
Abstract
Inflammatory myofibroblastic tumors arising in infants are rare, poorly investigated and mostly reported as isolated cases or as a part of larger series thus, their clinicopathological and molecular features are essentially unknown. Archival files from two large pediatric institutions and a tumor registry were queried for pediatric inflammatory myofibroblastic tumors. Available material from patients ≤12 months of age was reviewed. Additional immunostains (ALK-1, D240, WT1) and ALK-FISH studies were performed as needed. Targeted anchored multiplex PCR with next-generation sequencing was done in all cases. A total of 12 of 131 infantile cases (mean 5.5 months) were identified (M:F of 2:1). Anatomic locations included intestinal/mesenteric (n = 6), head/neck (n = 3), and viscera (n = 3). Half of tumors showed a hypocellular myxoid pattern, perivascular condensation, and prominent vasculature with vague glomeruloid structures present in four of them. The remaining cases exhibited a more cellular pattern with minimal myxoid component. ALK-1 immunohistochemistry was positive in most cases (11/12) with cytoplasmic-diffuse (n = 6), cytoplasmic-granular (n = 2), and dot-like (n = 3) staining patterns. ALK fusion partners identified in five cases included EML4, TPM4, RANBP2, and a novel KLC1. Three inflammatory myofibroblastic tumors showed fusions with other kinases including TFG-ROS1 and novel FN1-ROS1 and RBPMS-NTRK3 rearrangements. Favorable outcome was documented in most cases (10/11) with available follow-up (median 17 months) while three patients were successfully treated with crizotinib. In summary, infantile inflammatory myofibroblastic tumors are rare and can exhibit paucicellular, extensively myxoid/vascular morphology with peculiar immunophenotype mimicking other mesenchymal or vascular lesions. All tumors harbored kinase fusions involving ALK, ROS1, and NTRK3 including three novel fusion partners (KLC1, FN1, and RBPMS, respectively). A favorable response to crizotinib seen in three cases supports its potential use in infants as seen in older patients. Awareness of these unusual morphologic, immunophenotypic, and molecular features is critical for appropriate diagnosis and optimized targeted therapy.
Similar content being viewed by others
Introduction
Inflammatory myofibroblastic tumors are rare mesenchymal myofibroblastic lesions of intermediate malignant potential with tendency for local recurrence and rare metastases usually seen within the first and second decades of life [1], though infantile cases have also been reported [2,3,4]. Although inflammatory myofibroblastic tumors may arise at any site, these tumors preferentially involve the abdominal cavity, lung, head and neck, and mediastinum. In children, there is a predilection for the abdominal cavity, especially omentum, mesentery, retroperitoneum, pelvis, or intrabdominal viscera [3,4,5,6,7,8].
Histologically, inflammatory myofibroblastic tumors are composed of plump or elongated myofibroblasts admixed with a characteristic inflammatory infiltrate of lymphocytes, plasma cells, and eosinophils [3, 4] embedded in a variably myxoid to collagenous stroma [2]. Although inflammatory myofibroblastic tumors exhibit a wide morphologic spectrum, Coffin et al. [1] described three basic histologic patterns that occasionally can be present even within the same tumor in variable proportions. The first pattern displays loose spindle cells in a myxoid and edematous stroma along with a network of small blood vessels (myxoid-vascular pattern). The second pattern shows a denser proliferation of spindle cells within an inflammatory background resembling tumors such as fibrous histiocytoma, fibromatosis, or smooth muscle neoplasms (compact spindle cell pattern). The third pattern is paucicellular with extensive areas of plate-like collagen resembling desmoid fibromatosis or scar tissue and relatively sparse inflammation (hypocellular fibrous pattern) [1, 7]. The distinction of inflammatory myofibroblastic tumors displaying the latter pattern from IgG4-related sclerosing disease may be challenging [9].
A subset of inflammatory myofibroblastic tumors exhibiting a strikingly diffuse myxoid-vascular morphology has been recently described in infants [10]. These tumors overlap with the so-called omental mesenteric myxoid hamartoma of infancy described by González-Crussi in 1983 [11]. As a matter of fact, the current World Health Organization (WHO) classification of soft tissue tumors [12] considers omental mesenteric myxoid hamartoma within the spectrum of inflammatory myofibroblastic tumors, likely representing an infantile variant [1].
Up to 60% of inflammatory myofibroblastic tumors harbor ALK gene rearrangements on chromosome 2p23 leading to the formation of a chimeric fusion protein and overexpression of the ALK-1 protein, which can be detected by ancillary techniques including immunohistochemistry, revealing particular staining patterns according to the specific partner gene involved [4,5,6]. Numerous ALK fusion partner genes are now recognized including NPM, TPM3, tropomyosin 4 (TPM4), TFG, ATIC, CLTC, MSN, RAN binding protein 2 (RANBP2), CARS, SE31L1, NUMA1, and echinoderm microtubule-associated protein-like 4 (EML4), among others [2, 4, 8, 13]. Subsequently, Lovly et al. [5] demonstrated the presence of alternative fusions involving other kinases different from ALK (ROS1 or PDGFRB) by next-generation sequencing. More recently, Alassiri et al. [14] identified two inflammatory myofibroblastic tumors harboring ETV6-NTRK3 and other NTRK rearrangements—one of them arising in a young adult—in a series of ALK negative inflammatory myofibroblastic tumors, while a rare RET-rearranged inflammatory myofibroblastic tumor was reported by Antonescu et al. [4] in an adult patient.
Despite several large series of pediatric inflammatory myofibroblastic tumors available in the literature, infantile cases have been poorly investigated, mainly due to their rarity. To the best of our knowledge, most infantile inflammatory myofibroblastic tumors have been described as isolated case reports with only one small series while their clinicopathological features remain largely unknown [1, 2, 4,5,6, 8,9,10,11, 15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63]. The identification of a dot-like ALK-1 immunohistochemical staining pattern in a recent series [10] as well as the identification of a chimeric A2M-ALK rearrangement in two infantile pulmonary inflammatory myofibroblastic tumors with peculiar microcystic morphology [20] might suggest distinct clinicopathological and biologic features of these entities when occurring within the first year of life. Our aim is to investigate the frequency of inflammatory myofibroblastic tumors in children ≤12 months of age and to characterize their clinical, histologic, and molecular features.
Materials and methods
Institutional and consultation files from two large pediatric institutions (UPMC Children’s Hospital of Pittsburgh and the Children’s Hospital of Philadelphia) were searched for cases of inflammatory myofibroblastic tumors, following institutional review board approval. Additional cases were also ascertained from the Italian Association of Pediatric Hematology and Oncology registry for soft tissue tumors. Only those patients diagnosed with inflammatory myofibroblastic tumors ≤12 months of age were selected for further review. Two of the cases included in this study were previously reported as individual case reports (cases 1 and 8) [8, 21].
