
Abstract
Targeting the programmed death 1 (PD-1)/programmed death ligand 1 (PD-L1) pathway represents a milestone in cancer therapy. However, the biologic features of diffuse large B-cell lymphoma (DLBCL) with PD-L1 expression remains unknown. We evaluated the correlation between pSYK and PD-L1 mRNA levels with RNAscope in situ hybridization and protein levels with immunohistochemistry in 108 cases of DLBCL, 25 of which featured loss of B-cell receptor (BCR), and investigated the effects of BCR signaling and MYC on PD-L1 mRNA and protein level with qPCR, immunoblotting and flow cytometery in DLBCL cell lines. PD-L1 amplification was detected with fluorescent in situ hybridization. Animal studies were applied to validate the in vitro findings. pSYK and MYC correlated with both PD-L1 mRNA and protein level. Genetic aberrations involving PD-L1 were rare in DLBCL. BCR signaling and MYC increased PD-L1 mRNA and protein expression. Inhibition of BCR signaling and BCR knockdown down-regulated PD-L1. DLBCL with a loss of loss of BCR showed low levels of PD-L1 mRNA and protein. PD-L1 was down-regulated by ibrutinib in a xenograft mouse model and correlated with slower tumor growth. In conclusion, this study demonstrates that DLBCL with PD-L1 expression features an activated B-cell receptor signal pathway, and that BCR inhibition and PD-L1 blockage may potentially synergize to targeting DLBCL.
Similar content being viewed by others


Introduction
Diffuse large B-cell lymphoma (DLBCL), the most common form of lymphoma in adults, exhibits a striking heterogeneity at the clinical, genetic and molecular levels, and accounts for nearly over one third of non-Hodgkin lymphomas (NHL). While the prognosis of DLBCL has improved due to wider use of immunochemotherapy, patients may develop relapse disease that is refractory to current available therapies, and there is an urgent need to develop new treatment options for these patients. One of the most exciting recent developments in cancer therapy is the introduction of immune checkpoint blockade. Antibodies targeting the (programmed death 1) PD-1–(programmed death Ligand-1) PD-L1 immune checkpoint pathway have shown promising results in the treatment of a series of malignancies [1]. In DLBCL, PD-L1 was reported to be expressed in a subset of cases, mainly activated B-cell-like (ABC) or non-germinal center B-cell-like (non-GCB) [2,3,4]. Clinical trials evaluating therapies targeting PD-L1-PD1 immune checkpoint and developing companion diagnosis for these therapies are ongoing [5]. The frequency of PD-L1 positivity in DLBCL (PD-L1 + DLBCL) varied in previous studies, depending on the assays, antibodies, positive cut-off values and cohorts [1, 5]. Some studies revealed that genetic abnormalities in PD-L1 gene resulted in over-expression of PD-L1 protein [6]. However, these may only account for a small fraction of PD-L1 + DLBCL [7], and other mechanisms remains to be explored.
B-cell receptor (BCR), a transmembrane receptor protein located on the outer surface of B-cells, is expressed on normal mature B-cells and their neoplastic counterparts. Upon BCR activation, effector kinases SYK, LYN and BTK are recruited and phosphorylated, which will in turn activate downstream signaling pathways [8]. Recognition of the pathogenic role of BCR signaling in B-cell lymphoma, such as ABC subtype DLBCL, mantle cell lymphoma and chronic lymphocytic leukemia paved the way for therapeutic strategies targeting the BCR signaling pathway [9, 10]. Previous studies from us and others showed that BCR signaling promotes MYC protein stability and drives malignant behavior in B-cell lymphoma [11, 12]. As BCR and BCR signaling plays a critical role in antigen capture and presenting in normal B-cell, and PD-L1 is expressed on antigen-presenting cells, including B-cell, to control immune interaction between B-cell and effector T-cell, we hypothesized that BCR signaling may regulate PD-L1, undermining anti-tumor immune response.
In the present study, we found the correlation of BCR activity, MYC expression level with PD-L1 mRNA and protein in primary DLBCL, demonstrated BCR signaling and MYC regulating surface PD-L1 level in BCR-dependent DLBCL cell lines, and validated the findings in vivo.
