
Abstract
Oral-facial-digital syndrome type 1 (OFD1) is a ciliopathy characterized by oral, facial, and digital malformations that are often accompanied by polycystic lesion of the kidney and central nervous involvement. OFD1 shows an X-linked recessive inheritance caused by mutation in the OFD1 gene (Xp22.2). The disease is generally considered embryonic lethal for hemizygous males. However, males with OFD1 mutations were recently reported. Here, we report four additional Japanese male patients with OFD1 variants and describe the variable clinical manifestation and disease severity among the four patients. Patient 1 with pathogenic indels including a 19-bp deletion and 4-bp insertion (c.2600–18_2600delinsACCT) had end-stage renal disease (ESRD) with bilateral cystic kidneys and sensory hearing loss. He showed neither intellectual disability nor facial or digital dysmorphism. Patient 2 with a missense variant in exon 7 (c.539 A > T, p.Asp180Val) presented head circumference enlargement, brachydactyly, high-arched palate, micropenis, severe global developmental delay, and ESRD. Patient 3 had a single base substitution at the splice donor site of intron 16 (c.2260 + 2 T > G) causing a 513-bp deletion at the transcript level. The patient had chronic kidney disease and speech delay, but no oral, facial, or digital dysmorphism. His uncle (patient 4) carried the same OFD1 variant and showed ESRD with extra-renal malformations including obesity and micropenis, which was previously diagnosed as Bardet-Biedl syndrome. The OFD1 mutations were not lethal in these four male patients, likely because the three mutations were in-frame or missense. This report provided insights into the onset mechanism and phenotype-genotype association in patients with OFD1 mutations.
Similar content being viewed by others


Introduction
Oral-facial-digital syndrome type 1 (OFD1; MIM #311200) is a rare X-linked congenital disorder characterized by oral, facial, and digital abnormalities and is mostly seen in females. The incidence of OFD1 is approximately 1 in 50,000 live births [1]. This disorder is often accompanied with polycystic lesion of the kidney and a broad range of central nervous involvement. Cystic changes in the liver and pancreas have also been described in patients with OFD1. Central nervous involvement, which may include intellectual disability, hydrocephalus, cerebellar anomalies, porencephaly, and corpus callosum defects, is seen in 40% of patients with OFD1 [2]. Polycystic lesion of the kidney occurs in approximately 15–50% of female patients, most of whom progress to end-stage renal disease (ESRD) [1, 2]. In contrast, those without polycystic lesion of the kidney have normal renal function [3]. Some patients without external features are diagnosed by visceral features such as polycystic kidney in adulthood [4].
OFD1 is located on the short arm of the X chromosome (Xp22.2), and its mutations are responsible for OFD1 [5]. To date, at least 157 OFD1 mutation sites have been cataloged in the Human Gene Mutation Database (HGMD) (http://www.hgmd.cf.ac.uk/). OFD1 encodes a centrosome/basal body protein necessary for the assembly of primary cilia and left-right axis determination [6]. Mouse mutants of Ofd1 have absent or malformed cilia in various tissues [7]. Thus, OFD1 is considered a form of ciliopathy. Phenotypic variability is often seen in affected females including those within the same family, which may be partly due to the varying degree of somatic mosaicism resulting from random X inactivation [8].
OFD1 is generally thought to cause lethality in males during the fetal period, and almost all affected patients are females. However, recently, males with OFD1 mutation have been described. As in classical OFD1, there are four phenotypes of OFD1 mutation in males: X-linked Joubert syndrome with polydactyly and retinal involvement (JBTS10), Simpson-Golabi-Behmel syndrome type 2 (SGBS2), retinitis pigmentosa 23 (RP23), and unclassified malformation syndromes [9]. However, male patients are so rare that very little is currently known about the phenotype-genotype correlation for OFD1 in males. We now report four Japanese male patients from three families who had novel OFD1 mutations. They all had chronic kidney diseases including ESRD, but with varying degrees of extra-renal malformations and central nervous system involvement. Our findings may elucidate the phenotype-genotype correlation in OFD1.
