
Abstract
Reduction of expression and activity of sirtuin 3 (SIRT3) contributes to the pathogenesis of cardiomyopathy via inducing mitochondrial injury and energy metabolism disorder. However, development of effective ways and agents to modulate SIRT3 remains a big challenge. In this study we explored the upstream suppressor of SIRT3 in angiotensin II (Ang II)-induced cardiac hypertrophy in mice. We first found that SIRT3 deficiency exacerbated Ang II-induced cardiac hypertrophy, and resulted in the development of spontaneous heart failure. Since miRNAs play crucial roles in the pathogenesis of cardiac hypertrophy, we performed miRNA sequencing on myocardium tissues from Ang II-infused Sirt3−/− and wild type mice, and identified microRNA-214 (miR-214) was significantly up-regulated in Ang II-infused mice. Similar results were also obtained in Ang II-treated neonatal mouse cardiomyocytes (NMCMs). Using dual-luciferase reporter assay we demonstrated that SIRT3 was a direct target of miR-214. Overexpression of miR-214 in vitro and in vivo decreased the expression of SIRT3, which resulted in extensive mitochondrial damages, thereby facilitating the onset of hypertrophy. In contrast, knockdown of miR-214 counteracted Ang II-induced detrimental effects via restoring SIRT3, and ameliorated mitochondrial morphology and respiratory activity. Collectively, these results demonstrate that miR-214 participates in Ang II-induced cardiac hypertrophy by directly suppressing SIRT3, and subsequently leading to mitochondrial malfunction, suggesting the potential of miR-214 as a promising intervention target for antihypertrophic therapy.
Similar content being viewed by others


Introduction
Pathological cardiac hypertrophy, an adaptive response of the heart to increased workload, occurs in various cardiovascular conditions, such as hypertension, myocardial infarction and valvular disease. Although it initially helps to normalize wall tension and maintain output, prolonged hypertrophy deteriorates cardiac function, eventually leading to heart failure [1, 2]. During the last decade, accumulating evidence has demonstrated that the development and progression of pathological cardiac hypertrophy is driven by an energy substrate switch from fatty acids to glucose, reductions in mitochondrial biogenesis, and dysregulation of enzymes associated with mitochondrial energy transduction and respiratory pathways [1, 3]. These abnormalities result in energy deficiency and compromise contractile ability, facilitating the transition from the compensatory to the decompensatory stage. Moreover, mitochondrial impairment induces oxidative stress and promotes apoptotic cell death, which further exacerbates cardiac injury [4, 5]. These findings suggest that mitochondrial malfunction may be a valuable target for therapy to improve myocardial energy metabolism and ameliorate the detrimental consequences of pathological hypertrophy.
Sirtuin 3 (SIRT3) is a key member of the sirtuin family, which belongs to the group of NAD+-dependent class III histone deacetylases. It localizes mainly within mitochondria and plays essential roles in modulating multiple aspects of mitochondrial biology [6, 7]. Proteomics and bioinformatic analyses have identified abundant mitochondrial proteins that are deacetylated by SIRT3, including long-chain acyl-CoA dehydrogenase (LCAD), ATP synthase (ATP5O), forkhead box O3a (FoxO3a), manganese-dependent superoxide dismutase (MnSOD), optic atrophy 1 (OPA1) and others [8,9,10]. These substrates of SIRT3 are tightly associated with fatty acid and glucose metabolism, electron transport in the respiratory chain, redox homeostasis and mitochondrial integrity. It has been observed that the content and activity of SIRT3 are significantly reduced in human failing hearts and that these reductions are accompanied by extensive mitochondrial protein hyperacetylation and energy metabolism dysfunction [11, 12]. Activation of SIRT3 by supplementation with the NAD+ precursor nicotinamide mononucleotide has been demonstrated to restore energy metabolism, thereby alleviating hypertrophic cardiomyopathy [13, 14]. These findings indicate the protective roles of SIRT3 against mitochondrial impairments and the potential of SIRT3 as a promising drug target for cardiac hypertrophy and heart failure. Nevertheless, the causes of the reductions in SIRT3 expression and function in the setting of pathological cardiac hypertrophy remain to be clarified. More importantly, previous efforts to develop direct SIRT3 agonists have been hampered due to the allosteric activation mechanism of SIRT3 and the lack of knowledge regarding the binding sites of available compounds [15]. Thus far, only a handful of small-molecule SIRT3 activators have been reported, such as honokiol and resveratrol, which universally exhibit low specificity and potency [16]. It is therefore necessary to identify alternative methods of SIRT3 modulation as therapeutic treatments for hypertrophy-related cardiomyopathy.
MicroRNAs (miRNAs) are small endogenous noncoding RNAs that are 21–24 nucleotides in length. In mammalian cells, they exert crucial biological functions mainly by binding with the 3’ untranslated regions (3’ UTRs) of target mRNAs, resulting in translational suppression or mRNA degradation [17, 18]. It is estimated that miRNAs influence up to approximately 30% of the protein-coding genes in the human genome [19]. Dysregulation of miRNAs has been demonstrated to participate in the pathogenesis of various cardiovascular disorders, especially pathological hypertrophy and heart failure [20, 21]. Among the numerous miRNA molecules, microRNA-214 (miR-214) is highly conserved across species and abundantly expressed in the heart. MiR-214 was initially identified as a promoter of tumor formation and progression, but accumulating evidence has revealed that it is also an important epigenetic regulator in cardiac diseases [22, 23]. In an acute myocardial infarction rat model, miR-214 has been found to inhibit left ventricular remodeling by attenuating apoptosis [24]. On the other hand, miR-214 promotes collagen synthesis in cardiac fibroblasts and induces angiogenesis impairment in mice infused with isoproterenol (ISO) [25, 26]. These facts suggest that miR-214 plays dual roles in different cardiac pathological circumstances, but further research is required before miR-214 can be used as a diagnostic factor or therapeutic target. Moreover, the influence of miR-214 on myocardial energy metabolism and mitochondrial biology has not yet been determined. In this study, we provide the first evidence that miR-214 acts as an upstream suppressor of SIRT3 and contributes to Ang II-induced cardiac hypertrophy. MiR-214 directly targets SIRT3 and downregulates its expression, which causes mitochondrial dysfunction and energy metabolism abnormalities, resulting in the onset of hypertrophy. Therefore, miR-214-based intervention might be a promising new strategy for antihypertrophic treatment.
Materials and methods
Study animals
Male SIRT3-knockout (129-Sirt3tm1.1Fwa/J) mice and their respective wild-type (WT) control (129/SvlmJ) mice were purchased from Jackson Laboratory (Bar Harbor, ME, USA). C57B/L6 mice (male, age 10–12 weeks, certification No. 44007200064634) were obtained from the Experimental Animal Center of Guangzhou University of Chinese Medicine (Guangzhou, China). All surgical and care procedures were approved by the Research Ethics Committee of Sun Yat-sen University and were in accordance with the Guide for the Care and Use of Laboratory Animals (NIH Publication No. 85-23, revised 1996). The mice were housed in individually ventilated cages in a specific pathogen-free facility with 12 h daylight/dark cycles at 21–23 °C. The animals were fed standard laboratory chow and water ad libitum.
Cardiac hypertrophy was induced in mice (10–12 weeks old) by chronic subcutaneous infusion of Ang II (2 mg·kg−1·d−1, No. A9525, Sigma-Aldrich, Saint Louis, MO, USA) using Alzet osmotic pumps (model 2002, Durect, Cupertino, CA, USA) for two weeks. The control animals were infused with normal saline (NS). A specific miR-214 agomir and antagomir were purchased from GenePharma (Shanghai, China). The sequence information is shown in Supplementary Table S1. To modulate miR-214 in vivo, the miR-214 agomir (5 OD) or antagomir (8 OD) or an equal dose of negative control sequence (Nc agomir or Nc antagomir, respectively) was administered via tail vein once every two days. The mice were randomly divided into the following groups: the Nc-agomir + NS group, the Nc-agomir + Ang II group, the miR-214-agomir + NS group, the miR-214-agomir + Ang II group, the Nc-antagomir + NS group, the Nc-antagomir + Ang II group, the miR-214-antagomir + NS group, and the miR-214-antagomir + Ang II group. Each group contained 8–12 animals.
Echocardiography and histological analysis
Mice were anaesthetized with 3% (v/v) isoflurane, and two-dimensional-guided M-mode echocardiography was performed with a Technos MPX ultrasound system (Esaote, Genoa, Italy) as described in our previous reports [27, 28]. M-mode images and 2D parasternal short-axis images were taken. Cardiac function indexes, including the ejection fraction (EF), fractional shortening (FS), left ventricular posterior wall thickness in the diastolic and systolic phases (LVPW,d and LVPW,s), left ventricular internal dimensions (LVID,d and LVID,s), and left ventricular volume (LV Vol,d and LV Vol,s), were measured. Afterwards, all animals were sacrificed. The hearts were arrested with 0.1 mM KCl solution at end-diastole, fixed in 4% paraformaldehyde, and then embedded in paraffin for morphometric measurement.
