
Abstract
Marfan syndrome (MFS) is an autosomal dominant connective tissue disorder. Diagnostic criteria of neonatal MFS (nMFS), the most severe form, are still debated. The aim of our study was to search for clinical and molecular prognostic factors that could be associated with length of survival. Probands ascertained via the framework of the Universal Marfan database-FBN1, diagnosed before the age of 1 y and presenting with cardiovascular features (aortic root dilatation or valvular insufficiency) were included in this study. Clinical and molecular data were correlated to survival. Among the 60 individuals, 38 had died, 82% died before the age of 1 y, mostly because of congestive heart failure. Three probands reached adulthood. Valvular insufficiencies and diaphragmatic hernia were predictive of shorter life expectancy. Two FBN1 mutations were found outside of the exon 24–32 region (in exons 4 and 21). Mutations in exons 25–26 were overrepresented and were associated with shorter survival (p = 0.03). We report the largest genotyped series of probands with MFS diagnosed before 1 y of life. In this population, factors significantly associated with shorter survival are presence of valvular insufficiencies or diaphragmatic hernia in addition to a mutation in exons 25 or 26.
Similar content being viewed by others


Main
Marfan syndrome (MFS) is an autosomal dominant connective tissue disorder (MIM 154700), the skeletal features of which were first described by Marfan (1) in 1896. The cardinal features of MFS involve the cardiovascular, ocular, and skeletal systems. The skin, lung, and dura may also be involved (2). The prevalence of MFS has been estimated at 2–3 per 10,000 of the population, and ∼25% of cases are sporadic (3). Mutations in the gene encoding fibrillin-1, FBN1, are known to cause MFS and are widely distributed along the entire FBN1 gene. MFS is notable for its variability in age of onset, tissue distribution, and severity of clinical manifestations. neonatal Marfan syndrome (nMFS) is considered the most severe end of the spectrum of type I fibrillinopathies, but diagnostic criteria are still debated. Indeed, Booms et al. (4) proposed to reserve the term of nMFS for patients diagnosed at birth or within the first 3 mo of life, presenting with pronounced atrioventricular valve dysfunction, with death often occurring within the first year of life because of congestive heart failure. Pulmonary emphysema, joint contractures, crumpled ears, and loose skin are often associated. They also suggested, like others, that all mutations causing nMFS cluster in a specific region between exons 24 and 32 (4–6). According to the most recent comment on the subject by Hennekam (7), children with nMFS show severe mitral and/or tricuspid valvular insufficiency and infantile pulmonary emphysema, in addition to the common symptoms of ectopia lentis, arachnodactyly, joint contractures, and loose skin. Death may occur within the first 2 y of life from congestive heart failure (7). Life expectancy seems to be the central issue when managing a patient with early diagnosed MFS with cardiovascular involvement. To search for prognostic factors associated with early death, we studied the clinical and molecular features of 60 probands carrying mutations in the FBN1 gene and with cardiovascular involvement before the age of 1 y.
METHODS
Patients.
Patients were ascertained through the Universal Marfan database-FBN1 (UMD-FBN1; http://www.umd.be) (8), which collects molecular data of patients carrying an FBN1 mutation from 38 countries in five continents and registered between 1995 and 2008 and patients with a pathogenic FBN1 mutation unpublished were included (9,10). Inclusion criteria were as follows: i) clinical features allowing a diagnosis of MFS or type I fibrillinopathy in a child before the age of 1 y and ii) presence of cardiovascular manifestations including mitral and/or tricuspid valvular insufficiency and/or aortic root dilatation. These criteria were designed to exclude children with some skeletal features of the MFS spectrum but without cardiovascular involvement that could be diagnosed early based on a positive family history. They were also designed to exclude children with clinical features of MFS but mutated in an other gene such as TGFBR1 or TGFBR2 and to permit molecular description of the series.
Sixty children diagnosed as MFS before the age of 1 y were identified, designated as nMFS or not by the clinician. Fifty-three of the probands were part of the 320 children described by Faivre et al. (11), whose study was designed for another purpose, and the remaining patients were issued from personal observations or recent publications. Informed consent has been obtained by each clinician. The required clinical information included a range of qualitative and quantitative clinical parameters, among them cardiovascular, ophthalmological, skeletal, skin, and lung manifestations. The age and cause of death or the age at last follow-up and the characteristics of FBN1 mutations were also collected. Patients still alive at last follow-up were designated as “survivors.”
