
Abstract
Background
Soft tissue sarcomas (STS) are generally considered non-immunogenic, although specific subtypes respond to immunotherapy. Antitumour response within the tumour microenvironment relies on a balance between inhibitory and activating signals for tumour-infiltrating lymphocytes (TILs). This study analysed TILs and immune checkpoint molecules in STS, and assessed their prognostic impact regarding local recurrence (LR), distant metastasis (DM), and overall survival (OS).
Methods
One-hundred and ninety-two surgically treated STS patients (median age: 63.5 years; 103 males [53.6%]) were retrospectively included. Tissue microarrays were constructed, immunohistochemistry for PD-1, PD-L1, FOXP3, CD3, CD4, and CD8 performed, and staining assessed with multispectral imaging. TIL phenotype abundance and immune checkpoint markers were correlated with clinical and outcome parameters (LR, DM, and OS).
Results
Significant differences between histology and all immune checkpoint markers except for FOXP3+ and CD3−PD-L1+ cell subpopulations were found. Higher levels of PD-L1, PD-1, and any TIL phenotype were found in myxofibrosarcoma as compared to leiomyosarcoma (all p < 0.05). The presence of regulatory T cells (Tregs) was associated with increased LR risk (p = 0.006), irrespective of margins. Other TILs or immune checkpoint markers had no significant impact on outcome parameters.
Conclusions
TIL and immune checkpoint marker levels are most abundant in myxofibrosarcoma. High Treg levels are independently associated with increased LR risk, irrespective of margins.
Similar content being viewed by others


Introduction
The tumour microenvironment (TME), consisting of several cell types (e.g. immune cells, fibroblasts) and extracellular components (e.g. hormones, cytokines, growth factors) [1], plays a crucial role in hosts’ immune response to neoplasms [2]. The magnitude of antitumour response relies on a balance between inhibitory and activating signals for T cells, predominantly mediated through immune checkpoint molecules [2, 3]. Within the TME, however, aberrant expression of specific immune checkpoint markers, including programmed cell death protein 1 (PD-1) and its ligand PD-L1, usually promotes immune tolerance and tumour cells’ immune escape [4, 5]. For example, increased expression of PD-1 on the surface of T cells together with consecutive overactivation of the PD-L1/PD-1 pathway mitigates apoptosis of regulatory T cells (Tregs) whilst enhancing apoptosis of antigen-specific T cells [6,7,8].
A rough classification of >50 histological soft tissue sarcoma (STS) subtypes differentiates between “simple” and “complex” karyotypes [9]. Whilst in “simple” karyotypes, specific mutations or genetic translocations can be identified, unspecific genetic alterations are seen in STS subtypes with “complex” karyotypes [10]. Representatives of the latter group include leiomyosarcomas (LMS), undifferentiated pleomorphic sarcomas (UPS), and myxofibrosarcomas (MFS) [9, 10]. These genetically heterogeneous STS subtypes may provide multiple potential targets for the host’s immune system, resulting in an increased amount of tumour-infiltrating lymphocytes (TILs), as well as aberrant expression of immune checkpoint markers [11,12,13]. In line with this, distinct STS subtypes like UPS seem to respond to immune checkpoint inhibitors pembrolizumab [14] and nivolumab plus ipilimumab [15]. Yet, overall response rates are lower than observed in non-small cell lung cancer, renal cell carcinoma, or melanoma [14, 15]. This can be explained by the generally low immunogenicity of STS, resulting in an immune cell-poor TME, with a consecutive lack of targetable molecules [16,17,18].
So far, only a few studies investigated the abundance and prognostic impact of immune checkpoint molecules and TILs in STS [18,19,20,21,22]. Therefore, this study aimed at thoroughly investigating the abundance of immune checkpoint markers, as well as one of TILs in STS of extremities and trunk. Furthermore, their potential impact as prognostic markers for local recurrence (LR), distant metastasis (DM), and overall survival (OS), was analysed.
Materials and methods
Overall, 192 patients treated with curative intent for localised STS at a single centre between 1998 and 2016 were retrospectively included in this study, after exclusion of patients receiving neoadjuvant treatment (neoadjuvant radiotherapy: n = 4; neoadjuvant chemotherapy: n = 10). The median patient age was 63.5 years (interquartile range [IQR]: 49.5–76.0 years), and 103 patients were male (53.6%). The median follow-up was 45.5 months (IQR: 19.0–99.0 months).