Cell culture
Primary cultures of neonatal mouse cardiomyocytes (NMCMs) and neonatal rat cardiomyocytes (NRCMs) were isolated from myocardial tissues of 1- to 3-day-old C57B/L6 mice and Sprague–Dawley (SD) rats as described previously [29]. The purified cardiomyocytes were maintained in Dulbecco’s modified Eagle’s medium (DMEM, Gibco, Grand Island, NY, USA) supplemented with 10% fetal bovine serum (FBS, Invitrogen, Carlsbad, CA, USA) and 0.1 mM 5-bromodeoxyuridine (BrdU, Thermo Fisher Scientific, Rockford, IL, USA) and cultured in an incubator at 37 °C with 5% CO2. The AC16 human cardiomyocyte-like cell line was purchased from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China) and was cultured under the same conditions.
MiRNA mimic/inhibitor transfection and adenovirus infection
A miR-214 mimic and inhibitor and negative control miRNA (Nc) were synthesized by RiboBio (Guangzhou, China). Cardiomyocytes were transfected with the miR-214 mimic and inhibitor using TransIntro EL transfection reagent (TransGen Biotech, Beijing, China) following the manufacturer’s protocol for 24–48 h before harvest. Adenoviruses expressing SIRT3 (Ad-SIRT3) and GFP (Ad-GFP) were constructed by Vigene Biosciences (Shandong, China). Cells were exposed to Ad-SIRT3 or the negative control vector Ad-GFP for 48 h, and then the expression of SIRT3 was measured by Western blot analysis.
RNA extraction and quantitative real-time PCR (qRT-PCR)
Total RNA was extracted from cultured cells or mouse cardiac tissues using TRIzol Reagent (Takara Biotechnology, Dalian, China) according to the manufacturer’s instructions. For mRNA expression analysis, 1–5 μg of total RNA was reverse transcribed into first-strand cDNA using a Revert Aid First Strand cDNA Synthesis Kit (Thermo Fisher Scientific). The mRNA levels of the target genes were determined using a SYBR Green Quantitative PCR kit (Toyobo Life Science, Osaka, Japan) with an iCycler iQ system (Bio-Rad, Hercules, CA, USA). β-Actin served as an endogenous control. Mouse-specific primers (listed in Supplementary Table S2) were synthesized by Sangon Biotech (Shanghai, China). For miRNA expression analysis, qRT-PCR was performed using a Bulge-Loop miRNA qRT-PCR Starter Kit (RiboBio). The U6 gene served as the internal reference. RT primers and specific primers for miRNAs were purchased from RiboBio. The relative expression level was determined by the 2-ΔΔCt method.
MiRNA sequencing analysis
MiRNA sequencing was performed by KangChen Bio-tech using the Illumina Small RNA Sequencing Platform (San Diego, CA, USA). Each RNA sample was quantified with a spectrophotometer (NanoDrop 1000, Thermo Fisher Scientific), and then RNA samples were used to prepare the miRNA sequencing library, which included the following steps: (1) 3′-adaptor ligation, (2) 5′-adaptor ligation, (3) cDNA synthesis, (4) PCR amplification, and (5) size selection of ~135–155 bp PCR amplified fragments (corresponding to ~15–35 nt small RNAs). The sequencing library was determined by an Agilent 2100 Bioanalyzer using an Agilent DNA 1000 chip kit (part # 5067-1504, Agilent, Santa Clara, CA, USA). The libraries were denatured as single-stranded DNA molecules, captured on Illumina flow cells (Illumina), amplified in situ as clusters and finally sequenced for 50 cycles on an Illumina NextSeq per the manufacturer’s instructions. After sequencing, the data were processed and used for bioinformatics analysis (KangChen Bio-tech, Shanghai, China).
Mitochondrial protein isolation
Mitochondrial proteins were isolated from NMCMs with a mitochondrial isolation kit (Beyotime, Shanghai, China) according to the manufacturer’s instructions. Isolated mitochondria were lysed in lysis buffer. After quantification by a BCA Protein Assay Kit (Thermo Fisher Scientific), the mitochondrial proteins were used for Western blot assays.
Western blot and immunoprecipitation (IP) assays
Cardiomyocytes or mouse heart tissues were rinsed in PBS three times and then incubated for 10 min in RIPA lysis buffer (Beyotime). The lysates were centrifuged at 12000 × g for 15 min. The proteins were then extracted and measured with a BCA Protein Assay Kit (Thermo Fisher Scientific). Equal amounts of protein (30 μg) were loaded onto 8%–12% SDS-PAGE gels for separation and transferred to polyvinylidene fluoride (PVDF) membranes (Millipore, Billerica, MA, USA). After they were blocked in 5% defatted milk powder for 1 h, the membranes were incubated with primary antibodies and then incubated with secondary antibodies. The signals indicating the protein levels were visualized with High-sig ECL Western blotting substrate (Tanon, Shanghai, China) and quantified by ImageJ software (NIH, Bethesda, MD, USA).
For the immunoprecipitation assay, at least 200 μg of protein was extracted with IP buffer (Beyotime). Lysates were incubated with primary antibodies or corresponding IgG overnight at 4 °C and incubated with 20 μL of protein A/G-agarose beads (Cat# 20241, Thermo Fisher Scientific) at 4 °C for 3–4 h. Then, the beads were washed with IP washing buffer three times and supplemented with 25 μL of 2× loading buffer. The immunoprecipitated proteins were detected by Western blot analysis. The primary antibodies are listed in the materials.
Immunofluorescence assay
Cardiomyocytes were seeded and cultured on glass coverslips. Cells were incubated with the MitoTracker Red (M7512) fluorescence probe (Invitrogen) for 30 min at 37 °C to label the mitochondria. After that, the cells were washed with warm PBS, fixed with 4% paraformaldehyde for 10 min, and then permeabilized with 0.3% Triton X-100 (Sangon Biotech) for 10 min at room temperature. After washing with PBS three times, the cells were incubated with 10% goat serum (Boster, Wuhan, China) for 1 h at room temperature. Then, the cells were treated with a primary antibody against SIRT3 (diluted 1:100) overnight at 4 °C and subsequently incubated with an Alexa Fluor 488-conjugated anti-rabbit IgG secondary antibody (diluted 1:200, Cell Signaling Technology, Danvers, MA, USA). DAPI (Cat# 4083, Cell Signaling Technology) staining was performed to visualize the nuclei. Images were captured with a laser scanning microscope and analyzed (Olympus Corporation, Tokyo, Japan).
Measurement of cell surface area
Cardiomyocytes seeded in 48-well plates were fixed with 4% paraformaldehyde for 15 min at room temperature and then treated with 0.3% Triton X-100 for 10 min. After incubation with 0.1% rhodamine phalloidin (No. MAN0001777, Invitrogen) for 1 h, the cells were washed with PBS and further stained with DAPI. Then, the cells were assessed with a High Content Screening System (Arrayscan VTi 600 Plus, Thermo Fisher Scientific), and the cell surface area from randomly selected fields (50 for each group) was determined by built-in image analysis software.
Dual-luciferase reporter assay
HEK293 T cells were seeded and cotransfected with the miR-214 mimic or mimic Nc and wt-sirt3-3’ UTR or mut-sirt3-3’ UTR plasmids (GeneCopoeia, Rockville, MD, USA) by using TransIntro EL transfection reagent. The cells were harvested after 36 h, and a luciferase activity assay was performed using a Luc-Pair Duo-Luciferase Assay Kit (GeneCopoeia). Luciferase activity was measured by a microplate reader (Molecular Devices, San Jose, CA, USA). The relative luciferase ratio of firefly luciferase/renilla luciferase was calculated as the normalized luciferase activity.
Measurement of mitochondrial oxygen consumption rate (OCR)
OCR measurement was performed using an XF96 Extracellular Flux Analyzer (Seahorse Bioscience, North Billerica, MA, USA) as described previously [30]. Briefly, AC16 cells were seeded into Seahorse XF96 microplates (Seahorse Bioscience) at a density of 5 × 104 cells/well in a humidified 37 °C incubator with 5% CO2 (DMEM) for 24 h. Then, the normal medium of the cells was replaced with XF assay medium containing 1 nM pyruvate, 2 mM glutamine and 10 mM glucose. A sensor cartridge was loaded with various compounds as follows: oligomycin (2 μM), carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (FCCP, 1 μM), and rotenone/antimycin A (1 μM). After the sensor cartridge was calibrated in the analyzer, the cell plates were incubated in a 37 °C/non-CO2 incubator for 60 min prior to the start of the OCR assay. The cell count in each well was used to normalize the OCR. The data were analyzed with Wave software, and respiratory parameters were quantified. The experiment was repeated at least in triplicate.
