205:51–75.
Mulla H . Understanding developmental pharmacodynamics: importance for drug development and clinical practice. Paediatr Drugs 2010;12:223–33.
Tayman C, Rayyan M, Allegaert K . Neonatal pharmacology: extensive interindividual variability despite limited size. J Pediatr Pharmacol Ther 2011;16:170–84.
Loebstein R, Koren G . Clinical pharmacology and therapeutic drug monitoring in neonates and children. Pediatr Rev 1998;19:423–8.
Koren G . Therapeutic drug monitoring principles in the neonate. National Academy of CLinical Biochemistry. Clin Chem 1997;43:222–7.
Skinner AV . Neonatal pharmacology. Anaesth Intensive Care Med 2011;12:79–84.
Visscher M, Narendran V . The Ontogeny of Skin. Adv Wound Care (New Rochelle) 2014;3:291–303.
Kearns GL . Impact of developmental pharmacology on pediatric study design: overcoming the challenges. J Allergy Clin Immunol 2000;106:Suppl 3:S128–38.
Tom-Revzon C . Erratic absorption of intramuscular antimicrobial delivery in infants and children. Expert Opin Drug Metab Toxicol 2007;3:733–40.
American Academy of Pediatrics, Committee on Drugs. Alternative routes of drug administration—advantages and disadvantages (subject review). Pediatrics 1997;100:143–52.
Allegaert K, Verbesselt R, Naulaers G, et al. Developmental pharmacology: neonates are not just small adults. Acta Clin Belg 2008;63:16–24.
Warner A . Drug use in the neonate: interrelationships of pharmacokinetics, toxicity, and biochemical maturity. Clin Chem 1986;32:721–7.
Lorenz JM, Kleinman LI, Ahmed G, Markarian K . Phases of fluid and electrolyte homeostasis in the extremely low birth weight infant. Pediatrics 1995;96(3 Pt 1):484–9.
Watt KM, Benjamin DK Jr, Cheifetz IM, et al. Pharmacokinetics and safety of fluconazole in young infants supported with extracorporeal membrane oxygenation. Pediatr Infect Dis J 2012;31:1042–7.
Aranda JV, Sitar DS, Parsons WD, Loughnan PM, Neims AH . Pharmacokinetic aspects of theophylline in premature newborns. N Engl J Med 1976;295:413–6.
Routledge PA . Pharmacokinetics in children. J Antimicrob Chemother 1994;34:Suppl A:19–24.
Seyberth HW, Kauffman RE . Basics and dynamics of neonatal and pediatric pharmacology. Handb Exp Pharmacol 2011;205:3–49.
Strazielle N, Ghersi-Egea JF . Physiology of blood-brain interfaces in relation to brain disposition of small compounds and macromolecules. Mol Pharm 2013;10:1473–91.
Ek CJ, Dziegielewska KM, Habgood MD, Saunders NR . Barriers in the developing brain and Neurotoxicology. Neurotoxicology 2012;33:586–604.
Liu X, Chen C, Smith BJ . Progress in brain penetration evaluation in drug discovery and development. J Pharmacol Exp Ther 2008;325:349–56.
Takashima T, Yokoyama C, Mizuma H, et al. Developmental changes in P-glycoprotein function in the blood-brain barrier of nonhuman primates: PET study with R-11C-verapamil and 11C-oseltamivir. J Nucl Med 2011;52:950–7.
Vieux R, Hascoet JM, Merdariu D, Fresson J, Guillemin F . Glomerular filtration rate reference values in very preterm infants. Pediatrics 2010;125:e1186–92.
Blake MJ, Castro L, Leeder JS, Kearns GL . Ontogeny of drug metabolizing enzymes in the neonate. Semin Fetal Neonatal Med 2005;10:123–38.
Hines RN . Developmental expression of drug metabolizing enzymes: impact on disposition in neonates and young children. Int J Pharm 2013;452:3–7.
Hines RN, McCarver DG . The ontogeny of human drug-metabolizing enzymes: phase I oxidative enzymes. J Pharmacol Exp Ther 2002;300:355–60.
Hines RN . The ontogeny of drug metabolism enzymes and implications for adverse drug events. Pharmacol Ther 2008;118:250–67.
Yanni SB, Smith PB, Benjamin DK Jr, Augustijns PF, Thakker DR, Annaert PP . Higher clearance of micafungin in neonates compared with adults: role of age-dependent micafungin serum binding. Biopharm Drug Dispos 2011;32:222–32.
Blake MJ, Abdel-Rahman SM, Pearce RE, Leeder JS, Kearns GL . Effect of diet on the development of drug metabolism by cytochrome P-450 enzymes in healthy infants. Pediatr Res 2006;60:717–23.
Brodie MJ, Mintzer S, Pack AM, Gidal BE, Vecht CJ, Schmidt D . Enzyme induction with antiepileptic drugs: cause for concern? Epilepsia 2013;54:11–27.
Yanni SB, Annaert PP, Augustijns P, et al. Role of flavin-containing monooxygenase in oxidative metabolism of voriconazole by human liver microsomes. Drug Metab Dispos 2008;36:1119–25.