Clinical data
Maximal tumour diameter was taken from pathological reports or preoperative magnetic resonance imaging (MRI) scans. Resection margins were defined according to the Union International Contre le Cancer guidelines [23]. R0 defines negative margins with at least 1 mm of healthy tissue between surface and tumour, R1 define marginal margins with <1 mm of healthy tissue separating surface and tumour, and R2 contaminated margins with tumour cells directly at the surface. R1 and R2 margins were grouped together against R0 margins for statistical analyses. Six histological categories based on their frequency were formed, i.e. MFS, synovial sarcoma (SS), UPS, liposarcoma (LPS), LMS, and “other” (for a complete list of rare histological subtypes see Supplementary Table 1). Histologies were defined according to the World Health Organisation Classification of Tumours of Soft Tissue and Bone 2013 [24]. The French Federation of Cancer Centres Sarcoma Group (FNCLCC) grading system was used to differentiate low grade (i.e. G1), intermediate (i.e. G2), and high-grade (i.e. G3) STS. Depending on the location of the tumour in relation to the fascia, tumours were either classified as superficial (above fascia), deep (beneath fascia), or superficial and deep (tumour mass above and beneath fascia, with fascia breached).
Institutional review board (IRB) approval was obtained prior to study initiation (IRB approval number: 29-205 ex 16/17). Written informed consent has been obtained from all patients included.
Regular clinical and radiographic (chest X-ray or chest computed tomography [CT] scan; local MRI) follow-up appointments were scheduled after surgery, with 3-monthly check-ups for the first 3 postoperative years, 6-monthly check-ups for years 4 and 5, and yearly check-ups thereafter.
LR was defined as recurrent tissue at the original tumour site, either diagnosed on MRI scans or upon histopathological analysis of re-resection specimens. DMs were defined as tumour seeding remotely from the original tumour site, as detected by MRI, CT scans, or x-rays. Time to LR and DM was defined as the time interval from surgery to either diagnosis of LR or DM, or last follow-up. Time to the last follow-up was defined as the time from surgery to last follow-up or death.
Construction of tissue microarrays
All initially included tumour samples (n = 206) were independently re-evaluated by two specialised soft tissue tumour pathologists (IB and BL-A). After marking representative tumour areas, paraffin blocks were punched at the regions of interest into a total of 1621 cores of 4 µm thickness. Subsequently, cores were transferred to five recipient paraffin blocks (containing between 235 and 420 cores) and 3–5 µm sections were cut from the tissue microarray (TMA)-paraffin blocks to perform multiplex immunohistochemistry (MP-IHC). Notably, 541 cores (between 25 and 88 per TMA block) were lost and/or damaged during cutting and other pre-processing steps. The remaining 1080 cores, equivalent to seven cores per tumour on average, were finally analysed and evaluated.
Multiplex immunohistochemistry
MP-IHC with six fluorescence-conjugated antibodies and 4′,6-diamidino-2-phenylindole (DAPI) was performed, using the following reagents: PD-1 (clone NAT105, Abcam plc, Cambridge, UK, ab234444), PD-L1 (clone 22C3, Dako, M3653), FOXP3 (clone D6O8R, part of Opal 7 TIL Kit from Akoya Biosciences, OP7TL3001KT), CD3 (clone LN10, Leica Biosystems Inc., Vienna, Austria, NCL-L-CD3-565), CD4 (clone EPR6855, Abcam plc, Cambridge, UK, ab133616), CD8 (C8/144B, Abcam plc, Cambridge, UK, ab75129). Tyramide signal amplification (TSA) Kit (Akoya Biosciences, Marlborough, USA) technique was used for MP-IHC.
Staining, scanning, and image analysis
Prior to MP-IHC, all monoclonal-specific primary antibodies were first tested as monoplex staining on positive control tissues according to the recommendations given by the manufacturers. In addition, appropriate negative control staining for the secondary antibodies was applied on human tissue to avoid non-specific binding and false-positive results. The final multiplex panel was tested on sarcoma tissue (formal-fixed paraffin-embedded full section) before applying it on the sarcoma TMA sections.
All TMA slides were stained with the autostainer system Bond RX (Leica Biosystems Inc., Vienna, Austria). The Vectra® 3 (Akoya Biosystems, Marlborough, USA; software version 3.0.7) microscope was subsequently used to scan the slides. To detect individual TMA scores, whole-slide scans at ×4 magnification were taken. At ×20 magnification, multispectral images of identified TMA cores were subsequently recorded. This resulted in one image colour channel for every antibody stained. The inForm software (Akoya Biosystems, Marlborough, USA; software version 2.4.8) was applied for image processing, spectral unmixing, and removal of autofluorescence. The HALO® Image Analysis Platform (Indica Labs, Albuquerque, NM, USA; version 3.1.1076.342) was used for the evaluation of the multispectral images. For this purpose, single recorded images at ×20 magnification were stitched together to obtain a continuous field of view covering the entire TMA slide (Fig. 1). By defining a threshold for DAPI-stained nucleus size, signal intensity, and roundness, individual cells were identified. Thresholds for the analysed fluorescence-labelled markers were set based on the staining intensity of the entire cell.

