
Abstract
Pro-survival stress-inducible chaperone HSP110 is the only HSP for which a mutation has been found in a cancer. Multicenter clinical studies demonstrated a direct association between HSP110 inactivating mutation presence and excellent prognosis in colorectal cancer patients. Here, we have combined crystallographic studies on human HSP110 and in silico modeling to identify HSP110 inhibitors that could be used in colorectal cancer therapy. Two molecules (foldamers 33 and 52), binding to the same cleft of HSP110 nucleotide-binding domain, were selected from a chemical library (by co-immunoprecipitation, AlphaScreening, Interference-Biolayer, Duo-link). These molecules block HSP110 chaperone anti-aggregation activity and HSP110 association to its client protein STAT3, thereby inhibiting STAT3 phosphorylation and colorectal cancer cell growth. These effects were strongly decreased in HSP110 knockdown cells. Foldamer’s 33 ability to inhibit tumor growth was confirmed in two colorectal cancer animal models. Although tumor cell death (apoptosis) was noted after treatment of the animals with foldamer 33, no apparent toxicity was observed, notably in epithelial cells from intestinal crypts. Taken together, we identified the first HSP110 inhibitor, a possible drug-candidate for colorectal cancer patients whose unfavorable outcome is associated to HSP110.
Similar content being viewed by others

Introduction
Colorectal cancer (CRC) is the third most common cancer worldwide with almost 1.4 million newly diagnosed patients annually [1]. Surgery constitutes the first therapy together with chemotherapy for patients with primary metastatic CRC (stage II and III). Over the last decades, therapeutic strategies have emerged to hamper malignant neoplastic development with the recent breakthrough of immunotherapy with anti-PD-1/PD-L1 or CTLA-4 mAbs (pembrolizumab, atezolizumab, or ipilumumab) [2, 3]. The complex nature of tumorigenesis coupled to a modest success rate of immunotherapy in CRCs [2, 3], or immunotherapy backfire effects in some cases [4], prompts us to find new molecularly targeted therapies. One such new lead is through inhibition of Heat Shock Proteins (HSP).
HSPs participate in the correct folding, activity, transport and stability of proteins. HSPs have intracellular (pro-survival) and extracellular (danger signal) functions [5, 6]. When a cancer cell accumulates mutations, it violates the physiological laws and acquires sets of hallmarks requiring a constitutively high level of HSPs to ensure its survival/maintenance. Traditionally, HSPs are classified by their molecular weight: HSP110 (also called HSPH), HSP90 (HSPD), HSP70 (HSPA), HSP60 (HSPD/E), and the small HSPs (HSPB). A number of these proteins have been correlated to cancer aggressiveness, and to cancer resistance to cell death induced by radiotherapy and adjuvant chemotherapy [7,8,9] and, for HSP90, some inhibitors are already in advanced clinical trials with encouraging results [10].
We previously described in CRCs with microsatellite instability, the presence of a truncated HSP110 mutant, caused by the skipping of exon 9. This inactive HSP110 was formed at the expense of wild-type HSP110. Patients with high expression levels of this inactive HSP110 mutant (and therefore low levels of HSP110 wild type) show extraordinary disease-free survival rates [11]. This result in itself is a powerful lead to a new strategy against cancer. Unfortunately, no molecules targeting HSP110 have been described so far.
HSP110 is overexpressed in stress conditions and prevents aggregation of misfolded/unfolded proteins [12, 13]. Strongly expressed in most CRC tumors, HSP110 is involved in stabilization of oncogenic proteins promoting cancer cells proliferation, metastasis, and poor prognosis [14,15,16,17]. HSP110 inhibits apoptosis and enhances certain signaling pathways and transcription factors, notably the proliferative Wnt/β-catenin [18] and the transcription factor STAT3 pathway. Indeed, recently our team demonstrated that HSP110 favored STAT3 phosphorylation in the cytosol, thereby promoting cell proliferation [5, 6]. Colon cancer cells in which HSP110 was knocked down had an impaired STAT3 phosphorylation and hardly proliferate. Moreover, STAT3 phosphorylation and cell proliferation were restored by HSP110 re-expression. Accordingly, in CRC mice models and patients, HSP110 levels correlated with that of phosphorylated STAT3 and tumor growth [5, 17].