Methods and materials
Case selection
We retrospectively reviewed consecutive flow cytometry (FC) archives maintained at the Department of Pathology, Fudan University Shanghai Cancer Center from 2010 through 2013. All the FC assays were performed with lymphoid tissue biopsies. All cases included in current work received histological confirmation as de novo DLBCL, not otherwise specified (DLBCL, NOS) by two independent pathologists (W-G W and X-Q L) in compliance with WHO classification. Especially, specified subtypes of DLBCL, such as EBV + DLBCL, primary mediastinal large B-cell lymphoma, T-cell/histiocyte-rich large B-cell lymphoma and primary cutaneous DLBCL, leg type, were excluded. BCR negative DLBCL (BCR-DLBCL) was defined with loss of surface immunoglobulin of DLBCL tumor cell population according to our reported methods [13], and BCR positive DLBCL (BCR + DLBCL) was defined with detection of restricted immunoglobulin light chain in DLBCL tumor cells. The use of human samples were approved by the ethical committee of Fudan University Shanghai Cancer Center.
Morphology and immunohistochemistry
Formalin-fixed, paraffin-embedded (FFPE) tissues were obtained and cut into 4-μm sections for morphological examination via H&E staining. PD-L1 antibody (clone number: 28-8, Abcam, Cambridge, MA) was applied in 1:200 dilution on BOND-III automated immunostainer (Leica Biosystems, Melbourne, Australia). The following antibodies (Ventana Medical Systems, Tucson, Arizona, USA) were applied on BenchMark XT automated immunostainer (Ventana Medical Systems, Tucson, Arizona, USA) with Cell Conditioning 1 heat retrieval solution (Ventana Medical Systems, Tucson, Arizona, USA): MYC, CD10, BCL6, and MUM1. Antibody of phospho (pY323) SYK (Epitomics, Burlingame, CA, USA) in 1:100 dilution was applied manually using Envision Method (Dako, Glostrup, Denmark) according to the manufacturer’s protocol. For all stainings, tonsils with reactive hyperplasia were served as external controls, and the reactive lymphocytes as internal controls. The cut-off value of positivity for CD10, BCL6 and MUM1 was set at 30%. IHC results of pSYK, MYC and PD-L1 were calculated as IHC score by multiply the percentage of positive cells (0–100, recorded in the increment by 5%) with mean intensity (0, no staining; 1, weak staining; 2, moderate staining; 3, strong staining), and given a range from 0 to 300. Cases were designated as GCB or non-GCB, using the algorithm specified by Hans et al. [14]. Two pathologists (D S and W-G W) were independently responsible for evaluating the morphological and IHC results.
Genetics
Interphase FISH technique was used to detect PD-L1 gene breaks, and gain of PD-L1 gene was performed with PDL1 Break Apart FISH Probe (Empire Genomics, Buffalo, NY, USA) and CD274(PD-L1)/CEN9q FISH Probe (Abnova, Taipei City, Taiwan District), respectively, using FISH-Tissue Implementation Kit (ZytoVision, Bremerhaven, Germany) according to the protocol specified by the manufacturer. 50 interphase nuclei were counted for each tested probe. Nuclei with a target:control probe ratio of ≥2.0 were defined as amplification. Break apart signal in >5% nuclei was defined as translocation.
mRNA in situ hybridization
PD-L1 mRNA in FFPE samples were examined with RNAscope assay (Advanced Cell Diagnostics, Hayward, CA) following the manufacturer’s manual instructions. Briefly, 4-μm sections were deparaffinized, processed with pretreatment reagents. Tissue sections were hybridized with Hs-CD274-probes (Advanced Cell Diagnostics, Hayward, CA) at 40 °C for 2 h. Hybridization signals were amplified and visualized with RNAscope 2.0 HD detection kit (Advanced Cell Diagnostics, Hayward, CA). Positive control probes targeting the PPIB housekeeping gene (Advanced Cell Diagnostics, Hayward, CA) and negative control probes targeting the bacterial DapB gene (Advanced Cell Diagnostics, Hayward, CA) were used to test mRNA integrity in FFPE tissue and exclude unspecific staining. In situ hybridization (ISH) results of PD-L1 mRNA was calculated as ISH score by multiply the percentage of positive cells (0 to 100, recorded in the increment by 5%) with mean intensity (0, no staining; 1, weak staining; 2, moderate staining; 3, strong staining), and given a range from 0 to 300.