Statistical analysis.
For descriptive purposes, time-to-event analysis techniques were used for survival analyses, and calculation of cumulative probability of a clinical feature when the age at diagnosis was collected. Subjects for whom the age at diagnosis of a specific manifestation was not available were excluded from these analyses. Subjects who did not manifest the feature during the follow-up course were censored at their last follow-up. The baseline date (time zero) was the date of birth. In particular, the Kaplan-Meier method (12) was used to estimate the cumulative probabilities of clinical manifestations of the disease.
To search for prognostic factors associated with shorter survival, comparison of prevalence of different clinical features was made between the group of patients deceased before the age of 1 y and the group of patients alive at this age. Only clinical features present before the age of 1 y were considered. Only patients with available follow-up beyond the age of 1 y were included in the analysis. The χ2 or Fisher exact test were used.
Genotype-phenotype correlation analyses were difficult because of the small sample size. It was not possible to compare the type of mutations because the large majority of mutation was private missense mutations. We questioned whether the substitution of a cysteine residue was more likely to have phenotypic consequences than mutations affecting another amino acid. Because of the small sample size, it was not possible to compare the phenotypic data for each exon of the region with the others. Therefore, we evaluated whether the phenotypic consequences of a mutation in the exons preferentially involved (exons 25 and 26) were different to the consequences of mutations in the other exons. Differences among the different types of mutation groups were tested using the nonparametric log-rank test.
We used SAS software version 9.2 (SAS Institute, Inc., Cary, NC) and Stata software version 9 (Stata Corp, College Station, TX) for all statistical analyses. The p values <0.05 were considered significant. The study has been approved by the Assistance Publique Hôpitaux de Paris.
RESULTS
Clinical description of the population.
Of the 60 probands, 58% (34/59) were males and 42% (25/59) were females. All cases were sporadic except for three siblings born to a father with somatic and germline mosaicism (13). Therefore, 95% (57/60) of cases were de novo. Mean age at diagnosis was 0.05 ± 0.2 y.
The median age at last follow-up was 4 mo [interquartile ratio (IQR), 2:66, extremes 0–26 y; Fig. 1]. Thirty-eight patients (63%) died during follow-up; among them 31 (82%) before the age of 1 y and 34 (89%) before the age of 2 y. The cause of death was congestive heart failure in 32 children (85%), during or directly after cardiac surgery in three (8%), respiratory failure in two (5%), and in utero death in the remaining patient. Among patients who died of congestive heart failure, all had valvular insufficiencies.

Kaplan-Meier analysis for survival in patients diagnosed as MFS or type 1 fibrillinopathy before the age of 1 y. (A) Overall survival (solid line): 50% of patients were alive at 4 mo, IQR = 2:66. Area between dashed lines show pointwise confidence bands (95%). (B) Survival in patients with a mutation in FBN1 exons 25–26 (dashed line) vs patients with a mutation in other exons (solid line): 50% of patients with mutation within exons 25–26 were alive at 3 mo, whereas 50% of patients with mutation located in other exons were alive at 12 mo.
The median age of the 22 survivors was 24 mo (IQR, 0:48). They included 14 males and 8 females (NS). Among them, 21 (95%) had aortic root dilatation and 16 (73%) had valvular insufficiencies. Notably, seven survivors (32%) have had a valvular surgery.
Clinical features of the overall study population are summarized in Table 1. Ectopia lentis found in 24 of 52 patients (46%) was not always present at the time of diagnosis but evolved during follow-up in 11 of 24 patients. Spontaneous pneumothorax was reported in five patients. Emphysema was recorded in two autopsy reports. Six patients had diaphragmatic hernia and all died in the first months of life (range, 0–3.5 mo). The presence of senile appearance, loose skin, and crumpled ears that has been described as characteristic of nMFS was noted in less than a third of patients. Dural ectasia was checked only for five patients and was present in two.
The most frequent cardiac manifestation was aortic root dilatation (56/60, 93%), with a mean age at diagnosis of dilatation of 0.2 ± 0.4 y (Fig. 2). Only two children (females) had aortic dissection, one child shortly after birth and another at the age of 12 y. In the study population, six probands (four males and two females) underwent aortic surgery during infancy at a mean age of 5.4 ± 6.4 y (range, 0.7–15 y). Two of them died after surgery at the age of 10 mo and 4 y, respectively. The remaining patients were 2, 6, 20, and 26 y old, respectively, at last follow-up.