In this work, upon screening of a chemical library, we selected two abiotic foldamers (33 and 52) that bound directly to the nucleotide-binding domain (NBD) of HSP110, thereby blocking HSP110 chaperone function and colorectal cancer growth. The anti-tumor properties of molecule 33 was subsequently confirmed in in vivo mouse models. This foldamer, the first experimental therapeutic HSP110 inhibitor described, may pave the way to a new type of chemical molecules against CRC.
Results
HSP110 crystallographic studies and chemical library screening
To obtain crucial information on the druggability of the nucleotide-binding domain (NBD) of HSP110, we first performed crystallographic studies. Since no structural information was available to date for mammalian HSP110, we solved the structure of human HSP110 NBD using X-ray crystallography (Table S1 for data refinement and reduction).
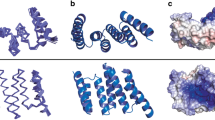
When compared with yeast HSP110 (Sse1p) and HSP70 members, for which the structure have been already solved [19, 20], human HSP110 NBD presents a similar organization in four subdomains (IA, IB, IIA, IIB) shaping two similar lobes (I and II; Fig. 1a). The ATP binding site is located in a deep cleft at the interface of the four subdomains (zoomed view in Fig. 1a). Structural comparison of HSP70’s and HSP110’s counterpart reveals superimposable structures with a Cα root mean square deviation (RMSD) value inferior to 2 Å (Table S2). The only noticeable differences are located on amino acids 186–297 and 287–297 where HSP70 exhibits protuberant loops (Fig. 1b). HSP70 and HSP110 members present also some sequence identity noticeably in the ATP binding pocket, which seems to be the most conserved part of the NBD (Fig. 1c). However, the overall structural and sequence similarity between HSP110 and HSP70 ATP binding pocket is shaded by the fact that some residues present in the cleft are different. Amongst the 26 residues involved in ATP binding in HSP70, only 8 are conserved in the selected HSP members while others are mainly substituted with similar amino acids (e.g., G→S, S→A) (Fig. 1d). This ATP-binding site divergence emphasizes HSP70 and HSP110 functional differences.

Structural studies of human NBD HSP110. a HSP110 NBD tri-dimensional structure, based on our crystallographic studies (Table S1), is represented. NBD exhibits classical HSP’s binding site lobes, indicated in the figure as IA, IIA, IB, and IIB. The presence of ATP was identified in the solved structure. The density corresponding to ATP is depicted on the right panel. b Structural alignment of ATP bound NBD from human HSP110 with human HSP70 (PDB ID 3ATU), E. coli HSP70 (DnaK. PDB ID 4B9Q), and yeast HSP110 (Sse1p. PDB ID 2QXL). Structures are represented in loop and, respectively, colored in light blue, pink, green, and yellow. Protuberant loops present in HSP70 are circled in magenta. c Interface conservation of human HSP110 modeling using ConSurf server [50]. Variable residues are colored in blue shaded whereas conserved amino acids are colored in purple shaded. ATP binding site is circled in yellow. d Sequence alignment of the NBD of HSP70 from E. coli (DnaK) and human, and of HSP110 from yeast (Sse1p) and human. Amino acids involved in ATP binding are highlighted in yellow
We next screened a library of protein–protein interaction inhibitors (PPIs) called foldamers, for which the design was based on pyridyl scaffolds mimicking α-helix twist [21] and tested their capacity to inhibit the intrinsic chaperone activity of HSP110 (Suppl. Fig. 1a) [22]. Among 132 compounds, 3 molecules were able to significantly inhibit HSP110 anti-aggregating activity with IC50 values estimated at 87.8 ± 13, 125 ± 31, and 152 ± 22 μM for compounds 33, 52, and 61, respectively (Fig. 2b, c). Interestingly, the three molecules carry a similar scaffold (Fig. 2b). Since HSP110 has structural homologies with HSP70, we tested whether these compounds affected also HSP70 chaperone activity. As shown in Suppl Fig. 1b, in contrast to HSP110, no effect was observed for HSP70 chaperone activity, suggesting a certain specificity.