Cell lines and reagents
BCR-dependent DLBCL cell lines (TMD8, HBL1) and BCR-non-dependent/OxPhos-DLBCL cell lines (Toledo, LY4) [10, 15] were used in this study. TMD8 and HBL1 were kindly provided by Dr. Lynn Y. Wang (University of Chicago, Chicago, IL, USA). Toledo and LY4 were purchased from American Tissue Culture Collection (ATCC). All cells were maintained at 37 °C in a 5% CO2 incubator and grown in 1640 medium (Gibco, Grand Island, NE, USA) supplemented with 10% FBS (Gibco, Grand Island, NE, USA). For BCR cross-linking and inhibition, 3 × 106 cells pre-treated with 4 μM R406 (Santa Cruz Biotechnology, Dallas, Texas, USA) or 1 uM ibrutinib (Selleck, Houston, TX, USA) for 60 min or equivalent DMSO vehicle, were stimulated by 10 μg/mL rabbit anti-human IgM (Jackson ImmunoResearch Laboratories, West Grove, PA USA) and goat anti-human IgG (Jackson ImmunoResearch Laboratories, West Grove, PA USA). MYC inhibitor 10058-F4 (Selleck, Houston, TX, USA) was used at 50uM.
Immunoblotting
Cells and tissues were lysed in RIPA Lysis Buffer (Cwbiotech, Beijing, China) containing pan protease and phosphatase inhibitors (Roche, Basel, Switzerland). Total protein was estimated using the Pierce BCA protein assay kit (Thermo Scientific, Rockford, IL, USA). Protein samples of 20 μg were separated by 10.5% SDS-PAGE gel and transferred onto a polyvinylidene fluoride membrane (Millipore, Billerica, MA, USA) using Wet Transfer Cell (Bio-Rad, Hercules, CA, USA). The membrane was blocked in Tris-buffered saline containing 5% bovine serum albumin and 0.1% Tween at room temperature for 1 h, incubated with diluted primary antibody overnight at 4 °C with gentle shaking, and then incubated with goat anti-rabbit IgG and goat anti-mouse IgG secondary antibody (Cell Signaling, Beverley, MA, USA) for 1 h at room temperature. The following primary antibodies were used for analysis: PD-L1 (clone number: 28-8, Abcam, Cambridge, MA, USA) in 1:2000 dilution, MYC (Epitomics, Burlingame, CA, USA) in 1:1000 dilution. GAPDH in 1:100,000 dilution (Proteintech Group, Chicago, IL, USA) was used as a loading control. Signals were developed with enhanced chemiluminescence substrates (Thermo Scientific, Rockford, IL, USA) and visualized by Image Quant LAS 4000 (GE Healthcare Biosciences AB, Uppsala, Sweden). Immunoblotting was quantified using Gel-Pro Analyzer V4.0 (Media Cybernetics, Rockville, MD, USA). The relative expression level of PD-L1 protein was calculated as the integral optical density (IOD) of PD-L1 to IOD of loading control GAPDH protein ratio.
siRNA transfections and adenovirus infection
The small interfering RNA (siRNA) pool against MYC was a custom commercial order from Dharmacon (Lafayette, CO, USA). A non-targeting control siRNA of scrambled nucleotide sequence, used as a negative control, was produced by the same manufacturer. Delivery of MYC siRNA into the TMD8 cell line was performed using nucleofection technology with the Amaxa Nucleofector (Lonza, Basel, Switzerland) following the manufacturer’s instructions. The Fiber-Modified Ad5 adenovirus producing full-length MYC was generated and purchased from Hanbio (Shanghai, China). The TMD8 cell line was infected with MYC adenovirus and control empty adenovirus at a multiplicity of infection (MOI) of 25 and 48 h prior to experiments.
Surface flow cytometry analysis of PD-L1
DLBCL cells were stained with Anti-PD-L1-Alexa Fluor 647 antibody without any permeabilization on ice for 20 min and analyzed by flow cytometry using Accuri C6 (BD, Biosciences, San Jose, CA, USA). Data were analyzed using FlowJo software (Tree Star). Experiments were performed at least twice for each cell line.
RNA extraction and qPCR
Total RNA was extracted using RNeasy mini kit (Qiagen, Hilden, Germany), according to the manufacturer’s instructions. Extracted mRNA was reversed into PrimeScript RT Master Mix (Takara, Dalian, China). Gene expression was quantified by qPCR using the One Step SYBR PrimeScript RT-PCR Kit II (Takara, Dalian, China) and specific primers to analyzed transcripts:
PD-L1: forward 5′-GGTGGTGCCGACTACAA GCGA-3′, reverse 5′-TGACTTCGGCCTTGGGGTAGC-3′;
β-actin forward, 5′-AGCGGGAAATCGTGCGTGAC-3′ and reverse, 5′-ACTCCTGCTTGCTGATCCACATC-3′.
Relative quantitation of the transcript was performed using β-Actin as internal controls for normalization. Each experiment was performed in triplicate and analyzed using the 2−∆CT method of quantitation.
