
Abstract
Advances in technology and software have radically expanded the scope of metabolomics studies and allow us to monitor a broad transect of central carbon metabolism in routine studies. These increasingly sophisticated tools have shown that many human diseases are modulated by microbial metabolism. Despite this, it remains surprisingly difficult to move beyond these statistical associations and identify the specific molecular mechanisms that link dysbiosis to the progression of human disease. This difficulty stems from both the biological intricacies of host-microbiome dynamics as well as the analytical complexities inherent to microbiome metabolism research. The primary objective of this review is to examine the experimental and computational tools that can provide insights into the molecular mechanisms at work in host–microbiome interactions and to highlight the undeveloped frontiers that are currently holding back microbiome research from fully leveraging the benefits of modern metabolomics.
Similar content being viewed by others
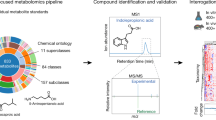

Introduction
Metabolism plays a foundational role in essentially all aspects of life and disruptions in metabolism can affect a wide range of basic functions including nutrition, athletic performance, immune function, pain perception, and the progression of both chronic and infectious diseases1,2,3,4,5,6,7. Although mammalian metabolism has been intensively studied for over a century, it has primarily been investigated through the lens of host metabolic function. However, over the last 20 years we have become increasingly aware of the role that microbial communities play in modulating the availability of nutrients and how these microbial modulations can impact homeostasis5. Everything that mammals eat enters the gastrointestinal (GI) tract, where the metabolism of the gut microbiome can transform these molecules and directly influence the complement of nutrients that are passed along to the host3.
Disruptions in the microbiome caused by antibiotics, diet, and disease can alter these intricate host-microbiome metabolic exchanges and affect biological functions throughout the body1,2,3,5,6,8,9,10,11,12. A few examples of diseases that are modulated by microbial metabolism include colitis (e.g., irritable bowel syndrome (IBS) and Crohn’s disease)10,13,14,15,16,17, immune diseases (e.g., multiple sclerosis)1,18,19, neurodegenerative diseases (e.g., amyotrophic lateral sclerosis and Alzheimer’s disease)20,21,22, psychological conditions (e.g., depression)8,23, cystic fibrosis24,25,26,27,28,29, cancer30,31,32, and cardiovascular disease33. Microbial metabolism can also play a role in the pharmacokinetics of drugs9,34,35. These surprising associations have led researchers to investigate the microbiome as a vehicle for stimulating specific metabolic activities21,36,37,38 and as a tool to modulate inflammation39,40,41.
The links between microbial metabolism and human diseases are now well-defined thanks to large-scale efforts, such as the >15,000 feces samples collected by the American Gut Project42. These efforts have helped demonstrate links between the microbiome and diverse conditions including IBS, cystic fibrosis, diabetes mellitus, and cancer31,43,44,45. While identifying the specific molecular mechanisms contributing to these diseases remains challenging, recent advances in metabolomics technologies allow us to capture a broad swath of central carbon metabolism and track the microbial metabolism of carbon chains as they are passed through networks of over 5000 reactions46,47,48,49. Metabolomics technology, when coupled to animal models, isotope tracing studies, and in vitro organ models allows researchers to probe the complexities of host-microbiome metabolic dynamics with a greater degree of experimental control than was previously possible (Fig. 1, Table 1). The main objective of this review is to examine the unique challenges encountered in microbiome-metabolism studies and discuss how these emerging tools and techniques can provide insights into the molecular mechanisms at work behind complex host–microbiome interactions. We highlight examples of how these tools can be used to study host-microbiota and microbe-microbe interactions, as well as interactions of the microbiome with diet and pharmaceuticals (Table 1; see Supplementary Table 1 for more examples).

Figure created with BioRender, available at Biorender.com.
Biological complexities in microbiome metabolic studies
Complex microbial ecosystems are found throughout the integument, GI tract, airways, mucosa, and urogenital tract5. The environmental conditions that microbes encounter at these sites vary dramatically with respect to pH, oxygen, bicarbonate, and nutrient availability and these site-to-site differences can have a profound impact on which microbes inhabit the niche50. Since the metabolic capacity of microbes differs considerably species to species, these differences in microbial community composition can have a profound impact on the metabolic capacity of the overall system51,52,53,54,55. Moreover, the ensemble action of host and microbial enzymes creates a more complex metabolic network than exists in any individual species5,6.
Microbiome metabolism is further complicated by the multi-species pathways that nutrients can take through the microbial ecosystem. The metabolic waste products of some microbes are the preferred carbon sources for others. Succinate, for example, is a primary waste product of Enterobacteriales56 but is also one of the preferred carbon sources of Pseudomonas aeruginosa57,58,59. These differences in nutritional strategies enable microbial cross-feeding interactions that create multi-species metabolic networks in the microbiome50,60. Cross-feeding can significantly alter the metabolic capacity of systems24,25,27,28,52,61 and modulate microbial phenotypes (e.g., their sensitivity to antibiotics)35,53,54. Cross-feeding interactions also enable some species to survive in otherwise inhospitable environments23,35,62. For example, a previously unreported genus of bacteria (KLE1738) was recently found to depend on γ-aminobutyric acid (GABA)-producing Bacteroides fragilis for growth23. Cross-feeding interactions are also thought to play a role in a range of health issues, including periodontal health62, the clinical progression of pulmonary infections in cystic fibrosis patents24,25,28,29, and may contribute to the overgrowth of pathobionts in response to undernutrition63.
Tracking metabolism through multi-species networks
Decoupling the complex flow of molecules organism-to-organism is a non-trivial challenge and to study cross-feeding, researchers have employed a variety of methods including genome-scale metabolic reconstruction28,64, computational modeling with in vitro data28,65,66,67,68, and in vitro checkerboard assays53,54 (Table 1; Supplementary Table 1). Recently, metabolomics has played a larger role in dissecting these dynamics, largely via the use of isotope tracing experiments69,70,71. Isotope tracing approaches track the flow of stable isotope-labeled nutrients (typically 13C, 15N or 2H) through microbial communities and their exchange with the host23,24,61,72. This strategy has been used to identify microbe-specific biomarkers23,33,61, demonstrate the exchange of nutrients from microbes to the host72, and demonstrate syntrophic relationships between microbes that overcome nutrient imbalance in the diet61. Though powerful, isotope labeling approaches are a serious analytical undertaking, especially in the context of untargeted and semi-targeted metabolomics studies. The multi-species metabolic networks can scramble isotope labeling and make it difficult to predict which molecules and which isotopologues (i.e., the number of isotopically labeled atoms present in a molecule) will be produced from a microbiome-linked processing of a precursor. This uncertainty can create computational challenges because it requires all possible metabolites to be screened for all possible isotopologues. This dramatically increases the search space and thereby the likelihood of misidentifying metabolites in the context of untargeted/semi-targeted studies. Although this can be partially mitigated via high-resolution mass spectrometry73, additional care must be given to the molecular assignment process since mass and retention time alone may not be sufficient to unambiguously identify metabolites in isotopically complex microbiome extracts.
Organ models of microbiome metabolism
In vitro bioreactors and organ models are well-established systems for reducing the complexity of microbiome analyses and provide a path for identifying specific molecular interactions between cell types26,27,74,75,76,77,78,79. One of the best-established bioreactor systems is the Simulator of the Human Intestinal Microbial Ecosystem (SHIME®), which simulates the entire human GI tract74,75. The SHIME® system simulates digestion by pumping contents into a series of chambers74. This system can be primed with fecal suspensions or engineered with specific microbes to enable specific molecular interactions to be studied under controlled conditions. This model has been used to elucidate the effects of diet and pre- or probiotics on organic acid and short-chain fatty acid (SCFA) production in different GI compartments36,38,80,81,82. Other organ models include the Winogradsky column system, which has been used to study the microbial community interactions underlying pulmonary infections25,26,27, and a range of single compartment chemostat reactors that have been used to simulate specific microbiome ecosystems (e.g., the human colon)37,76,77,79,83,84,85,86.
Although each of these established in vitro systems enable detailed molecular studies to be conducted under well-controlled conditions, they lack human cell interactions87 and thus do a poor job of simulating the significant host contributions to the environment, such as the absorption of nutrients associated with mammalian host cells27,74,83,88. To address this, a range of new in vitro organ models have been developed. Organoids, organ-on-a-chip, and related in vitro human biomimetic models allow researchers to simulate specific compartments of the human body while manipulating parameters to simulate disease pathogenesis, host-cell responses, and drug interactions89,90,91. Organoid culture is a well-established tool for studying a multitude of organs and disease models, but most current approaches use a microfluidic organ-on-a-chip approach to mimic complex interactions between the microbiome and host or other microbial cells. The human Colon Chip was used to determine the human microbiome-associated metabolites that mediated susceptibility to enterohemorrhagic E. coli infection, which is not common in mice and therefore cannot be studied in a murine model92. The human gut-on-a-chip, comprised of two microfluidic channels which are separated by a flexible membrane lined with human epithelial cells, is a useful tool to manipulate different factors in the gut microbiome, such as the presence of immune cells and pathogenic microbes, to determine the factors that contribute most to intestinal inflammation and bacterial overgrowth40,93. Similarly, the simplified human microbiota (SIHUMIx) model consists of three parallel bioreactors inoculated with a standard mix of eight bacterial species that are dominant in human feces and improves the reproducibility of results from prior bioreactor systems94.