Screening of chemical compounds targeting HSP110. a Screening of foldamers library. Cut-off represents the standard deviation of all values obtained in the screening. b Structure of compounds displaying HSP110 inhibition properties. c Dose–response curves of the hits’ anti-aggregation activity (compound 33: R2 = 0.97; IC50 = 58.3 ± 1.7 µM, compound 52: R2 = 0.98, IC50 = 86.0 ± 1.5 µM, compound 61: R2 = 0.97, IC50 = 227.5 ± 1.9 µM). d Molecular docking of compound 33, 52, and 61. The three best docking positions were used as starting pose to perform molecular dynamics analysis. e Energy monitoring. Color code represents the monitoring of energy over the time of the best docking solutions. f Interaction of foldamers 33, 52, and 61 with HSP110 immobilized in a SSA biosensor by biolayer interferometry (BLI, Octet). One representative experiment is shown (n = 3)
Study of the interaction of compounds 33, 52, and 61 with HSP110
Molecular docking modeling suggests that compounds 33 and 52 bind to the same cleft within the ATP binding pocket of the HSP110 NBD (Fig. 2d). Molecular dynamics experiments revealed that this pose was stable (>20 ns, Fig. 2e), suggesting that docking might be relevant. Compound 61, although docked to the same area, was completely buried in the ATP site.
A dose-dependent HSP110 direct interaction with the foldamers 33, 52, and 61 was determined by biolayer interferometry (BLI) with an Octet device (Fig. 2f), using purified recombinant HSP110. A dose-dependent binding could also be observed when using a truncated protein carrying only the NBD of HSP110 (amino acids 1–384), demonstrating that the foldamers can bind to this domain of HSP110 (Suppl Fig. 2a, for the foldamer 33).
HSP110-targetting compounds interfere with STAT3 binding, a HSP110 client protein
STAT3 is a well-known partner of HSP110, as this chaperone binds to STAT3 thereby favoring its phosphorylation [6]. The docking of STAT3/HSP110 (Suppl. Fig. 2b), as well as our experiments of BLI (Suppl. Figure 2c) and immunoprecipitation (Fig. 3e) using HSP110 truncated proteins carrying or not the NBD (amino acids 1–384) indicate that STAT3 may bind to the NBD of HSP110.

Foldamers 33 and 52 disrupt HSP110:STAT3 interaction. a Interaction of HSP110 with STAT3-GST immobilized in antiGST-biosensor by BLI (n = 2). b Disruption of HSP110-STAT3 interaction with growing concentrations of HSP110 not biotinylated (n = 2) using alphascreen technology®. c AlphaScreen dose–response curves of the effect of the foldamers dissociating HSP110:STAT3 association (in blue, compound 33: R2 = 0.94; IC50 = 35.9 ± 1.1 µM, in red, compound 52: R2 = 0.90; IC50 = 8.5 ± 1.3 µM and in green, compound 61). d Immunoprecipitation with a STAT3 (IP: STAT3) or non-relevant antibody (IP: IgCtrl) in SW480 cells treated with the indicated compounds at 10 µM for 48 h, was followed by HSP110 immunoblotting. e Recombinant STAT3 (1 ng) was incubated with HA-tagged truncated HSP110 bearing either the NBD (1–84 amino acids) or the peptide binding domain PBD (amino acids 384–858). Immunoprecipitation of STAT3 was followed by immunoblotting for the indicated proteins. f Immunofluorescence analysis of HSP110:STAT3 association visualized by PLA in SW480 cells, treated or not with the indicated compounds at 10 µM for 96 h. Nuclei were stained with DAPI. Images were taken randomly and obtained using an Axio Imager 2. Right panel, the number of interaction foci in each cell was counted using the spot detector plugin. Statistical analysis (Mann-Whitney test) was carried out using GraphPad. ****P < 0.0001
We next studied STAT3/HSP110 interaction, in the presence or absence of compound 33, 52, or 61 (Fig. 3). STAT3/HSP110 Kd (1.5 ± 0.2 μM) was estimated through competitive BLI assays using non-biotinylated HSP110 (Fig. 3a). Using AlphaScreen® technology (Fig. 3b, c), based on an amplified luminescent proximity assay capable of detecting the proximity of two molecules, we demonstrated that, in presence of the foldamers 33 and 52, HSP110/STAT3 complex was disrupted with IC50 values of 35.9 ± 1.1 and 8.5 ± 1.3 μM, respectively, whereas foldamer 61 was unable to affect this interaction (Fig. 3c). The interference of compounds 33 and 52 with HSP110/STAT3 association, might be explained by the fact that both compounds docked at the interaction area between STAT3 and HSP110. Compound 61 was completely buried within the ATP site (Fig. 2a), which might explain why it was unable to disturb HSP110 association to STAT3.