Hematoxylin-eosin stained sections and available immunohistochemical and molecular material were reviewed. Additional molecular studies and/or FISH for ALK rearrangements were reviewed as needed. Four pediatric and soft tissue pathologists (RA, IJ, SR, LS) reviewed all stains. The pertinent clinical information was retrieved from electronic medical records. For each case, the following histologic features were reviewed: (1) Cellularity (hypocellular or hypercellular); (2) amount of inflammatory component (scant or brisk); (3) degree of nuclear atypia (focal or diffuse); (4) number of mitoses per 10 high-power fields; (5) degree of vascularization (scant or prominent); and (6) type/amount of stroma (collagenized or myxoid/scant or prominent).
Immunohistochemistry for ALK-1 (clone ALK01, Ventana Roche, Tucson, AZ or monoclonal mouse anti-human CD246, clone ALK1, DAKO UK), D240 (podoplanin, mouse monoclonal Covance; Cat # 3730, Dil 1:50) and WT1 (mouse monoclonal, clone 6F-H2, Cell marque; Cat # 760.4397, Predilute) were performed on 3-µm thick formalin-fixed, paraffin-embedded sections when not available for review. Neoplasms showing any positive cytoplasmic and/or membranous staining in the tumor cells were considered to be ALK-1 positive. For each case, the ALK-1 immunohistochemistry was qualified as membranous, smooth-cytoplasmic, granular cytoplasmic, or paranuclear dot-like staining pattern. WT1 and D240 were considered positive when more than 10% of cells showed nuclear and membranous staining, respectively.
When tissue was available, targeted anchored multiplex PCR was performed followed by next-generation sequencing (Archer DX, Boulder, CO) as previously described [64]. Briefly, RNA or total nucleic acid was extracted from fresh or formalin-fixed, paraffin-embedded tissue, respectively. RNA was reverse-transcribed to generate cDNA and molecular barcode adapters were ligated to cDNA followed by two rounds of target-specific PCR. All libraries were sequenced on Illumina MiSeq or HiSeq (Illumina, San Diego, CA). Data analysis was performed using Archer analysis software. In most instances, identified fusions were then confirmed with Sanger sequencing. When tissue was inadequate for sequencing, FISH was performed on formalin-fixed, paraffin-embedded tissue with ALK (2p23) and receptor tyrosine kinase (ROS1) (6q22) break apart FISH probes (Leica Biosystems, Amsterdam, The Netherlands) using standard protocols.
Results
Clinical findings
In total, 12 infantile inflammatory myofibroblastic tumors were identified. The median age at diagnosis was 6 months (range 0–12 months) with a male-to-female ratio of 2:1. The most common site of involvement was intestinal/mesenteric (n = 6), followed by head and neck (n = 3), and viscera (n = 3; one each in lung, liver, and adrenal gland, respectively). No evidence of any remarkable syndromic association was identified except for one patient who developed a pulmonary inflammatory myofibroblastic tumor in the setting of multiple congenital anomalies, including heterotaxia, asplenia, imperforate anus, micrognathia, and right-sided sequestration with esophageal fistula (case 10). No recurrences were documented in nine cases with available follow-up information. The clinical presentation was available for review in ten cases and included isolated abdominal distention (n = 3), gastrointestinal (n = 1), and site-specific symptoms (n = 3). In addition, one patient had a prenatal diagnosis while two patients were asymptomatic. In three cases there was documented treatment with crizotinib with adequate response (cases 4, 8, and 12). The median follow-up was 17 months with complete remission for most patients (11/12). One patient died due to numerous congenital anomalies (case 10). The clinicopathological features of the inflammatory myofibroblastic tumors in this series are summarized in Table 1.
Pathologic findings
Macroscopic descriptions were available in eight cases. Overall, the tumors presented as lobulated and firm nodules with a homogeneous tan-white to tan-pink cut surface and focal translucent or fibrotic areas. Only focal areas of hemorrhage were described, and no evidence of necrosis was reported. The tumor size ranged from 1 to 10 cm with an average of 5.2 cm. There was no evidence of multifocality. Morphologic, immunophenotypic, and molecular findings are summarized in Table 2.
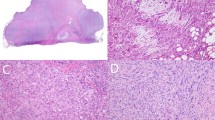
Two main histologic patterns were identified. Half of inflammatory myofibroblastic tumors (cases 1–6) showed a predominantly diffuse myxoid or edematous stroma with variably collagenized areas and low cellularity, with focal perivascular condensation (hypocellular myxoid pattern, Fig. 1a). A prominent vascular network characterized by irregularly shaped and dilated vessels was consistently identified in all tumors being particularly prominent in myxoid areas. Numerous vessels also exhibited perivascular hyalinosis. All inflammatory myofibroblastic tumors displayed relatively bland, plump to stellate cells with small nucleoli. An intra-abdominal inflammatory myofibroblastic tumor (case 2) showed focal epithelioid-like cellular areas with vesicular nuclei and occasional small conspicuous nucleoli (Fig. 2). Four cases showed multifocal condensation of myofibroblastic cells forming glomeruloid structures (cases 1, 2, 4, and 5) (Fig. 1a, inset) whereas one case presented with few isolated cartilage-like nodules. In case 4, the unusual vascular pattern and expression of podoplanin (D240) initially prompted a diagnosis of a vascular tumor (i.e., kaposiform hemangioendothelioma, Fig. 1c).

a, inset: Hypocellular myxoid inflammatory myofibroblastic tumor detected in-utero (case 4, EML4-ALK) with prominent glomeruloid-like features. b Diffuse cytoplasmic expression of ALK-1. c Multifocal cytoplasmic expression of D240. d Diffuse cytoplasmic and focal nuclear expression of WT1

a Hypocellular myxoid inflammatory myofibroblastic tumor presenting as a rapidly growing intrabdominal mass (case 2, RANBP2-ALK). b Scant inflammation with only rare stromal neutrophils. c Focally denser areas containing small to medium sized oval cells with inconspicuous nucleoli. d Subtle, dot-like expression of ALK-1
The remaining six cases (cases 7–12) exhibited more classic features with higher cellularity, dense collagenized stroma with variably sclerotic areas and a minimal to focal myxoid component (cellular collagenized pattern, Fig. 3a). In this particular group, the inflammatory component was moderate to prominent (brisk) with a consistent number of lymphocytes and plasma cells and variable presence of neutrophils, eosinophils and macrophages. No significant atypia was observed other than occasional cells with multinucleation and prominent nucleoli. The mitotic activity was low, with less than 2 mitosis per 10 high-power fields. No atypical mitotic figures or necrosis were identified in any case regardless histologic pattern.