Percentage of different cardiac manifestations in probands during infancy.
Among valvular insufficiencies, the most frequent was mitral insufficiency in 81% of patients, with a mean age of 0.2 ± 0.4 y. Tricuspid insufficiency was found in 78% of patients when mentioned. Aortic insufficiency was found in 40% of patients, with a mean age of 0.1 ± 0.5 y. Eight patients had aortic dilatation without valvular involvement. Thirteen patients (seven males and six females) had valvular surgery at a mean age of 1.7 ± 1.5 y (range, 0.2 and 6 y). Among them, six died (46%) at a mean age of 2.2 ± 2.1 y, including four patients shortly after surgery. The mean age at surgery was not predictive of survival. Bicuspid aortic valve and congenital cardiomyopathy were each reported in three patients. Three patients reached adulthood, two of them with multiple cardiac and orthopedic surgeries.
Comparison of probands who died before 1 year of age and those who survived.
The three features more frequently present in probands who died before 1 y of age were aortic dilatation (90 versus 73%, NS), valvular insufficiencies (94 versus 64%, p = 0.01), diaphragmatic herniae (21 versus 0%, p = 0.03; Table 1).
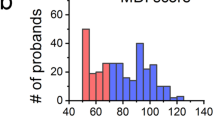
The deleterious effect of identified sequence variants was assessed by various algorithms [among which are Polymorphism Phenotyping (PolyPhen); Sorting Intolerant From Tolerant (SIFT), Biochemical values, and BLOSUM 62], which were developed to predict whether a nucleotide variation is likely to affect protein function or not. All, except PolyPhen, are implemented in the new version of the UMD-LSDB software in the “UMD-Predictor” tool. Forty-seven different pathogenic FBN1 mutations were found (Table 2), seven of which were recurrent. There were 46 missense mutations (77%), nine splice-site mutations (15%), three inframe deletions or insertions (5%), and two frameshift mutations (3%). As no nonsense mutations were found, only two mutations led to a premature truncation of translation. Of the missense mutations, 63% involved a cysteine residue. Most mutations (98%) were located in an EGF domain, and only one was located in a TGFBP-like domain. Fifty-eight mutations (97%) were found in exons 24–32. One patient had a mutation in exon 4 (386G>A) and one in exon 21 (2552_2566del; Table 2). Notably, 58% of mutations were clustered in exons 25 and 26 (Fig. 3). This overrepresentation of exons 25 and 26 was not found in other clinical manifestations (14). No compound heterozygosity was found.

Percentage of mutations in the “exon 24–32 region” when compared with the percentage of mutations found in other MFS phenotypes: for each exon, black bars represent the percentage of patients diagnosed as MFS before the age of 1 year (n = 60) and white bars the percentage of mutations found whatever the phenotype (n = 155). **p < 0.0001 and *p = 0.002.
We detected a significant difference for survival at 1 year between patients carrying a mutation in exons 25–26 (27%; 95% CI = 13–44%) versus patients carrying a mutation elsewhere in the gene (48%; 95% CI = 27–66%; Fig. 1B). Survival before 1 y was not significantly different in patients carrying a missense mutation involving a cysteine versus another missense mutation.
The two mutations leading to premature truncation are frameshift mutations located in exon 31 (3908_3909del) and 32 (4085_4091del; Table 2). The two patients had different survivals. One patient died within the first year of life from congestive heart failure and the other was still alive at age 20 y after valvular and aortic surgeries. Similarly, among the two patients with a mutation located outside of exons 24–32, one female died after surgery for aortic dilatation at 4 y (exon 21) and one male was alive at 20 y and had not yet required surgery (exon 4).