Materials and methods
Patients
Patient 1
Patient 1 was an 8-year-old boy who was born to non-consanguineous parents at 36 weeks and 5 days of gestation. His birth weight was 2881 g. During the neonatal period, he was diagnosed with an acute kidney injury due to a severe upper-pole primary ureteropelvic junction obstruction of the left kidney and non-functional right kidney (Fig. 1a). Ultrasound examination showed bilateral cystic kidneys with a multicystic dysplastic right kidney. After an emergency nephrostomy, his kidney function improved temporarily but then progressively declined. At the age of 8, he progressed to ESRD and underwent a kidney transplant. He also had bilateral sensory hearing loss, but no other abnormalities including facial, oral, or digital deformities. He had no failure of growth (height 116.9 cm, body weight 20.3 kg) or intellectual disability, and brain magnetic resonance imaging did not show abnormalities, including molar tooth sign. He is now 11 years old, and his transplanted renal function is preserved.

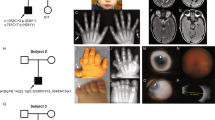
Renal imaging of patient 1 (a), patient 2 (b), and patient 3 (c and d). a Left renal hydronephrosis and right multicystic dysplastic kidney shown by abdominal computed tomography in patient 1. White arrow indicates left nephrostomy catheter. b Magnetic resonance image showing bilateral enlarged kidney and small cysts in patient 2. c, d Bilateral hyperechogenic kidney (c, left; d, right) in patient 3
Patient 2
Patient 2 was a 4-year-old boy. He was also born to non-consanguineous parents at 37 weeks and 1 day of gestation. His birth weight was 3160 g. A leptomeningeal cyst and hydrocephalus were detected during the neonatal period. He presented progressively worsening kidney dysfunction at the age of 1 and has been on peritoneal dialysis due to ESRD since the age of 2. Magnetic resonance imaging showed bilateral enlarged kidneys with numerous cysts (Fig. 1b), and kidney biopsy indicated nephronophthisis. He had congenital deformations including head circumference enlargement, brachydactyly, high-arched palate, micropenis, and optic nerve hypoplasia. He also displayed a severe intellectual disability and motor developmental delay. He is now 6 years old. He undergoes peritoneal dialysis and shows growth failure (height 82.0 cm, weight 12.3 kg).
Patient 3
Patient 3 was a 3-year-old boy. He was also born to non-consanguineous parents at 38 weeks and 4 days of gestation. His birth weight was 2638 g. At the age of 1, bilateral kidney enlargement with high intensity was detected by ultrasound (Figs. 1c, d). At that time, his estimated glomerular filtration rate was 62.2 ml/min/1.73 m2, although his urinalysis findings were normal, and he was diagnosed with chronic kidney disease. He displayed a speech delay and autistic features with hyperactivity, but no facial, oral, or digital abnormality. He exhibited short stature (85.6 cm) at the age of 3. He had a family history of renal disease, and his pedigree is shown in Fig. 2. One of his uncles (II-1 in Fig. 2) also had ESRD and died at a young age. His mother (II-4) and grandmother (I-2) had normal urinalysis findings, with no oral and dental symptoms.

Family tree of patient 3 and patient 4
Patient 4
Patient 4 was a 29-year-old male and a maternal uncle of patient 3 (II-5 in Fig. 2). He had ESRD and underwent a renal transplantation at the age of 14. At the age of 13, he also had extra-renal malformations such as obesity (height 156 cm and body weight 91.7 kg), borderline intellectual disability (his intelligence quotient was 76 at the age of 12), and micropenis. His serum levels of follicle stimulating hormone and luteinizing hormone were normal (3.56 mIU/ml and 4.06 mIU/ml), and testosterone was low (0.91 ng/dl). He had no retinal disorder or hearing loss. He was diagnosed with Bardet-Biedl syndrome based on the clinical manifestations.
Analysis of OFD1 mutations
Genetic analysis was performed after informed consent was received. This study was approved by the institutional review board of Kobe University School of Medicine. Genomic DNA was isolated from peripheral blood leukocytes of the patients and their family using the QuickGene whole blood kit S (Kurabo, Osaka, Japan). Additionally, total RNA was extracted from blood leukocytes using the NucleoSpin RNA Blood (Macherey-Nagel, Hoerdt, France) and an RNA stabilization agent (RNAlater; Thermo Fisher Scientific, Waltham, MA, USA). Total RNA was reverse-transcribed to cDNA using SuperScript III First-Standard Synthesis SuperMix (Thermo Fisher Scientific) for cDNA analysis.