Determination of mitochondrial membrane potential (ΔΨm)
After being seeded in 48-well plates, NMCMs were transfected with miR-214 mimic or inhibitor for 24 h. Then, ΔΨm was measured with a JC-1 mitochondrial membrane potential assay kit (Beyotime). Under normal physiological conditions, in cells with high mitochondrial membrane potential, JC-1 spontaneously forms aggregates and yields red fluorescence (excitation wavelength: 585 nm; emission wavelength: 590 nm). At low ΔΨm, JC-1 remains in a monomeric form that yields green fluorescence (excitation wavelength: 514 nm; emission wavelength: 529 nm). The fluorescence intensity was measured by a High Content Screening System. Images (25 for each well) were captured with an automated cell imaging system.
Transmission electron microscopy (TEM)
Left ventricle myocardial tissues of mice were cut into approximately 1 mm3 sized pieces and fixed in 2.5% glutaraldehyde solution (Ted Pella, Redding, CA, USA). After washing with Hank’s modified salt solution (HMSS), the tissues were postfixed in 2% OsO4 (Electron Microscopy Sciences, Hatfield, PA, USA), dehydrated in graded ethanol and infiltrated with epoxy resin. After embedding, ultrathin sections were cut with an ultracryomicrotome (EM UC6, Leica, Wetzlar, Germany), transferred to 200-mesh carbon-coated copper grids, and stained with 2% uranyl acetate (Merck, Darmstadt, Germany) and lead citrate (Sigma-Aldrich). The mitochondrial ultrastructure was observed under a JEM 1400 transmission electron microscope (JEOL, Tokyo, Japan) at 120 kV.
Materials
Primary antibodies against SIRT3 (Cat# 5490), COX-IV (Cat# 4850), Drp1 (Cat# 14647) and FoxO3a (Cat# 12829) were obtained from Cell Signaling Technology. An ANF antibody (Cat# sc-80686) was purchased from Santa Cruz Biotechnology (Dallas, TX, USA). Beta-MHC antibody was obtained from Sigma-Aldrich. GAPDH (Cat# 60004-1-AP), Mfn2 (Cat# 12186-1-AP), OPA1 (Cat# 27733-1-AP) and Uqcrc1 (Cat# 21705-1-AP) antibodies were purchased from Proteintech Group (Manchester, UK). An ATP5O (Cat# A8103) antibody was purchased from Abcam (Cambridge, UK). An acetylated lysine antibody (ICP0380) was purchased from Immunechem (Burnaby, Canada).
Statistical analysis
The data are expressed as the mean ± SEM of multiple independent replicates. The statistical analyses included two-tailed, unpaired Student’s t tests for experiments with two groups or one-way ANOVA with Bonferroni posttests for multiple groups (GraphPad Prism 7.0 software, San Diego, CA, USA). Statistical significance was defined as P < 0.05.
Results
SIRT3 knockout exacerbates pathological cardiac hypertrophy in Ang II-infused mice
To investigate the role of SIRT3 in cardiac hypertrophy, male Sirt3−/− mice and their WT littermates were subcutaneously infused with Ang II (2 mg·kg−1·d−1 for 2 weeks). The pathological changes associated with hypertrophy were determined by gross morphologic examination, hematoxylin-eosin (H&E) staining, Masson staining and echocardiography (Fig. 1a–e). The hearts of mice treated with Ang II were significantly larger than those of animals that received normal saline, and the hypertrophy was accompanied by histological abnormalities, including disarrangement of cardiomyocytes, infiltration of inflammatory cells and deposition of collagen. These typical hypertrophic responses triggered by Ang II were more obvious in Sirt3−/− mice than in WT animals. Moreover, the heart weight-to-body weight (HW/BW) ratio increased following Ang II administration (Fig. 1f). The echocardiography results showed that EF, FS and LVPW were elevated, while LVID and LV Vol were reduced, in the Ang II-treated groups (Fig. 1g–j and Supplementary Table S3). Depletion of SIRT3 provoked further changes in the above echocardiographic parameters, indicating exacerbated cardiac malfunction. Ang II-induced mitochondrial damage was also visualized by using TEM (Fig. 1k), and Ang II promoted mitochondrial swelling, cristae rupture and vacuolization, which were aggravated in Sirt3−/− mice. In addition, the levels of the hypertrophic markers ANF, BNP and β-MHC were upregulated in response to Ang II infusion and were further elevated by SIRT3 deficiency (Fig. 1l). The expression of SIRT3 was obviously reduced following Ang II infusion (Fig. 1m–o). Notably, although the cardiac function parameters of Sirt3−/− and WT mice had no obvious differences at 10 weeks of age, Sirt3−/− mice were more likely to be stricken with spontaneous heart failure than WT mice at 28 weeks of age. As shown in Supplementary Fig. S1, the HW/BW ratio, LV Vol and LVID were markedly augmented in 28-week-old Sirt3−/− mice, while the EF, FS and LVPW were reduced. These data indicate that a lack of SIRT3 not only exacerbated pathological cardiac hypertrophy induced by Ang II but also accelerated the deterioration of cardiac contractile function with age.

Wild-type (WT) and Sirt3−/− mice were subcutaneously infused with Ang II (2mg·kg−1·d−1) or an equal volume of normal saline (NS) for 2 weeks. a Gross morphologic examination of the hearts. Hematoxylin-eosin staining of cardiac sections (b scale bar = 1 mm; c scale bar = 100 μm). d Masson staining for collagen deposition (scale bar = 100 μm). e Representative images from echocardiography. f Heart weight-to-body weight (HW/BW) ratio. g–j The ejection fraction (EF), fractional shortening (FS) and left ventricular posterior wall thickness in the diastolic and systolic phases (LVPW,d and LVPW,s) were measured by echocardiography. The data are presented as the mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001; n = 10–12. k The ultrastructure of mitochondria was visualized by transmission electron microscopy (scale bar = 500 nm). l The mRNA levels of hypertrophic biomarkers (ANF, BNP and β-MHC) were detected by qRT-PCR. The data are presented as the mean ± SEM. *P < 0.05, **P < 0.01, ****P < 0.0001 vs. the WT+NS group; ###P < 0.001, ####P < 0.0001 vs. the Sirt3−/− + NS group; n = 4. m–o The expression of SIRT3 was detected by Western blot analysis and qRT-PCR. The data are presented as the mean ± SEM. **P < 0.01, ****P < 0.0001 vs. the WT+NS group; n = 4.
MiR-214 expression is upregulated by Ang II treatment
In this study, Illumina deep sequencing was used to characterize miRNA expression profiles in left ventricular tissues of Sirt3−/− and WT mice following Ang II treatment. We performed filtering between the groups with and without Ang II treatment using the following criteria: a fold change (FC) ≥ 1.5 (or ≤0.67) and a P value ≤ 0.05 (unpaired). Volcano plots of the aberrant miRNAs, including 64 upregulated miRNAs in WT mice and 27 upregulated miRNAs and 1 downregulated miRNA in Sirt3−/− mice, are shown in Fig. 2a, b. Unsupervised hierarchical clustering analysis further revealed that the myocardial tissue samples of Ang II-treated mice could be distinguished clearly from those of animals that received normal saline based on their patterns of significantly higher expression of certain miRNAs (Supplementary Fig. S2a, b). Among these miRNAs, some of the most differentially expressed miRNAs, including miR-224, miR-411, miR-214, miR-21a, miR-199a, miR-23b and miR-185 (Fig. 2c, d), were abundant in cardiac tissue, and the changes in their levels were confirmed by real-time PCR assay (Fig. 2e, f). It is well known that most miRNAs negatively regulate gene expression at the posttranscriptional level by directly binding to the 3’ UTRs of mRNAs. Thus, bioinformatics analysis of putative targets of these candidate miRNAs was performed using web-based tools, including TargetScan (http://www.targetscan.org/) and miRDB (http://www.mirdb.org), which consistently suggested that SIRT3 is a potential target of miR-214. Furthermore, the level of miR-214 was significantly elevated by Ang II in hypertrophic hearts of WT and Sirt3−/− mice (4.88- and 4.62-fold increases, respectively, compared with the levels in the group that only received normal saline). Moreover, Ang II (100 nM) treatment led to enhanced miR-214 expression in a time-dependent manner in cultured NMCMs (Fig. 2g).