Newer models are allowing researchers to study more complex interactions including co-culture and multi-organ systems41,87,89. The microfluidics-based HuMix (human-microbial crosstalk) model consists of gaskets divided into three co-laminar microchannels (medium perfusion microchamber, human epithelial cell culture chamber, microbial culture microchamber) with cell-covered membranes used for the extraction of intracellular metabolites87. This provides a means to continually monitor the effect of co-culture on individual co-cultured cell contingents. The authors validated HuMix with human intestinal epithelial cells co-cultured with Lactobacillus rhamnosus GG grown under anaerobic conditions, which induced the intracellular accumulation of GABA in the epithelial cells. Another model connected the human gut, liver, and circulating immune cells (T regulatory and T helper 17 cells) to simulate ulcerative colitis ex vivo using an integrated co-culture system of two fluidically communicating human micro-physiological systems41. In this system, the authors tested the immune response to microbiome-derived SCFAs, which was dependent on the involvement of effector CD4 T cells.
Each of these in vitro organ models provides a mechanism for investigating the molecular underpinnings of host-microbiome interactions. However, all bioreactors and organ models are sensitive to subtle changes in pH, temperature, nutrients, and oxygen levels which can have a dramatic impact the metabolic phenotypes observed in these model systems27,74,83,95. Although these platforms provide a controlled environment for testing metabolic hypotheses about interactions of individual species, they generally must be combined with metabolic profiling or in vivo approaches to verify the physiological relevance of the findings (see Table 1 for examples).
Animal model strategies for investigating molecular interactions
Gnotobiotic animal models, which have been extensively reviewed elsewhere96,97,98, are one of the most effective tools for investigating the molecular underpinnings of host-microbe molecular interactions99,100,101. Germ-free mice (and other species) can be colonized with defined collections of microbes, which enables direct comparisons between germ-free (GF; free of all microorganisms), specific pathogen free (SPF; contain a microbiota that is free of specified pathogens), and monocolonized/polycolonized animals. For human studies, microbial communities linked to specific metabolic phenotypes can be mapped via metagenomic sequencing102 and the taxonomic structure of populations can be linked to disease states. These phylogenetic mapping efforts can be effective when combined with fecal microbiota transplantation studies to separate host versus microbiome contributions to complex diseases103.
These model systems present a powerful framework for integrating hypothesis testing into metabolomics studies of the microbiome and have been used to investigate diverse biological processes including aging104, reproduction105,106, and metabolism13,14,16,107,108. This strategy has been very successful in providing molecular insights into diverse diseases including colitis (both IBS and Crohn’s disease)15,17, neurodegenerative disease20,21, breast cancer32, diabetes109, the biological response to toxin exposure9,10,34,110,111,112,113, and the role that the microbiome plays in immunity30,114. Though powerful, gnotobiotic models have some limitations. Human physiology and our microbial communities differ from those found in model organisms100 and this can present challenges in translating findings back to human disease115,116. Furthermore, rodents are coprophagic, a behavior that is not common in people, and this can have a direct impact on the metabolic composition of the GI tract, including microbial catabolites of bile acids115. Despite these shortcomings, gnotobiotic models are currently the best tools available for testing specific microbiome metabolic hypotheses and, if carefully coupled with in vitro organ models or human studies, provide the most direct path for unraveling the molecular underpinnings of host-microbiome metabolic dynamics.
Sampling considerations
Metabolic associations can be established through the analysis of non-invasive samples (analyses of feces, serum, and urine), but these samples are indirect reporters of microbial metabolism, are diluted significantly after they leave their microenvironment, and can undergo significant biological or chemical degradation before they can be sampled from these distal sites44,117,118,119,120,121,122. Although the obvious solution to this problem is to collect samples directly from microbial communities, this approach is not always practical (i.e., sampling the GI tract in humans is invasive and not all sites can be reached via endoscopy123). Moreover, the metabolites produced by microbes in one site can affect a wide range of other organs11,20,21,113,122,124,125,126,127. Microbially-derived trimethylamine N-oxide and phenylacetylglutamine produced in the gut, for example, are linked to elevated risks of cardiovascular disease and pancreatic cancer31,33. In addition, a growing body of literature has shown that the microbiome influences both local and systemic immune function2,127. Microbial inosine, for example, plays a direct role in the activation of antitumor T cells30. Resolving these indirect modes of action is critical for understanding the molecular mechanisms that contribute to disease but poses significant challenges to study designs. At present, the best strategy is to sample broadly from both the local microbial communities (wherever possible) and from distal sites around the body.
The choice of metabolite extraction solvents will also directly affect the scope of metabolites that can be observed in a study46,121. The merits of diverse sample preparation methods, the timing of collection, transport and storage conditions of the samples, homogenization, and pretreatment strategies (e.g., use of preservatives) have been discussed at length elsewhere117,118,119,128,129. Briefly, some general principles that need to be considered are (1) the extraction solvent must match the downstream analysis (i.e., aqueous extractions should be matched with analyses of hydrophilic molecules and vice versa), (2) metabolism needs to be quenched to prevent samples from degrading, (3) solvent/solute ratios should be adjusted according to the target molecules to maximize extraction efficiencies, (4) freezing samples will cause some metabolites to precipitate out of solution and can affect quantification, and (5) every sample extraction method excludes certain groups of molecules and introduces biases46,121,130. Consequently, the primary objective of any extraction should be to capture the target transect of molecules with the least technical error. With this objective in mind, we have increasingly favored extractions in 4 °C 50% methanol:H2O (at a 1:50 or 1:20 volume:volume dilution or 50 mg tissue/mL) for general studies involving central carbon metabolism131,132,133, extracellular metabolome analyses134,135,136, and other projects involving the analysis of polar metabolites137,138,139 (see Section S1 in the SI file for detailed extraction protocols). We have shown that this simple extraction, when coupled with hydrophilic interaction liquid chromatography (HILIC) mass spectrometry (MS), can reproducibly capture metabolites over thousands of samples with minimal technical error (coefficient of variation <0.15 over 1000+ injections)140. We find that this method is a good first choice for analyzing polar compounds when the molecular targets are poorly defined; however, other methods can be better optimized for specific compound classes such as SCFAs. Samples containing SCFAs should be immediately frozen (at −20 °C or preferably −80 °C) and can then undergo extraction using solvents such as an acetonitrile:H2O blend or specialized cleanup steps like solid-phase microextraction141,142,143,144. Appropriate sample preparation steps for the sample matrix and the classes of molecules analyzed help ensure reproducible and meaningful results in these complex microbiome-metabolomics studies.
New frontiers in human sampling
Gnotobiotic animals are a powerful platform for studying disease but will always be imprecise re-creations of human diseases. Validating these findings requires human studies, which are generally limited to blood, urine, feces, and other non-invasive samples. Although some researchers have employed surgical biopsy145 and mucosal endoscopic lavage17,123, these medical procedures are difficult to organize for most studies. To address this, several groups are working to develop ingestible GI sample collection devices146,147. These new tools allow insights into physiology that were previously only practical in animal models.
Three examples of this emerging platform are the CapScan® (Envivo Bio)146, the Ingestible Osmotic Pill147, and the Small Intestine MicroBiome Aspiration Capsule (SIMBA™) (Small Intestine MicroBiome Aspiration Capsule (2022) at https://www.nimblesci.com/technology). The CapScan® consists of a collapsed collection bladder, capped by a one-way valve inside a dissolvable capsule with an enteric coating146. Once ingested, the device moves down the GI tract until it reaches a pre-set pH level (e.g., pH 7–8 in the ileum), where the enteric coating dissolves and the collection bladder draws in the luminal contents. The one-way valve prevents further entrance of liquid into the capsule, which is later recovered from the stool. These researchers tested the device on 240 intestinal samples from 15 healthy patients and found significant differences in microbes and metabolites present in the intestines compared to the stool and determined that bile acid profiles varied along the intestines, as found previously in animal models. Other devices follow similar principles, although the mechanism of action for the 3D-printed Osmotic Pill involves a pressure differential created across the semipermeable membrane, which induces a passive pumping action as it moves down the GI tract147. The pill is embedded with a small neodymium magnet, and thus can be held in a precise location inside the GI tract for sampling of specific locations. These ingestible sampling devices have just recently been introduced and their applications into metabolomics have just started to come online. One consideration in applying this emerging technology to metabolomics is that samples collected via these capsules cannot be metabolically quenched until after the capsule has been collected (potentially a day or more after sampling the microbiome)46,121,148. Consequently, these tools will be most amenable to analyzes of metabolic phenotypes that are biologically and chemically stable.
Analytical complexities in microbiome metabolic studies
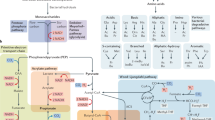
Microbiome metabolomics studies present significant analytical challenges because of the exceptional breadth of chemical diversity and the high degree of metabolic complexity that can be found in metabolic extracts from microbial communities (Fig. 2)44,46,47. Complex carbohydrates, lipids, bile acids, SCFAs, peptides, amino acids, nucleotides, vitamins and cofactors, and a stunning diversity of secondary metabolites are a few examples of small molecule classes that are metabolized by the microbiome1,2,3,6,8 and can modulate host-microbiome dynamics (see Table 1 for examples). Unfortunately, no single analytical technique can capture this full spectrum of molecules in a single analysis46,48,49. Consequently, each study must designate a target range of molecules and select an extraction method and analytical framework that are compatible with the chemical properties of the target analytes.

Figure created with BioRender, available at Biorender.com.
As discussed in “Sampling considerations”, there are dozens of commonly used sample preparation and analytical workflows for metabolomics analyses. These have generally consolidated around methods for the three core instrumentation platforms used in metabolomics: liquid chromatography-mass spectrometry (LC-MS)10,13,21,45,142, gas chromatography-mass spectrometry (GC-MS)110,149,150, and nuclear magnetic resonance (NMR)12,15,120. Although the general merits and pitfalls of these platforms are thoroughly described elsewhere46, there are some special considerations that these platforms have in the context of microbiome research.