Róka A, Melinda KT, Vásárhelyi B, Machay T, Azzopardi D, Szabó M . Elevated morphine concentrations in neonates treated with morphine and prolonged hypothermia for hypoxic ischemic encephalopathy. Pediatrics 2008;121:e844–9.
de Zwart L, Scholten M, Monbaliu JG, et al. The ontogeny of drug metabolizing enzymes and transporters in the rat. Reprod Toxicol 2008;26:220–30.
Anderson BJ . Pharmacology in the very young: anaesthetic implications. Eur J Anaesthesiol 2012;29:261–70.
Jadcherla SR, Berseth CL . Effect of erythromycin on gastroduodenal contractile activity in developing neonates. J Pediatr Gastroenterol Nutr 2002;34:16–22.
Ali BH . Gentamicin nephrotoxicity in humans and animals: some recent research. Gen Pharmacol 1995;26:1477–87.
Sokol PP, Huiatt KR, Holohan PD, Ross CR . Gentamicin and verapamil compete for a common transport mechanism in renal brush border membrane vesicles. J Pharmacol Exp Ther 1989;251:937–42.
Dutt A, Priebe TS, Teeter LD, Kuo MT, Nelson JA . Postnatal development of organic cation transport and mdr gene expression in mouse kidney. J Pharmacol Exp Ther 1992;261:1222–30.
Rose K, Della Pasqua O . Development of paediatric medicines: concepts and principles. Handb Exp Pharmacol 2011;205:111–24.
Howie SR . Blood sample volumes in child health research: review of safe limits. Bull World Health Organ 2011;89:46–53.
McBeth CL, McDonald RJ, Hodge MB . Antibiotic sampling from central venous catheters versus peripheral veins. Pediatr Nurs 2004;30:200–2.
Ramasethu J . Complications of vascular catheters in the neonatal intensive care unit. Clin Perinatol 2008;35:199–222, x.
Food and Drug Administration. Guidance for industry—Population pharmacokinetics, 1999. http://www.fda.gov/downloads/Drugs/.../Guidances/UCM072137.pdf. Accessed 09 June 2014.
Cohen-Wolkowiez M, Ouellet D, Smith PB, et al. Population pharmacokinetics of metronidazole evaluated using scavenged samples from preterm infants. Antimicrob Agents Chemother 2012;56:1828–37.
Wade KC, Wu D, Kaufman DA, et al.; National Institute of Child Health and Development Pediatric Pharmacology Research Unit Network. Population pharmacokinetics of fluconazole in young infants. Antimicrob Agents Chemother 2008;52:4043–9.
Cohen-Wolkowiez M, Benjamin DK Jr, Ross A, et al. Population pharmacokinetics of piperacillin using scavenged samples from preterm infants. Ther Drug Monit 2012;34:312–9.
Cohen-Wolkowiez M, Sampson M, Bloom BT, et al.; Best Pharmaceuticals for Children Act–Pediatric Trials Network. Determining population and developmental pharmacokinetics of metronidazole using plasma and dried blood spot samples from premature infants. Pediatr Infect Dis J 2013;32:956–61.
Suyagh M, Collier PS, Millership JS, et al. Metronidazole population pharmacokinetics in preterm neonates using dried blood-spot sampling. Pediatrics 2011;127:e367–74.
Patel P, Mulla H, Kairamkonda V, et al. Dried blood spots and sparse sampling: a practical approach to estimating pharmacokinetic parameters of caffeine in preterm infants. Br J Clin Pharmacol 2013;75:805–13.
Cohen-Wolkowiez M, Watt KM, Hornik CP, Benjamin DK Jr, Smith PB . Pharmacokinetics and tolerability of single-dose daptomycin in young infants. Pediatr Infect Dis J 2012;31:935–7.
Bell MJ, Shackelford P, Smith R, Schroeder K . Pharmacokinetics of clindamycin phosphate in the first year of life. J Pediatr 1984;105:482–6.
Koren G, Zarfin Y, Maresky D, Spiro TE, MacLeod SM . Pharmacokinetics of intravenous clindamycin in newborn infants. Pediatr Pharmacol (New York) 1986;5:287–92.
Li Z, Chen Y, Li Q, et al. Population pharmacokinetics of piperacillin/tazobactam in neonates and young infants. Eur J Clin Pharmacol 2013;69:1223–33.
Cohen-Wolkowiez M, Watt KM, Zhou C, et al. Developmental pharmacokinetics of piperacillin and tazobactam using plasma and dried blood spots from infants. Antimicrob Agents Chemother 2014;58:2856–65.
van Enk JG, Touw DJ, Lafeber HN . Pharmacokinetics of meropenem in preterm neonates. Ther Drug Monit 2001;23:198–201.
Bradley JS, Sauberan JB, Ambrose PG, Bhavnani SM, Rasmussen MR, Capparelli EV . Meropenem pharmacokinetics, pharmacodynamics, and Monte Carlo simulation in the neonate. Pediatr Infect Dis J 2008;27:794–9.
van den Anker JN, Pokorna P, Kinzig-Schippers M, et al. Meropenem pharmacokinetics in the newborn. Antimicrob Agents Chemother 2009;53:3871–9.