WT and Sirt3−/− mice were subcutaneously infused with 2 mg·kg−1·d−1 Ang II for 2 weeks. The control animals received normal saline (NS). Illumina deep sequencing was performed to detect miRNA expression profiles in left ventricular tissues (3 samples/group). a, b Volcano plot of differentially expressed miRNAs between the Ang II and NS groups. The x-axis shows the log2 fold change in the expression of miRNAs between the Ang II and NS groups, while the y-axis shows the -log10 P value for each miRNA. c, d Heatmap showing differentially expressed miRNAs between the Ang II treatment and NS groups as calculated from the regularized log transformation. Each column represents a sample. The color scale shown at the top illustrates the relative expression levels of a miRNA in different groups. Red represents an expression level above the mean, and green represents an expression level below the mean. e, f The levels of miRNAs were validated by qRT-PCR. The data are presented as the mean ± SEM. *P < 0.05, **P < 0.01 vs. the WT+NS group; #P < 0.05, ##P < 0.01, ###P < 0.001 vs. the Sirt3−/− + NS group. n = 4. g Neonatal mouse cardiomyocytes (NMCMs) were isolated and stimulated with Ang II (100 nM) for the indicated time periods. The mRNA expression of miR-214 was measured by qRT-PCR. The data are presented as the mean ± SEM. *P < 0.05, **P < 0.01 vs. the control group. n = 4.
MiR-214 promotes hypertrophic responses in vitro and in vivo
Previous studies have proven that dysregulation of miR-214 facilitates the development of cancer and various cardiovascular diseases [31]. Here, gain-of-function approaches were used to further investigate the role of miR-214 in cardiac hypertrophy in vitro and in vivo. Cultured NMCMs were transfected with a miR-214 mimic to increase miR-214 expression. The transfection efficiency was validated by measuring miR-214 levels (Supplementary Fig. S3a, b). Our results showed that forced expression of miR-214 with its mimic elevated the protein levels of hypertrophic markers, including β-MHC and ANF (Fig. 3a), and markedly increased the cell surface area (Fig. 3b). To further confirm the prohypertrophic effect of miR-214 in vivo, mice were injected by tail vein with a miR-214 agomir. Overexpression of miR-214 in cardiac tissues with agomir administration was confirmed by qRT-PCR. In addition, treatment with Ang II increased miR-214 expression, which was consistent with the above result (Supplementary Fig. S3c). Ang II infusion and miR-214 agomir administration provoked the development of representative hypertrophic abnormalities, including enlarged cardiomyocytes, disorganized myocardial architecture and decreased intercellular space, as indicated by H&E staining and wheat germ agglutinin (WGA) staining (Fig. 3c–e). Obvious accumulation of collagen fibers was also detected by picrosirius red (PSR) staining, suggesting that Ang II and the miR-214 agomir had profibrotic effects (Fig. 3f, g). The HW/BW ratio and echocardiographic parameters, such as EF, FS and LVPW, were prominently increased by Ang II (Fig. 3i–m), while the LVID and LV Vol were attenuated (Supplementary Fig. S3d–g). MiR-214 agomir injection alone had no significant influence on the HW/BW ratio or echocardiographic parameters. Furthermore, the protein and mRNA levels of hypertrophic markers were determined in cardiac tissues (Fig. 3n–p). The results showed that the miR-214 agomir significantly enhanced the expression of ANF, BNP and β-MHC. Taken together, these findings suggested that the upregulation of miR-214 was sufficient to induce cardiac hypertrophy.

NMCMs were transfected with a miR-214 mimic (miR-214-m) or negative control (Nc-m) and incubated with 100 nM Ang II for 36 h. a The protein expression of ANF and β-MHC was detected by Western blot analysis. b The surface area of NMCMs was measured. Scale bar = 100 μm. The data are presented as the mean ± SEM. *P < 0.05, ***P < 0.001, ****P < 0.0001 vs. the Nc-m group. n = 4. C57B/L6 mice were subcutaneously infused with Ang II or normal saline (NS) for 2 weeks. To overexpress miR-214 in vivo, a specific agomir (5 OD) and its negative control (Nc-agomir) were administered via the tail vein. The pathological changes of the myocardium were observed by c gross morphologic examination, d H&E staining (scale bar = 1.5 mm), e WGA staining (scale bar = 50 μm), f, g PSR staining (top: white light, scale bar = 400 μm; bottom: polarized light, scale bar = 50 μm), and h echocardiography. i The HW/BW ratio was calculated. j–m Echocardiographic parameters were measured. The data are presented as the mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. n = 8–11. n–p The protein and mRNA levels of β-MHC, ANF and BNP in cardiac tissues were measured by Western blot analysis and qRT-PCR. The data are presented as the mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 vs. the NS+Nc-agomir group. n = 4.
Inhibition of miR-214 alleviates pathological hypertrophy induced by Ang II
NMCMs were transfected with a miR-214 inhibitor, and the inhibition efficiency was validated by measuring miR-214 levels (Supplementary Fig. S4a, b). Ang II stimulation led to upregulation of β-MHC and ANF expression, which could be attenuated by downregulation of miR-214 (Fig. 4a). The miR-214 inhibitor also suppressed the enlargement of NMCMs induced by Ang II (Fig. 4b). Next, we explored whether miR-214 interference could be utilized to combat cardiac hypertrophy in vivo. In the mouse model of Ang II-induced cardiac hypertrophy, a miR-214 antagomir, a synthetic antisense oligonucleotide with chemical modifications, was administered through the tail vein to specifically inhibit the expression of endogenous miR-214. The inhibitory efficacy was confirmed by qRT-PCR assay (Supplementary Fig. S4c). Morphologic and histological examinations showed that the miR-214 antagomir attenuated Ang II-stimulated pathological changes associated with hypertrophy (Fig. 4c–g). It also suppressed the increase in the HW/BW ratio following Ang II treatment (Fig. 4i). In addition, treatment with the miR-214 antagomir alleviated Ang II-induced abnormalities in cardiac structure and function, including elevations in EF, FS and LVPW (Fig. 4j–m), and reduced LVID and LV Vol (Supplementary Fig. S4d–g). In cardiac tissues from Ang II-infused mice, the increases in the expression of hypertrophic markers, including ANF, BNP, and β-MHC, were ameliorated by the miR-214 antagomir (Fig. 4n–p). These results demonstrated that inhibition of miR-214 could protect against Ang II-triggered cardiac hypertrophy.

NMCMs were transfected with a miR-214 inhibitor (miR-214-in) or negative control (Nc-in) and incubated with 100 nM Ang II for 36 h. a The protein expression of ANF and β-MHC was detected by Western blot analysis. b The surface area of NMCMs was measured. Scale bar = 100 μm. The data are presented as the mean ± SEM. ***P < 0.001 vs. the Nc-in group; ###P < 0.001 vs. the Ang II+Nc-in group; ns not significant vs. the Nc-in group. n = 4. C57B/L6 mice were subcutaneously infused with Ang II or normal saline (NS) for 2 weeks. To inhibit miR-214 in vivo, a specific antagomir (8 OD) and its negative control (Nc antagomir) were administered via the tail vein. The pathological changes of the myocardium were observed by c gross morphologic examination, d H&E staining (scale bar = 1.5 mm), e WGA staining (scale bar = 50 μm), f, g PSR staining (top: white light, scale bar = 400 μm; bottom: polarized light, scale bar = 50 μm), and h echocardiography. i The HW/BW ratio was calculated. j–m Echocardiographic parameters were measured. The data are presented as the mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. n = 8–11. n–p The protein and mRNA levels of β-MHC, ANF and BNP in cardiac tissues were measured by Western blot analysis and qRT-PCR. The data are presented as the mean ± SEM. *P < 0.05, **P < 0.01, ****P < 0.0001 vs. the NS+Nc-antagomir group; #P < 0.05, ##P < 0.01, ####P < 0.0001 vs. the Ang II+Nc-antagomir group. n = 4.
SIRT3 is directly targeted and negatively regulated by miR-214
Our previous study has demonstrated that SIRT3 exerts a protective effect and that its expression significantly declines during Ang II-induced cardiac hypertrophy both in vitro and in vivo [14]. Here, we also observed a time-dependent downregulation of SIRT3 protein content in response to Ang II treatment in NMCMs, which was accompanied by increased levels of the hypertrophic markers ANF, BNP and β-MHC (Fig. 5a–c). The inverse correlation between miR-214 and SIRT3 expression in hypertrophy supports a targeting relationship between them. The predicted binding sites of miR-214 are located at positions 200–206 of the mmu-Sirt3 3’ UTR and are highly conserved in humans (258–264 of the hsa-Sirt3 3’ UTR) and rats (227–233 of the rno-Sirt3 3’ UTR). To further validate SIRT3 as a direct target of miR-214, HEK293 cells were transfected with dual-luciferase vectors containing intact or mutated miR-214 binding sites in the SIRT3 3’ UTR (Fig. 5d). A dual-luciferase reporter assay revealed that the miR-214 mimic efficiently suppressed luciferase activity in cells expressing the wild-type SIRT3 3’ UTR but not in cells expressing the mutated form (Fig. 5e). The expression of SIRT3 was significantly decreased by the miR-214 mimic in NMCMs, while treatment with the miR-214 inhibitor enhanced SIRT3 protein levels (Fig. 5f, g). Similar results were observed in primary cultures of NRCMs as well as in AC16 cells, a human cardiomyocyte cell line (Supplementary Fig. S5). In addition, immunofluorescence analysis showed that mitochondrial fragmentation was increased after overexpression of miR-214. The colocalization between mitochondria and SIRT3 was decreased in response to downregulation of SIRT3 by the miR-214 mimic, while the cell surface area was augmented. In contrast, inhibition of miR-214 enhanced the colocalization between SIRT3 and mitochondria (Fig. 5h, i).