One of the most intensively studied classes of molecules in microbiome research are SCFAs, which are microbially-derived compounds that are implicated in a vast range of biological processes ranging from colitis and immune function to pain perception10,13,15,21,22,85,110,151,152. Although these molecules can be detected on any of the three core analytical platforms, they are surprisingly difficult to accurately quantify. NMR methods can detect them directly but cannot accurately quantify them without selective or multidimensional methods130,153. Although SCFAs are small and polar, they are difficult to resolve by liquid or gas chromatography without derivatization154. To address this, a range of specialized SCFA analysis techniques have been developed10,21,45,142. Of these, we prefer SCFA Quantification Using Aniline Derivatization, which is an isotope-based LC-MS strategy that enables robust absolute quantification of SCFAs in complex samples142.
Another important class of metabolites for microbiome research are bile acids, which encompass a rich collection of cholesterol-derived molecules and whose conjugated derivatives are secreted from the liver into the gut and converted into secondary bile acids via microbial catabolism1,11,44,113,122,155,156. A selection of conjugated and unconjugated bile acids reach the circulatory system, where they interact with bile acid receptors1,11,113,156,157. These molecules are primarily detected in the cecum or the feces and are best detected using reverse-phase LC-MS44,113,158 or via targeted analyses using chemical labeling kits that are now commercially available (e.g., Biocrates)11.
Beyond these intensively studied classes of compounds, a range of aqueous central carbon metabolites, including amino acids (e.g., neurotransmitters, choline derivatives, and tryptophan derivatives), nucleotides, and energy intermediates are emerging as important regulators of the interplay between host and microbiome1,8,20,21,111,124,159,160. These metabolites are more commonly associated with systemic effects (i.e., are found in the bloodstream and tissues such as the brain and liver)12,20,21,111,124,159, but can also serve as important immune regulators in the GI tract30,122,161,162. These hydrophilic compounds are most easily analyzed by LC-MS using HILIC140,163,164,165 but can also be derivatized and analyzed by GC-MS14,109,112. Our preferred strategy for aqueous analyzes of the GI tract and feces uses a zwitterionic HILIC (Thermo Fisher Syncronis™) stationary phase combined with a short linear ammonium formate (aqueous phase)/ acetonitrile with formic acid (organic phase) gradient to capture amino acids, carbohydrates, nucleotide derivatives, and other common compounds that are found outside the cell140. We have recently shown that the uptake and secretion of these compounds is a sensitive predictor of microbial taxa56.
Phosphate-containing metabolites (e.g., ATP, NADH, glucose-6P, and carbamoyl phosphate) and organic acids (e.g., citrate and α-ketoisovalerate) play a critical role in central carbon metabolism, energy transfer, and redox166. Although a range of HILIC methods have been developed to chromatographically resolve these compounds, they tend to ionize poorly by electrospray ionization. This problem can be mitigated through the use of ion pairing agents (e.g., tributylamine; TBA), which improve their chromatographic properties and stabilizes their negative charges167,168. The TBA-metabolite complexes formed through this method enhance LC-MS sensitivity by orders of magnitude for these compounds. One of the most effective of these methods is C18 reverse phase ion pairing (RPIP) that was developed by the Rabinowitz group169. This method can be challenging to set up and involves spraying 15 mM TBA into an instrument, which is very difficult to clean out of the system and effectively commits the LC-MS system to negative mode analyses. For labs with the technical expertise to establish this method and resources to commit an instrument to this setup, the RPIP method offers a robust and high-sensitivity mechanism for quantifying phosphate-containing metabolites and organic acids.
In summary, the vast diversity of microbial metabolites produced by the microbiome cannot be captured using a single analytical assay and the selection of analytical method(s) will have a direct impact on the transect of metabolic pathways that will be observable for any given study. Thus, the analytical approach must be tailored to each investigation and multiple methods are generally required to capture a comprehensive picture of host-microbiome metabolic dynamics.
Data normalization in microbiome metabolic studies
The variability of microbiome samples (e.g., fecal water content and variation in urine water content) along with the frequently large scale of many microbiome studies creates significant complexities with regards to normalizing metabolomics datasets31,33,42,43,56,170. Data normalization is a complex task that is affected by both the analytical platform and sample type. Whereas NMR data can be normalized post-acquisition, mass spectrometry data are very challenging to correct post-acquisition because each molecule follows its own unique ionization properties that are nonlinearly affected by the composition of the matrix171,172,173,174. Thus, no single normalization constant can be used to correct for sample-to-sample differences in composition. To address this, a range of computational strategies have been proposed including normalization to a constant sum175, probabilistic quotient normalization176, metabolic ratio correction171, median fold change177, and normalization to MS total useful signal173. However, in our experience, none of these computational strategies completely control for variability and all of these mathematical operations contribute to undesirable propagation of error. Additionally, normalization can produce significant artefacts—especially in untargeted analyses (partially due to missing signals in analytes approaching the limit of detection). Consequently, our preferred approach, wherever possible, is to prepare homogeneous samples or otherwise correct the sample extractions prior to analysis to minimize the need for post-analysis data normalization. We employ a range of analytical strategies depending on the sample type: for tissue and fecal samples we prefer weighing, for fluids we normalize to initial sample volume, and for microbial samples and cell cultures we correct to optical density or colony counts; alternatively, we introduce isotope-labeled reference metabolites into the extraction mixture142 (see SI Section S1 for details).
In addition to sample-to-sample variability, metabolomics studies must also contend with the inherent instability of the analytical platforms. LC-MS response factors drift day-to-day and thus preclude the direct comparison of raw signal intensities from batch-to-batch. This problem can be addressed by collecting a common reference sample in every batch and expressing observed intensity relative to the common refence140. This common sample can be prepared as a mixture of all of the representative samples (i.e., a “super mix”) which is ideal in untargeted analyses that may capture unusual metabolites. Alternatively, a mixture of analytical reference standards in a representative sample matrix can be collected along with each batch of samples140,173. These calibration reference samples can then be used to compute the absolute concentration of target metabolites and thereby sidestep the data normalization pitfalls. Naturally, the standards-based approach limits analysis to compounds that can be matched to commercial standards and to signals that are present within the linear range of the calibration reference mixtures.
Data analysis complexities in microbiome metabolic studies
Untargeted metabolomics studies capture thousands of individual features from each sample, but conventional experimental designs involve cohorts of hundreds or fewer subjects178. This creates an inherent mismatch between data size and biological replication, which is a recipe for driving false discovery, statistical overfitting, and a variety of computational problems. Consequently, data processing steps, including removal of noise, peak detection, identification, quantification, and missing value imputation can play pivotal role in the quality of the resulting dataset178,179,180. Many of these challenges are inherent to all systems-level analysis and there are well-established statistical tools such as dimensionality reduction approaches (e.g., principal component analysis)12,16,32,105,124,181,182, correction for multiple-testing (e.g., Bonferroni correction)183, the use of linear models (e.g., ridge regression)184, and data visualization strategies (e.g., volcano or Manhattan plots)185 that can identify statistically significant correlations and avoid common pitfalls related to false discovery and statistical overfitting. Recently, new computational approaches have been developed, including machine learning and mediation analysis, that provide a powerful new approach for unlocking the molecular underpinnings of host-microbiome dynamics186,187,188,189,190,191.
Machine learning strategies for identifying molecular mechanisms
Microbiome-metabolomics computational approaches must decipher a complex mix of interactions to reveal the microbial composition, the interactions between its components, the interaction with the host, as well as time dependencies in the sample. In recent years, researchers have increasingly used machine learning (ML) approaches to resolve these complexities188,190,192,193,194. ML techniques compress high-dimensional data into models via a recursive learning (or fitting) process. Once trained, these models can then be used to predict, classify, or transform new data. A branch of ML called explainable ML focuses on the interpretability of such models and is useful for microbiome research because of its ability to model to highly complex functions, and to identify important combinations of features while simultaneously incorporating confounding variables189,190,195,196,197. Some examples of successful applications of ML are high-capacity models (e.g., Random Forest190 and Gradient Boosting189), deep artificial neural networks that can model arbitrarily complex functions196,197, and the SHAP algorithm that uses concepts from cooperative game theory to analyze trained models and their predictions195. Each of these tools can return a human interpretable “importance” score of the original features and these scores can be used to help drive research into discrete molecular mechanisms. Recently, identifying these causal relationships has been taken a step forward with AutoEncoders, which are neural networks that were developed to identify causal relationships using a latent variable model187. Though effective in skilled hands, neural networks often require expert knowledge to be implemented effectively and other approaches including mediation analysis may provide a means to interpreting complex relationships in metabolomics data.
Mediation analysis in microbiome studies
Mediation analysis is a valuable tool that can estimate causality in relationships between study variables191,198. Mediation models were first employed in the study of psychosocial predictors of human health in order to identify potential causal relationships by decomposing the direct effect of a predictor versus a treatment or outcome or its indirect effect through a mediator199,200,201,202. A mediation model has three types of variables, the predictor (X), the outcome (Y), and the mediator (M). X could be the state of a patient (age, gender, comorbidities, etc.), Y the severity of a disease, and M the clinical interventions. Tests of association often use another variable called the confounding factor or variable (C), which does not mediate the association between X and Y but is an alternate (biasing) explanation for it. The goal is to quantify direct effects (caused by X) and indirect effects (caused by M) on (Y) and their statistical significances. Explanations for a potentially-causal association between a predictor (X) and an outcome (Y) almost always involve at least one mediator variable (M)203.