Note the difference between the left (low abundance) and right sample (high abundance) regarding immune cell and checkpoint marker abundance.
Positivity of each cell to immune checkpoint markers PD-1, PD-L1, and FOXP3 was automatically counted on TMAs. Moreover, the combination of PD-1 and PD-L1 expression on each cell was independently assessed, resulting in three additional phenotypes (i.e. PD-1+PD-L1+; PD-1+PD-L1−; PD-1PD-L1+). Based on immune cell markers CD3, CD4, and CD8, 4 TIL phenotypes were differentiated in all TMA cores: T cells (CD3+), helper T cells (CD3+CD4+), cytotoxic T cells (CD3+CD8+), and Treg (CD3+CD4+FOXP3+; Supplementary Table 2). The combination of TIL phenotypes with immune checkpoint markers resulted in five additional subtypes automatically counted on TMAs: PD-L1+CD3+ T cells, PD-1+CD3+ T cells, PD-1+CD3+CD4+ helper T cells, PD-1+CD3+CD8+ cytotoxic T cells, and PD-1+CD3+CD4+FOXP3+ Tregs. Furthermore, CD3− cells, positive for either PD-1 (CD3−PD-1+) or PD-L1 (CD3−PD-L1+), were differentiated.
All phenotypes were automatically counted in each TMA core individually and later combined for cores of the same patient. Percentage of immune checkpoint marker and TIL phenotype abundances were calculated by aggregating all cells of all TMA cores of the same patient. Through this approach, large local fluctuations in cell counts or pooling of individual cell types within one core can be accounted for. For descriptive analyses, “low” and “high” densities of respective TIL phenotype and immune checkpoint marker abundances were defined as <1% or >1% of the entire cell count (Table 1). Notably, due to non-normal distribution of phenotype abundance across the entire patient cohort, statistical analyses were performed on median values rather than means.
Statistical analysis
Means and medians with corresponding standard deviations (SDs) and IQRs were provided for demographic, tumour, and treatment-related variables, as well as phenotype abundances. We analysed quantitative differences in phenotype abundances between patient groups and binominal or categorical variables using Wilcoxon’s rank-sum and Kruskal–Wallis tests, respectively. Post hoc Dunn tests (using Benjamini–Hochberg adjustment for multiple comparisons [25]) were used. Spearman’s rank correlation coefficients were calculated to assess potential associations between immune checkpoint markers and TILs.
Time-dependent effects of prognostic variables on the development of LR and DM were assessed with univariate and multivariate Fine and Gray models, with death as the competing event. The influence of variables on OS was analysed with univariate and multivariate Cox regression models. All factors with a p value of <0.1 in the univariate analyses were included in the multivariate models as long as the “one in ten rule” [26] was not violated. For all results, a p value of <0.05 was considered statistically significant.
Results
The mean tumour size was 8.5 ± 5.2 cm, and most STS were located in the lower limb (n = 125; 65.5%). The majority of STS was classified as G3 (n = 132; 72.5%), and most tumours were located deep to the muscular fascia (n = 111; 57.8%). CD3+ T cells were the most abundant TIL subpopulation with 1.02% (IQR: 0.30–3.15%), whereas FOXP3 was the most abundant marker with 2.6% (IQR: 0.38–16.85%). In other words, 50.5% of tumour samples contained >1% CD3+ T cells, and 63.5% of samples >1% FOXP3+ cells. Further quantitative and qualitative immune checkpoint marker and TIL phenotype abundances, as well as clinical, pathological, and treatment-related variables, are listed in Table 1.
Correlation between immune checkpoint markers and TILs
There was a strong positive correlation between presence of PD-1+ cells and FOXP3+ cells (rho = 0.737), as well as a weak positive correlation between PD-1+ cells and Tregs (rho = 0.311; Supplementary Fig. 1). A moderate positive correlation was found for PD-L1+ cells and all T cell subtypes. As expected, a strong correlation between CD3+ T cells and CD3+CD4+ helper T cells (rho = 0.761), CD3+CD8+ cytotoxic T cells (rho = 0.880), and CD3+CD4+FOXP3+ Tregs was present (rho = 0.742; Supplementary Fig. 1).