a A more conventional/cellular inflammatory myofibroblastic tumor presenting as an adrenal gland mass (case 7, TPM4-ALK). b Diffuse cytoplasmic expression of ALK-1. c Diffuse cytoplasmic expression of D240. d Diffuse nuclear and cytoplasmic expression of WT1
Immunohistochemistry
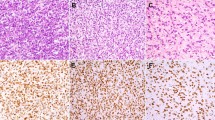
ALK-1 immunostaining was positive in 11 out of 12 cases. Hypocellular myxoid inflammatory myofibroblastic tumors exhibited a dot-like (n = 3) (Figs. 2d and 4b), diffuse cytoplasmic (n = 2) (Figs. 1b and 3b), or granular cytoplasmic (n = 1) staining pattern. Most inflammatory myofibroblastic tumors with a predominantly cellular collagenized morphology had either a diffuse cytoplasmic (n = 4) or granular cytoplasmic (n = 1) staining pattern for ALK-1 with one negative case (case 11).

a Hypocellular myxoid inflammatory myofibroblastic tumor presenting as an intrabdominal mass (case 1, ALK-FISH positive). b, arrows: Subtle, dot-like expression of ALK-1. c Diffuse cytoplasmic expression of D240. d Predominantly nuclear expression of WT1
Smooth muscle actin was variably positive in all cases except by one (case 2). Desmin was positive in 5/12 inflammatory myofibroblastic tumors and cytokeratin AE1/AE3 was focally positive in 2/12 cases. All inflammatory myofibroblastic tumors with available myogenin immunohistochemistry were negative (cases 1, 2, 8, 10, and 12). Additional immunohistochemistry in nine inflammatory myofibroblastic tumors with available material showed cytoplasmic positivity for podoplanin (D240) in 89% (8/9) of cases (Figs. 1c, 3c, and 4c) as well as cytoplasmic and nuclear positivity for WT1 in 66% (6/9) of cases (Figs. 1d, 3d, and 4d).
Molecular cytogenetics and RNA sequencing
ALK-FISH studies were performed in nine inflammatory myofibroblastic tumors with the majority of them being positive (n = 7) and two negative cases (cases 10 and 11). In addition, targeted RNA sequencing was attempted in ten inflammatory myofibroblastic tumors (cases 1–5, 7–11) confirming ALK rearrangements in five of them including the following fusion partners: TPM4-ALK (n = 2), EML4-ALK (n = 1), RANBP2-ALK (n = 1), and Kinesin light chain 1(KLC1)-ALK (n = 1, Fig. 5a). The two ALK-FISH negative cases (cases 10 and 11) demonstrated FN1-ROS1 (Fig. 5b) and TFG-ROS1 rearrangements, respectively. In one case (case 3) an RBPMS-NTRK3 rearrangement was identified (Fig. 5c), whereas in two inflammatory myofibroblastic tumors the targeted RNA sequencing failed (cases 1 and 5). The remaining cases had no material available for further ancillary testing (cases 6 and 12). Detailed molecular findings are summarized in Table 3.

a An ALK-rearranged inflammatory myofibroblastic tumor presenting as a tongue mass (case 8) harboring a novel KLC1 fusion partner b An ROS1-rearranged inflammatory myofibroblastic tumor presenting as lung mass (case 10) harboring a novel FN1 fusion partner c An NTRK3-rearranged inflammatory myofibroblastic tumor presenting as an intrabdominal mass (case 3) harboring a novel RBPMS fusion partner. T1 fibronectin type 1 domain, T2 fibronectin type 2 domain, T3 fibronectin type 3 domain, TM transmembrane domain, MAM meprin/A-5 protein/receptor protein-tyrosine phosphatase mu domain
Discussion
Infantile inflammatory myofibroblastic tumors are rare, most of them included as part of larger pediatric series or as isolated case reports. Our review of the literature identified 85 infantile inflammatory myofibroblastic tumors; most of them with limited morphologic/immunophenotypic descriptions and minimal to absent molecular characterization [1, 2, 4,5,6, 8,9,10,11, 15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63] (Table 4) with only one clinicopathological study describing four so-called omental mesenteric myxoid hamartomas [11] and ALK-1 dot-like staining pattern with unusual expression of calretinin, podoplanin, and WT1 [10].
The morphologic heterogeneity of inflammatory myofibroblastic tumors is well recognized across all age groups. In our study, 50% of inflammatory myofibroblastic tumors showed a hypocellular myxoid pattern. A rich vascular network was consistently present in all cases, but it was particularly evident within myxoid areas. Peculiar multifocal vascular structures showing a vague glomeruloid arrangement were seen in 33% (4/12) of inflammatory myofibroblastic tumors and were prominent in one case (case 4) prompting an initial diagnosis of kaposiform hemangioendothelioma. Remarkably, the inflammatory component was never a dominant finding in any of the hypocellular myxoid inflammatory myofibroblastic tumors, especially in case 4. The reasons for this minor inflammatory component are unclear, but we can speculate that the immature inflammatory milieu typical of newborn infants might be responsible for the limited number of inflammatory cells [20, 36]. Based on our findings, we propose that the hypocellular myxoid appearance is, at least in part, age-related as it appears to be less predominant in older children; however, these observations require further confirmation in additional series. The remaining inflammatory myofibroblastic tumors in our series (6/12) exhibited a denser cellular collagenized pattern. Morphologic atypia including necrosis, prominent fascicular architecture, focal herringbone pattern, multinucleation, pleomorphism, abundant large ganglion-like cells, and atypical mitoses [45], do not appear to be part of the morphologic spectrum seen in infantile inflammatory myofibroblastic tumors. Our series also highlights diagnostic pitfalls in those predominantly hypocellular myxoid inflammatory myofibroblastic tumors. Morphologically, the differential diagnosis in this group of tumors may be challenging without an appropriate immunohistochemical panel including ALK-1 antibody. This is well demonstrated by case 4, originally diagnosed as a kaposiform hemangioendothelioma, in absence of ALK-1 staining and presence of diffuse expression of D240. This case was subsequently reclassified as inflammatory myofibroblastic tumor only after next-generation sequencing studies were performed. However, the glomeruloid areas observed in infantile inflammatory myofibroblastic tumors (in contrast to kaposiform hemangioendothelioma), are composed of myofibroblastic cells rather than endothelial cells/pericytes and they are negative for arterial/venous endothelial markers (i.e., CD34 and CD31), which can be a helpful aid to avoid misinterpretations. Another differential includes primitive myxoid mesenchymal tumor of infancy, which is a myxoid lesion composed of immature mesenchymal cells and potentially aggressive behavior arising in children within the first year of life [65]. Although both entities overlap, careful searching for inflammatory cells, prominent non-arciform vascularity, and identification of denser myofibroblastic areas should be indicative of inflammatory myofibroblastic tumor. Desmoid fibromatosis with extensive myxoid stroma may also be a mimicker; generally showing more elongated thin vessels paralleling myofibroblastic fascicles with presence of scattered mast cells.