a–c NMCMs were incubated with 100 nM Ang II for the indicated durations. The protein levels of hypertrophic biomarkers and SIRT3 were measured by Western blot analysis. The data are presented as the mean ± SEM. **P < 0.01, ***P < 0.001 vs. the control group. n = 4. d Schematic representation of the predicted binding sites for miR-214 and the mutated binding sites in the 3’ UTR of SIRT3. e HEK293 T cells were transfected with dual luciferase reporter vectors containing the wild-type or mutant SIRT3 3’ UTR. The cells were further treated with a miR-214 mimic (miR-214-m) or Nc mimic (Nc-m), and luciferase activity was detected. The data are presented as the mean ± SEM. **P < 0.01 vs. the Nc-m group. n = 3. f, g NMCMs were transfected with a miR-214 mimic or inhibitor (miR-214-in). The protein and mRNA levels of SIRT3 were determined. The data are presented as the mean ± SEM. *P < 0.05, **P < 0.01 vs. the Nc-m group; ####P < 0.0001 vs. the Nc-in group. n = 4. h, i The intracellular colocalization of SIRT3 (green) and mitochondria (red) was visualized by confocal immunofluorescence microscopy, and the immunofluorescence intensity of SIRT3 was quantified. Representative images of four independent tests are shown (scale bar = 40 μm). The data are presented as the mean ± SEM. *P < 0.05 vs. the Nc-m group. n = 4.
Upregulation of SIRT3 attenuates miR-214-induced cardiomyocyte hypertrophy
The above findings suggested that miR-214 might contribute to pathological cardiac hypertrophy by negatively regulating SIRT3. To validate this hypothesis, NMCMs were cotransfected with an adenovirus encoding SIRT3 (Ad-SIRT3) and the miR-214 mimic. As shown in Fig. 6a–c, overexpression of SIRT3 attenuated the miR-214-triggered increases in β-MHC and ANF levels. Furthermore, upregulation of the expression of SIRT3 attenuated the augmentation of cardiomyocyte surface area induced by the miR-214 mimic, while overexpression of SIRT3 alone had no effect on cell surface area (Fig. 6d, e).

a–c NMCMs were cotransfected with an adenovirus encoding SIRT3 (Ad-SIRT3) and a miR-214 mimic or their corresponding negative controls, an adenovirus encoding green fluorescent protein (Ad-GFP) and an Nc mimic, for 48 h. The protein levels of SIRT3, ANF and β-MHC were measured by Western blot analysis. The data are presented as the mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 vs. the Ad-GFP+Nc-m group; ####P < 0.0001 vs. the Ad-SIRT3+Nc-m group; &P < 0.05, &&P < 0.01 vs. the Ad-GFP+miR-214 mimic group. n = 4. d, e The surface area of NMCMs was measured. Scale bar = 100 μm. The data are presented as the mean ± SEM. **P < 0.01 vs. the Ad-GFP+Nc-m group; &P < 0.05 vs. the Ad-GFP+miR-214 mimic group. n = 3.
MiR-214 impairs mitochondrial homeostasis and respiratory activity
SIRT3 is a mitochondria-localized deacetylase and plays important roles in regulating mitochondrial energy metabolism [32]. Considering our finding that SIRT3 was directly targeted and modulated by miR-214, the potential effects of miR-214 on mitochondrial homeostasis and respiratory activity were subsequently investigated. Our results revealed that upregulation of miR-214 obviously decreased the mitochondrial membrane potential (ΔΨm) in NRCMs, as indicated by JC-1 staining, but inhibition of miR-214 had no significant effect on ΔΨm (Fig. 7a, b). Mitochondrial respiratory activity was further determined in AC16 cells by using a Seahorse XF96 Extracellular Flux Analyzer. Measurement of respiration parameters revealed that basal respiration (−33%), ATP production (−41%) and maximal respiration (−40%) were all decreased in miR-214-overexpressing cells (Fig. 7c, d). In contrast, inhibition of miR-214 promoted mitochondrial respiratory activity, as indicated by elevated respiration parameters (Fig. 7e, f). The expression of SIRT3 was downregulated in cardiac tissues with Ang II infusion or miR-214 agomir treatment (Fig. 7g–i), which was consistent with our previous observation in NMCMs. In addition, the expression of mitochondrial fusion/fission proteins was determined. Mitofusin 2 (Mfn2) expression was decreased, while dynamin-related peptide 1 (Drp1) expression was increased by the miR-214 agomir (Fig. 7g, j, k). Besides, the Ang II-induced reduction in SIRT3 expression was effectively eliminated following miR-214 antagomir treatment. The increase in Drp1 expression and reduction in Mfn2 expression were also attenuated by the miR-214 antagomir (Fig. 7m–q). The levels of OPA1 and mitochondrial fission protein 1 (Fis1) showed no evident differences after miR-214 intervention (Supplementary Fig. S6). Additionally, the influences of miR-214 on mitochondrial morphology were detected in cardiac tissues of mice by TEM. Our results demonstrated that administration of the miR-214 agomir provoked mitochondrial swelling, cristae rupture and vacuolization. These abnormalities were exacerbated when the mice were concurrently subjected to Ang II infusion (Fig. 7l). In contrast, suppression of miR-214 by antagomir treatment significantly alleviated the changes in mitochondrial morphology induced by Ang II (Fig. 7r).

a, b The mitochondrial membrane potential (ΔΨm) in NRCMs was detected via JC-1 staining after transfection with a miR-214 mimic, inhibitor or relevant negative control for 24 h. Scale bar = 200 μm. The data are presented as the mean ± SEM. **P < 0.01 vs. the Nc-m group. n = 4. c–f AC16 cells were transfected with a miR-214 mimic, inhibitor or negative control for 24 h. The oxygen consumption rate (OCR) and respiration parameters, including basal respiration, ATP production, maximal respiration and H+ leak, were determined by using a Seahorse XF96 Analyzer. The data are presented as the mean ± SEM. *P < 0.05, **P < 0.01 vs. the Nc-m or Nc-in group. n = 3. C57B/L6 mice were subcutaneously infused with Ang II or normal saline (NS) for 2 weeks and administered a miR-214-agomir or Nc-agomir via the tail vein. g–k The protein and mRNA levels of SIRT3, Mfn2 and Drp1 in cardiac tissues were measured by Western blot analysis and qRT-PCR. The data are presented as the mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001 vs. the NS+Nc-agomir group. n = 4. l The mitochondrial ultrastructure of cardiac tissues was detected by TEM. Representative images of four independent samples are shown (scale bar = 1 μm). C57B/L6 mice were given the same treatment with Ang II and administered a miR-214 antagomir or Nc antagomir via the tail vein. m–q The protein and mRNA levels of SIRT3, Mfn2 and Drp1 in cardiac tissues were measured by Western blot analysis and qRT-PCR. The data are presented as the mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001 vs. the NS+Nc-antagomir group; #P < 0.05 vs. the Ang II+Nc-antagomir group; &P < 0.05 vs. the NS+miR-214-antagomir group. n = 4. r The mitochondrial ultrastructure of cardiac tissues was detected by TEM. Representative images of four independent samples are shown (scale bar = 1 μm).
MiR-214 downregulates Mfn2 by suppressing SIRT3/FoxO3a signaling
In NMCMs, intervention with miR-214 influenced the total expression of Mfn2 but not the expression of Drp1 and OPA1 (Fig. 8a–c and Supplementary Fig. S7a). Because SIRT3 and Mfn2 are located mainly in the mitochondria and are involved in manipulating energy metabolism and mitochondrial dynamics [8, 33], we further detected whether their subcellular protein levels could be affected by miR-214. In the mitochondrial fraction, the levels of both SIRT3 and Mfn2 were markedly elevated after transfection with the miR-214 inhibitor but were decreased after transfection with the miR-214 mimic (Fig. 8d–f). Previous studies have demonstrated that SIRT3 positively regulates the expression of FoxO3a. Moreover, SIRT3 deacetylates FoxO3a to promote its transcriptional activity, thereby facilitating FoxO3a-dependent Mfn2 expression [34, 35]. Here, we observed that overexpression of SIRT3 increased Mfn2 protein levels and could also reverse the decline in Mfn2 expression induced by the miR-214 mimic. The protein level of FoxO3a was decreased after treatment with the miR-214 mimic (Fig. 8g, h). In addition, the miR-214 mimic enhanced the acetylation of total protein in NMCMs (Fig. 8i). Immunoprecipitation assays further revealed that it diminished the interaction between SIRT3 and FoxO3a and then increased FoxO3a acetylation (Fig. 8j, k). These data suggested the involvement of suppression of SIRT3/FoxO3a signaling in miR-214-mediated Mfn2 downregulation.