To determine whether the HSP110-targetting compounds were also able to interfere with HSP110/STAT3 association in cells, we have used human colorectal cancer cells (SW480) and two different approaches: co-immunoprecipitation and a proximity ligation assay (PLA). Based on our toxicity tests (Suppl. Figs. 3a, b and 4), we decided to use the compounds at a 10 µM concentration, which was either not toxic or had a toxicity lower than 20% (just for compound 52 in the SW480 cells, Suppl. Fig. 3). Co-immunoprecipiation of endogenous proteins showed that HSP110 interaction with STAT3 was strongly reduced upon cell treatment with compounds 33 or 52 (Fig. 3d), whereas compound 61 had no effect. Compound 51 (a compound that did not target HSP110 in our first screening) was used here as a negative control. Interestingly, reduction of HSP110/STAT3 association observed with molecules 33 and 52 was similar to that observed upon HSP110 (shRNA) cell depletion (Fig. 3d). Similar results were obtained when immunoprecipitations were performed in vitro, using recombinant STAT3 and HSP110 truncated proteins (Fig. 3e). In agreement with our previous results, STAT3 bound to HSP110 truncated protein carrying only the 1–384 amino acid region containing the NBD, but not to the truncated protein carrying amino acids 385–858. Furthermore, molecules 33 and 52 inhibited this interaction (Fig. 3e).
We next confirmed by PLA that compounds 33 and 52 were able to interfere with the association of cellular endogenous HSP110 to STAT3. As shown in Fig. 3f, the number of HSP110/STAT3 foci strongly decreased upon cell treatment with the compounds.
Effect of the HSP110-targetting compounds 33, 52, and 61 in colorectal cancer cell proliferation
HSP110 has been reported to affect proliferation, at least in colorectal cancer cells, through its effect favoring STAT3 phosphorylation [6]. We therefore tested whether our selected compounds (33, 52, and 61) affected STAT3 phosphorylation in synchronized human colorectal cancer cells (SW480 and HCT116 cell lines). As expected, depletion of HSP110 blocked STAT3 phosphorylation (Fig. 4a). Interestingly, decrease in STAT3 phosphorylation was also observed after cell treatment with compounds 33 and 52, but not with compound 61 (Fig. 4a). This is in agreement with our previous results demonstrating that while compounds 33 and 52 interfered with HSP110 association to STAT3, compound 61 did not affect STAT3 binding to the chaperone (Fig. 3c–e).

HSP110-targetting foldamers 33 and 52 inhibit SW480 colorectal cancer cell proliferation. a Immunoblot analysis of HSP110, P-STAT3, and STAT3 in SW480 cells treated with indicated compounds at 10 µM for 48 h (compound 52 was tested at 5 and 10 µM). b Real-time cell density assay (Xcelligence) of SW480 cells either left untreated (black line) or treated with the foldamers 33 (blue line), 52 (red line), or 61 (green line) at 10 µM. c Relative quantification of cell proliferation in SW480 cells transfected with shRNA for HSP110-silencing (sh110) or shRNA control (shCtrl) and treated or not with the indicated foldamers at 10 µM for 96 h. Insert, western blot of HSP110 in the shRNA-silenced and control cells
We then determined the effect of foldamers (33, 52, and 61) in human colorectal SW480 and HCT116 cancer cell proliferation, in real time. In agreement with our previous results, treatment with foldamers 33 and 52 (but not 61) induced a decrease in cell density (measured using the xCELLigence technology), indicating lower proliferation rates in both cell lines (Fig. 4b and Suppl. Fig. 5a). To confirm the ability of these drugs to decrease cell proliferation and to test whether this effect was dependent of HSP110, we traced and quantified the number of viable cells generated in each cell division during 96 h in SW480 and HCT116 cells depleted (with both a shRNA and a siRNA approach) or not for HSP110 (Fig. 4c and Suppl. Fig. 5b). First, we found that the rate of cell proliferation inhibition obtained by treatment of control cells (Ctrl) with compounds 33 or 52 was comparable with that obtained by HSP110 depletion (sh110 or si110) (Fig. 4c, Suppl. Fig. 5b. FACS data in Suppl. Fig. 6). Second, we demonstrated that in HSP110-depleted cells, the inhibitors did not have any significant effect (Fig. 4c and Suppl. Fig. 5b). In sharp contrast, in cells transfected with a control shRNA or siRNA (and therefore expressing HSP110), compounds 33 and 52 were able to hamper cell proliferation by ~60 and 82% in SW480 cells and by ~40% in HCT116 cells. Altogether, these results strongly suggested that colorectal cancer cell growth inhibition by foldamers was HSP110-dependent.