The ALK-1 immunohistochemical expression pattern varies, at least partially, due to fusion partner genes. Different partners in some instances correspond to specific subcellular locations of ALK, leading to distinct patterns of expression by the ALK-1 antibody [66]. We identified seven cases harboring ALK alterations with corresponding ALK-1 immunohistochemical expression. Remarkably, our three cases showing a dot-like staining pattern demonstrated a predominantly hypocellular myxoid morphology in keeping with the omental mesenteric myxoid hamartoma reported by Ludwig et al. [10]. This single paranuclear dot-like pattern may be easily overlooked or wrongly considered a staining artifact, representing another potential diagnostic pitfall within this group of tumors. The consistent expression of WT1 and D240 in the majority of our inflammatory myofibroblastic tumors is similar to the observations rendered by Ludwig et al. and should be considered in the diagnostic work up to minimize misdiagnoses, as shown in case 4. Interestingly, while in their series all infantile inflammatory myofibroblastic tumors were consistently positive for these markers (but negative in conventional inflammatory myofibroblastic tumors in older children), in our study only WT1 was predominantly expressed by myxoid hypocellular inflammatory myofibroblastic tumors [10], while D240 was invariably positive in all inflammatory myofibroblastic tumors regardless their morphologic pattern.
Besides their variable morphologic and immunophenotypic patterns, inflammatory myofibroblastic tumors also show a significant molecular heterogeneity with more than ten different ALK fusion partners identified, all of them leading to its oncogenic activation [4]. We identified ALK rearrangements in four inflammatory myofibroblastic tumors in association with previously reported transcripts [4, 5] including EML4 in case 4, TPM4 in cases 7 and 9 as well as RANBP2 in case 2. In addition, one inflammatory myofibroblastic tumor (case 8) revealed a novel ALK-KLC1 rearrangement.
The RANBP2 encodes for a GTP-binding nuclear pore protein of the RAS superfamily. The RANBP2-ALK fusion has been reported within a wide age range including one confirmed infantile case [43]. These tumors display epithelioid-to-round cell morphology, conspicuous nucleoli, predominantly neutrophilic inflammatory infiltrate, and a diffuse myxoid stroma. They show a nuclear membrane ALK-1 staining pattern and strong desmin expression. This variant is associated with aggressive behavior and unfavorable outcome with a proposed designation as epithelioid inflammatory myofibroblastic sarcoma [2, 66]. Our RANBP2-ALK case (case 2), presented as a rapidly growing, large intrabdominal mass in a male infant. Although we identified several hypercellular clusters of desmin positive, round ganglion-like cells within a diffuse myxoid stroma reminiscent of epithelioid inflammatory myofibroblastic sarcoma, it also displayed other unusual features: (1) predominantly hypocellular pattern, (2) scant inflammatory component with only rare scattered neutrophils, (3) small to medium sized oval cells with inconspicuous nucleoli and (4) a peculiar ALK-1 dot-like immunohistochemical staining pattern. Although these features are at least, atypical for epithelioid inflammatory myofibroblastic sarcoma, this diagnosis cannot be entirely excluded, particularly in the setting of an RANBP2-ALK fusion. After a review of the literature, we identified 21 RANBP2-ALK rearranged [2, 43, 66,67,68,69,70,71,72,73,74,75,76,77,78] epithelioid inflammatory myofibroblastic sarcomas with only one infantile case reported in a 7-month-old male [43]. In addition, Marino-Enriquez et al. [2] reported an epithelioid inflammatory myofibroblastic tumor in a 7-month-old male who ultimately died of disease; however, no additional morphologic details were provided and the underlying molecular alteration remains unknown. Based on the features seen in our case, we could speculate that epithelioid inflammatory myofibroblastic sarcomas arising within the first year of life may display unconventional morphology in contrast to cases seen in older children; however, this hypothesis deserves further investigation in larger series; a challenging endeavor considering their rarity. We also identified two TPM4-ALK rearranged inflammatory myofibroblastic tumors presenting as an adrenal mass (case 7) and neck mass (case 9), respectively. These cases exhibited a cellular pattern with minimal myxoid component and diffuse cytoplasmic ALK-1 expression. The presence of TPM4-ALK has been consistently reported in other series of inflammatory myofibroblastic tumors, most of them children [5, 18, 63]. Nonetheless, only one infantile inflammatory myofibroblastic tumor harboring this fusion has been reported thus far [63]. Remarkably, a KLC1-ALK rearranged inflammatory myofibroblastic tumor in the tongue of a 10-month-old male (case 8) was identified. This lesion showed a cellular pattern with diffuse cytoplasmic ALK-1 expression. KLC1 encodes the light chain component of kinesin, a protein involved in organelle transport [79]. The KLC1-ALK is a rare driving fusion gene that has been reported only in five adult patients in the setting of lung carcinomas [79,80,81,82]. Our case is unique, as the KLC1-ALK fusion gene has never been identified either in inflammatory myofibroblastic tumors or in any other pediatric neoplasm.
The ROS proto-oncogene 1, ROS1 gene is considered the second most common alteration following ALK fusions in pediatric thoracic inflammatory myofibroblastic tumors [4, 18, 83]. We identified a TFG-ROS1 (case 11) and a fibronectin 1 (FN1)-ROS1 (case 10) rearranged inflammatory myofibroblastic tumor involving liver and lung, respectively. Both of these demonstrated a more cellular and collagenized pattern. Although several neoplasms harboring FN1-ALK rearrangements have been described [84, 85]; the presence of FN1-ROS1 rearrangement in an inflammatory myofibroblastic tumor is a novel finding not yet reported in any other neoplasm or age group. FN1 is located in chromosome 2q34 and encodes fibronectin, a surface glycoprotein also present within the extracellular matrix of various cell types that provides a strong promoter for its fusion partner gene [85]. It is possible that the oncogenicity of FN1-ROS1 parallels the mechanisms involved in FN1-ALK rearranged inflammatory myofibroblastic tumors expanding the group of already known partners that can overlap between different actionable kinases in similar way as TFG partners with ALK, ROS1, and NTRK genes [86]. Intriguingly, this case (as well as case 3) showed ALK-1 expression despite lacking ALK rearrangements. ALK-1 immunohistochemical expression has been described in association with mechanisms other than rearrangements, including isolated mutations [87] and increases in copy number of both rearranged and native ALK [88]. Remarkably, ROS1 rearrangements can be co-expressed with other mutated oncogenic drivers in the setting of non-small cell lung cancer [89]. In our series, an increased native ALK copy number can be ruled out by FISH studies; therefore, we hypothesize alternative activation mechanisms co-existing with other kinase rearrangements that remain to be explored.