NMCMs were transfected with a miR-214 mimic, inhibitor or relevant negative control for 24 h. a–c The total protein levels of SIRT3 and Mfn2 were measured by Western blot analysis. d–f The SIRT3 and Mfn2 levels in the mitochondrial and cytosol fractions were determined. The data are presented as the mean ± SEM. *P < 0.05, **P < 0.01 vs. the Nc-m group; #P < 0.05, ##P < 0.01 vs. the Nc-in group. n = 4. g, h NMCMs were cotransfected with an adenovirus encoding SIRT3 (Ad-SIRT3), the miR-214 mimic and their corresponding negative controls, Ad-GFP and an Nc mimic, for 48 h. The protein expression levels of FoxO3a and Mfn2 were measured by Western blot analysis. The data are presented as the mean ± SEM. **P < 0.01, ***P < 0.001 vs. the Ad-GFP+Nc-m group; ##P < 0.01, ####P < 0.0001 vs. the Ad-SIRT3+Nc-m group; &P < 0.05 vs. the Ad-GFP+miR-214 mimic group. n = 4. i NMCMs were transfected with the miR-214 mimic or Nc mimic and incubated with 100 nM Ang II for 36 h. The global protein acetylation level was measured by Western blot analysis. Representative bands from three independent experiments are shown. j, k Immunoprecipitation analysis showed the interaction of SIRT3 and FoxO3a and the acetylation levels of FoxO3a in NMCMs after treatment with the miR-214 mimic. The data are presented as the mean ± SEM. *P < 0.05, **P < 0.01 vs. the Nc-m group. n = 3.
Discussion
Pathological cardiac hypertrophy has been well recognized as a precursor and risk factor for heart failure. Sustained cardiac hypertrophy results in myocardial remodeling, metabolic dysfunction and consequent cardiac decompensation [36]. Thus, early prevention of hypertrophy can not only slow down disease progression towards heart failure but also improve clinical outcomes. In recent years, great advances have been made in understanding the molecular basis of cardiac hypertrophy. It is now known that miRNAs, which form an important group of epigenetic regulators, play crucial roles in the pathogenesis of cardiac hypertrophy. For instance, miR-22, miR-217 and miR-29 have been determined to act as prohypertrophic miRNAs by targeting phosphatase and tensin homolog (PTEN) and activating the phosphatidyl-3 kinase (PI3K)/Akt/mammalian target of the rapamycin (mTOR) system [37,38,39]. Conversely, miR-1 can ameliorate cardiac hypertrophy by decreasing the expression of the transcription factor nuclear factor of activated T cells (NFATC3) and mitochondrial calcium uniporter (MCU), which restrains the prohypertrophic calcium signaling pathway [40, 41]. These findings support the idea that miRNAs may serve as potential biomarkers or therapeutic targets for cardiac hypertrophy.
In the present work, we performed deep miRNA sequencing and observed that miR-214 expression was remarkably upregulated in the hearts of mice suffering from Ang II-induced cardiac hypertrophy (Fig. 2). MiR-214 overexpression was sufficient to induce cardiomyocyte hypertrophy in vitro and could further aggravate the hypertrophic phenotype of mice infused with Ang II. Moreover, suppression of miR-214 expression significantly mitigated the hypertrophic responses stimulated by Ang II in cultured NMCMs and in mice (Figs. 3 and 4). These findings validate miR-214 as a prohypertrophic miRNA and suggest that miR-214 interference has potential cardioprotective benefits. To date, a variety of pathophysiological factors, such as hemodynamic overload, neurohumoral stimuli, oxidative stress, inflammation and aging, have been identified to orchestrate the initiation and development of cardiac hypertrophy [1, 3]. Nevertheless, the current pharmaceutical agents used in the clinic, including β-adrenergic receptor blockers, angiotensin-converting enzyme inhibitors and angiotensin II type 1 receptor antagonists, yield limited effects, and the prognosis of the disease remains poor [42]. Considering their extensive influences on gene expression and critical roles in regulating cellular biological processes, miRNAs may present alternative and promising targets for the prevention and treatment of cardiac hypertrophy [21, 43]. Here, our study revealed that miR-214 suppression by antagomir delivery alleviated the histological changes associated with pathological hypertrophy, restored heart function and attenuated mitochondrial impairments in Ang II-infused mice (Figs. 4 and 7). Although quite preliminary, the data provide a rationale for a new intervention strategy involving suppression of miR-214.
To investigate the mechanism underlying the prohypertrophic role of miR-214, the potential targets of this miRNA were predicted using bioinformatic analysis, which suggested SIRT3 as a key downstream effector of miR-214. A dual-luciferase reporter assay proved that miR-214 could directly bind with the complementary region of the SIRT3 3’ UTR and suppress the expression of SIRT3 (Fig. 5). In response to Ang II treatment, SIRT3 levels declined both in NMCMs and in hypertrophic mouse hearts, whereas miR-214 inhibitor and antagomir treatment reversed the decreases in SIRT3 levels caused by Ang II (Figs. 4 and 7). Previously, it has been reported that reduced expression of SIRT3 contributes to contractile dysfunction in failing hearts [44, 45]. In this study, we observed that SIRT3 knockout exacerbated Ang II-induced cardiac hypertrophy (Fig. 1). Moreover, Sirt3−/− mice incurred hyperacetylation of total protein and developed a spontaneous heart failure phenotype at the age of 28 weeks (Supplementary Fig. S1). However, reintroduction of SIRT3 attenuated the miR-214-mediated increases in hypertrophic marker expression and cell surface area (Fig. 6). Therefore, the prohypertrophic and detrimental effects of miR-214 may be at least partially attributable to miR-214-mediated negative regulation of SIRT3.
Thus far, it has been recognized that mitochondrial malfunction is a key event contributing to the development and progression of hypertrophy [46, 47]. As the most metabolically active organ, the heart accounts for approximately 8% of total ATP consumption. Mitochondria are the central organelles for ATP generation, and the energy supply in cardiomyocytes is heavily dependent on mitochondrial oxidative phosphorylation [46, 48]. SIRT3, as a pivotal deacetylase in mitochondria, is tightly associated with energy metabolism, oxidative stress and apoptosis [6, 49, 50]. It can promote efficient electron transport to optimize ATP generation by deacetylating a diversity of mitochondrial enzymes [51]. Malfunction of SIRT3 leads to disturbance of fatty acid β-oxidation, amino acid metabolism and the electron transport chain (ETC), which has been recognized to trigger cardiac hypertrophy. In our study, miR-214 overexpression inhibited SIRT3 expression, and this effect was accompanied by obvious declines in mitochondrial membrane potential, basal respiration, ATP production and maximal respiration; in contrast, miR-214 inhibition enhanced SIRT3 expression and rescued mitochondrial respiratory activity (Fig. 7a–f). Collectively, these results indicate that miR-214-induced SIRT3 inhibition may contribute to impairments in mitochondrial function.
Mitochondria are highly dynamic organelles, and abnormal morphologic changes can lead to grievous impairments of their function. Mitochondrial morphology is maintained by the balance between fusion and fission. In mammals, mitochondrial fusion is regulated mainly by Mfn2 and OPA1, two GTPases located on the outer and inner membranes of mitochondria, respectively, whereas the fission process is tightly controlled by Drp1 and Fis1 [5, 33]. In this study, we found that the protein levels of Mfn2 and OPA1 were reduced during Ang II-induced cardiac hypertrophy in vitro and in vivo, while Drp1 expression was increased (Fig. 7, Supplementary Fig. S7b). Similar results were observed following miR-214 overexpression, but the levels of OPA1 and Fis1 were not significantly affected by miR-214 (Fig. 7, Supplementary Fig. S6, S7). Aberrant expression of miR-214 damaged mitochondrial structural integrity, as visualized by TEM (Fig. 7l, r). In contrast, inhibition of miR-214 rectified the abnormal expression of mitochondrial fusion/fission proteins and ameliorated morphologic injuries (Figs. 7 and 8). SIRT3 is known to physically interact with FoxO3a in mitochondria and to deacetylate FoxO3a to increase its DNA-binding activity, which promotes the transcriptional expression of peroxisome proliferator-activated receptor γ coactivator 1α (PGC-1α) [9, 52]. Several studies have well described that SIRT3 inhibition leads to inactivation of FoxO3a and subsequent downregulation of PGC-1α, which further suppresses Mfn2 expression and thereby impacts on mitochondrial dynamics and function [33, 53, 54]. Our data showed that the miR-214 mimic decreased the expression and transcriptional activity of FoxO3a by disrupting its association with SIRT3 and increasing FoxO3a acetylation levels. SIRT3 overexpression increased Mfn2 protein levels and reversed the decrease in Mfn2 expression induced by the miR-214 mimic (Fig. 8g–k). These findings raise the possibility that miR-214 may cause impairment of mitochondrial homeostasis by suppressing Mfn2 via a SIRT3/FoxO3a-dependent cascade, suggesting the potential protective effects of miR-214-targeted intervention against pathological cardiac hypertrophy.