Targeted sequencing using next-generation sequencing (NGS) was conducted for 91 genes in patient 1 (Supplementary Table 1) and 128 genes in patient 2 (Supplementary Table 2) associated with congenital anomalies of the kidney and urinary tract or nephronophthisis-related ciliopathies as cataloged in the OMIM database (http://www.omim.org/) or PubMed (http://www.ncbi.nlm.nih.gov/pubmed). NGS samples were prepared using the HaloPlex target enrichment system kit (Agilent Technologies, Santa Clara, CA, USA) according to the manufacturer’s instructions. The captured DNA samples were amplified by PCR and sequenced using the MiSeq platform (Illumina, San Diego, CA, USA). Analysis of the NGS data was performed using the SureCall software (Agilent Technologies). The mutations detected by NGS were confirmed by standard Sanger direct sequencing using the 3130 Genetic Analyzer (Thermo Fisher Scientific). For patient 3, the OFD1 mutation was directly detected by standard Sanger sequencing without NGS because the patient’s family tree indicated the mutation. The direct sequencing data were analyzed by CLC Main Workbench version 6.7.1 (CLC bio, Aarhus, Denmark). For variant descriptions, NM_003611.2 was used as a reference sequence of OFD1.
For prioritized candidate novel exonic variants, wANNOVAR (http://wannovar.wglab.org/) was used as an in silico evaluation tool to predict whether the mutations were pathogenic.
Results
We identified three different mutations by NGS and direct sequencing of OFD1 in the four male patients. In patients 3 and 4, their pedigree indicated an X-linked dominant ciliopathy; therefore, their mutations were analyzed only by direct sequencing of OFD1.
In patient 1, we identified a novel 19-bp deletion and 4-bp insertion (c.2600–18_2600delinsACCT) in exon 20 of OFD1 and its splice acceptor site, resulting in an in-frame mutation (p.Ser867_Asp869delinsAsn) (Figs. 3a, b, left panels). His mother was heterozygous for this aberration.

Gene mutations identified in the patients. a Confirmation of the genetic analysis by Sanger sequencing. b cDNA analysis and schematic representation of the changes in OFD1 mRNA
In patient 2, we identified a novel missense variant in exon 7 of OFD1 (c. 539 A > T, p.Asp180Val) (Fig. 3a, middle panel). His mother did not carry this mutation; thus, the mutation was likely de novo in the patient. This variant was estimated to be pathogenic by in silico analysis using wANNOVAR. The analysis showed a CADD score of 27.7 and predicted the mutation as deleterious by PROVEAN and SIFT algorithms, probably damaging by PolyPhen2, and disease-causing by Mutation Taster.
In patient 3 and 4, a single base substitution at the splice donor site of intron 16 (c.2260 + 2 T > G) was detected. The result of the cDNA analysis showed that this aberration caused a 513-bp in-frame deletion in exon 16 and 17 (Fig. 3a, b, right panels).
Discussion
We presented two familial and two sporadic male patients with novel mutations in OFD1. All patients had severe renal dysfunction, but they presented different extra-renal phenotypes.
Phenotype-genotype correlation in female OFD1 patients has been shown to be dependent on the location of the mutation. Previous reports indicated that the location of mutations causing typical OFD1 in female patients extends only to exon 17 out of 23 coding exons in OFD1 [1, 10]. Additionally, mutations in exon 3, 8, 9, 13, and 16 have been associated with intellectual disability [5], while those in exon 12 have been associated with renal cysts [1]. Phenotypic variability is often seen in affected females even within the same family. Thauvin-Robinet et al. suggested that skewed × inactivation is partially involved in the pathogenesis leading to the intrafamiliar clinical variability [2]. Bisschoff et al. reported that the direction of skewing is not correlated with disease severity [11]. Nevertheless, nearly all reports on the phenotype-genotype correlation in OFD1 were on female patients. Male OFD1 patients are exceedingly rare, and therefore, very little is currently known on the phenotype-genotype correlation of OFD1 mutations in male patients.
The phenotypic spectrum associated with OFD1 mutations has been recently extended to include JBTS10, SGBS2, RP23, and unclassified malformation syndromes as noted above. JBTS is characterized by a unique cerebellar and brain stem malformation, hypotonia, developmental delay, and unusual breathing pattern. Several male patients presenting symptoms of classical JBTS who have mutations in OFD1 have been reported [12, 13]. SGBS is characterized by overgrowth, coarse face, and other congenital anomalies including heart defect. SGBS2 with an OFD1 mutation of a male patient has also been reported [14]. The patient had a frameshift mutation in exon 16 and showed macrocephaly, severe intellectual disability, low-set ears, digital malformations, and obesity. Except for the patient, other affected males in his family died in their early postnatal period. In both JBTS10 and SGBS2, females are clinically inconspicuous. Webb et al. reported a deep intronic mutation in intron 9 of OFD1 that produced an aberrant transcript and reduced level of correctly spliced transcript, causing X-linked RP [15]. Sharma et al. reported an atypical presentation of OFD1 mutation in a male patient without the classical OFD1 phenotype. This patient presented ESRD without any evidence of polycystic kidney disease [16]. The spectrum of OFD1 mutations in males reported previously is summarized in Table 1. The findings demonstrate phenotypic variability of patients with OFD1 mutation. We were the first to report a patient with OFD1 mutation presenting a Bardet-Biedl syndrome-like phenotype (patient 4).