An NTRK fusion (RBPMS-NTRK3) was present in case 3. Various neoplasms with NTRK3 alterations have been described including infantile fibrosarcoma, a tumor that characteristically harbors the ETV6-NTRK3 fusion [90]. So far, eight cases of ETV6-NTRK3 positive inflammatory myofibroblastic tumors are reported involving various organs within a widespread age range (2–61 years) and a spectrum of morphologic patterns [14, 18, 83, 91, 92]. The RBPMS-NTRK3 fusion has been reported rarely [83, 90] and no infantile inflammatory myofibroblastic tumors (or other infantile neoplasms) harboring this genetic alteration have ever been documented. This case, as well as several other fibroblastic tumors with NTRK rearrangements, embodies the issue of morphologic versus molecular characterization of tumors. Morphologically, its hypocellular myxoid pattern and location (small bowel wall) made this tumor indistinguishable from the omental mesenteric myxoid hamartoma reported by Gonzales-Crussi [11] and the subsequent cases described by Ludwig et al. [10]. It is intriguing that it also overlaps with a brain tumor described by Bale et al. as strongly reminiscent of low-grade fibromyxoid sarcoma [93]. Whether these tumors should be classified as inflammatory myofibroblastic tumors or as a subset of infantile fibrosarcoma, is an issue that remains to be explored. Perhaps gene expression profiles may help to find a more definitive answer, although the similar prognosis of these two entities and the possibility of a common targeted treatment make this a matter of semantics. Nevertheless, the morphologic-to-molecular correlation represents an important step in a systematic diagnostic approach, such as that proposed in Fig. 6, until molecular diagnostic panels become widely available in clinical practice.

Proposed diagnostic approach for infantile myofibroblastic and/or myxo-inflammatory lesions. ALK anaplastic lymphoma kinase, IHC immunohistochemistry, FISH fluorescence in situ hybridization, NGS next-generation sequencing, IMT inflammatory myofibroblastic tumor, ROS1 ROS proto-oncogene 1, receptor tyrosine kinase, PDGFRβ platelet-derived growth factor receptor beta, NTRK3 neurotrophic receptor tyrosine kinase 3
Inflammatory myofibroblastic tumors have intermediate prognosis, with potential for local relapse and rare metastases. Interestingly, those occurring in infants seem to have a favorable clinical behavior. Our literature review of infantile inflammatory myofibroblastic tumors with available outcome showed that 9% died as a direct consequence of their underlying neoplasm while 86% were in complete remission with a median follow-up of 24 months (Table 4). In the current series, 91% (10/11) of inflammatory myofibroblastic tumors with available follow-up had a favorable outcome with no recurrences and a median follow-up of 17 months. Although one of our patients died, this was attributed to various associated congenital anomalies.
The use of ALK tyrosine kinase inhibitors (ALK-TKIs), particularly crizotinib, is increasingly reported in the setting of inflammatory myofibroblastic tumors [68] and the National Comprehensive Cancer Network supports its use in inflammatory myofibroblastic tumors harboring ALK translocations [94]. Nonetheless, their effects in children younger than 1 year are still largely unknown, as infants were not included in the Children’s Oncology Group phase 1 consortium study [95]. Furthermore, the identification of targetable fusions other than ALK has raised interest in other TKIs (i.e., ROS1, NTRK, and RET inhibitors), among patients with inflammatory myofibroblastic tumors. However, caution is warranted in infants, as the major pharmacokinetic processes are significantly different due to their immaturity and continuous organ development [96]. To the best of our knowledge, only one patient with an inflammatory myofibroblastic tumor arising within the first year of life was successfully treated with crizotinib [21]. Our series identified three additional infantile inflammatory myofibroblastic tumors successfully treated with crizotinib (cases 4, 8, and 12) with sequence information available in two of them (EML4-ALK and KLC1-ALK fusion genes, respectively) and ALK-FISH positivity only in one. Our results highlight the importance of molecular characterization of inflammatory myofibroblastic tumors as treatment responses might be affected based upon the presence of specific fusion partners and lack of specificity of ALK-1 immunohistochemistry [86]. This also paves the way for a more customized and less invasive therapeutic approach, becoming particularly relevant among infants presenting with recurrent and/or unresectable tumors in whom a conservative approach is not an option.
In summary, inflammatory myofibroblastic tumors are rare among infants. When they occur, they can display cellular morphology with variable collagenized stroma and inflammation as seen in most inflammatory myofibroblastic tumors across all ages, but they can also show a paucicellular, extensively myxoid pattern potentially leading to misdiagnoses. Our series showed that most inflammatory myofibroblastic tumors in this age group harbored kinase fusions involving ALK, ROS1, and NTRK including three novel fusion partners (KLC1, FN1, and RBPMS, respectively). Due to the limited simple size of our study, it is difficult to draw clear-cut morphologic, immunophenotypic, and molecular correlations. We also identified favorable response to crizotinib among three infants, highlighting the potential use of TKIs in this population of patients as an alternative therapeutic approach that deserves further investigation. Awareness of these unusual morphologic features and molecular alterations is critical for appropriate diagnosis and optimized targeted therapy.
References
Coffin CM, Watterson J, Priest JR, Dehner LP. Extrapulmonary inflammatory myofibroblastic tumor (inflammatory pseudotumor). A clinicopathologic and immunohistochemical study of 84 cases. Am J Surg Pathol. 1995;19:859–72.
Marino-Enriquez A, Wang WL, Roy A, et al. Epithelioid inflammatory myofibroblastic sarcoma: An aggressive intra-abdominal variant of inflammatory myofibroblastic tumor with nuclear membrane or perinuclear ALK. Am J Surg Pathol. 2011;35:135–44.
Coffin CM, Alaggio R. Fibroblastic and myofibroblastic tumors in children and adolescents. Pediatr Dev Pathol. 2012;15:127–80.
Antonescu CR, Suurmeijer AJ, Zhang L, et al. Molecular characterization of inflammatory myofibroblastic tumors with frequent ALK and ROS1 gene fusions and rare novel RET rearrangement. Am J Surg Pathol. 2015;39:957–67.
Lovly CM, Gupta A, Lipson D, et al. Inflammatory myofibroblastic tumors harbor multiple potentially actionable kinase fusions. Cancer Discov. 2014;4:889–95.
Ghosh M, Islam N, Saha H, et al. Cytodiagnosis of inflammatory myofibroblastic tumor: A report of three cases in infants. Diagn Cytopathol. 2018;46:776–81.
Cook JR, Dehner LP, Collins MH, et al. Anaplastic lymphoma kinase (ALK) expression in the inflammatory myofibroblastic tumor: a comparative immunohistochemical study. Am J Surg Pathol. 2001;25:1364–71.
Buccoliero AM, Ghionzoli M, Castiglione F, et al. Inflammatory myofibroblastic tumor: clinical, morphological, immunohistochemical and molecular features of a pediatric case. Pathol Res Pract. 2014;210:1152–5.
Saab ST, Hornick JL, Fletcher CD, Olson SJ, Coffin CM. IgG4 plasma cells in inflammatory myofibroblastic tumor: inflammatory marker or pathogenic link? Mod Pathol. 2011;24:606–12.
Ludwig K, Alaggio R, Dall'Igna P, et al. Omental mesenteric myxoid hamartoma, a subtype of inflammatory myofibroblastic tumor? Considerations based on the histopathological evaluation of four cases. Virchows Arch. 2015;467:741–7.