DISCUSSION
The goal of this study was to identify prognostic factors associated with early death in a large population of genotyped patients diagnosed with MFS or type I fibrillinopathy before 1 year of age gathered through an international collaboration. We chose to include only those probands carrying a mutation in an FBN1 for two reasons. First, to avoid overlapping phenotypes, e.g. Beals syndrome associated with an FBN2 gene mutation (15), MFS type 2 and Loeys-Dietz syndrome linked to TGFBR1 or 2 gene mutations (16–18), and second, to allow for description of the molecular characteristics and search for genotype-phenotype correlations. We chose to avoid the term of neonatal MFS because the dividing line between neonatal MFS and severe infantile MFS is difficult to define. In an editorial, Hennekam (7) proposed that the term nMFS should be restricted to neonates who show severe mitral and/or tricuspid valvular insufficiency and infantile pulmonary emphysema. The presence or absence of pulmonary emphysema was rarely ascertained in our series; therefore, we could not apply this definition. Although rare events, two cases who died before 1 y of age from respiratory distress did not have valvular insufficiency. The presence of ascending aortic dilatation was not part of the definition of nMFS; however, in our series, it was as common as valvular insufficiency, especially in patients who died before 1 y of age. Nevertheless, the presence or absence of ascending aortic dilatation was not predictive of early death. A significant result was only found for the presence of valvular insufficiencies and diaphragmatic hernia, indicating that both features could be considered as adverse prognostic criteria. Morse et al., (19) reported their own series of 22 patients severely affected infants diagnosed with MFS in the first 3 mo of life, together with a review of 32 additional infants described in the literature. At the time, none of the patients had been genotyped. They found serious cardiac pathology, including mitral valve prolapse, valvular regurgitation, and aortic root dilatation in 82% of their patients, and congenital contractures in 64%. Nevertheless, prognosis was better because only three of 22 infants died during the first year of life. Our results and this report emphasize the difficulty in differentiating nMFS from severe infantile MFS, and therefore, in giving a prognosis to families. This point is further highlighted by the long-term outcome of three patients in our series who reached adulthood. Even if intensive cardiac surgeries contributed to survival for two of them the third patient did not need cardiac surgery.
This study analyzes for the first time the FBN1 mutation spectrum in a large series of neonatal/infantile onset cases. Previous studies of numerous child report nongenotyped cases. We found no mutation characteristic that is predictive of precocious death in individual cases, but the type and distribution of mutations differ from those described in the general MFS population (20,21). We confirmed that the hot spot region for MFS diagnosed before 1 y of age is located between exons 24 and 32; however, we newly discovered that exons 25 and 26 are most often involved (58% of mutations) whatever clinical presentation. Because two mutations were found outside the exons 24–32 region, it remains justifiable to sequence the whole FBN1 gene when no mutation is found in a first-step analysis of exons 24–32. Only two mutations leading to premature truncation were found and there were no nonsense mutations. These results are in agreement with previous results from Faivre et al. in patients mutated in exons 24–32 whatever the severity of the disease (nMFS, severe MFS, classical MFS, probable MFS, or other type I fibrillinopathy). Indeed, the authors showed that premature truncated codons (PTCs) were underrepresented in patients with severe phenotypes with an absence of nonsense mutations, whereas missense mutations were overrepresented. It is not known whether this could be explained by early lethality or by a milder phenotypic effect of mutations leading to premature truncations within the exon 24–32 region. Few data are available to assess the true effect of mutations leading to premature truncations and whether they are subjected to nonsense-mediated RNA decay or give rise to truncated peptides of various sizes (22). The difference in clinical manifestation between both patients with truncating mutations could be explained by a different pathogenesis, dominant negative effect versus nonsense-mediated decay. Until more information is available, the true impact on microfibril formation from mutations leading to premature truncations remains speculative. In their study, Schrijver et al. (23) found that the position of a substituted cysteine residue within an EGF-like domain of fibrillin 1 has an effect on the severity of symptoms in MFS. In our population, we have compared the mutation position in probands who died before the age of 1 y and those alive after this age, but we could not find any significant difference, probably because of the size of our sample.
In conclusion, we report the largest genotyped series of probands with MFS diagnosed before 1 y of life. We found that the presence of valvular insufficiencies and diaphragmatic herniae were predictive of shorter life expectancy in this population. Such phenotypes are most often secondary to a de novo mutation in FBN1 exons 25 and 26. These data should add to the understanding, care, prognosis, and counseling of patients and their families.