Coene et al. reported a case of JBTS with OFD1 mutation and proposed that the inverse correlation between the length of OFD1 mutant protein and phenotypic severity is due to differences in the binding of the mutant protein to functionally interacting proteins and disruption of ciliary localization [12]. They also suggested that variable degrees of RNA degradation could contribute to the differences in phenotype. Thus, the location and type of the mutation may be associated with the disease severity. To the best of our knowledge, there have been three reports to date on perinatal death of OFD1 male patients [9,17,20] with three different OFD1 mutations: a missense mutation in exon 2 (ref. 17), a splice site mutation in intron 17 (ref. 20), and a frameshift mutation in exon 21 (ref. 9). Most OFD1 mutations in living male patients were located at the 3′ side of exon 7 or downstream and were non-truncating mutations (Fig. 4). Male patients with OFD1 mutation in exon 21 are alive despite the mutation producing a truncated OFD1 protein. Our patients survived past their perinatal period, which may be due to them having a non-truncating OFD1 mutation downstream of exon 7. Three of the four patients reported here displayed intellectual disability, and all had severe renal disease. Patient 1, who had an in-frame OFD1 mutation in exon 20, did not have intellectual disability. In contrast, patient 2 with a missense mutation in exon 7 had severe intellectual and psychomotor disabilities. Although there may be a strict genotype-phenotype correlation in psychomotor development, this may not be the case with renal disease in male patients with OFD1 mutation.

Known disease-related variants of male patients with OFD1 mutation. JS Joubert syndrome, RP retinitis pigmentosa, SGBS2 Simpson-Golabi-Behmel syndrome type 2; †: perinatal death
In conclusion, the phenotypic spectrum of OFD1 mutations is very broad; thus, evaluation of OFD1 mutation should not be restricted to patients presenting the classical OFD1 phenotype. A comprehensive genetic analysis using NGS is also useful to determine the presence of OFD1 mutation in patients with no family history of OFD1. Further investigations are needed to clarify the mechanisms of such phenotypic diversity of OFD1 mutations.
References
Prattichizzo C, Macca M, Novelli V, Giorgio G, Barra A, Franco B, et al. Mutational spectrum of the oral-facial-digital type I syndrome: a study on a large collection of patients. Hum Mutat. 2008;29:1237–46.
Thauvin-Robinet C, Cossée M, Cormier-Daire V, Van Maldergem L, Toutain A, Alembik Y, et al. Clinical, molecular, and genotype-phenotype correlation studies from 25 cases of oral-facial-digital syndrome type 1: a French and Belgian collaborative study. J Med Genet. 2006;43:54–61.
Saal S, Faivre L, Aral B, Gigot N, Toutain A, Van Maldergem L, et al. Renal insufficiency, a frequent complication with age in oral-facial-digital syndrome type I. Clin Genet. 2010;77:258–65.
Adeva MM, Bisschoff IJ, Castro E, Mouriño D, Morris-Rosendahl DJ. Polycystic kidney disease and orofaciodigital syndrome type 1. Br J Ren Med. 2014;19:9–14.
Ferrante MI, Giorgio G, Feather SA, Bulfone A, Wright V, Ghiani M, et al. Identification of the gene for oral-facial-digital type I syndrome. Am J Hum Genet. 2001;68:569–576.
Toriello HV. Are the oral-facial-digital syndromes ciliopathies? Am J Med Genet A. 2009;149A:1089–95.
Ferrante MI, Zullo A, Barra A, Bimonte S, Messaddeq N, Studer M, et al. Oral-facial-digital type I protein is required for primary cilia formation and left-right axis specification. Nat Genet. 2006;38:112–7.
Morleo M, Franco B. Dosage compensation of the mammalian X chromosome influences the phenotypic variability of X-linked dominant male-lethal disorders. J Med Genet. 2008;45:401–8.