Gonzalez-Crussi F, deMello DE, Sotelo-Avila C. Omental-mesenteric myxoid hamartomas. Infantile lesions simulating malignant tumors. Am J Surg Pathol. 1983;7:567–78.
Fletcher CDM, World Health Organization., International Agency for Research on Cancer. WHO classification of tumours of soft tissue and bone. In: World Health Organization classification of tumours. 4th edn, Lyon:IARC Press;2013, p. 468.
Bridge JA, Kanamori M, Ma Z, et al. Fusion of the ALK gene to the clathrin heavy chain gene, CLTC, in inflammatory myofibroblastic tumor. Am J Pathol. 2001;159:411–5.
Alassiri AH, Ali RH, Shen Y, et al. ETV6-NTRK3 is expressed in a subset of ALK-negative inflammatory myofibroblastic tumors. Am J Surg Pathol. 2016;40:1051–61.
Alaggio R, Cecchetto G, Bisogno G, et al. Inflammatory myofibroblastic tumors in childhood: a report from the Italian Cooperative Group studies. Cancer. 2010;116:216–26.
Dalton BG, Thomas PG, Sharp NE, et al. Inflammatory myofibroblastic tumors in children. J Pediatr Surg. 2016;51:541–4.
Fragoso AC, Eloy C, Estevao-Costa J, et al. Abdominal inflammatory myofibroblastic tumor a clinicopathologic study with reappraisal of biologic behavior. J Pediatr Surg. 2011;46:2076–82.
Yamamoto H, Yoshida A, Taguchi K, et al. ALK, ROS1 and NTRK3 gene rearrangements in inflammatory myofibroblastic tumours. Histopathology. 2016;69:72–83.
Hornick JL, Sholl LM, Dal Cin P, Childress MA, Lovly CM. Expression of ROS1 predicts ROS1 gene rearrangement in inflammatory myofibroblastic tumors. Mod Pathol. 2015;28:732–9.
Tanaka M, Kohashi K, Kushitani K, et al. Inflammatory myofibroblastic tumors of the lung carrying a chimeric A2M-ALK gene: report of 2 infantile cases and review of the differential diagnosis of infantile pulmonary lesions. Hum Pathol. 2017;66:177–82.
Caporalini C, Moscardi S, Tamburini A, et al. Inflammatory Myofibroblastic Tumor of the Tongue. Report of a Pediatric Case and Review of the Literature. Fetal Pediatr Pathol. 2018;37:117–25.
Tian JT, Cheng LC, Yung TC. Multiple cardiac inflammatory myofibroblastic tumors in the right ventricle in an infant. Ann Thorac Surg. 2006;82:1531–5.
Gandy KL, Burtelow MA, Reddy VM, Silverman NH. Myofibroblastic tumor of the heart: a rare intracardiac tumor. J Thorac Cardiovascular Surg. 2005;130:888–9.
Li L, Cerilli LA, Wick MR. Inflammatory pseudotumor (myofibroblastic tumor) of the heart. Ann Diagn Pathol. 2002;6:116–21.
Swain RS, Tihan T, Horvai AE, et al. Inflammatory myofibroblastic tumor of the central nervous system and its relationship to inflammatory pseudotumor. Hum Pathol. 2008;39:410–9.
Pucci A, Valori A, Muscio M, et al. Asymptomatic inflammatory myofibroblastic tumor of the heart: immunohistochemical profile, differential diagnosis, and review of the literature. Cardiovasc Pathol. 2009;18:187–90.
Sebire NJ, Ramsay A, Sheppard M, et al. Intravascular inflammatory myofibroblastic tumors in infancy. Pediatr Dev Pathol. 2002;5:400–4.
Bao M, Zheng C, Zhang H, et al. Inflammatory myofibroblastic tumor of the left atrium in infant. Int J Cardiol. 2016;222:965–7.
Shukla S, Singh SK, Pujani M, Pujani M, Chowdhury SR. Omental myxoid hamartomas–a case report with review of literature. Trop Gastroenterol. 2009;30:49–50.
Dhua AK, Garg M, Sen A, Chauhan DS. Inflammatory myofibroblastic tumor of parotid in infancy–a new entity. Int J Pediatr Otorhinolaryngol. 2013;77:866–8.
Henriquez A, Latorre JJ, Gonzalez S. Mesocolic myxoid hamartoma showing neural differentiation: an ultrastructural and immunohistochemical study. Pediatr Pathol. 1989;9:559–66.
Alobeid B, Beneck D, Sreekantaiah C, Abbi RK, Slim MS. Congenital pulmonary myofibroblastic tumor: a case report with cytogenetic analysis and review of the literature. Am J Surg Pathol. 1997;21:610–4.
Asanuma H, Nakai H, Shishido S, et al. Inflammatory pseudotumor of the bladder in neonates. Int J Urol. 2000;7:421–4.
Houben CH, Knüchel-Clarke R. Neonatal umbilical inflammatory myofibroblastic tumor. Ann Pediatr Surg. 2017;13:160–2.
Klein AM, Schoem SR, Altman A, Eisenfeld L. Inflammatory myofibroblastic tumor in the neonate: a case report. Otolaryngol–Head Neck Surg. 2003;128:145–7.
Owusu-Brackett N, Johnson R, Schindel DT, Koduru P, Cope-Yokoyama S. A novel ALK rearrangement in an inflammatory myofibroblastic tumor in a neonate. Cancer Genet. 2013;206:353–6.
Ma L, Wang K, Liu WK, Zhang YK. Is radical surgery necessary to head and neck inflammatory myofibroblastic tumor (IMT) in children? Childs Nerv Syst. 2009;25:285–91.
Oscoz Lizarbe M, Hualde Olascoaga J, Rúperez García E, et al. Tumor miofibroblástico en la edad pediátrica. A propósito de 3 casos. An de Pediatría. 2009;71:331–5.
Pearson PJ, Smithson WA, Driscoll DJ, Banks PM, Ehman RL. Inoperable plasma cell granuloma of the heart: spontaneous decrease in size during an 11-month period. Mayo Clin Proc. 1988;63:1022–5.
Tawfik HA, Raslan AO. Infantile inflammatory myofibroblastic tumor of the orbit with apical bone involvement. Ophthalmic Plast Reconstr Surg. 2013;29:e44–6.
Pang R, Merritt NH, Shkrum MJ, Tijssen JA. Febrile Illness in an infant with an intracardiac inflammatory myofibroblastic tumor. Pediatrics. 2016;137:e20143544.
Nagarajan S, Jayabose S, McBride W, et al. Inflammatory myofibroblastic tumor of the liver in children. J Pediatr Gastroenterol Nutr. 2013;57:277–80.