Animal study
Female SCID mice (18–22 g) from Charles River Labs (Beijing, China) were used for this study. Tumor growth was initiated by subcutaneous injection of 1 × 107 TMD-8 cells in a 1:1 mixture of serum-free growth medium and Matrigel (BD Bioscience, San Jose, CA, USA) in the right flank of each subject animal. When tumor volume reached approximately 100 mm3 in size, after removing animals with extreme tumor size and body weight, the remaining mice were randomized into treatment and control groups (6 animals in each group). Either 0.5% methylcellulose or ibrutinib at 10 mg/kg of body weight was administered by oral gavage daily. Tumor size and body weight were recorded twice a week. Animals were euthanatized on Day 25 after treatment, and tumor tissues were removed and embedded in paraffin.
Statistical analyses
Comparisons of continuous variables were performed using unpaired t-test or Mann-Whitney test. Linear correlation and linear regression were used to reveal the relationship between IHC or ISH markers. A P value below 0.05 was considered significant. All analyses were performed using Stata program (V11.0, StataCorp LP, College Station, TX, USA).
Results
PD-L1 expression is increased in ABC-DLBCL but not as a result of amplification or rearrangement
Eighty-three BCR-positive DLBCL (BCR + DLBCL) and twenty-five BCR-negative DLBCL (BCR-DLBCL) were identified and included for subsequent analysis. Clinicopathologic features of 83 BCR + DLBCL and 25 BCR-DLBCL were previously reported [12]. Briefly, of the 83 BCR + DLBCL cases, 40 were males and 43 females with the median age of 57 years (range 19–81 years). Fifty cases had an International prognostic index (IPI) above 2. Germinal center B cell-like (GCB) immunophenotype was seen in 36 (43%) cases, and remaining 47(57%) cases were non-GCB.
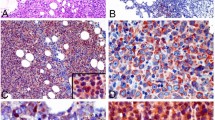
To determine the level of PD-L1 expression in the DLBCL samples, we first analyzed the mRNA and protein level of PD-L1 through in situ hybridization and immunohistochemistry (IHC), respectively. The level of PD-L1 mRNA and protein varied from cases to cases in the current cohort, however the expression pattern in an individual case was quite homogeneous instead of the heterogeneous pattern seen in solid malignancies. As the cut-off value to define PD-L1 positivity is undetermined, we applied H-score method to analyze PD-L1 mRNA and protein level as continuous parameters. PD-L1 mRNA (range: 0–200) and protein (range: 0–300) expression across the samples are summarized in Fig. 1a–c. Overall, PD-L1 mRNA level correlated well with the protein level (Fig. 1a and Fig. S1, r2 = 0.29, P < 0.01), though a small fraction of DLBCL cases with low PD-L1 mRNA level showed high PD-L1 protein expression (Fig. 1a). Since increased PD-L1 expression could be a result of genetic aberrations, we performed FISH to check for PD-L1 rearrangement or amplification events in the samples. Interestingly, PD-L1 gene break-apart and arrangement was seen in only one case (1.2%, Fig. 1a and Fig. S2), and amplification of PD-L1 gene locus was seen in five cases (6%, Fig. 1a and Fig. S2), demonstrating that PD-L1 gene rearrangements or amplifications are rare and do not account for PD-L1 expression in the majority of DLBCL cases. In concurrence with previous studies [7], non-GCB cell of origin (COO) immunophenotype was enriched in cases with high level of both PD-L1 mRNA and protein (Fig. 1a). Compared with GCB cases, non-GCB cases showed a significant higher level of both PD-L1 mRNA (median score 40 vs. 0, P < 0.01) and protein (median score 80 vs. 20, P < 0.01).

Spectrum of PD-L1 genetic status, PD-L1 mRNA expression, PD-L1 protein expression in DLBCL, NOS. a Grid plot of PD-L1 amplification, translocation, PD-L1 mRNA expression and PD-L1 protein expression in DLBCL, NOS; b Representative pictures of PD-L1 mRNA high expression (left) and low expression (right); c Representative pictures of PD-L1 protein high expression (left) and low expression (right)
BCR signaling as demonstrated by pSYK expression is associated with PD-L1 expression in situ
One of the biologic features of ABC (non-GCB) DLBCL is the configuration of chronic active BCR signaling [10], thus we speculated that the expression of PD-L1 is regulated by BCR signaling. Previous studies [16] have shown that pSYK level determined by immunohistochemistry is a representative marker of BCR signaling in DLBCL, thus we examined pSYK expression in IHC in the samples, and as we expected, BCR signaling, as demonstrated by pSYK IHC level correlated well with both PD-L1 mRNA level (Fig. S3 A-C, r2 = 0.22, P < 0.01) and protein level (Fig. S3 A, B, D, r2 = 0.27, P < 0.01). This supported our hypothesis that PD-L1 expression may be regulated by BCR signaling in DLBCL.