Abbreviations
- IQR:
-
interquartile ratio
- MFS:
-
Marfan syndrome
- nMFS:
-
neonatal MFS
References
Marfan A 1896 [A case of congenital deformation of four members more pronounced in the extremities characterized by the elongation of bones with a certain degree of thinning down]. French. Bull Mem Soc Med Hop Paris 13: 220–226
De Paepe A, Devereux RB, Dietz HC, Hennekam RC, Pyeritz RE 1996 Revised diagnostic criteria for the Marfan syndrome. Am J Med Genet 62: 417–426
Judge DP, Dietz HC 2005 Marfan's syndrome. Lancet 366: 1965–1976
Booms P, Cisler J, Mathews KR, Godfrey M, Tiecke F, Kaufmann UC, Vetter U, Hagemeier C, Robinson PN 1999 Novel exon skipping mutation in the fibrillin-1 gene: two ‘hot spots' for the neonatal Marfan syndrome. Clin Genet 55: 110–117
Putnam EA, Cho M, Zinn AB, Towbin JA, Byers PH, Milewicz DM 1996 Delineation of the Marfan phenotype associated with mutations in exons 23–32 of the FBN1 gene. Am J Med Genet 62: 233–242
Tiecke F, Katzke S, Booms P, Robinson PN, Neumann L, Godfrey M, Mathews KR, Scheuner M, Hinkel GK, Brenner RE, Hovels-Gurich HH, Hagemeier C, Fuchs J, Skovby F, Rosenberg T 2001 Classic, atypically severe and neonatal Marfan syndrome: twelve mutations and genotype-phenotype correlations in FBN1 exons 24–40. Eur J Hum Genet 9: 13–21
Hennekam RC 2005 Severe infantile Marfan syndrome versus neonatal Marfan syndrome. Am J Med Genet A 139: 1
Collod-Béroud G, Le Bourdelles S, Ades L, Ala-Kokko L, Booms P, Boxer M, Child A, Comeglio P, De Paepe A, Hyland JC, Holman K, Kaitila I, Loeys B, Matyas G, Nuytinck L, Peltonen L, Rantamaki T, Robinson P, Steinmann B, Junien C, Béroud C, Boileau C 2003 Update of the UMD-FBN1 mutation database and creation of an FBN1 polymorphism database. Hum Mutat 22: 199–208
Frédéric MY, Boileau C, Hamroun D, Claustres M, Béroud C, Collod-Béroud G 2009 UMD-predictor, a new prediction tool for nucleotide substitution pathogenicity—application to four genes: FBN1, FBN2, TGFBR1, and TGFBR2. Hum Mutat 30: 952–959
Desmet FO, Hamroun D, Lalande M, Collod-Beroud G, Claustres M, Beroud C 2009 Human splicing finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res 37: e67
Faivre L, Masurel-Paulet A, Collod-Beroud G, Callewaert BL, Child AH, Stheneur C, Binquet C, Gautier E, Chevallier B, Huet F, Loeys BL, Arbustini E, Mayer K, Arslan-Kirchner M, Kiotsekoglou A, Comeglio P, Grasso M, Halliday DJ, Beroud C, Bonithon-Kopp C, Claustres M, Robinson PN, Ades L, De Backer J, Coucke P, Francke U, De Paepe A, Boileau C, Jondeau G 2009 Clinical and molecular study of 320 children with Marfan syndrome and related type I fibrillinopathies in a series of 1009 probands with pathogenic FBN1 mutations. Pediatrics 123: 391–398
Kaplan EM 1958 Non parametric estimation from incomplete observations. J Am Stat Assoc 53: 457–481
Tekin M, Cengiz FB, Ayberkin E, Kendirli T, Fitoz S, Tutar E, Ciftci E, Conba A 2007 Familial neonatal Marfan syndrome due to parental mosaicism of a missense mutation in the FBN1 gene. Am J Med Genet A 143A: 875–880
Faivre L, Collod-Beroud G, Callewaert B, Child A, Binquet C, Gautier E, Loeys BL, Arbustini E, Mayer K, Arslan-Kirchner M, Stheneur C, Kiotsekoglou A, Comeglio P, Marziliano N, Wolf JE, Bouchot O, Khau-Van-Kien P, Beroud C, Claustres M, Bonithon-Kopp C, Robinson PN, Ades L, De Backer J, Coucke P, Francke U, De Paepe A, Jondeau G, Boileau C 2009 Clinical and mutation-type analysis from an international series of 198 probands with a pathogenic FBN1 exons 24–32 mutation. Eur J Hum Genet 17: 491–501
Frédéric MY, Monino C, Marschall C, Hamroun D, Faivre L, Jondeau G, Klein HG, Neumann L, Gautier E, Binquet C, Maslen C, Godfrey M, Gupta P, Milewicz D, Boileau C, Claustres M, Béroud C, Collod-Béroud G 2009 The FBN2 gene: new mutations, locus-specific database (Universal Mutation Database FBN2), and genotype-phenotype correlations. Hum Mutat 30: 181–190