Smith PB, Cohen-Wolkowiez M, Castro LM, et al.; Meropenem Study Team. Population pharmacokinetics of meropenem in plasma and cerebrospinal fluid of infants with suspected or complicated intra-abdominal infections. Pediatr Infect Dis J 2011;30:844–9.
Padari H, Metsvaht T, Kõrgvee LT, et al. Short versus long infusion of meropenem in very-low-birth-weight neonates. Antimicrob Agents Chemother 2012;56:4760–4.
Cohen-Wolkowiez M, Poindexter B, Bidegain M, et al.; Meropenem Study Team. Safety and effectiveness of meropenem in infants with suspected or complicated intra-abdominal infections. Clin Infect Dis 2012;55:1495–502.
Pfizer Labs, Division of Pfizer Inc. Metronidazole injection, solution, 2010. http://dailymed.nlm.nih.gov/dailymed/lookup.cfm?setid=9db2ddc3-8193-4efc-bd39-f532ccec47c5. Accessed 09 June 2014.
Cardinal Health. Piperacillin and tazobactam (piperacillin sodium, tazobactam sodium) injection, powder, for solution, 2013. http://dailymed.nlm.nih.gov/dailymed/lookup.cfm?setid=7b687747-bf51-4366-9277-29bc82fc665a. Accesssed 09 June 2014.
AstraZeneca Pharmaceuticals, LP. Merrem IV (meropenem) injection, 2013. http://dailymed.nlm.nih.gov/dailymed/lookup.cfm?setid=c15e88d3-d903-4e7a-f683-e29f51afa848. Accessed 09 June 2014.
Benjamin DK Jr, Smith PB, Arrieta A, et al. Safety and pharmacokinetics of repeat-dose micafungin in young infants. Clin Pharmacol Ther 2010;87:93–9.
Heresi GP, Gerstmann DR, Reed MD, et al. The pharmacokinetics and safety of micafungin, a novel echinocandin, in premature infants. Pediatr Infect Dis J 2006;25:1110–5.
Hope WW, Smith PB, Arrieta A, et al. Population pharmacokinetics of micafungin in neonates and young infants. Antimicrob Agents Chemother 2010;54:2633–7.
Smith PB, Walsh TJ, Hope W, et al. Pharmacokinetics of an elevated dosage of micafungin in premature neonates. Pediatr Infect Dis J 2009;28:412–5.
Astellas Pharma US, Inc. Mycamine (micafungin sodium) injection, powder, lyophilized, for solution, 2013. http://dailymed.nlm.nih.gov/dailymed/lookup.cfm?setid=a064c4a7-25ec-4a2c-afc2-703491a4a38b. Accessed 09 June 2014.
Watt K, Manzoni P, Cohen-Wolkowiez M, et al. Triazole use in the nursery: fluconazole, voriconazole, posaconazole, and ravuconazole. Curr Drug Metab 2013;14:193–202.
Turner K, Manzoni P, Benjamin DK, Cohen-Wolkowiez M, Smith PB, Laughon MM . Fluconazole pharmacokinetics and safety in premature infants. Curr Med Chem 2012;19:4617–20.
Wade KC, Benjamin DK Jr, Kaufman DA, et al. Fluconazole dosing for the prevention or treatment of invasive candidiasis in young infants. Pediatr Infect Dis J 2009;28:717–23.
Piper L, Smith PB, Hornik CP, et al. Fluconazole loading dose pharmacokinetics and safety in infants. Pediatr Infect Dis J 2011;30:375–8.
Watterberg K . Evidence-based neonatal pharmacotherapy: postnatal corticosteroids. Clin Perinatol 2012;39:47–59.
Patel P, Tanna S, Mulla H, Kairamkonda V, Pandya H, Lawson G . Dexamethasone quantification in dried blood spot samples using LC-MS: The potential for application to neonatal pharmacokinetic studies. J Chromatogr B Analyt Technol Biomed Life Sci 2010;878:3277–82.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Ku, L., Smith, P. Dosing in neonates: special considerations in physiology and trial design. Pediatr Res 77, 2–9 (2015). https://doi.org/10.1038/pr.2014.143
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/pr.2014.143
This article is cited by
-
Pre- and Postnatal Maturation are Important for Fentanyl Exposure in Preterm and Term Newborns: A Pooled Population Pharmacokinetic Study
Clinical Pharmacokinetics (2022)
-
“How can a drug to treat claudication in adults save preterm newborns?”
European Journal of Pediatrics (2020)
-
Preterm Physiologically Based Pharmacokinetic Model. Part II: Applications of the Model to Predict Drug Pharmacokinetics in the Preterm Population
Clinical Pharmacokinetics (2020)
-
Two decades of off-label prescribing in children: a literature review
World Journal of Pediatrics (2018)
-
Safety of Enalapril in Infants Admitted to the Neonatal Intensive Care Unit
Pediatric Cardiology (2017)