PD-L1 expression is regulated by BCR signaling in vitro
We next tested whether PD-L1 expression can be modulated by BCR signaling using in vitro assays on four DLBCL cell lines, including two that were defined as BCR-dependent type (TMD8 and HBL1), and two that were BCR-independent or oxidative phosphorylation (OxPhos) type (Toledo and Ly4) for control. We found that PD-L1 mRNA level was increased in the BCR-dependent cell lines TMD1 and HBL1 after BCR stimulation through surface immunoglobulin cross-linking, but the effect was abolished when BCR signaling was inhibited either by the SYK inhibitor R406 (4 uM) or BTK inhibitor ibrutinib (1 uM) (Fig. S4). In contrast, PD-L1 mRNA level was not changed by BCR stimulation in the OxPhos type DLBCL cell lines Toledo and Ly4 (Fig. S4). Accordingly, the effect of BCR stimulation on PD-L1 expression was also seen at the protein level as demonstrated by immunoblotting experiment (Fig. 2a, b). Our results demonstrate that BCR signaling is necessary and sufficient to drive PD-L1 expression in BCR-dependent DLBCL cells.

BCR signaling regulating PD-L1 total protein level in BCR-dependent DLBCL cell lines instead of BCR-non-dependent/ OxPhos-DLBCL cell lines. a PD-L1 and MYC protein was increased by BCR stimulation and decreased by SYK inhibitor R406 and BTK inhibitor ibrutinib in BCR-dependent DLBCL cell lines (TMD8 and HBL1) instead of OxPhos-DLBCL cell lines (Toledo and Ly4); b Densitometry plots were summarized and analyzed from gel band from triplicates. *P < 0.05; ***P < 0.001, NS not significant
Since surface PD-L1 on cancer cell interacts with its receptor PD-1 in tumor microenvironment to induce immune suppression, surface PD-L1 is considered to be biologically functional. To show that BCR signaling specifically drives PD-L1 surface expression, we performed flow cytometry to analyze surface PD-L1 expression in our four DLBCL cell lines. To our expectation, surface expression of PD-L1 is increased by BCR stimulation, but the effect was abolished by BCR inhibition in BCR-dependent cell lines (TMD8 and HBL1) but not in OxPhos type cell lines (Fig. 3). The results show that BCR signaling drives not only PD-L1 expression at the mRNA and protein level in DLBCL, but also PD-L1 expressed at the cell surface, indicating biological functionality.

BCR signaling regulating surface PD-L1 in BCR-dependent DLBCL cell lines instead of BCR-non-dependent/ OxPhos- DLBCL cell lines. Flow cytometery demonstrated that surface PD-L1 protein was increased by BCR stimulation and decreased by BCR inhibition in BCR-dependent DLBCL cell lines (TMD8 and HBL1) instead of OxPhos-DLBCL cell lines (Toledo and Ly4). Mean fluorescent insensitivities from triplicates were summarized and analyzed in the right panel. *P < 0.05; **P < 0.01; ***P < 0.001, NS not significant
PD-L1 expression is regulated by BCR signaling via increasing MYC expression
Previous studies from us [12] and others [11] revealed that BCR stabilizes and increases MYC protein in B-cell malignancies including DLBCL. Meanwhile, MYC can directly bind to the promoter of PD-L1 gene and promote PD-L1 transcript [17]. We thus hypothesized that PD-L1 expression is regulated by BCR through increased MYC expression. We first confirmed that BCR stimulation does indeed increase MYC expression in BCR-dependent DLBCL cell lines but not OxPhos type cell lines (Fig. S5A). To determine if MYC is required for PD-L1 expression in DLBCL, we treated HBL1 and TMD8 cells with the MYC inhibitor 10058-F4. Concurrent with our hypothesis, MYC inhibition significantly decreased PD-L1 mRNA level in the cells (Fig. S5B). Similarly, PD-L1 mRNA levels decreased with siRNA knockdown in TMD8 cells (Fig. S5C), but increased when MYC was overexpressed through adenovirus transfection (Fig. S5D). The effect of MYC inhibition or expression on PD-L1 were also evident at the cell surface expression level, as demonstrated by flow cytometry (Fig. 4b, c).