Ma Z, Hill DA, Collins MH, et al. Fusion of ALK to the Ran-binding protein 2 (RANBP2) gene in inflammatory myofibroblastic tumor. Genes Chromosomes Cancer. 2003;37:98–105.
Castanon M, Saura L, Weller S, et al. Myofibroblastic tumor causing severe neonatal distress. Successful surgical resection after embolization. J Pediatr Surg. 2005;40:e9–12.
Coffin CM, Hornick JL, Fletcher CD. Inflammatory myofibroblastic tumor: comparison of clinicopathologic, histologic, and immunohistochemical features including ALK expression in atypical and aggressive cases. Am J Surg Pathol. 2007;31:509–20.
Sirvent N, Hawkins AL, Moeglin D, et al. ALK probe rearrangement in a t(2;11;2)(p23;p15;q31) translocation found in a prenatal myofibroblastic fibrous lesion: toward a molecular definition of an inflammatory myofibroblastic tumor family? Genes Chromosomes Cancer. 2001;31:85–90.
Thompson RJ, Barrett AM, Dildey P. Congenital multifocal inflammatory pseudotumor: a case report. J Pediatr Surg. 2003;38:E17–9.
Kube S, Vokuhl C, Dantonello T, et al. Inflammatory myofibroblastic tumors-A retrospective analysis of the Cooperative Weichteilsarkom Studiengruppe. Pediatr Blood Cancer. 2018;65:e27012.
Shukla SK, Parashar S. Intra-abdominal inflammatory myofibroblastic tumour (IMT) mimicking gonadal mass in a male infant with undescended testes. BMJ Case Rep. 2017;2017:pii: bcr-2017-219745.
Di Maria M, N Campbell D, B Mitchell M, et al. Successful orthotopic heart transplant in an infant with an inflammatory myofibroblastic tumor of the left ventricle. J Heart Lung Transplant. 2008;27:792–6.
Hill KA, Gonzalez-Crussi F, Chou PM. Calcifying fibrous pseudotumor versus inflammatory myofibroblastic tumor: a histological and immunohistochemical comparison. Mod Pathol. 2001;14:784–90.
de Montpreville VT, Serraf A, Aznag H, et al. Fibroma and inflammatory myofibroblastic tumor of the heart. Ann Diagn Pathol. 2001;5:335–42.
Syed AU, Al Mutairi MB, Al Muhaya M, et al. Inflammatory myofibroblastic tumor of heart in a 4-month-old infant. Asian Cardiovasc Thorac Ann. 2018;26:47–9.
Jenkins PC, Dickison AE. Flanagan MFJPC. Cardiac Inflammatory Pseudotumor: rapid appearance in an infant with congenital heart disease. Pediatr Cardiol. 1996;17:399–401.
Elkiran O, Karakurt C, Erdil N, Disli OM, Dagli AF. An unexpected cause of respiratory distress and cyanosis: cardiac inflammatory myofibroblastic tumor. Congenit Heart Dis. 2013;8:E174–7.
Hirschburger M, Enders J, Alzen G, Padberg W, Wagner HJ. An inflammatory myofibroblastic tumor of the stomach as a rare cause of gastric outlet obstruction in an 8-month-old infant. Klin Padiatr. 2010;222:192–3.
Qiu X, Montgomery E, Sun B. Inflammatory myofibroblastic tumor and low-grade myofibroblastic sarcoma: a comparative study of clinicopathologic features and further observations on the immunohistochemical profile of myofibroblasts. Hum Pathol. 2008;39:846–56.
Mizia-Malarz A, Sobol-Milejska G, Buchwald J, Wos H. Inflammatory Myofibroblastic Tumor of the Heart in the Infant: Review of the Literature. J Pediatr Hematol Oncol. 2016;38:e298–e302.
Chun YS, Wang L, Nascimento AG, Moir CR, Rodeberg DA. Pediatric inflammatory myofibroblastic tumor: anaplastic lymphoma kinase (ALK) expression and prognosis. Pediatr Blood Cancer. 2005;45:796–801.
Mergan F, Jaubert F, Sauvat F, et al. Inflammatory myofibroblastic tumor in children: clinical review with anaplastic lymphoma kinase, Epstein-Barr virus, and human herpesvirus 8 detection analysis. J Pediatr Surg. 2005;40:1581–6.
Soyer T, Talim B, Karnak I, et al. Surgical treatment of childhood inflammatory myofibroblastic tumors. Eur J Pediatr Surg. 2017;27:319–23.
Tavora F, Shilo K, Ozbudak IH, et al. Absence of human herpesvirus-8 in pulmonary inflammatory myofibroblastic tumor: immunohistochemical and molecular analysis of 20 cases. Mod Pathol. 2007;20:995–9.
Lawrence B, Perez-Atayde A, Hibbard MK, et al. TPM3-ALK and TPM4-ALK oncogenes in inflammatory myofibroblastic tumors. Am J Pathol. 2000;157:377–84.
Surrey LF, MacFarland SP, Chang F, et al. Clinical utility of custom-designed NGS panel testing in pediatric tumors. Genome Med. 2019;11:32.
Alaggio R, Ninfo V, Rosolen A, Coffin CM. Primitive myxoid mesenchymal tumor of infancy: a clinicopathologic report of 6 cases. Am J Surg Pathol. 2006;30:388–94.
Li J, Yin WH, Takeuchi K, et al. Inflammatory myofibroblastic tumor with RANBP2 and ALK gene rearrangement: a report of two cases and literature review. Diagn Pathol. 2013;8:147.
Patel AS, Murphy KM, Hawkins AL, et al. RANBP2 and CLTC are involved in ALK rearrangements in inflammatory myofibroblastic tumors. Cancer Genet Cytogenet. 2007;176:107–14.
Butrynski JE, D'Adamo DR, Hornick JL, et al. Crizotinib in ALK-rearranged inflammatory myofibroblastic tumor. N Engl J Med. 2010;363:1727–33.
Huang YH, Tian YF, Li CF. Inflammatory myofibroblastic tumor with RANBP2 and ALK gene rearrangement with bland cytological features mimicking desmoid-type fibromatosis: a case report and review of the literature. Oncol Lett. 2016;11:1429–34.
Lee JC, Li CF, Huang HY, et al. ALK oncoproteins in atypical inflammatory myofibroblastic tumours: novel RRBP1-ALK fusions in epithelioid inflammatory myofibroblastic sarcoma. J Pathol. 2017;241:316–23.
Kozu Y, Isaka M, Ohde Y, Takeuchi K, Nakajima T. Epithelioid inflammatory myofibroblastic sarcoma arising in the pleural cavity. Gen Thorac Cardiovasc Surg. 2014;62:191–4.
Chen ST, Lee JC. An inflammatory myofibroblastic tumor in liver with ALK and RANBP2 gene rearrangement: combination of distinct morphologic, immunohistochemical, and genetic features. Hum Pathol. 2008;39:1854–8.