Difference in immune checkpoint markers between patient subpopulations
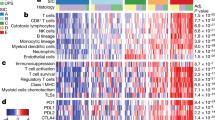
Higher amounts of PD-L1+ cells were found in STS of older patients (≥63.5 vs. <63.5 years: 0.71% vs. 0.46%; p = 0.047), as were increased levels of PD-L1+PD-1+ cells (0.03 vs. 0.00%; p = 0.038). Also, CD3−PD-L1+ cell amounts were higher in patients >63.5 years of age (0.28 vs. 0.16%; p = 0.014), whereas no difference for CD3−PD-1+ cells depending on age was present (p = 0.233). Corresponding to this, no difference depending on patient age was found for PD-1+ cells (p = 0.215). Neither there was a difference for FOXP3+ cells (p = 0.860), PD-L1+PD-1− cells (p = 0.074), or PD-L1−PD-1+ cells (p = 0.211) between age groups. Abundance of all immune checkpoint marker-positive cells (i.e. PD-L1+; PD-1+; PD-L1+PD-1−; PD-L1−PD-1+; PD-L1+PD-1+; CD3−PD-1+), except for FOXP3+ and CD3−PD-L1+ subpopulations, were significantly different depending on histological subtype (p < 0.05; Fig. 2a). Of note, significantly higher immune checkpoint marker levels were found in MFS in comparison to LMS (Fig. 2a). No significant difference in immune checkpoint markers between gender, grading, tumour size, depth, or tumour location was found.

Difference in immune checkpoint markers and CD3− subpopulations (a), as well as TIL phenotype abundance depending on histological subtype (b). Significant differences (p < 0.05) were highlighted with bars and asterisks. UPS undifferentiated pleomorphic sarcoma.
Difference in TIL abundance between patient subpopulations
The amount of CD3+ T cells was significantly higher in G3 STS in comparison to G1 or G2 STS (G3 vs. G1/2: 1.31 vs. 0.78%; p = 0.047). The abundance of the other TILs did not significantly differ depending on grading.
In older patients, higher amounts of CD3+CD4+ helper T cells (0.14 vs. 0.07%; p = 0.030), PD-1+CD3+ T cells (0.06 vs. 0.01%; p = 0.013), PD-1+CD3+CD4+ helper T cells (0.01 vs. 0.00%; p = 0.010), CD3+CD4+FOXP3+ Tregs (0.06 vs. 0.02%; p = 0.010), and PD-1+CD3+CD8+ cytotoxic T cells (0.03 vs. 0.00%; p = 0.019) were found. On the other hand, CD3+ T cells (p = 0.072), CD3+CD8+ cytotoxic T cells (p = 0.106), PD-L1+CD3+ T cells (p = 0.195), PD-1+CD3+CD4+FOXP3+ Tregs (p = 0.116), and PD-L1+ T cells (p = 0.195) did not significantly differ depending on patient age.
All TIL phenotypes did significantly differ depending on histological subtype (p < 0.05). Notably, for any TIL phenotype analysed, the levels were significantly higher in MFS in comparison to SS and LMS (Fig. 2b). For the remaining histological subtypes, except for UPS, significant differences in comparison to MFS, depending on TIL phenotype, were present. Moreover, in UPS, we found significantly higher levels of CD3+ T cells, PD-1+CD3+ T cells, CD3+CD8+ cytotoxic T cells, and PD-1+CD3+CD8+ cytotoxic T cells in comparison to SS, and higher levels of CD3+CD8+ cytotoxic T cells compared to LPS (Fig. 2b).
There was no significant difference between TIL abundance and gender, tumour size, depth, or tumour location.
Prognostic impact of clinical variables and phenotype abundances on LR risk
In the univariate analysis, the only TIL phenotype significantly associated with increased risk for LR were high levels of Tregs (p = 0.016; Table 2). There was a trend towards increased LR risk in case of high levels of PD-1+ Tregs (p = 0.061), PD-L1+PD-1+ cells (p = 0.070), and CD3−PD-1+ cells (p = 0.078), as well as a decreased LR risk for high helper T cell abundancy (p = 0.062), which, however, did not reach statistical significance (Table 2). All other markers and TIL phenotypes showed no statistically significant association (Table 2). Margin status (p = 0.063) was not significantly associated with LR risk. Other clinical parameters such as tumour size (p = 0.343) and patient age (p = 0.251) did not reach statistical significance (Table 2). In the multivariate model, high levels of Tregs remained significantly associated with increased risk of LR (p = 0.006), irrespective of margin status (p = 0.072; Table 3).
Prognostic impact of clinical variables and phenotype abundances on DM risk
In the univariate Fine and Gray model for DM, the only factors significantly associated with higher risk for metastatic spread were large tumour size (p = 0.010), G3 in comparison to G1 STS (p = 0.042), and the histological subtype UPS (p = 0.020) compared to MFS. None of the immune checkpoint markers or TIL phenotype abundances was significantly associated with higher DM risk in the univariate analysis (Table 2). In the multivariate model, a large tumour size (p = 0.003), and histological subtypes UPS (p = 0.008) and LMS (p = 0.031) in comparison to MFS, was associated with lower DM risk, irrespective of grading (Table 3).