MYC regulating PD-L1 level. a Surface PD-L1 protein was decreased by MYC inhibitor 10058-F4. b or MYC siRNA knockdown, c and was increased by MYC over-expression
Finally, to determine if our in vitro findings were also relevant in a clinical context, we retrospectively examined PD-L1 expression in our primary DLBCL cohort previously used to identify an association between BCR signaling and MYC expression [12]. Indeed, the expression spectrum of MYC was similar to that of PD-L1 mRNA and PD-L1 protein in our DLBCL cohort (Fig. S6A), and MYC protein level correlated well with both PD-L1 mRNA and PD-L1 protein levels (Fig. S6B and S6C). These observations together with the in vitro findings supported our hypothesis that BCR signaling regulated PD-L1 via increasing MYC expression.
BCR-DLBCL features low level of PD-L1 expression
We next analyzed the association between BCR signaling and MYC and PD-L1 expression in our BCR-DLBCL samples. As previous reported, BCR-DLBCL featured a low level of pSYK (Fig. S7A) and MYC (Fig. S7B), indicating a lack of BCR-MYC signaling in this entity [13]. Interestingly, both PD-L1 mRNA level (Fig. S7C and S7E) and PD-L1 protein level (Fig. S7D and S7F) were homogeneous lower than those in BCR + DLBCL. As the BCR-non-dependent/OxPhos type DLBCL cell lines (Toledo and Ly4) used in current study were also negative for surface immunoglobulin/BCR [18], and their PD-L1 and MYC levels did not respond to either BCR stimulation or inhibition, the observations in primary BCR-DLBCL further supported that PD-L1 was specifically regulated by BCR signaling via MYC.
PD-L1 is down-regulated by ibrutinib in xenograft mouse model and is correlated with slower tumor growth
To demonstrate that PD-L1 is regulated by BCR signaling in vivo, we established a xenograft model using the TMD8 cell line. Concurrent with our in vitro findings, oral administration of 50 mg/kg of ibrutinib dramatically suppressed the tumor growth in the TMD8-transplanted SCID mice (Fig. 5a). As shown in Fig. 5b, c, this is accompanied by significantly reduced levels of PD-L1 protein in xenograft tumor tissues compared to mice administered with vehicle (Fig. 5b, c). The results of PD-L1 staining of 6 tissue samples from either ibrutinib-administered or control-administered mice were summarized in Fig. 5d. Our results lends further support that BCR signaling regulates PD-L1 expression and reveals a previously unexplored mechanism of BCR inhibition treatment in DLBCL.

BCR inhibition down-regulates PD-L1 protein in vivo. a Ibrutinib treatment resulted in growth inhibition of TMD8 xenografts. Tumor-bearing animals were administered 10 mg/kg Ibrutinib PO (n = 6) or vehicle consisting of 0.5% methylcellulose (n = 6). b Immunostaining of PD-L1 of TMD8 xenografts treated with vehicle or Ibrutinib. c Quantification of of PD-L1 staining in six xenografts treated with either vehicle or ibrutinib. *P < 0.05; **P < 0.01. d Western blotting of PD-L1 of TMD8 xenografts treated with vehicle or Ibrutinib
Discussion
PD-L1 is ligand molecule expressed on the surface of antigen-presenting cells. When it interacts with its receptor PD-1 on effector T-cells such as CD8 + cytotoxic T-cell, it transmits a negative regulatory signal resulting in T-cell suppression and anergy [19]. Various malignancies have been shown to overexpress PD-L1 and escape T cell–mediated surveillance and killing [1, 20]. Successful treatment of patients with malignancies using antibodies against PD-1 or PD-L1 highlighted the critical importance of PD-1-PD-L1 axis in immune escape during cancer pathogenesis and progression. However, the mechanism for this immune escape has not been fully elucidated in lymphoma, with the exception of PD-L1 expression by gene amplification in classic Hodgkin’s lymphoma [6] and by structural variations disrupting the 3′-untranslated region (UTR) PD-L1 gene mainly in adult T-cell leukaemia/lymphoma [21]. Georgiou K et al. reported that amplification and translocation of PD-L1/PD-L2 locus are rare genetic aberrations in DLBCL that may lead to PD-L1 expression, and involves 5% of primary DLBCL including EBV + DLBCL in a Chinese cohort [7]. In the Cancer Genome Atlas database, an insertion mutation at the 3′-UTR is the only PD-L1 mutation identified in one out of the 37 DLBCL cases [22] (TCGA, https://portal.gdc.cancer.gov/ssms/c694714a-9650-5c93-8285-db05de46d0c7). These observation led us to pursue other mechanisms that may account for PD-L1 over-expression in DLBCL. As PD-L1 is commonly expressed on the surface of antigen-presenting cells such as B-cells and macrophages to modulate the immune-inhibitory network [23], and BCR signaling is critical for antigen presentation in B-cells [24], we hypothesized that DLBCL with PD-L1 expression featured an activated B-cell receptor signal pathway.