Frederic MY, Hamroun D, Faivre L, Boileau C, Jondeau G, Claustres M, Beroud C, Collod-Beroud G 2008 A new locus-specific database (LSDB) for mutations in the TGFBR2 gene: UMD-TGFBR2. Hum Mutat 29: 33–38
Mizuguchi T, Collod-Beroud G, Akiyama T, Abifadel M, Harada N, Morisaki T, Allard D, Varret M, Claustres M, Morisaki H, Ihara M, Kinoshita A, Yoshiura K, Junien C, Kajii T, Jondeau G, Ohta T, Kishino T, Furukawa Y, Nakamura Y, Niikawa N, Boileau C, Matsumoto N 2004 Heterozygous TGFBR2 mutations in Marfan syndrome. Nat Genet 36: 855–860
Loeys BL, Chen J, Neptune ER, Judge DP, Podowski M, Holm T, Meyers J, Leitch CC, Katsanis N, Sharifi N, Xu FL, Myers LA, Spevak PJ, Cameron DE, De Backer J, Hellemans J, Chen Y, Davis EC, Webb CL, Kress W, Coucke P, Rifkin DB, De Paepe AM, Dietz HC 2005 A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat Genet 37: 275–281
Morse RP, Rockenmacher S, Pyeritz RE, Sanders SP, Bieber FR, Lin A, MacLeod P, Hall B, Graham JM Jr 1990 Diagnosis and management of infantile marfan syndrome. Pediatrics 86: 888–895
Faivre L, Collod-Beroud G, Loeys BL, Child A, Binquet C, Gautier E, Callewaert B, Arbustini E, Mayer K, Arslan-Kirchner M, Kiotsekoglou A, Comeglio P, Marziliano N, Dietz HC, Halliday D, Beroud C, Bonithon-Kopp C, Claustres M, Muti C, Plauchu H, Robinson PN, Ades LC, Biggin A, Benetts B, Brett M, Holman KJ, De Backer J, Coucke P, Francke U, De Paepe A, Jondeau G, Boileau C 2007 Effect of mutation type and location on clinical outcome in 1,013 probands with Marfan syndrome or related phenotypes and FBN1 mutations: an international study. Am J Hum Genet 81: 454–466
Stheneur C, Collod-Beroud G, Faivre L, Buyck JF, Gouya L, Le Parc JM, Moura B, Muti C, Grandchamp B, Sultan G, Claustres M, Aegerter P, Chevallier B, Jondeau G, Boileau C 2009 Identification of the minimal combination of clinical features in probands for efficient mutation detection in the FBN1 gene. Eur J Hum Genet 17: 1121–1128
Schrijver I, Liu W, Odom R, Brenn T, Oefner P, Furthmayr H, Francke U 2002 Premature termination mutations in FBN1: distinct effects on differential allelic expression and on protein and clinical phenotypes. Am J Hum Genet 71: 223–237
Schrijver I, Liu W, Brenn T, Furthmayr H, Francke U 1999 Cysteine substitutions in epidermal growth factor-like domains of fibrillin-1: distinct effects on biochemical and clinical phenotypes. Am J Hum Genet 65: 1007–1020
Author information
Authors and Affiliations
Corresponding author
Additional information
Supported by a Grant from the French Ministry of Health (PHRC 2004), GIS maladies rares 2004, Bourse de la Société Française de Cardiologie, Fédération Française de Cardiologie 2005, Assistance Publique Hôpitaux de Paris (CIRC 2007), and ANR-05-PCOD-014.
Rights and permissions
About this article
Cite this article
Stheneur, C., Faivre, L., Collod-Béroud, G. et al. Prognosis Factors in Probands With an FBN1 Mutation Diagnosed Before the Age of 1 Year. Pediatr Res 69, 265–270 (2011). https://doi.org/10.1203/PDR.0b013e3182097219
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/PDR.0b013e3182097219
This article is cited by
-
Ocular, cardiovascular, and genetic characteristics and their associations in children with Marfan syndrome and related fibrillinopathies
Graefe's Archive for Clinical and Experimental Ophthalmology (2023)
-
Marfan syndrome
Nature Reviews Disease Primers (2021)
-
Aggressive Aortopathy in neonatal Marfan syndrome
Journal of Congenital Cardiology (2019)
-
A novel fibrillin-1 gene missense mutation associated with neonatal Marfan syndrome: a case report and review of the mutation spectrum
BMC Pediatrics (2016)
-
Variable severity of cardiovascular phenotypes in patients with an early-onset form of Marfan syndrome harboring FBN1 mutations in exons 24–32
Heart and Vessels (2016)