Although we have presented evidence concerning the participation of miR-214 in Ang II-induced cardiac hypertrophy via suppression of SIRT3, there are still some limitations of our study. It is known that an individual miRNA may regulate a broad range of transcripts [55, 56]. Several target genes of miR-214 have been previously identified, such as X-box binding protein 1 (XBP1), zeste homologue 2 (EZH2) and myocyte-specific enhancer factor 2C (MEF2C). These molecules also participate in miR-214-mediated effects in various cardiovascular conditions [25, 26, 57, 58]. Thus, we cannot rule out the involvement of other downstream effectors besides SIRT3 in the prohypertrophic role of miR-214. On the other hand, the development of cardiac hypertrophy is driven by multiple factors and signaling pathways. It is not known whether the same conclusions can be drawn in the settings of cardiac hypertrophy caused by stimuli other than Ang II, such as pressure overload, diabetes and aging. The involvement of miR-214 in the pathogenesis of cardiac hypertrophy should be further investigated.
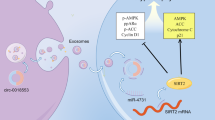
In summary, our data demonstrated that miR-214 participated in Ang II-induced cardiac hypertrophy, and SIRT3 was identified as a direct target of miR-214. Overexpression of miR-214 reduced SIRT3 expression through posttranscriptional regulation, thereby leading to hypertrophic responses and mitochondrial dysfunction (Fig. 9). In contrast, inhibition of miR-214 attenuated the detrimental effects of Ang II by restoring SIRT3. These findings add new information regarding the biological functions of miR-214 and suggest that modulation of miR-214 may be a potential treatment strategy for cardiac hypertrophy.

When cells are exposed to Ang II stimulation, the expression of miR-214 is significantly upregulated. MiR-214 directly targets and suppresses SIRT3 and subsequently influences the interaction of SIRT3 with FoxO3a, leading to downregulation of Mfn2. Collectively, these effects result in mitochondrial impairment and malfunction, which promote the development of cardiac hypertrophy.
References
Nakamura M, Sadoshima J. Mechanisms of physiological and pathological cardiac hypertrophy. Nat Rev Cardiol. 2018;15:387–407.
Shimizu I, Minamino T. Physiological and pathological cardiac hypertrophy. J Mol Cell Cardiol. 2016;97:245–62.
Oldfield CJ, Duhamel TA, Dhalla NS. Mechanisms for the transition from physiological to pathological cardiac hypertrophy. Can J Physiol Pharmacol. 2020;98:74–84.
Gottlieb RA, Bernstein D. Mitochondrial remodeling: rearranging, recycling, and reprogramming. Cell Calcium. 2016;60:88–101.
Shirihai OS, Song M, Dorn GW. How mitochondrial dynamism orchestrates mitophagy. Circ Res. 2015;116:1835–49.
Tang X, Chen XF, Chen HZ, Liu DP. Mitochondrial Sirtuins in cardiometabolic diseases. Clin Sci. 2017;131:2063–78.
Li P, Ge J, Li H. Lysine acetyltransferases and lysine deacetylases as targets for cardiovascular disease. Nat Rev Cardiol. 2020;17:96–115.
Yang W, Nagasawa K, Münch C, Xu YJ, Satterstrom K, Jeong SM, et al. Mitochondrial sirtuin network reveals dynamic SIRT3-dependent deacetylation in response to membrane depolarization. Cell. 2016;167:985–1000.
Sundaresan NR, Gupta M, Kim G, Rajamohan SB, Isbatan A, Gupta MP. Sirt3 blocks the cardiac hypertrophic response by augmenting Foxo3a-dependent antioxidant defense mechanisms in mice. J Clin Invest. 2009;119:2758–71.
Samant SA, Zhang HJ, Hong ZG, Pillai VB, Sundaresan NR, Wolfgeher D, et al. SIRT3 deacetylates and activates OPA1 to regulate mitochondrial dynamics during stress. Mol Cell Biol. 2014;34:807–19.
Zhang XK, Ji RP, Liao XH, Castillero E, Kennel PJ, Brunjes DL, et al. MicroRNA-195 regulates metabolism in failing myocardium via alterations in sirtuin 3 expression and mitochondrial protein acetylation. Circulation. 2018;137:2052–67.
Hu DX, Liu XB, Song WC, Wang JA. Roles of SIRT3 in heart failure: from bench to bedside. J Zhejiang Univ Sci B. 2016;17:821–30.
Pillai VB, Sundaresan NR, Kim G, Gupta M, Rajamohan SB, Pillai JB, et al. Exogenous NAD blocks cardiac hypertrophic response via activation of the SIRT3-LKB1-AMP-activated kinase pathway. J Biol Chem. 2010;285:3133–44.
Yue ZB, Ma YZ, You J, Li ZM, Ding YQ, He P, et al. NMNAT3 is involved in the protective effect of SIRT3 in Ang II-induced cardiac hypertrophy. Exp Cell Res. 2016;347:261–73.
Dai H, Sinclair DA, Ellis JL, Steegborn C. Sirtuin activators and inhibitors: promises, achievements, and challenges. Pharmacol Ther. 2018;188:140–54.
Gertz M, Steegborn C. Using mitochondrial sirtuins as drug targets: disease implications and available compounds. Cell Mol Life Sci. 2016;73:2871–96.
Lai EC. Micro RNAs are complementary to 3’ UTR sequence motifs that mediate negative post-transcriptional regulation. Nat Genet. 2002;30:363–4.
Winter J, Jung S, Keller S, Gregory RI, Diederichs S. Many roads to maturity: microRNA biogenesis pathways and their regulation. Nat Cell Biol. 2009;11:228–34.
Felekkis K, Touvana E, Stefanou C, Deltas C. MicroRNAs: a newly described class of encoded molecules that play a role in health and disease. Hippokratia. 2010;14:236–40.
Duygu B, de Windt LJ, da Costa Martins PA. Targeting microRNAs in heart failure. Trends Cardiovasc Med. 2016;26:99–110.
Katz MG, Fargnoli AS, Kendle AP, Hajjar RJ, Bridges CR. The role of microRNAs in cardiac development and regenerative capacity. Am J Physiol Heart Circ Physiol. 2016;310:H528–H541.
Katoh M. Cardio-miRNAs and onco-miRNAs: circulating miRNA-based diagnostics for non-cancerous and cancerous diseases. Front Cell Dev Biol. 2014;2:61.
Zhou SS, Jin JP, Wang JQ, Zhang ZG, Freedman JH, Zheng Y, et al. miRNAS in cardiovascular diseases: potential biomarkers, therapeutic targets and challenges. Acta Pharmacol Sin. 2018;39:1073–84.
Yang XW, Qin YJ, Shao SX, Yu YQ, Zhang CY, Dong H, et al. MicroRNA-214 inhibits left ventricular remodeling in an acute myocardial infarction rat model by suppressing cellular apoptosis via the phosphatase and tensin homolog (PTEN). Int Heart J. 2016;57:247–50.
Sun M, Yu HY, Zhang YY, Li ZJ, Gao W. MicroRNA-214 mediates isoproterenol-induced proliferation and collagen synthesis in cardiac fibroblasts. Sci Rep. 2015;5:18315.
Duan QL, Yang L, Gong W, Chaugai S, Wang F, Chen C, et al. MicroRNA-214 is upregulated in heart failure patients and suppresses XBP1-mediated endothelial cells angiogenesis. J Cell Physiol. 2015;230:1964–73.
Li JY, Huang JY, Lu J, Guo Z, Li ZM, Gao H, et al. Sirtuin 1 represses PKC‐ζ activity through regulating interplay of acetylation and phosphorylation in cardiac hypertrophy. Br J Pharmacol. 2019;176:416–35.
Wang PX, Wang LP, Lu J, Hu YH, Wang QQ, Li ZZ, et al. SESN2 protects against doxorubicin-induced cardiomyopathy via rescuing mitophagy and improving mitochondrial function. J Mol Cell Cardiol. 2019;133:125–37.
Fu JJ, Gao J, Pi RB, Liu PQ. An optimized protocol for culture of cardiomyocyte from neonatal rat. Cytotechnology. 2005;49:109–16.
Winer LSP, Wu M. Rapid analysis of glycolytic and oxidative substrate flux of cancer cells in a microplate. PLoS One. 2014;9:e109916.