Foldamer 33 displays an anti-tumor effect in mice bearing a colorectal cancer
Because foldamer 33, in contrast to foldamer 52, at the doses tested (10–30 µM) did not seem to display any toxicity in vitro (Suppl. Fig. 3 and Suppl. Fig. 4), we have chosen this compound for our in vivo experiments. Two different colorectal mouse models were used, a syngeneic model for which mouse colon cancer CT26 cells were injected into Balb/c mice and a NOD/SCID model, in which mice were implanted with human colorectal cancer SW480 cells (Fig. 5a, b). When tumor size reached about 0.9 mm3, mice were treated i.p. with foldamer 33 at 5 mg/kg (average concentration used for other chemical HSP inhibitors), every 3 days until the end of the experiment (determined for ethical reasons by the size of the tumor in the control group). Treatment by compound 33 induced a decrease in tumor growth of 40% and 60% in the Balb/c and NOD mice, respectively (Fig. 5a, b). This effect was consistent with the reduced expression level of Ki-67, a proliferation marker, observed on tumor slides (~40%; Fig. 5c), and with the reduced cell proliferation observed when cells were treated with foldamer 33 (~ 40% for both cells lines CT26 and SW480, Suppl. Fig. 7 and Fig. 4, respectively). In agreement with our previous results, compound 33 treatment also led to a reduction in the amount of phosphorylated STAT3 detected in tumor slides (Fig. 5d).

HSP110-targeting compound 33 induces tumor volume reduction in mice. a Tumor volume monitoring of CT26 cells in Balb/c mice control-treated (Ctrl—black lines, non-relevant foldamer) and treated with the foldamer 33 (5 mg/kg—blue lines). Animals were treated (i.p.) every 3 days. Mean volume (± SD) is represented (n = 6) (p = 0.0053). b Mean tumoral volume (±SD) of SW480 cells grown in NOD/SCID animals either treated with a non-relevant foldamer (Ctrl) or foldamer 33 (5 mg/kg, injected i.p. every 3 days, six animals per group, p = 0.0053). c Ki-67 labeling was studied by IF on dissected tumors from Balb/c mice control- and compound 33-treated. Corresponding normalized fluorescence over DAPI of Ki-67 (p = 0.0065) is shown. One representative image is shown (n = 10). d IF assay on dissected syngeneic tumors of pSTAT3 (n = 10). Scale bar = 50 µm. p = 0.0019. e Anchorage-independent growth of SW480 cells silenced (shRNA) or not for HSP110 (see Fig. 3d) and treated or not with compound 33 (10 μM). Colony number was evaluated after 2–3 weeks of culture (n = 3)
Our clonogenicity studies in colorectal cancer cells, grown in soft agar, confirmed these results. Whereas the compound strongly reduced the number of colonies in cells expressing HSP110, it hardly had any effect in HSP110 depleted cells (Fig. 5e).