More recently, there has been a growing interest in studying the role of human gut microbiota as a “mediating” biological pathway in the association between diet, a medical intervention or an environmental exposure, and adult health200,201,202. Several publications have also explored the role of early-life microbiota by testing the role of gut microbiota during infancy in mediating associations between a variety of early life exposures such as maternal pre-pregnancy weight, cesarean section delivery, and household cleaning product use, and comparing these to future health outcomes204,205,206. Mediation analysis can be used to identify microbiota metabolic pathways for breastfeeding in promoting gut immunity. For example, γ-Proteobacteria and its metabolite lactate have been shown experimentally to promote mucosal immunity and aid maturation of gut microbiota by stimulating Immunoglobulin A responses and enhancing intestinal cell activity of dendritic cells207,208.
In a multiple mediator pathway model (Fig. 3), we demonstrate the effects of two sequential mediators, gut γ-Proteobacteria (Mediator 1) and lactate levels (Mediator 2), in the pathway between breastfeeding (BF) status (X) and fecal secretory Immunoglobulin A (sIgA) levels, a marker of gut immunity, after 3 months of breastfeeding (Y). At this young infant age, breast milk is the sole source of sIgA and the infant gut only produces small amounts. In this example, the predictor variable (X) is divided into two categories, X1 (partially-breastfed infants) and X2 (non-breastfed infants), and compared to the reference category, exclusively breastfed infants. The mediating path (or indirect effect) being tested is the γ-Proteobacteria – lactate pathway in the association between the extent of non-breastfeeding and fecal sIgA levels. This path shows statistical significance for partial breastfeeding [Path 3 × 1: −0.05, 95% (−0.10, −0.01)] and no breastfeeding [Path 3 × 2: −0.06, 95%CI (−0.13, −0.02)], indicating that limiting breast milk intake can lower infant sIgA levels through a pathway of reduced abundance of γ-Proteobacteria and its metabolite lactate. As expected, the model also shows a substantial direct effect for lack of any breastfeeding in lowering sIgA levels [X2, −4.34]. Importantly, the microbiota-lactate path is a separate path to the direct effects of breast milk in supplying sIgA to the infant, suggesting that reduced availability of fecal lactate due to limited breastfeeding may lower sIgA production in the infant gut. Experimentally, it has been shown that lactate stimulates sIgA production208. By identifying multiple metabolic paths or consequences of reduced milk intake, this model underscores the importance of breastfeeding in not only providing passive IgA immunization to the infant but also its role in promoting the immuno-stimulatory activity of γ-Proteobacteria and lactate during early infancy when mucosal immunity is poorly developed209,210.

a We establish the variables in the causal diagram, showing the association between causal agent X (breastfeeding status) and presumed effect Y (infant fecal sIgA levels at 3 months). b Using a sequential mediation model, we establish the direct effects of breastfeeding status on fecal sIgA levels, where breastfeeding status is the categorical variable and exclusive-BF is the reference group. c We then calculate the indirect and total effects in the causal diagram. β-coefficients are shown with 95% Confidence Intervals (CI) and significant differences (p < 0.05) are indicated in red.
Although classical mediation approaches may not model nonlinear effects well, new approaches including parametric models and ML mediation analysis show promising results in establishing high-dimensional data without pre-selection of control variables186,191,192,194,211. These exciting new methodologies will enable researchers to handle the ever-increasing amounts and complexity of future datasets generated through large-scale metabolomics studies.
Examples of host-microbiome dynamics that are modulated through metabolism
Advances in metabolomics technologies have greatly expanded our ability to dissect the molecular underpinnings of complex host-microbiome interactions. Recently, there has been a major expansion of the literature in this area. Our small selection of examples provided here (Table 1) can serve as a starting point for exploring this exciting new body of literature.
Host-microbe metabolic dynamics encompass a wide range of biological functions including chronic and infectious disease, immunity, aging, neurology, and physiology10,13,15,18,22,30,33,104,212. One classic example of these host-microbe dynamics relates to SCFAs, which are produced by the gut microflora and have protective effects against colitis and IBS10,13,15. SCFAs also modulate the maturation of immune cells, including microglia, and shape the properties of the visceral pain signaling pathways10,22. Recent studies have shown that SCFAs have radioprotective effects by stimulating repair processes in the gastrointestinal tract and by reducing proinflammatory responses in the host22,110.
Other select examples of host-microbe interactions include the production of inosine by Bifidobacterium pseudolongum, which aids in the activation of host responses to colorectal cancer in immune checkpoint blockade therapy (with anti-CLTA-4 treatment)30. In addition, phenylacetyl-glutamine and phenylacetyl-glycine, produced by Clostridium sporogenes, are associated with cardiovascular disease through modulation of G-protein coupled receptors33. Lactobacillus and Bacteroides microbiome members are correlated with enhanced neurobiological functions like improved memory or alleviated depression symptoms23,159. Bacteroides possess the ability to produce GABA, a key neurotransmitter for mood and memory regulation23, while Lactobacillus spp. enhance GABA production in a lactate-dependent manner159. Lactobacillus members also metabolize dietary tryptophan into indole compounds (e.g., indoxyl-3-sulfate, indole 3-propionic acid, indole 3-aldehyde) which have a protective effect against host inflammation via the activation of aryl hydrocarbon receptors in both experimental autoimmune encephalomyelitis (EAE; representative of multiple sclerosis)18 and colorectal cancer models212.
Microbiota also participate in microbe-to-microbe interactions that impact host homeostasis and affect the ability of specific pathogens to cause infection. In cystic fibrosis infections, a range of in vitro, computational, and human metabolic profiling approaches have collectively established that dominance by the pathogen Pseudomonas aeruginosa is driven by its ability to cross-feed on amino acids, organic acids, and alcohols produced by facultative anaerobes in the environment24,25,28,29. Phascolarctobacterium spp. have a protective effect against Clostridium difficile infections by consuming the succinate needed for Clostridium difficile growth39 while Bacteroides vulgatus produce SCFAs and trimethylamines, which have a protective effect against Vibrio cholerae infections151.
Diet plays a large role in microbiota-mediated effects in the host. Probiotic and prebiotic (e.g., inulin and stachyose) treatments can be used to stimulate specific strains in the gut microbiome to produce higher levels of SCFAs21,36,37,38. Host nutrition can also be corrected through cross-feeding, where in flies, Lactobacillus and Acetobacter establish a syntrophic relationship to overcome nutrient scarcity due to an imbalanced diet61. Diet can also have a negative impact on the host, such as in the case of Chron’s disease, where catabolism of dietary serine by blooms of Escherichia coli and Citrobacter rodentium can worsen inflammatory responses39. Butyrate produced by Firmicutes members of the microbiome in response to a high carbohydrate diet is associated with the hyperproliferation of colon epithelial cells in a colorectal cancer model213. While current technologies have enabled us to unravel many of these host-microbiome interactions along multiple axes of interaction, new technologies will allow us to elucidate even more of the complex interactions underlying community metabolism, disease pathology, and dysbiosis at a molecular level.
Conclusion
Over the last two decades metabolomics has matured from a largely descriptive activity into a tool for probing the molecular underpinnings of biology. Host-microbiome dynamics are some of the most complex biological systems where these tools have been applied and the biological, logistical, analytical, and computational challenges inherent to this field of research make it difficult to move beyond correlational statistics. However, recent advances in model systems, experimental strategies, analytics, and computational tools have opened the door to mechanistic insights into microbiome-mediated biological phenomena. As these approaches mature, we anticipate that we will quickly gain molecular understanding of the myriad mechanisms that the microbiome uses to modulate the host immune system and other important phenomena.
References
Fettig, N. M. & Osborne, L. C. Direct and indirect effects of microbiota-derived metabolites on neuroinflammation in multiple sclerosis. Microbes Infect. 23, 104814 (2021).
Blacher, E., Levy, M., Tatirovsky, E. & Elinav, E. Microbiome-modulated metabolites at the interface of host immunity. J. Immunol. 198, 572–580 (2017).
Thursby, E. & Juge, N. Introduction to the human gut microbiota. Biochem. J. 474, 1823–1836 (2017).
Pittayanon, R. et al. Gut microbiota in patients with irritable bowel syndrome—a systematic review. Gastroenterology 157, 97–108 (2019).
Gilbert, J. A. et al. Current understanding of the human microbiome. Nat. Med. 24, 392–400 (2018).
Baümler, A. J. & Sperandio, V. Interactions between the microbiota and pathogenic bacteria in the gut. Nature 535, 85–93 (2016).
Rothhammer, V. & Quintana, F. J. The aryl hydrocarbon receptor: an environmental sensor integrating immune responses in health and disease. Nat. Rev. Immunol. 19, 184–197 (2019).
Groen, R. N., de Clercq, N. C., Nieuwdorp, M., Hoenders, H. J. R. & Groen, A. K. Gut microbiota, metabolism and psychopathology: a critical review and novel perspectives. Crit. Rev. Clin. Lab. Sci. 55, 283–293 (2018).
Flannigan, K. L. et al. An intact microbiota is required for the gastrointestinal toxicity of the immunosuppressant mycophenolate mofetil. J. Hear. Lung Transplant. 37, 1047–1059 (2018).
Esquerre, N. et al. Colitis-induced microbial perturbation promotes postinflammatory visceral hypersensitivity. Cmgh 10, 225–244 (2020).
Behr, C. et al. Analysis of metabolome changes in the bile acid pool in feces and plasma of antibiotic-treated rats. Toxicol. Appl. Pharmacol. 363, 79–87 (2019).
Fröhlich, E. E. et al. Cognitive impairment by antibiotic-induced gut dysbiosis: analysis of gut microbiota-brain communication. Brain. Behav. Immun. 56, 140–155 (2016).
Zhang, J. D. et al. Berberine alleviates visceral hypersensitivity in rats by altering gut microbiome and suppressing spinal microglial activation. Acta Pharmacol. Sin. 42, 1821–1833 (2021).
Kong, C. et al. Ketogenic diet alleviates colitis by reduction of colonic group 3 innate lymphoid cells through altering gut microbiome. Signal Transduct. Target. Ther. 6, 1–12 (2021).