Prognostic impact of clinical variables and phenotype abundances on OS
Advanced patient age (p < 0.001), high CD3−PD-L1+ levels (p = 0.021) and low levels of FOXP3+ cells (p = 0.023) were associated with worse OS in the univariate Cox regression analysis. Furthermore, a large tumour size (p = 0.051) was marginally associated with worse OS (Table 2). Other clinical variables, immune checkpoint markers, or TIL phenotype abundances showed no statistically significant association with OS (Table 2). In the multivariate analysis, advanced patient age (p < 0.001) remained significantly associated with worse OS, irrespective of tumour size (p = 0.076), FOXP3+ levels (p = 0.076), and CD3−PD-L1+ levels (p = 0.087; Table 3).
Discussion
In the present study, a positive correlation between PD-L1 expression and abundance of TILs in STS was found. Higher numbers of PD-L1+ cell levels were found in older patients and patients with MFS compared to LMS. CD3+ T cell abundancy increased with tumour dedifferentiation, as indicated by higher levels being present in G3 in comparison to G1/G2 STS. Specific TIL phenotypes, namely CD3+CD4+ helper T cells, PD-1+CD3+ T cells, CD3+CD4+FOXP3+ Tregs, PD-1+CD3+CD4+ helper T cells, and PD-1+CD3+CD8+ cytotoxic T cells were more abundant in older STS patients compared with younger ones. High FOXP3+ cell levels were significantly associated with improved OS in the univariate analysis, while none of the other investigated immune checkpoint markers had prognostic significance regarding LR risk, DM risk, or OS. Moreover, Tregs were the only TIL phenotype of independent negative prognostic significance for LR. None of the other TIL phenotypes was significantly associated with LR risk, DM risk, or OS.
Some limitations regarding the present study have to be considered. First, the study comprises a limited and heterogeneous cohort of patients, owing to the rarity of STS in general as well as the numerous histological subtypes with different biological behaviour. Second, due to the limited number of patients, known prognostic factors as age, margin status, grading, and tumour size were only in part significant in the current study. Third, the retrospective design of the current study naturally led to various (adjuvant) treatment approaches that could not be totally adjusted for in the present study despite multivariate analyses. Fourth, IHC only was used to analyse the presence and phenotype of TILs, as well as immune checkpoint marker expression. Further studies, e.g. by applying gene expression analyses, are thus warranted to eventually confirm the herein described preliminary findings. Fifth, rather than defining cut-off values for individual immune checkpoint markers and TIL abundances, they were measured quantitatively. Although this approach may not allow for direct transfer of observed results into clinical practice, it avoids information loss for statistical analysis.
Differences in the microenvironment of STS subtypes, together with immune checkpoint marker expression and TIL abundance, may be an explanation for varying response rates to immunotherapies. For example, Boxberg et al. [18] reported on higher PD-L1 expression in UPS compared with angiosarcoma, LMS, or SS. Likewise, Pollack et al. [13] discovered that both PD-L1 and PD-1 levels are significantly higher in UPS in comparison to well- or dedifferentiated LPS, LMS, or SS, and that UPS and SS show the highest and lowest amounts of T cell infiltration, respectively [13]. In line with this, we observed significantly higher levels of various TIL phenotypes (T cells, PD-1+ T cells, cytotoxic T cells, and PD-1+ cytotoxic T cells) in UPS in comparison to SS. Furthermore, a higher prevalence of all immune checkpoint markers, except for FOXP3, was observed in MFS in comparison to LMS, indicating a strong immunogenicity of this specific histological subtype.
Yet, the immune checkpoint marker expression levels on TILs in STS are generally lower than that observed in other solid tumours, with 5.7 and 10.4% of STS samples in the current study containing high levels of CD3+PD-L1+ TILs and CD3+PD-1+ TILs, defined as density >1%. As a comparison, PD-L1 expression levels on ≥1% of TILs are observed in up to 48% of gastric cancers [27], 37.3% of small cell lung cancers [28], and 25.1% of triple-negative breast cancers [29]. Moreover, in small cell lung cancer, PD-1 expression on >1% of TILs is found in 40.2% of tissue samples [28].
In the current study, specific TIL phenotypes were more abundant in older patients, corresponding to previous observations [30]. This might explain a potentially enhanced response to immune checkpoint inhibitors in elderly patients [31, 32]. However, experimental evidence using mouse models suggests that despite a high abundance of immune checkpoint markers on aged T cells, restoration of their activity by immune checkpoint blockade is less effective than in T cells of young mice [33].