Zhao YF, Ponnusamy M, Zhang L, Zhang Y, Liu CY, Yu WP, et al. The role of miR-214 in cardiovascular diseases. Eur J Pharmacol. 2017;816:138–45.
Rardin MJ, Newman JC, Held JM, Cusack MP, Sorensen DJ, Li B, et al. Label-free quantitative proteomics of the lysine acetylome in mitochondria identifies substrates of SIRT3 in metabolic pathways. Proc Natl Acad Sci U S A. 2013;110:6601–6.
Ong SB, Hausenloy DJ. Mitochondrial dynamics as a therapeutic target for treating cardiac diseases. Handb Exp Pharmacol. 2017;240:251–79.
Tseng AHH, Shieh SS, Wang DL. SIRT3 deacetylates FOXO3 to protect mitochondria against oxidative damage. Free Radic Biol Med. 2013;63:222–34.
Jacobs KM, Pennington JD, Bisht KS, Aykin-Burns N, Kim HS, Mishra M, et al. SIRT3 interacts with the daf-16 homolog FOXO3a in the Mitochondria, as well as increases FOXO3a dependent gene expression. Int J Biol Sci. 2008;4:291–9.
Bisping E, Wakula P, Poteser M, Heinzel FR. Targeting cardiac hypertrophy: toward a causal heart failure therapy. J Cardiovasc Pharmacol. 2014;64:293–305.
Xu XD, Song XW, Li Q, Wang GK, Jing Q, Qin YW. Attenuation of microRNA-22 derepressed PTEN to effectively protect rat cardiomyocytes from hypertrophy. J Cell Physiol. 2012;227:1391–8.
Nie X, Fan JH, Li HP, Yin ZW, Zhao YR, Dai BB, et al. miR-217 promotes cardiac hypertrophy and dysfunction by targeting PTEN. Mol Ther Nucleic Acids. 2018;12:254–66.
Shi JY, Chen C, Xu X, Lu Q. miR-29a promotes pathological cardiac hypertrophy by targeting the PTEN/AKT/mTOR signalling pathway and suppressing autophagy. Acta Physiol. 2019;227:e13323.
Yin HL, Zhao LH, Zhang SL, Zhang Y, Lei SY. MicroRNA-1 suppresses cardiac hypertrophy by targeting nuclear factor of activated T cells cytoplasmic 3. Mol Med Rep. 2015;12:8282–8.
Zaglia T, Ceriotti P, Campo A, Borile G, Armani A, Carullo P, et al. Content of mitochondrial calcium uniporter (MCU) in cardiomyocytes is regulated by microRNA-1 in physiologic and pathologic hypertrophy. Proc Natl Acad Sci U S A. 2017;114:E9006–15.
Tham YK, Bernardo BC, Ooi JYY, Weeks KL, McMullen JR. Pathophysiology of cardiac hypertrophy and heart failure: signaling pathways and novel therapeutic targets. Arch Toxicol. 2015;89:1401–38.
Pinti MV, Hathaway QA, Hollander JM. Role of microRNA in metabolic shift during heart failure. Am J Physiol Heart Circ Physiol. 2017;312:H33–H45.
Koentges C, Pfeil K, Schnick T, Wiese S, Dahlbock R, Cimolai MC, et al. SIRT3 deficiency impairs mitochondrial and contractile function in the heart. Basic Res Cardiol. 2015;110:36.
Wei T, Huang GJ, Gao J, Huang CL, Sun MW, Wu J, et al. Sirtuin 3 deficiency accelerates hypertensive cardiac remodeling by impairing angiogenesis. J Am Heart Assoc. 2017;6:e006114.
Brown DA, Perry JB, Allen ME, Sabbah HN, Stauffer BL, Shaikh SR, et al. Expert consensus document: Mitochondrial function as a therapeutic target in heart failure. Nat Rev Cardiol. 2017;14:238–50.
Dai DF, Rabinovitch PS, Ungvari Z. Mitochondria and cardiovascular aging. Circ Res. 2012;110:1109–24.
Balaban RS. Domestication of the cardiac mitochondrion for energy conversion. J Mol Cell Cardiol. 2009;46:832–41.
Parodi-Rullán RM, Chapa-Dubocq X, Rullán PJ, Jang S, Javadov S. High sensitivity of SIRT3 deficient hearts to sschemia-reperfusion is associated with mitochondrial abnormalities. Front Pharmacol. 2017;8:275.
Dikalova AE, Pandey A, Xiao L, Arslanbaeva L, Sidorova T, Lopez MG, et al. Mitochondrial deacetylase Sirt3 reduces vascular dysfunction and hypertension while Sirt3 depletion in essential hypertension is linked to vascular inflammation and oxidative stress. Circ Res. 2020;126:439–52.
van de Ven RAH, Santos D, Haigis MC. Mitochondrial sirtuins and molecular mechanisms of aging. Trends Mol Med. 2017;23:320–31.
Borniquel S, García-Quintáns N, Valle I, Olmos Y, Wild B, Martínez-Granero F, et al. Inactivation of Foxo3a and subsequent downregulation of PGC-1 alpha mediate nitric oxide-induced endothelial cell migration. Mol Cell Biol. 2010;30:4035–44.
Soriano FX, Liesa M, Bach D, Chan DC, Palacín M, Zorzano A. Evidence for a mitochondrial regulatory pathway defined by peroxisome proliferator-activated receptor-gamma coactivator-1 alpha, sstrogen-related receptor-alpha, and mitofusin 2. Diabetes. 2006;55:1783–91.
Seo AY, Joseph AM, Dutta D, Hwang JCY, Aris JP, Leeuwenburgh C. New insights into the role of mitochondria in aging: mitochondrial dynamics and more. J Cell Sci. 2010;123:2533–42.
Baek D, Villén J, Shin C, Camargo FD, Gygi SP, Bartel DP. The impact of microRNAs on protein output. Nature. 2008;455:64–71.
Selbach M, Schwanhäusser B, Thierfelder N, Fang Z, Khanin R, Rajewsky N. Widespread changes in protein synthesis induced by microRNAs. Nature. 2008;455:58–63.
Yang T, Zhang GF, Chen XF, Gu HH, Fu SZ, Xu HF, et al. MicroRNA-214 provokes cardiac hypertrophy via repression of EZH2. Biochem Biophys Res Commun. 2013;436:578–84.
Tang CM, Liu FZ, Zhu JN, Fu YH, Lin QX, Deng CY, et al. Myocyte-specific enhancer factor 2C: a novel target gene of miR-214-3p in suppressing angiotensin II-induced cardiomyocyte hypertrophy. Sci Rep. 2016;6:36146.
Acknowledgements
This research was supported by grants from the National Natural Science Foundation of China (81872860, 81673433), the Local Innovative and Research Teams Project of Guangdong Pearl River Talents Program (2017BT01Y093), the National Major Special Projects for the Creation and Manufacture of New Drugs (2019ZX09301104), the National Engineering and Technology Research Center for New Drug Druggability Evaluation (Seed Program of Guangdong Province, 2017B090903004), the Special Program for Applied Science and Technology of Guangdong Province (2015B020232009), the Guangdong Basic and Applied Basic Research Foundation (2020A1515011512) and the Young Teacher Training Program of Sun Yat-sen University (18ykpy26).
Author information
Authors and Affiliations
Contributions
PQL, JTY and YQD were responsible for the experimental design and supervision of this project. YQD performed most of the experiments and was the main writer of the manuscript. YHZ, JL, ZBY and WJY contributed to animal experiments. YHH and PXW contributed to the cell culture and immunofluorescence analysis. JYL, BL and SDC contributed to the data analysis and collation. All authors made important suggestions regarding the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Supplementary information
Rights and permissions
About this article

Cite this article
Ding, Yq., Zhang, Yh., Lu, J. et al. MicroRNA-214 contributes to Ang II-induced cardiac hypertrophy by targeting SIRT3 to provoke mitochondrial malfunction. Acta Pharmacol Sin 42, 1422–1436 (2021). https://doi.org/10.1038/s41401-020-00563-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-020-00563-7
Keywords
This article is cited by
-
SZC-6, a small-molecule activator of SIRT3, attenuates cardiac hypertrophy in mice
Acta Pharmacologica Sinica (2023)
-
Non-coding RNAs regulating mitochondrial function in cardiovascular diseases
Journal of Molecular Medicine (2023)
-
The sirtuin family in health and disease
Signal Transduction and Targeted Therapy (2022)
-
GDH promotes isoprenaline-induced cardiac hypertrophy by activating mTOR signaling via elevation of α-ketoglutarate level
Naunyn-Schmiedeberg's Archives of Pharmacology (2022)
-
MicroRNAs in hypertrophic cardiomyopathy: pathogenesis, diagnosis, treatment potential and roles as clinical biomarkers
Heart Failure Reviews (2022)