Shute, A. et al. Cooperation between host immunity and the gut bacteria is essential for helminth-evoked suppression of colitis. Microbiome 9, 1–18 (2021).
Ye, J. et al. Metabolomics-guided hypothesis generation for mechanisms of intestinal protection by live biotherapeutic products. Biomolecules 11, 1–29 (2021).
Tong, M. et al. Reprograming of gut microbiome energy metabolism by the FUT2 Crohn’s disease risk polymorphism. ISME J. 8, 2193–2206 (2014).
Rothhammer, V. et al. Type i interferons and microbial metabolites of tryptophan modulate astrocyte activity and central nervous system inflammation via the aryl hydrocarbon receptor. Nat. Med. 22, 586–597 (2016).
Rothhammer, V. et al. Dynamic regulation of serum aryl hydrocarbon receptor agonists in MS. Neurol. Neuroimmunol. NeuroInflammation 4, 1–8 (2017).
Blacher, E. et al. Potential roles of gut microbiome and metabolites in modulating ALS in mice. Nature 572, 474–480 (2019).
Hoffman, J. D. et al. Dietary inulin alters the gut microbiome, enhances systemic metabolism and reduces neuroinflammation in an APOE4 mouse model. PLoS One 14, 1–22 (2019).
Erny, D. et al. Microbiota-derived acetate enables the metabolic fitness of the brain innate immune system during health and disease. Cell Metab. 33, 2260–2276.e7 (2021).
Strandwitz, P. et al. GABA-modulating bacteria of the human gut microbiota. Nat. Microbiol. 4, 396–403 (2019).
Gao, B. et al. Tracking polymicrobial metabolism in cystic fibrosis airways: pseudomonas aeruginosa metabolism and physiology are influenced by Rothia mucilaginosa-derived metabolites. mSphere 3, 1–6 (2018).
Silveira, C. B. et al. Multi-omics study of keystone species in a cystic fibrosis microbiome. Int. J. Mol. Sci. 22, 1–15 (2021).
Quinn, R. A. et al. Biogeochemical forces shape the composition and physiology of polymicrobial communities in the cystic fibrosis lung. MBio 5, 1–13 (2014).
Quinn, R. A. et al. A Winogradsky-based culture system shows an association between microbial fermentation and cystic fibrosis exacerbation. ISME J. 9, 1024–1038 (2015).
Henson, M. A., Orazi, G., Phalak, P. & O’Toole, G. A. Metabolic modeling of cystic fibrosis airway communities predicts mechanisms of pathogen dominance. mSystems 4, 1–20 (2019).
Whiteson, K. L. et al. Breath gas metabolites and bacterial metagenomes from cystic fibrosis airways indicate active pH neutral 2,3-butanedione fermentation. ISME J. 8, 1247–1258 (2014).
Mager, L. F. et al. Microbiome-derived inosine modulates response to checkpoint inhibitor immunotherapy. Science 369, 1481–1489 (2020).
Morgell, A. et al. Metabolic characterization of plasma and cyst fluid from cystic precursors to pancreatic cancer patients reveal metabolic signatures of bacterial infection. J. Proteome Res. 20, 2725–2738 (2021).
Paul, B. et al. Impact of genistein on the gut microbiome of humanized mice and its role in breast tumor inhibition. PLoS One 12, 1–20 (2017).
Nemet, I. et al. A cardiovascular disease-linked gut microbial metabolite acts via adrenergic receptors. Cell 180, 862–877.e22 (2020).
Taylor, M. R. et al. Vancomycin relieves mycophenolate mofetil–induced gastrointestinal toxicity by eliminating gut bacterial -glucuronidase activity. Sci. Adv. 5, 1–10 (2019).
Klünemann, M. et al. Bioaccumulation of therapeutic drugs by human gut bacteria. Nature 597, 533–538 (2021).
Chaikham, P., Apichartsrangkoon, A., Jirarattanarangsri, W. & Wiele, T. Van de Influence of encapsulated probiotics combined with pressurized longan juice on colon microflora and their metabolic activities on the exposure to simulated dynamic gastrointestinal tract. Food Res. Int. 49, 133–142 (2012).
Püngel, D. et al. Bifidobacterium breve UCC2003 exopolysaccharide modulates the early life microbiota by acting as a potential dietary substrate. Nutrients 12, 1–17 (2020).
Marzorati, M. et al. Treatment with a spore-based probiotic containing five strains of Bacillus induced changes in the metabolic activity and community composition of the gut microbiota in a SHIME® model of the human gastrointestinal system. Food Res. Int. 149, 110676 (2021).
Kitamoto, S. et al. Dietary l-serine confers a competitive fitness advantage to Enterobacteriaceae in the inflamed gut. Nat. Microbiol. 5, 116–125 (2020).
Kim, H. J., Li, H., Collins, J. J. & Ingber, D. E. Contributions of microbiome and mechanical deformation to intestinal bacterial overgrowth and inflammation in a human gut-on-a-chip. Proc. Natl Acad. Sci. U.S.A. 113, E7–E15 (2016).
Trapecar, M. et al. Gut-liver physiomimetics reveal paradoxical modulation of IBD-related inflammation by short-chain fatty acids. Cell Syst. 10, 223–239.e9 (2020).
McDonald, D. et al. American gut: an open platform for citizen science microbiome research. mSystems 3, e00031–18 (2018).
Melnik, A. V. et al. Coupling targeted and untargeted mass spectrometry for metabolome-microbiome-wide association studies of human fecal samples. Anal. Chem. 89, 7549–7559 (2017).
Quinn, R. A. et al. Global chemical effects of the microbiome include new bile-acid conjugations. Nature 579, 123–129 (2020).
Han, J., Lin, K., Sequeira, C. & Borchers, C. H. An isotope-labeled chemical derivatization method for the quantitation of short-chain fatty acids in human feces by liquid chromatography-tandem mass spectrometry. Anal. Chim. Acta 854, 86–94 (2015).
Lu, W. et al. Metabolite measurement: pitfalls to avoid and practices to follow. Annu. Rev. Biochem. 86, 277–304 (2017).
Fiehn, O. Metabolomics—the link between genotypes and phenotypes. Plant Mol. Biol. 48, 155–171 (2002).
Kuehnbaum, N. L. & Britz-Mckibbin, P. New advances in separation science for metabolomics: resolving chemical diversity in a post-genomic era. Chem. Rev. 113, 2437–2468 (2013).
Patti, G. J., Yanes, O. & Siuzdak, G. Innovation: Metabolomics: the apogee of the omics trilogy. Nat. Rev. Mol. Cell Biol. 13, 263–269 (2012).
Seth, E. C. & Taga, M. E. Nutrient cross-feeding in the microbial world. Front. Microbiol. 5, 1–6 (2014).
Hoek, M. J. A. V. & Merks, R. M. H. Emergence of microbial diversity due to cross-feeding interactions in a spatial model of gut microbial metabolism. BMC Syst. Biol. 11, 1–18 (2017).
Yang, D. D. et al. Fitness and productivity increase with ecotypic diversity among escherichia coli strains that coevolved in a simple, constant environment. Appl. Environ. Microbiol. 86, 1–17 (2020).
Adamowicz, E. M., Flynn, J., Hunter, R. C. & Harcombe, W. R. Cross-feeding modulates antibiotic tolerance in bacterial communities. ISME J. 12, 2723–2735 (2018).
Adamowicz, E. M. & Harcombe, W. R. Weakest-link dynamics predict apparent antibiotic interactions in a model cross-feeding community. Antimicrob. Agents Chemother. 64, 1–13 (2020).
Wyss, M. et al. Using precisely defined in vivo microbiotas to understand microbial regulation of IgE. Front. Immunol. 10, 1–14 (2020).
Rydzak, T. et al. Metabolic preference assay for rapid diagnosis of bloodstream infections. Nat. Commun. 13, 1–12 (2022).
Rojo, F. Carbon catabolite repression in Pseudomonas: optimizing metabolic versatility and interactions with the environment. FEMS Microbiol. Rev. 34, 658–684 (2010).
Riquelme, S. A. et al. CFTR-PTEN-dependent mitochondrial metabolic dysfunction promotes Pseudomonas aeruginosa airway infection. Sci. Transl. Med. 11, 1–27 (2019).
Riquelme, S. A., Wong Fok Lung, T. & Prince, A. Pulmonary pathogens adapt to immune signaling metabolites in the airway. Front. Immunol. 11, 1–14 (2020).
Faust, K. & Raes, J. Microbial interactions: from networks to models. Nat. Rev. Microbiol. 10, 538–550 (2012).
Henriques, S. F. et al. Metabolic cross-feeding in imbalanced diets allows gut microbes to improve reproduction and alter host behaviour. Nat. Commun. 11, 1–15 (2020).
Sakanaka, A., Kuboniwa, M., Takeuchi, H., Hashino, E. & Amano, A. Arginine-ornithine antiporter ArcD controls arginine metabolism and interspecies biofilm development of Streptococcus gordonii. J. Biol. Chem. 290, 21185–21198 (2015).
Huus, K. E. et al. Cross-feeding between intestinal pathobionts promotes their overgrowth during undernutrition. Nat. Commun. 12, 1–14 (2021).
Magnúsdóttir, S. et al. Generation of genome-scale metabolic reconstructions for 773 members of the human gut microbiota. Nat. Biotechnol. 35, 81–89 (2017).
Ponomarova, O. et al. Yeast creates a niche for symbiotic lactic acid bacteria through nitrogen overflow. Cell Syst. 5, 345–357.e6 (2017).
Konstantinidis, D. et al. Adaptive laboratory evolution of microbial co‐cultures for improved metabolite secretion. Mol. Syst. Biol. 17, 1–20 (2021).