Thauvin-Robinet C, Thomas S, Sinico M, Aral B, Burglen L, Gigot N, et al. OFD1 mutations in males: phenotypic spectrum and ciliary basal body docking impairment. Clin Genet. 2013;84:86–90.
Macca M, Franco B. The molecular basis of oral-facial-digital syndrome, type 1. Am J Med Genet C Semin Med Genet. 2009;151C:318–25.
Bisschoff IJ, Zeschnigk C, Horn D, Wellek B, Rieß A, Wessels M, et al. Novel mutations including deletions of the entire OFD1 gene in 30 families with type 1 orofaciodigital syndrome: a study of the extensive clinical variability. Hum Mutat. 2013;34:237–47.
Coene KL, Roepman R, Doherty D, Afroze B, Kroes HY, Letteboer SJ, et al. OFD1 is mutated in X-linked Joubert syndrome and interacts with LCA5-encoded lebercilin. Am J Hum Genet. 2009;85:465–81.
Field M, Scheffer IE, Gill D, Wilson M, Christie L, Shaw M, et al. Expanding the molecular basis and phenotypic spectrum of X-linked Joubert syndrome associated with OFD1 mutations. Eur J Hum Genet. 2012;20:806–9.
Budny B, Chen W, Omran H, Fliegauf M, Tzschach A, Wisniewska M, et al. A novel X-linked recessive mental retardation syndrome comprising macrocephaly and ciliary dysfunction is allelic to oral-facial-digital type I syndrome. Hum Genet. 2006;120:171–8.
Webb TR, Parfitt DA, Gardner JC, Martinez A, Bevilacqua D, Davidson AE, et al. Deep intronic mutation in OFD1, identified by targeted genomic next-generation sequencing, causes a severe form of X-linked retinitis pigmentosa (RP23). Hum Mol Genet. 2012;21:3647–54.
Sharma S, Kalish JM, Goldberg EM, Reynoso FJ, Pradhan M. An atypical presentation of a male with oral-facial-digital syndrome type 1 related ciliopathy. Case Rep Nephrol. 2016;2016:3181676.
Juric-Sekhar G, Adkins J, Doherty D, Hevner RF. Joubert syndrome: brain and spinal cord malformations in genotyped cases and implications for neurodevelopmental functions of primary cilia. Acta Neuropathol. 2012;123:695–709.
Wentzensen IM, Johnston JJ, Patton JH, Graham JM, Sapp JC, Biesecker LG. Exome sequencing identifies a mutation in OFD1 in a male with Joubert syndrome, orofaciodigital spectrum anomalies and complex polydactyly. Hum Genome Var. 2016;3:15069.
Zhang K, Meng C, Ma J, Gao M, Lv Y, Liu Y, et al. Novel OFD1 frameshift mutation in a Chinese boy with Joubert syndrome: a case report and literature review. Clin Dysmorphol. 2017;26:135–141.
Tsurusaki Y, Kosho T, Hatasaki K, Narumi Y, Wakui K, Fukushima Y, et al. Exome sequencing in a family with an X-linked lethal malformation syndrome: clinical consequences of hemizygous truncating OFD1 mutations in male patients. Clin Genet. 2013;83:135–44.
Acknowledgements
The authors thank all the study participants and their families. We are profoundly grateful to Mrs. Tetsuko Yamanouchi (Kobe University) for her technical assistance. We would like to thank Editage (www.editage.jp) for English language editing. This work was supported by the Health Labor Sciences Research Grant for the Research on Measures for Intractable Diseases (H24-nanchi-ippan-041 to K.I.; H29-nanchi-ippan-039 to N.M.) and Japan Society for the Promotion of Science (KAKENHI Grant Number JP15K09261 and 18K08243 to N.M.).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Kazumoto Iijima has received grant support from Daiichi Sankyo CO., Ltd. and Zenyaku Kogyo Co., Ltd. The remaining authors declare that they have no conflict of interest.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Sakakibara, N., Morisada, N., Nozu, K. et al. Clinical spectrum of male patients with OFD1 mutations. J Hum Genet 64, 3–9 (2019). https://doi.org/10.1038/s10038-018-0532-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s10038-018-0532-x
This article is cited by
-
Comprehensive genetic analysis using next-generation sequencing for the diagnosis of nephronophthisis-related ciliopathies in the Japanese population
Journal of Human Genetics (2022)
-
Clinical and genetic variability of PAX2-related disorder in the Japanese population
Journal of Human Genetics (2020)
-
Bardet–Biedl syndrome in two unrelated patients with identical compound heterozygous SCLT1 mutations
CEN Case Reports (2020)