In our DLBCL cohort, the spectrum of PD-L1 expression and genetic aberrations were concurrent with the findings reported by others [2,3,4, 7, 25, 26]. There was a broad spectrum of PD-L1 expression whereas translocation and amplification events involving PD-L1 were rare. In addition, PD-L1 expression was enriched in the non-GCB subtype. As non-GCB or ABC subtype of DLBCL represents a group of disease featuring abnormally active BCR signaling, suggesting a potential role of BCR signaling in PD-L1 expression, which may lead to immune escape. To validate the causal relation between BCR signaling and PD-L1 expression, we first performed in vitro assays using the BCR-dependent cell linesTMD8 and HBL1, and the BCR-independent cell lines Toledo and Ly4 as control. We showed that BCR signaling indeed up-regulated PD-L1 expression both at the transcription level and total/surface protein level in the BCR-dependent cell lines but not the BCR-independent ones. Importantly, the PD-L1 level can be decreased by the BCR inhibitor ibrutinib and R406, implicating the potential synergistic effect of anti-BCR and anti-PD-L1-PD1 therapies [27].
Previous studies have shown that PD-L1 transcription can be regulated by MYC, whereas BCR signaling is known to stabilize and up-regulate MYC protein. Our results is the first to clearly demonstrate the link between BCR, MYC and PD-L1 expression in DLBCL. While our findings have direct biological and clinical implications, we recognize possible limitations in our reductionist approach, which may preclude us from other important pathways. For example, the NF-kappaB pathway linked to BCR signaling is another possible mechanism which will be investigated in further study, since ABC DLBCL is characterized by NF-kappaB signaling [10] and NF-kappaB signaling may also promote the PD-L1 expression [28]. Meanwhile, MYC may confer a different effect on PD-L1 depending on the pathogenic driver of the malignancy. For example, Durand-Panteix S et al. reported that PD-L1 is down-regulated by MYC via repressing secretory lysosome migration and surface membrane export of PD-L1 in EBV-immortalized B cell [28]. On the other hand, EBV + DLBCL is driven by EBV latency III program, which may result in up-regulation of PD-L1 in this context [28]. Delineating the effect on MYC over PD-L1 in different disease contexts will be an important step in unveiling the full spectrum of the PD-L1 regulatory network and further aid in determining candidates for immune checkpoint therapy.
In conclusion, we demonstrated that BCR signaling up-regulates PD-L1 via MYC in DLBCL. We believe that tumor cells hijacking the BCR-MYC-PD-L1 axis may play an important role in escaping immunity, and by targeting abnormal BCR signaling we may be able to reset the immunosurveillance activity in the tumor microenvironment resulting in tumor clearance. Our findings may help select DLBCL patients that may better benefit from immune checkpoint inhibitors and also rationalizes the use of combination therapy with BCR inhibitors and checkpoint inhibitors.
References
Jelinek T, Mihalyova J, Kascak M, et al. PD-1/PD-L1 inhibitors in haematological malignancies: update 2017. Immunology. 2017;152:357–71.
Kiyasu J, Miyoshi H, Hirata A, et al. Expression of programmed cell death ligand 1 is associated with poor overall survival in patients with diffuse large B-cell lymphoma. Blood. 2015;126:2193–201.
Rossille D, Gressier M, Damotte D, et al. High level of soluble programmed cell death ligand 1 in blood impacts overall survival in aggressive diffuse large B-Cell lymphoma: results from a French multicenter clinical trial. Leukemia. 2014;28:2367–75.
Chen BJ, Chapuy B, Ouyang J, et al. PD-L1 expression is characteristic of a subset of aggressive B-cell lymphomas and virus-associated malignancies. Clin Cancer Res. 2013;19:3462–73.
Juarez-Salcedo LM, Sandoval-Sus J, Sokol L, et al. The role of anti-PD-1 and anti-PD-L1 agents in the treatment of diffuse large B-cell lymphoma: The future is now. Crit Rev Oncol Hematol. 2017;113:52–62.
Roemer MG, Advani RH, Ligon AH, et al. PD-L1 and PD-L2 Genetic Alterations Define Classical Hodgkin Lymphoma and Predict Outcome. J Clin Oncol. 2016;34:2690–7.
Georgiou K, Chen L, Berglund M, et al. Genetic basis of PD-L1 overexpression in diffuse large B-cell lymphomas. Blood. 2016;127:3026–34.