Zuñiga, C. et al. Environmental stimuli drive a transition from cooperation to competition in synthetic phototrophic communities. Nat. Microbiol. 4, 2184–2191 (2019).
Heinken, A., Sahoo, S., Fleming, R. M. T. & Thiele, I. Systems-level characterization of a host-microbe metabolic symbiosis in the mammalian gut. Gut Microbes 4, 28–40 (2013).
Date, Y. et al. New monitoring approach for metabolic dynamics in microbial ecosystems using stable-isotope-labeling technologies. J. Biosci. Bioeng. 110, 87–93 (2010).
Nakamura, A., Ooga, T. & Matsumoto, M. Intestinal luminal putrescine is produced by collective biosynthetic pathways of the commensal microbiome. Gut Microbes 10, 159–171 (2019).
Deng, P. et al. Untargeted stable isotope probing of the gut microbiota metabolome using 13C-labeled dietary fibers. J. Proteome Res. 20, 2904–2913 (2021).
Uchimura, Y. et al. Antibodies set boundaries limiting microbial metabolite penetration and the resultant mammalian host response. Immunity 49, 545–559.e5 (2018).
Junot, C., Fenaille, F., Colsch, B. & Bécher, F. High resolution mass spectrometry based techniques at the crossroads of metabolic pathways. Mass Spectrom. Rev. 33, 471–500 (2014).
Van de Wiele, T., Van den Abbeele, P., Ossieur, W., Possemiers, S. & Marzorati, M. The Simulator of the Human Intestinal Microbial Ecosystem (SHIME®). In Impact Food Bioact. Heal. Vitr. Ex Vivo Model. 305–317 https://doi.org/10.1007/978-3-319-16104-4 (2015).
Molly, K., Vande Woestyne, M., De Smet, I. & Verstraete, W. Validation of the simulator of the human intestinal microbial ecosystem (SHIME) reactor using microorganism-associated activities. Microb. Ecol. Health Dis. 7, 191–200 (1994).
Barry, J.-L. et al. Estimation of the fermentability of dietary fibre in vitro: a European interlaboratory study. Br. J. Nutr. 74, 303–322 (1995).
Cinquin, C., Le Blay, G., Fliss, I. & Lacroix, C. Immobilization of infant fecal microbiota and utilization in an in vitro colonic fermentation model. Microb. Ecol. 48, 128–138 (2004).
Aura, A. M., Härkönen, H., Fabritius, M. & Poutanen, K. Development of an in vitro enzymic digestion method for removal of starch and protein and assessment of its performance using rye and wheat breads. J. Cereal Sci. 29, 139–152 (1999).
Oliphant, K. et al. Effects of antibiotic pretreatment of an ulcerative colitis-derived fecal microbial community on the integration of therapeutic bacteria in vitro. mSystems 5, 1–13 (2020).
Marzorati, M. et al. High-fiber and high-protein diets shape different gut microbial communities, which ecologically behave similarly under stress conditions, as shown in a gastrointestinal simulator. Mol. Nutr. Food Res. 61, 1–13 (2017).
Selak, M. et al. Inulin-type fructan fermentation by bifidobacteria depends on the strain rather than the species and region in the human intestine. Appl. Microbiol. Biotechnol. 100, 4097–4107 (2016).
Grootaert, C. et al. Comparison of prebiotic effects of arabinoxylan oligosaccharides and inulin in a simulator of the human intestinal microbial ecosystem. FEMS Microbiol. Ecol. 69, 231–242 (2009).
Aura, A. M. & Maukonen, J. One compartment fermentation model. In Impact Food Bio-Actives Gut Heal. 281–292 (2015).
Aura, A. M. et al. Processing of rye bran influences both the fermentation of dietary fibre and the bioconversion of lignans by human faecal flora in vitro. J. Sci. Food Agric. 85, 2085–2093 (2005).
Aura, A. M. et al. Suitability of a batch in vitro fermentation model using human faecal microbiota for prediction of conversion of flaxseed lignans to enterolactone with reference to an in vivo rat model. Eur. J. Nutr. 45, 45–51 (2006).
Nordlund, E. et al. Formation of phenolic microbial metabolites and short-chain fatty acids from rye, wheat, and oat bran and their fractions in the metabolical in vitro colon model. J. Agric. Food Chem. 60, 8134–8145 (2012).
Shah, P. et al. A microfluidics-based in vitro model of the gastrointestinal human-microbe interface. Nat. Commun. 7, 1–15 (2016).
Wissenbach, D. K. et al. Optimization of metabolomics of defined in vitro gut microbial ecosystems. Int. J. Med. Microbiol. 306, 280–289 (2016).
Bein, A. et al. Microfluidic organ-on-a-chip models of human intestine. Cmgh 5, 659–668 (2018).
May, S., Evans, S. & Parry, L. Organoids, organs-on-chips and other systems, and microbiota. Emerg. Top. Life Sci. 1, 385–400 (2017).
Park, G. S. et al. Emulating host-microbiome ecosystem of human gastrointestinal tract in vitro. Stem Cell Rev. Rep. 13, 321–334 (2017).
Tovaglieri, A. et al. Species-specific enhancement of enterohemorrhagic E. coli pathogenesis mediated by microbiome metabolites. Microbiome 7, 1–20 (2019).
Kim, H. J., Huh, D., Hamilton, G. & Ingber, D. E. Human gut-on-a-chip inhabited by microbial flora that experiences intestinal peristalsis-like motions and flow. Lab Chip 12, 2165–2174 (2012).
Krause, J. L. et al. Following the community development of SIHUMIx–a new intestinal in vitro model for bioreactor use. Gut Microbes 11, 1116–1129 (2020).
Rohani, L. et al. Stirred suspension bioreactors maintain naïve pluripotency of human pluripotent stem cells. Commun. Biol. 3, 1–22 (2020).
Götz, J., Bodea, L. G. & Goedert, M. Rodent models for Alzheimer disease. Nat. Rev. Neurosci. 19, 583–598 (2018).
Wong, S. K., Chin, K. Y., Suhaimi, F. H., Fairus, A. & Ima-Nirwana, S. Animal models of metabolic syndrome: a review. Nutr. Metab. 13, 1–12 (2016).
Kersten, K., Visser, K. E., Miltenburg, M. H. & Jonkers, J. Genetically engineered mouse models in oncology research and cancer medicine. EMBO Mol. Med. 9, 137–153 (2017).
Tlaskalova-Hogenova, H. et al. Microbiome and colorectal carcinoma: Insights from germ-free and conventional animal models. Cancer J. 20, 217–224 (2014).
Park, J. C. & Im, S. H. Of men in mice: the development and application of a humanized gnotobiotic mouse model for microbiome therapeutics. Exp. Mol. Med. 52, 1383–1396 (2020).
Martín, R., Bermúdez-Humarán, L. G. & Langella, P. Gnotobiotic rodents: an in vivo model for the study of microbe-microbe interactions. Front. Microbiol. 7, 1–7 (2016).
Weinroth, M. D. et al. Considerations and best practices in animal science 16S ribosomal RNA gene sequencing microbiome studies. J. Anim. Sci. 100, 1–18 (2022).
Bokoliya, S. C., Dorsett, Y., Panier, H. & Zhou, Y. Procedures for fecal microbiota transplantation in murine microbiome studies. Front. Cell. Infect. Microbiol. 11, 1–14 (2021).
Lee, J. et al. Young versus aged microbiota transplants to germ-free mice: increased short-chain fatty acids and improved cognitive performance. Gut Microbes 12, 1–14 (2020).
Han, L. W. et al. Key hepatic metabolic pathways are altered in germ-free mice during pregnancy. PLoS One 16, 1–18 (2021).
Gnainsky, Y. et al. Systemic regulation of host energy and oogenesis by microbiome-derived mitochondrial coenzymes. Cell Rep. 34, 108583 (2021).
Kashyap, P. C. et al. Complex interactions among diet, gastrointestinal transit, and gut microbiota in humanized mice. Gastroenterology 144, 967–977 (2013).
Xi, M. et al. Microbiome-metabolomic analyses of the impacts of dietary stachyose on fecal microbiota and metabolites in infants intestinal microbiota-associated mice. J. Sci. Food Agric. 101, 3336–3347 (2021).
Liao, X. et al. Alteration of gut microbiota induced by DPP-4i treatment improves glucose homeostasis. EBioMedicine 44, 665–674 (2019).
Guo, H. et al. Multi-omics analyses of radiation survivors identify radioprotective microbes and metabolites. Science 370, 1–11 (2020).
Lim, J. J. et al. Gut microbiome critically impacts PCB-induced changes in metabolic fingerprints and the hepatic transcriptome in mice. Toxicol. Sci. 177, 168–187 (2020).
Li, C. Y. et al. Novel interactions between gut microbiome and host drug-processing genes modify the hepatic metabolism of the environmental chemicals polybrominated diphenyl ethers. Drug Metab. Dispos. 45, 1197–1214 (2017).
Li, C. Y. et al. PBDEs altered gut microbiome and bile acid homeostasis in male C57BL/6 mice. Drug Metab. Dispos. 46, 1226–1240 (2018).
Nagao-Kitamoto, H. et al. Interleukin-22-mediated host glycosylation prevents Clostridioides difficile infection by modulating the metabolic activity of the gut microbiota. Nat. Med. 26, 608–617 (2020).
Bogatyrev, S. R., Rolando, J. C. & Ismagilov, R. F. Self-reinoculation with fecal flora changes microbiota density and composition leading to an altered bile-acid profile in the mouse small intestine. Microbiome 8, 1–22 (2020).
Oh, J. H. & Rehermann, B. Natural versus laboratory world: incorporating wild-derived microbiota into preclinical rodent models. J. Immunol. 207, 1703–1709 (2021).