D’Angelo et al. discovered a positive association between high CD3+ and CD8+ T cell levels, and PD-L1 and PD-1 expression [19]. In our study, we likewise observed this positive correlation between PD-L1 and TILs, but not between PD-1 and TILs. This discrepancy may be explained by the fact that D’Angelo et al. used an arbitrary cut-off value of 5% to differentiate between “high-density” and “low-density” TILs, as well as 1% for “positive” vs. “negative” PD-L1 expression [19], whilst we analysed percentages of positively stained cells within total cells. Similar observations have been made by Boxberg et al. [18], with the authors further assuming that the varying results reported in the literature can be attributed to the different antibodies that were used, dynamic expression of immune checkpoint markers, influence of neoadjuvant treatments, and heterogeneous patient cohorts [18].
We further discovered a positive correlation between increased Treg levels and both PD-1 and PD-L1 expression, indicating PD-1/PD-L1 pathway activation, consecutively mitigating apoptosis of Tregs [6, 7]. In line with this, high levels of Tregs were the only TIL phenotype of independent negative prognostic impact on LR risk, whilst having no effect on DM risk or OS. Elevated Treg levels within the TME have been correlated with advanced disease and poor prognosis [34, 35]. In STS, the potential role of Tregs has been investigated by Que et al., reporting that high levels are associated with poor disease-free survival and OS [36]. Furthermore, they discovered a positive association between PD-L1 expression and Treg infiltration [36]. In the present study, a positive correlation between PD-L1 and Tregs was found. However, we were unable to demonstrate a significant impact of Treg infiltration on patients’ OS. Yet, high Treg levels were independently associated with higher risk for LR, irrespective of margin status. This implies a strong local effect of Tregs, suppressing the activity of TILs and thus mitigating the host’s antitumour immune response [34]. Notably, chemotherapeutics as cyclophosphamide and paclitaxel, agents also used in STS, can promote depletion of Tregs, thus eventually improving tumour control [37,38,39]. Therefore, administration of these agents in patients with high Treg levels may improve local control, particularly if a high LR risk is anticipated. On the other hand, targeted agents against FOXP3, a marker specific but not unique for Tregs, may not prove effective in STS, considering that we discovered a marginally significant (p = 0.076) protective effect of FOXP3+ abundance on patient OS. Our observations are in line with those made by Bae et al. on the genetic level, discovering that in STS patients with favourable OS, overexpression of FOXP3, amongst other immune-associated genes, is present [40]. Also, the seeming discrepancy between FOXP3 abundance within the TME and Tregs, in particular, has been previously observed in other tumours [41]. This contrast can be explained by the fact that FOXP3 is likewise expressed by other immune cell types apart from Tregs, including CD4+ and CD8+ cells [41,42,43].
In conclusion, Tregs seem to play a crucial role in the local progression of STS, being independently associated with higher LR risk, irrespective of margin status. Therefore, antitumour agents with the capability to affect Tregs may be administered in STS with high levels of tumour-infiltrating Tregs to improve local control. Yet, subsequent studies are warranted to further elucidate the impact and targetability of Tregs in STS.
Data availability
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
References
Wu T, Dai Y. Tumor microenvironment and therapeutic response. Cancer Lett. 2017;387:61–68.
Kim C, Kim EK, Jung H, Chon HJ, Han JW, Shin KH, et al. Prognostic implications of PD-L1 expression in patients with soft tissue sarcoma. BMC Cancer. 2016;16:434.
Darvin P, Toor SM, Sasidharan Nair V, Elkord E. Immune checkpoint inhibitors: recent progress and potential biomarkers. Exp Mol Med. 2018;50:1–11.
Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252–64.
Smolle MA, Calin HN, Pichler M, Calin GA. Noncoding RNAs and immune checkpoints-clinical implications as cancer therapeutics. FEBS J. 2017;284:1952–66.
Francisco LM, Sage PT, Sharpe AH. The PD-1 pathway in tolerance and autoimmunity. Immunol Rev. 2010;236:219–42.
McDermott DF, Atkins MB. PD-1 as a potential target in cancer therapy. Cancer Med. 2013;2:662–73.
Smolle MA, Prinz F, Calin GA, Pichler M. Current concepts of non-coding RNA regulation of immune checkpoints in cancer. Mol Asp Med. 2019;70:117–26.
Antonescu CR. The role of genetic testing in soft tissue sarcoma. Histopathology. 2006;48:13–21.
Heitzer E, Sunitsch S, Gilg MM, Lohberger B, Rinner B, Kashofer K, et al. Expanded molecular profiling of myxofibrosarcoma reveals potentially actionable targets. Mod Pathol. 2017;30:1698–709.
Tran E, Turcotte S, Gros A, Robbins PF, Lu YC, Dudley ME, et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science. 2014;344:641–5.
Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr., Kinzler KW. Cancer genome landscapes. Science. 2013;339:1546–58.