Fang H, Langstraat CL, Visscher DW, Folpe AL, Schoolmeester JK. Epithelioid inflammatory myofibroblastic sarcoma of the ovary with RANB2-ALK fusion: report of a case. Int J Gynecol Pathol. 2018;37:468–72.
Du X, Gao Y, Zhao H, et al. Clinicopathological analysis of epithelioid inflammatory myofibroblastic sarcoma. Oncol Lett. 2018;15:9317–26.
Liu Q, Kan Y, Zhao Y, He H, Kong L. Epithelioid inflammatory myofibroblastic sarcoma treated with ALK inhibitor: a case report and review of literature. Int J Clin Exp Pathol. 2015;8:15328–32.
Kimbara S, Takeda K, Fukushima H, et al. A case report of epithelioid inflammatory myofibroblastic sarcoma with RANBP2-ALK fusion gene treated with the ALK inhibitor, crizotinib. Jpn J Clin Oncol. 2014;44:868–71.
Kurihara-Hosokawa K, Kawasaki I, Tamai A, et al. Epithelioid inflammatory myofibroblastic sarcoma responsive to surgery and an ALK inhibitor in a patient with panhypopituitarism. Intern Med. 2014;53:2211–4.
Wu H, Meng YH, Lu P, et al. Epithelioid inflammatory myofibroblastic sarcoma in abdominal cavity: a case report and review of literature. Int J Clin Exp Pathol. 2015;8:4213–9.
Togashi Y, Soda M, Sakata S, et al. KLC1-ALK: a novel fusion in lung cancer identified using a formalin-fixed paraffin-embedded tissue only. PLoS ONE. 2012;7:e31323.
Wang P, Xiao P, Ye Y, et al. Rapid response of brain metastasis to crizotinib in a patient with KLC1-ALK fusion and MET gene amplification positive non-small cell lung cancer: a case report. Cancer Biol Med. 2017;14:183–6.
McLeer-Florin A, Duruisseaux M, Pinsolle J, et al. ALK fusion variants detection by targeted RNA-next generation sequencing and clinical responses to crizotinib in ALK-positive non-small cell lung cancer. Lung Cancer. 2018;116:15–24.
Zheng Q, Zheng M, Jin Y, et al. ALK-rearrangement neuroendocrine carcinoma of the lung: a comprehensive study of a rare case series and review of literature. Onco Targets Ther. 2018;11:4991–8.
Chang JC, Zhang L, Drilon AE, et al. Expanding the molecular characterization of thoracic inflammatory myofibroblastic tumors beyond ALK gene rearrangements. J Thorac Oncol. 2019;14:825–34.
Haimes JD, Stewart CJR, Kudlow BA, et al. Uterine inflammatory myofibroblastic tumors frequently harbor ALK fusions with IGFBP5 and THBS1. Am J Surg Pathol. 2017;41:773–80.
Ouchi K, Miyachi M, Tsuma Y, et al. FN1: a novel fusion partner of ALK in an inflammatory myofibroblastic tumor. Pediatr Blood Cancer. 2015;62:909–11.
Childress MA, Himmelberg SM, Chen H, et al. ALK fusion partners impact response to ALK inhibition: differential effects on sensitivity, cellular phenotypes, and biochemical properties. Mol Cancer Res. 2018;16:1724–36.
Wang YW, Tu PH, Lin KT, et al. Identification of oncogenic point mutations and hyperphosphorylation of anaplastic lymphoma kinase in lung cancer. Neoplasia. 2011;13:704–15.
Camidge DR, Skokan M, Kiatsimkul P, et al. Native and rearranged ALK copy number and rearranged cell count in non-small cell lung cancer: implications for ALK inhibitor therapy. Cancer. 2013;119:3968–75.
Lin JJ, Ritterhouse LL, Ali SM, et al. ROS1 fusions rarely overlap with other oncogenic drivers in non-small cell lung cancer. J Thorac Oncol. 2017;12:872–7.
Torre M, Jessop N, Hornick JL, Alexandrescu S. Expanding the spectrum of pediatric NTRK-rearranged fibroblastic tumors to the central nervous system: a case report with RBPMS-NTRK3 fusion. Neuropathology. 2018;38:624–30.
Takahashi A, Kurosawa M, Uemura M, Kitazawa J, Hayashi Y. Anaplastic lymphoma kinase-negative uterine inflammatory myofibroblastic tumor containing the ETV6-NTRK3 fusion gene: a case report. J Int Med Res. 2018;46:3498–503.
Pavlick D, Schrock AB, Malicki D, et al. Identification of NTRK fusions in pediatric mesenchymal tumors. Pediatr Blood Cancer 2017;64:e26433.
Bale TA, Oviedo A, Kozakewich H, et al. Intracranial myxoid mesenchymal tumors with EWSR1-CREB family gene fusions: myxoid variant of angiomatoid fibrous histiocytoma or novel entity? Brain Pathol. 2018;28:183–91.
Tsakiri K, Kotoula V, Lakis S, et al. Crizotinib failure in a TPM4-ALK–rearranged inflammatory myofibroblastic tumor with an emerging ALK kinase domain mutation. 2017;1:1–7.
Mosse YP, Lim MS, Voss SD, et al. Safety and activity of crizotinib for paediatric patients with refractory solid tumours or anaplastic large-cell lymphoma: a Children's Oncology Group phase 1 consortium study. Lancet Oncol. 2013;14:472–80.
O'Hara K, Wright IM, Schneider JJ, Jones AL, Martin JH. Pharmacokinetics in neonatal prescribing: evidence base, paradigms and the future. Br J Clin Pharm. 2015;80:1281–8.
Acknowledgements
The authors would like to thank Dr Pifu Luo (KingMed Diagnostics, Guangzhou, China) who provided unstained slides for immunohistochemical studies.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Lopez-Nunez, O., John, I., Panasiti, R.N. et al. Infantile inflammatory myofibroblastic tumors: clinicopathological and molecular characterization of 12 cases. Mod Pathol 33, 576–590 (2020). https://doi.org/10.1038/s41379-019-0406-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-019-0406-6
This article is cited by
-
Crizotinib combined with bronchoscopic interventional treatment in ALK-positive inflammatory myofibroblastic tumor of left main stem bronchus: a case report
Journal of Cardiothoracic Surgery (2023)
-
A novel TPD52L2-ROS1 gene fusion expanding the molecular alterations in inflammatory myofibroblastic tumor: case report and literature review
Diagnostic Pathology (2023)
-
Detection of sarcoma fusions by a next-generation sequencing based–ligation-dependent multiplex RT-PCR assay
Modern Pathology (2022)
-
The oncogenic fusion landscape in pediatric CNS neoplasms
Acta Neuropathologica (2022)
-
Calcified chondroid mesenchymal neoplasms with FN1-receptor tyrosine kinase gene fusions including FGFR2, FGFR1, MERTK, NTRK1, and TEK: a molecular and clinicopathologic analysis
Modern Pathology (2021)