Zubeldia-Varela, E., Barber, D., Barbas, C., Perez-Gordo, M. & Rojo, D. Sample pre-treatment procedures for the omics analysis of human gut microbiota: turning points, tips and tricks for gene sequencing and metabolomics. J. Pharm. Biomed. Anal. 191, 113592 (2020).
Matysik, S., Le Roy, C. I., Liebisch, G. & Claus, S. P. Metabolomics of fecal samples: a practical consideration. Trends Food Sci. Technol. 57, 244–255 (2016).
Deda, O., Gika, H. G., Wilson, I. D. & Theodoridis, G. A. An overview of fecal sample preparation for global metabolic profiling. J. Pharm. Biomed. Anal. 113, 137–150 (2015).
Gratton, J. et al. Optimized sample handling strategy for metabolic profiling of human feces. Anal. Chem. 88, 4661–4668 (2016).
Alseekh, S. et al. Mass spectrometry-based metabolomics: a guide for annotation, quantification and best reporting practices. Nat. Methods 18, 747–756 (2021).
Claus, S. P. et al. Systemic multicompartmental effects of the gut microbiome on mouse metabolic phenotypes. Mol. Syst. Biol. 4, 1–14 (2008).
Li, X. et al. A metaproteomic approach to study human-microbial ecosystems at the mucosal luminal interface. PLoS One 6, (2011).
Lai, Y., Liu, C. W., Chi, L., Ru, H. & Lu, K. High-resolution metabolomics of 50 neurotransmitters and tryptophan metabolites in feces, serum, and brain tissues using UHPLC-ESI-Q exactive mass spectrometry. ACS Omega 6, 8094–8103 (2021).
Wu, W. et al. Bioregional alterations in gut microbiome contribute to the plasma metabolomic changes in pigs fed with inulin. Microorganisms 8, 1–17 (2020).
Wikoff, W. R. et al. Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proc. Natl Acad. Sci. U.S.A. 106, 3698–3703 (2009).
Zheng, D., Liwinski, T. & Elinav, E. Interaction between microbiota and immunity in health and disease. Cell Res. 30, 492–506 (2020).
González-Domínguez, R., González-Domínguez, Á., Sayago, A. & Fernández-Recamales, Á. Recommendations and best practices for standardizing the pre-analytical processing of blood and urine samples in metabolomics. Metabolites 10, 1–18 (2020).
Diallo, A. F. et al. Metabolic profiling of blood and urine for exploring the functional role of the microbiota in human health. Biol. Res. Nurs. 22, 449–457 (2020).
Lewis, I. A., Shortreed, M. R., Hegeman, A. D. & Markley, J. L. Novel NMR and MS approaches to metabolomics. In Handbook of Metabolomics (Fan, T. W.-M., Lane, A. N. & Higashi, R. M.) 199–230 (Humana Press, Totowa, NJ, 2012). https://doi.org/10.1007/978-1-61779-618-0_7
Voigt, A. L. et al. Unique metabolic phenotype and its transition during maturation of juvenile male germ cells. FASEB J. 35, 1–22 (2021).
Giommi, C., Ladisa, C., Carnevali, O., Maradonna, F. & Habibi, H. R. Metabolomic and transcript analysis revealed a sex-specific effect of glyphosate in zebrafish liver. Int. J. Mol. Sci. 23, 1–16 (2022).
Lau, A. et al. Dipeptidase-1 governs renal inflammation during ischemia reperfusion injury. Sci. Adv. 8, 1–10 (2022).
Vicentini, F. A. et al. Colitis-associated microbiota drives changes in behaviour in male mice in the absence of inflammation. Brain. Behav. Immun. 102, 266–278 (2022).
van Tilburg Bernardes, E. et al. Intestinal fungi are causally implicated in microbiome assembly and immune development in mice. Nat. Commun. 11, 1–16 (2020).
Mager, L. F. et al. Microbiome-derived inosine modulates response to checkpoint inhibitor immunotherapy. Science 3421, eabc3421 (2020).
Riquelme, S. A. et al. Pseudomonas aeruginosa utilizes host-derived itaconate to redirect its metabolism to promote biofilm formation. Cell Metab. 31, 1091–1106 (2021).
Wong Fok Lung, T. et al. Klebsiella pneumoniae induces host metabolic stress that promotes tolerance to pulmonary infection. Cell Metab. 1–14 https://doi.org/10.1016/j.cmet.2022.03.009 (2022).
Michi, A. N., Yipp, B. G., Dufour, A., Lopes, F. & Proud, D. PGC-1α mediates a metabolic host defense response in human airway epithelium during rhinovirus infections. Nat. Commun. 12, 1–19 (2021).
Groves, R. A. et al. Methods for quantifying the metabolic boundary fluxes of cell cultures in large cohorts by high-resolution hydrophilic liquid chromatography mass spectrometry. Anal. Chem. 94, 8874–8882 (2022).
Fiori, J., Turroni, S., Candela, M. & Gotti, R. Assessment of gut microbiota fecal metabolites by chromatographic targeted approaches. J. Pharm. Biomed. Anal. 177, 112867 (2020).
Bihan, D. et al. Method for absolute quantification of short chain fatty acids via reverse phase chromatography mass spectrometry. PLoS One 17, 1–14 (2022).
Wu, J. et al. NMR-based metabolite profiling of human milk: a pilot study of methods for investigating compositional changes during lactation. Biochem. Biophys. Res. Commun. 469, 626–632 (2016).
Prentice, P. M. et al. Human milk short-chain fatty acid composition is associated with adiposity outcomes in infants. J. Nutr. 149, 716–722 (2019).
Loke, M. F. et al. Metabolomics and 16S rRNA sequencing of human colorectal cancers and adjacent mucosa. PLoS One 13, 1–20 (2018).
Shalon, D. et al. Profiling of the human intestinal microbiome and bile acids under 2 physiologic conditions using an ingestible sampling device. bioRxiv 1–42 (2022).
Rezaei Nejad, H. et al. Ingestible osmotic pill for in vivo sampling of gut microbiomes. Adv. Intell. Syst. 1, 1900053 (2019).
Pinu, F. R., Villas-Boas, S. G. & Aggio, R. Analysis of intracellular metabolites from microorganisms: quenching and extraction protocols. Metabolites 7, 1–20 (2017).
McGrath, L. T., Weir, C. D., Maynard, S. & Rowlands, B. J. Gas-liquid chromatographic analysis of volatile short chain fatty acids in fecal samples as pentafluorobenzyl esters. Anal. Biochem. 207, 227–230 (1992).
Zhao, G., Nyman, M. & Jönsson, J. Å. Rapid determination of short-chain fatty acids in colonic contents and faeces of humans and rats by acidified water-extraction and direct-injection gas chromatography. Biomed. Chromatogr. 20, 674–682 (2006).
You, J. S. et al. Commensal-derived metabolites govern Vibrio cholerae pathogenesis in host intestine. Microbiome 7, 1–18 (2019).
Ratajczak, W. et al. Immunomodulatory potential of gut microbiome-derived shortchain fatty acids (SCFAs). Acta Biochim. Pol. 66, 1–12 (2019).
Lewis, I. A. et al. Method for determining molar concentrations of metabolites in complex solutions from two-dimensional 1H-13C NMR spectra. Anal. Chem. 79, 9385–9390 (2007).
He, L. et al. Simultaneous quantification of straight-chain and branched-chain short chain fatty acids by gas chromatography mass spectrometry. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 1092, 359–367 (2018).
Chen, I. & Cassaro, S. Physiology, bile acids. In NCBI Bookshelf 1–6 (2022).
Shi, H. et al. Restructuring the gut microbiota by intermittent fasting lowers blood pressure. Circ. Res. 128, 1240–1254 (2021).
Duboc, H., Tache, Y. & Hofmann, A. F. The bile acid TGR5 membrane receptor: from basic research to clinical application. Dig. Liver Dis. 46, 302–312 (2014).
Ridlon, J. M. et al. The ‘in vivo lifestyle’ of bile acid 7α-dehydroxylating bacteria: comparative genomics, metatranscriptomic, and bile acid metabolomics analysis of a defined microbial community in gnotobiotic mice. Gut Microbes 11, 381–404 (2020).
Mao, J. H. et al. Genetic and metabolic links between the murine microbiome and memory. Microbiome 8, 1–14 (2020).
Kong, C. et al. Ketogenic diet alleviates colitis by reduction of colonic group 3 innate lymphoid cells through altering gut microbiome. Signal Transduct. Target. Ther. 6, 1–12 (2021).
Koistinen, V. M. et al. Contribution of gut microbiota to metabolism of dietary glycine betaine in mice and in vitro colonic fermentation. Microbiome 7, 1–14 (2019).
Moriya, T., Satomi, Y., Murata, S., Sawada, H. & Kobayashi, H. Effect of gut microbiota on host whole metabolome. Metabolomics 13, 1–12 (2017).
Bajad, S. U. et al. Separation and quantitation of water soluble cellular metabolites by hydrophilic interaction chromatography-tandem mass spectrometry. J. Chromatogr. A 1125, 76–88 (2006).
Buszewski, B. & Noga, S. Hydrophilic interaction liquid chromatography (HILIC)—a powerful separation technique. Anal. Bioanal. Chem. 402, 231–247 (2012).
Cubbon, S., Antonio, C., Wilson, J. & Thomas-Oates, J. Metabolomic applications of HILIC-LC-MS. Mass Spectrom. Rev. 29, 671–684 (2010).
Bar-Even, A., Flamholz, A., Noor, E. & Milo, R. Rethinking glycolysis: on the biochemical logic of metabolic pathways. Nat. Chem. Biol. 8, 509–517 (2012).