Pollack SM, He Q, Yearley JH, Emerson R, Vignali M, Zhang Y, et al. T-cell infiltration and clonality correlate with programmed cell death protein 1 and programmed death-ligand 1 expression in patients with soft tissue sarcomas. Cancer. 2017;123:3291–304.
Tawbi HA, Burgess M, Bolejack V, Van Tine BA, Schuetze SM, Hu J, et al. Pembrolizumab in advanced soft-tissue sarcoma and bone sarcoma (SARC028): a multicentre, two-cohort, single-arm, open-label, phase 2 trial. Lancet Oncol. 2017;18:1493–501.
D'angelo SP, Mahoney MR, Van Tine BA, Atkins J, Milhem MM, Jahagirdar BN, et al. Nivolumab with or without ipilimumab treatment for metastatic sarcoma (Alliance A091401): two open-label, non-comparative, randomised, phase 2 trials. Lancet Oncol. 2018;19:416–26.
Teng MW, Ngiow SF, Ribas A, Smyth MJ. Classifying cancers based on T-cell Infiltration and PD-L1. Cancer Res. 2015;75:2139–45.
Spranger S. Mechanisms of tumor escape in the context of the T-cell-inflamed and the non-T-cell-inflamed tumor microenvironment. Int Immunol. 2016;28:383–91.
Boxberg M, Steiger K, Lenze U, Rechl H, von Eisenhart-Rothe R, Wörtler K, et al. PD-L1 and PD-1 and characterization of tumor-infiltrating lymphocytes in high grade sarcomas of soft tissue - prognostic implications and rationale for immunotherapy. Oncoimmunology. 2018;7:e1389366.
D'angelo SP, Shoushtari AN, Agaram NP, Kuk D, Qin LX, Carvajal RD, et al. Prevalence of tumor-infiltrating lymphocytes and PD-L1 expression in the soft tissue sarcoma microenvironment. Hum Pathol. 2015;46:357–65.
Sorbye SW, Kilvaer T, Valkov A, Donnem T, Smeland E, Al-Shibli K, et al. Prognostic impact of lymphocytes in soft tissue sarcomas. PLoS ONE. 2011;6:e14611.
Sorbye SW, Kilvaer TK, Valkov A, Donnem T, Smeland E, Al-Shibli K, et al. Prognostic impact of peritumoral lymphocyte infiltration in soft tissue sarcomas. BMC Clin Pathol. 2012;12:5.
Fujii H, Arakawa A, Utsumi D, Sumiyoshi S, Yamamoto Y, Kitoh A, et al. CD8(+) tumor-infiltrating lymphocytes at primary sites as a possible prognostic factor of cutaneous angiosarcoma. Int J Cancer. 2014;134:2393–402.
Wittekind C, Compton CC, Greene FL, Sobin LH. TNM residual tumor classification revisited. Cancer. 2002;94:2511–6.
Fletcher, CD, Bridge, JA, Hodgendoorn, PCW, Mertens, F. WHO classification of tumours of soft tissue and bone. Lyon: IARC; 2013.
Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B. 1995;57:289–300.
Harrell FE, Lee KL, Califf RM, Pryor DB, Rosati RA. Regression modelling strategies for imporved prognostic prediction. Stat Med. 1984;3:143–52.
Ju X, Shen R, Huang P, Zhai J, Qian X, Wang Q, et al. Predictive relevance of PD-L1 expression with pre-existing TILs in gastric cancer. Oncotarget. 2017;8:99372–81.
Sun C, Zhang L, Zhang W, Liu Y, Chen B, Zhao S, et al. Expression of PD-1 and PD-L1 on tumor-infiltrating lymphocytes predicts prognosis in patients with small-cell lung cancer. OncoTargets Ther. 2020;13:6475–83.
Wang X, Liu Y. PD-L1 expression in tumor infiltrated lymphocytes predicts survival in triple-negative breast cancer. Pathol Res Pract. 2020;216:152802.
Kim JR, Moon YJ, Kwon KS, Bae JS, Wagle S, Kim KM, et al. Tumor infiltrating PD1-positive lymphocytes and the expression of PD-L1 predict poor prognosis of soft tissue sarcomas. PLoS ONE. 2013;8:e82870.
Elias R, Giobbie-Hurder A, McCleary NJ, Ott P, Hodi FS, Rahma O. Efficacy of PD-1 & PD-L1 inhibitors in older adults: a meta-analysis. J Immunother Cancer. 2018;6:26.
Kugel CH 3rd, Douglass SM, Webster MR, Kaur A, Liu Q, Yin X, et al. Age correlates with response to anti-PD1, reflecting age-related differences in intratumoral effector and regulatory T-cell populations. Clin Cancer Res. 2018;24:5347–56.