Rickert RC. New insights into pre-BCR and BCR signalling with relevance to B cell malignancies. Nat Rev Immunol. 2013;13:578–91.
Wilson WH, Young RM, Schmitz R, et al. Targeting B cell receptor signaling with ibrutinib in diffuse large B cell lymphoma. Nat Med. 2015;21:922–6.
Davis RE, Ngo VN, Lenz G, et al. Chronic active B-cell-receptor signalling in diffuse large B-cell lymphoma. Nature. 2010;463:88–92.
Varano G, Raffel S, Sormani M, et al. The B-cell receptor controls fitness of MYC-driven lymphoma cells via GSK3beta inhibition. Nature. 2017;546:302–6.
Wang WG, Liu ZB, Jiang XN, et al. MYC protein dysregulation is driven by BCR-PI3K signalling in diffuse large B-cell lymphoma. Histopathology. 2017;71:778–85.
Wang WG, Cui WL, Wang L, et al. Loss of B-cell receptor expression defines a subset of diffuse large B-cell lymphoma characterized by silent BCR/PI3K/AKT signaling and a germinal center phenotype displaying low-risk clinicopathologic features. Am J Surg Pathol. 2015;39:902–11.
Hans CP, Weisenburger DD, Greiner TC, et al. Confirmation of the molecular classification of diffuse large B-cell lymphoma by immunohistochemistry using a tissue microarray. Blood. 2004;103:275–82.
Caro P, Kishan AU, Norberg E, et al. Metabolic signatures uncover distinct targets in molecular subsets of diffuse large B cell lymphoma. Cancer Cell. 2012;22:547–60.
Bogusz AM, Baxter RH, Currie T, et al. Quantitative immunofluorescence reveals the signature of active B-cell receptor signaling in diffuse large B-cell lymphoma. Clin Cancer Res. 2012;18:6122–35.
Casey SC, Tong L, Li Y, et al. MYC regulates the antitumor immune response through CD47 and PD-L1. Science. 2016;352:227–31.
Chen L, Monti S, Juszczynski P, et al. SYK-dependent tonic B-cell receptor signaling is a rational treatment target in diffuse large B-cell lymphoma. Blood. 2008;111:2230–7.
Goodman A, Patel SP, Kurzrock R. PD-1-PD-L1 immune-checkpoint blockade in B-cell lymphomas. Nat Rev Clin Oncol. 2017;14:203–20.
Kythreotou A, Siddique A, Mauri FA, et al. PD-L1. J Clin Pathol. 2018;71:189–94.
Kataoka K, Shiraishi Y, Takeda Y, et al. Aberrant PD-L1 expression through 3’-UTR disruption in multiple cancers. Nature. 2016;534:402–6.
National cancer institute GDC data portal. https://portal.gdc.cancer.gov/ssms/c694714a-9650-5c93-8285-db05de46d0c7. Accessed on 8 Jul 2018.
Tangye SG, Ma CS, Brink R, et al. The good, the bad and the ugly—TFH cells in human health and disease. Nat Rev Immunol. 2013;13:412–26.
Jimenez DON, De Giovanni M, Fioravanti J, et al. Pathogen-specific B-cell receptors drive chronic lymphocytic leukemia by light-chain-dependent cross-reaction with autoantigens. EMBO Mol Med. 2017;9:1482–90.
Xing W, Dresser K, Zhang R, et al. PD-L1 expression in EBV-negative diffuse large B-cell lymphoma: clinicopathologic features and prognostic implications. Oncotarget. 2016;7:59976–86.
Nicolae A, Pittaluga S, Abdullah S, et al. EBV-positive large B-cell lymphomas in young patients: a nodal lymphoma with evidence for a tolerogenic immune environment. Blood. 2015;126:863–72.
Sagiv-Barfi I, Kohrt HE, Czerwinski DK, et al. Therapeutic antitumor immunity by checkpoint blockade is enhanced by ibrutinib, an inhibitor of both BTK and ITK. Proc Natl Acad Sci U S A. 2015;112:E966–E972.
Durand-Panteix S, Farhat M, Youlyouz-Marfak I, et al. B7-H1, which represses EBV-immortalized B cell killing by autologous T and NK cells, is oppositely regulated by c-Myc and EBV latency III program at both mRNA and secretory lysosome levels. J Immunol. 2012;189:181–90.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Wang, WG., Jiang, XN., Sheng, D. et al. PD-L1 over-expression is driven by B-cell receptor signaling in diffuse large B-cell lymphoma. Lab Invest 99, 1418–1427 (2019). https://doi.org/10.1038/s41374-019-0262-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41374-019-0262-5