Ivanisevic, J. et al. Toward’Omic scale metabolite profiling: a dual separation-mass spectrometry approach for coverage of lipid and central carbon metabolism. Anal. Chem. 85, 6876–6884 (2013).
Lu, W., Bennett, B. D. & Rabinowitz, J. D. Analytical strategies for LC-MS-based targeted metabolomics. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 871, 236–242 (2008).
Lu, W. et al. Metabolomic analysis via reversed-phase ion-pairing liquid chromatography coupled to a stand alone orbitrap mass spectrometer. Anal. Chem. 82, 3212–3221 (2010).
Yu, H. & Huan, T. MAFFIN: metabolomics sample normalization using maximal density fold change with high-quality metabolic features and corrected signal intensities. Bioinformatics 38, 3429–3437 (2022).
Yu, H. & Huan, T. Patterned signal ratio biases in mass spectrometry-based quantitative metabolomics. Anal. Chem. 93, 2254–2262 (2021).
Torgrip, R. J. O., Åberg, K. M., Alm, E., Schuppe-Koistinen, I. & Lindberg, J. A note on normalization of biofluid 1D 1H-NMR data. Metabolomics 4, 114–121 (2008).
Wu, Y. & Li, L. Sample normalization methods in quantitative metabolomics. J. Chromatogr. A 1430, 80–95 (2016).
Reisetter, A. C. et al. Mixture model normalization for non-targeted gas chromatography/mass spectrometry metabolomics data. BMC Bioinforma. 18, 1–17 (2017).
Craig, A., Cloarec, O., Holmes, E., Nicholson, J. K. & Lindon, J. C. Scaling and normalization effects in NMR spectroscopic metabonomic data sets. Anal. Chem. 78, 2262–2267 (2006).
Dieterle, F., Ross, A., Schlotterbeck, G. & Senn, H. Probabilistic quotient normalization as robust method to account for dilution of complex biological mixtures. Application in 1H NMR metabonomics. Anal. Chem. 78, 4281–4290 (2006).
Veselkov, K. A. et al. Profiles for improved information recovery. Anal. Chem. 83, 5864–5872 (2011).
Chen, M. X., Wang, S. Y., Kuo, C. H. & Tsai, I. L. Metabolome analysis for investigating host-gut microbiota interactions. J. Formos. Med. Assoc. 118, S10–S22 (2019).
Misra, B. B. New software tools, databases, and resources in metabolomics: updates from 2020. Metabolomics 17, 1–24 (2021).
Misra, B. B. & van der Hooft, J. J. J. Updates in metabolomics tools and resources: 2014-2015. Electrophoresis 37, 86–110 (2016).
O’Sullivan, V., Madrid-Gambin, F., Alegra, T., Gibbons, H. & Brennan, L. Impact of sample storage on the NMR fecal water metabolome. ACS Omega 3, 16585–16590 (2018).
Vanden Bussche, J., Marzorati, M., Laukens, D. & Vanhaecke, L. Validated high resolution mass spectrometry-based approach for metabolomic fingerprinting of the human gut phenotype. Anal. Chem. 87, 10927–10934 (2015).
Bonferroni, C. Teoria statistica delle classi e calcolo delle probabilita. Pubbl. del. R. Ist. Super. di Sci. Econ. e Commericiali di Firenze 8, 3–62 (1936).
Hoerl, A. E. & Kennard, R. W. Ridge regression: biased estimation for nonorthogonal problems. Technometrics 12, 55–67 (1970).
Li, W. Application of volcano plots in analyses of mRNA differential expressions with microarrays. J. Bioinform. Comput. Biol. 10, 1–30 (2012).
Jacob, D. CATE Meets ML - Conditional Average Treatment Effect and Machine Learning. arXiv 1–67 https://doi.org/10.2139/ssrn.3816558 (2021).
Louizos, C. et al. Causal effect inference with deep latent-variable models. arXiv 1–12 (2017).
Torrey, L. & Shavlik, J. Transfer learning. In: Handbook of Research on Machine Learning Applications 1–22 https://doi.org/10.1201/b17320 (2009).
Friedman, J. H. Greedy function approximation: a gradient boosting machine. Ann. Stat. 29, 1189–1232 (2001).
Breiman, L. Random forests. Mach. Learn. 45, 5–32 (2001).
VanderWeele, T. J. Mediation analysis: a practitioner’s guide. Annu. Rev. Public Health 37, 17–32 (2016).
Caron, A., Baio, G. & Manolopoulou, I. Estimating individual treatment effects using non‐parametric regression models: a review. J. R. Stat. Soc. Ser. A (Statistics Soc. 1–35 https://doi.org/10.1111/rssa.12824 (2022).
McInnes, L., Healy, J. & Melville, J. UMAP: uniform manifold approximation and projection for dimension reduction. arXiv 1–63 http://arxiv.org/abs/1802.03426 (2020).
Farbmacher, H., Huber, M., Lafférs, L., Langen, H. & Spindler, M. Causal mediation analysis with double machine learning. Econom. J. 00, 1–24 (2022).
Lundberg, S. M. & Lee, S.-I. A unified approach to interpreting model predictions. In Proc. 31st Conference on Neural Information Processing Systems 1–10 https://doi.org/10.1016/j.ophtha.2018.11.016 (2017).
Goodfellow, I., Bengio, Y. & Courville, A. Deep Learning. (MIT Press, 2016).at http://www.deeplearningbook.org
Pomyen, Y. et al. Deep metabolome: applications of deep learning in metabolomics. Comput. Struct. Biotechnol. J. 18, 2818–2825 (2020).
Lee, H., Herbert, R. D. & McAuley, J. H. Mediation analysis. JAMA 321, 697–698 (2019).
Baron, R. M. & Kenny, D. A. The moderator-mediator variable distinction in social psychological research: conceptual, strategic, and statistical considerations. J. Pers. Soc. Psychol. 51, 1173–1182 (1986).
Menni, C. et al. High intake of vegetables is linked to lower white blood cell profile and the effect is mediated by the gut microbiome. BMC Med. 19, 1–10 (2021).
Wang, D. et al. Characterization of gut microbial structural variations as determinants of human bile acid metabolism. Cell Host Microbe 29, 1802–1814.e5 (2021).
Brandao Gois, M. et al. Role of the gut microbiome in mediating lactose intolerance symptoms. Gut 71, 214–215 (2022).
Hayes, A. F. & Preacher, K. J. Quantifying and testing indirect effects in simple mediation models when the constituent paths are nonlinear. Multivar. Behav. Res. 45, 627–660 (2010).
Tun, H. M. et al. Roles of birth mode and infant gut microbiota in intergenerational transmission of overweight and obesity from mother to offspring. JAMA Pediatr. 172, 368–377 (2018).
Tun, M. H. et al. Postnatal exposure to household disinfectants, infant gut microbiota and subsequent risk of overweight in children. CMAJ 190, E1097–E1107 (2018).
Van Nimwegen, F. A. et al. Mode and place of delivery, gastrointestinal microbiota, and their influence on asthma and atopy. J. Allergy Clin. Immunol. 128, 948–955.e3 (2011).
Mirpuri, J. et al. Proteobacteria-specific IgA regulates maturation of the intestinal microbiota. Gut Microbes 5, 28–39 (2013).
Morita, N. et al. GPR31-dependent dendrite protrusion of intestinal CX3CR1 + cells by bacterial metabolites. Nature 566, 110–114 (2019).
Battersby, A. J. & Gibbons, D. L. The gut mucosal immune system in the neonatal period. Pediatr. Allergy Immunol. 24, 414–421 (2013).
Kawano, A. & Emori, Y. Changes in maternal secretory immunoglobulin a levels in human milk during 12 weeks after parturition. Am. J. Hum. Biol. 25, 399–403 (2013).
Knafl, G. J. et al. Incorporating nonlinearity into mediation analyses. BMC Med. Res. Methodol. 17, 1–21 (2017).
Hezaveh, K. et al. Tryptophan-derived microbial metabolites activate the aryl hydrocarbon receptor in tumor-associated macrophages to suppress anti-tumor immunity. Immunity 55, 324–340.e8 (2022).
Belcheva, A. et al. Gut microbial metabolism drives transformation of msh2-deficient colon epithelial cells. Cell 158, 288–299 (2014).
Acknowledgements
We thank K.A. Pittman for his critical review and editorial help in preparing this article. I.A.L. is supported by an Alberta Innovates—Health Solutions (AIHS, Translational Health Chair), the Natural Sciences and Engineering Research Council (NSERC) [DG 04547], and the International Microbiome Centre and IMPACTT Microbiome Research Core (CIHR IMC-161484). A.L.K. is supported by a Canadian Institutes of Health Research (CIHR) operating grant. S.L.B. is supported by a National Institutes of Health (NIH) grant [Project Number 1R01AI153521-01]. M.D. is supported by the IMPACTT Microbiome Research Core. S.W. is supported through a Genome Canada Large Scale Applied Research Project award. Y.Y.C. is supported through a Canadian Respiratory Research Network Postdoctoral Fellowship and a CIHR operating grant.
Author information
Authors and Affiliations
Contributions
I.A.L. and A.K. conceived of the review. S.L.B., M.D., S.W., and Y.Y.C. performed the literature review and wrote sections of the manuscript. S.L.B. wrote the primary draft of the manuscript. All authors contributed to revising and critically reviewing the manuscript and approved the final version.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Bishop, S.L., Drikic, M., Wacker, S. et al. Moving beyond descriptive studies: harnessing metabolomics to elucidate the molecular mechanisms underpinning host-microbiome phenotypes. Mucosal Immunol 15, 1071–1084 (2022). https://doi.org/10.1038/s41385-022-00553-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41385-022-00553-4
This article is cited by
-
To metabolomics and beyond: a technological portfolio to investigate cancer metabolism
Signal Transduction and Targeted Therapy (2023)