Lages CS, Lewkowich I, Sproles A, Wills-Karp M, Chougnet C. Partial restoration of T-cell function in aged mice by in vitro blockade of the PD-1/ PD-L1 pathway. Aging Cell. 2010;9:785–98.
Betts GJ, Clarke SL, Richards HE, Godkin AJ, Gallimore AM. Regulating the immune response to tumours. Adv Drug Deliv Rev. 2006;58:948–61.
Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10:942–9.
Que Y, Xiao W, Guan YX, Liang Y, Yan SM, Chen HY, et al. PD-L1 expression is associated with FOXP3+ regulatory T-cell infiltration of soft tissue sarcoma and poor patient prognosis. J Cancer. 2017;8:2018–25.
Pere H, Tanchot C, Bayry J, Terme M, Taieb J, Badoual C, et al. Comprehensive analysis of current approaches to inhibit regulatory T cells in cancer. Oncoimmunology. 2012;1:326–33.
Ercolini AM, Ladle BH, Manning EA, Pfannenstiel LW, Armstrong TD, Machiels JP, et al. Recruitment of latent pools of high-avidity CD8(+) T cells to the antitumor immune response. J Exp Med. 2005;201:1591–602.
Chen CA, Ho CM, Chang MC, Sun WZ, Chen YL, Chiang YC, et al. Metronomic chemotherapy enhances antitumor effects of cancer vaccine by depleting regulatory T lymphocytes and inhibiting tumor angiogenesis. Mol Ther. 2010;18:1233–43.
Baee JY, Choi KU, Kim A, Lee SJ, Kim K, Kim JY. Evaluation of immune-biomarker expression in high-grade soft-tissue sarcoma: HLA-DQA1 expression as a prognostic marker. Exp Ther Med. 2020;20:107–1.
deLeeuw RJ, Kost SE, Kakal JA, Nelson BH. The prognostic value of FoxP3+ tumor-infiltrating lymphocytes in cancer: a critical review of the literature. Clin Cancer Res. 2012;18:3022–9.
Miyara M, Yoshioka Y, Kitoh A, Shima T, Wing K, Niwa A, et al. Functional delineation and differentiation dynamics of human CD4+ T cells expressing the FoxP3 transcription factor. Immunity. 2009;30:899–911.
Thornton AM, Korty PE, Tran DQ, Wohlfert EA, Murray PE, Belkaid Y, et al. Expression of Helios, an Ikaros transcription factor family member, differentiates thymic-derived from peripherally induced Foxp3+ T regulatory cells. J Immunol. 2010;184:3433–41.
Acknowledgements
Not applicable.
Funding
This study was funded by the “Verein für Krebskranke” (Cancer Patient Society). Moreover, the current study was supported by the K1 COMET Competence Center CBmed (Center for Biomarker Research in Medicine), which is funded by the Federal Ministry of Transport, Innovation and Technology (BMVIT), Land Steiermark (Department 12, Business and Innovation), the Federal Ministry of Science, Research and Economy (BMWFW), the Styrian Business Promotion Agency (SFG), and the Vienna Business Agency. The COMET programme is executed by the Austrian Research Promotion Agency (FFG). LH was supported by CBmed via the PhD programme Advanced Medical Biomarker Research (AMBRA). The funding sources had no impact on the design of the study, its conduction, analysis, presentation of results or interpretation.
Author information
Authors and Affiliations
Contributions
JS, AL, and MAS were responsible for the study design. The literature search was performed by LH, MP, MT, BG, AG, MG, and AE-H. Figures were plotted by LH, BP, MAS, and PL-G. Data were ascertained by MAS, BG, MG, SS, LH, JR, MT, and PL-G. Tumour sample and TMA preparations were performed by IVAB, BL-A, MT, and BP. TMA analysis was performed by BP, LH, PL-G, and MT. LH, PL-G, AE-H, AL, JS, and MAS were responsible for data analysis. Data interpretation was performed by SS, IVAB, MB, MP, AG, CR, JR, JS, and MAS. MAS, LH, AL, PL-G, and JS wrote the primary draft of the manuscript. All authors were responsible for the setup, writing, and revision of the main manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
The current study was approved by the institutional review board (Ethics Commission of the Medical University of Graz, Austria; IRB approval number: 29-205 ex 16/17) of the primary study centre. Tissue samples were only included if patients’ informed consent to have their material analysed for research purposes was available.
Consent to publish
As no directly patient-identifying information is provided in the current study, consent for publication was not obtained from participants.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Smolle, M.A., Herbsthofer, L., Granegger, B. et al. T-regulatory cells predict clinical outcome in soft tissue sarcoma patients: a clinico-pathological study. Br J Cancer 125, 717–724 (2021). https://doi.org/10.1038/s41416-021-01456-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41416-021-01456-0