No apparent adverse effects of compound 33 were observed in in vivo experimental models
We also observed within tumors that treatment with compound 33 induced a 2.5-fold increase in tumor cell death, as assessed by analyzing cleaved caspase-3 staining in the tumor slides (Fig. 6a, b, for the Balb/c and NOD model, respectively). In sharp contrast, HSP110-targeting foldamer 33 did not seem to affect normal cells survival, since no apoptosis was detected in epithelial cells from animal intestinal crypts (Fig. 6c, d), cells known to be very sensitive to toxic insults [23]. Similarly, no difference in animal weight was detected (not shown). Moreover, no difference in spleen weight in treated vs control-treated animals was observed (Fig. 6e, f).

Absence of apparent toxicity in animals treated with HSP110-targetting compound 33. a, b IF assay of cleaved caspase-3 (C3C) in dissected tumors from Balb/c (a) or NOD SCID animals (b) either treated with a non-relevant foldamer (Ctrl) or foldamer 33 (5 mg/kg, injected i.p. every 3 days). Six animals per group. p = 0.0003, Scale bar = 50 µm. c, d C3C staining in intestinal crypts of tumor-bearing Balb/c (c) and NOD/SCID (d) mice treated or not with foldamer 33 (5 mg/kg, injected i.p. every 3 days until the end of the experiment). e, f Comparison of spleen weight between ctrl- and compound 33-treated for Balb-c mice (e) and NOD/SCID (f) mice
Discussion
Since cancer cells have to re-wire their metabolism, their survival requires a high content of stress-inducible chaperones such as HSPs. This cancer cells’ dependence on HSPs is the rationale for the use of HSP inhibitors in cancer therapy. Only HSP90 and HSP27 inhibitors are available for clinical development (in phase II/III).
HSP110 has long been a forgotten chaperone and until quite recently it was just considered a member of the HSP70 family playing a role as a nucleotide exchanging factor (NEF) for HSP70 proteins. However, ours and other recent studies, demonstrate that HSP110 has a chaperone activity by its own. For its chaperone activity, HSP110’s ability to hydrolyze ATP is still under debate and many contradictory studies have been published [22, 24,25,26,27]. For Yamagishi et al., human HSP110 is unable to present any detectable ATPase activity per se [27]. Nonetheless, steady-state assays demonstrated a substantial ATPase activity for yeast HSP110 (Sse1p) [24, 25]. The differences in the ATP binding site between human HSP110 and Sse1p may be responsible for this discrepancy. However, the fact that Sse1p has been co-crystallized with ATP (and not with ADP as HSP70 members) supports the hypothesis that Sse1p does not hydrolyze ATP. The results shown here confirm this hypothesis since HSP110 NBD co-crystalized with ATP. It would be nearly impossible to obtain crystals of Sse1p or human HSP110 with ATP if the protein presented a relevant ATPase activity. Moreover, a potent ATPase activity would be contradictory with a physiological NEF function [28].
A few years ago, we reported the first HSP mutation ever found associated to cancer. Indeed, HSP110 was systematically mutated in all microsatellite instable colorectal cancers [17, 29]. This discovery was followed by other studies confirming the presence of this HSP110 inactivating mutation in other microsatellite instable cancers (gastric, endometrial) [30, 31]. Remarkably, the expression of this HSP110 loss-of-function mutation, formed at the expense of wild-type HSP110, was always associated with an excellent patients’ outcome [11, 30]. Since microsatellite instable cancers only concern around 15% of patients, an attractive hypothesis is that we can render a microsatellite stable cancer (the majority, bad prognosis), microsatellite instable-like (good prognosis) by inactivating HSP110. This hypothesis is reinforced by the fact that HSP110 wild type expression in CRC (both microsatellite instable and stable types combined) has been associated with adverse clinical outcome [8, 14, 16, 31]. As this chaperone is involved in both cancer cell proliferation and survival, the rationale to inactivate HSP110 in cancer is reinforced. However, to date, no drug targeting HSP110 was described.
Foldamers, small PPIs molecules, have already been reported in cancer therapy and several are currently evaluated in phase II clinical trials. Moreover, a phase III clinical trial for acute myeloid leukemia treatment through inhibition of MDM2-p53 interaction is presently ongoing [32]. Herein, we screened a library of foldamers and discovered two molecules (foldamers 33 and 52) that bind to HSP110 and inhibit HSP110 interaction with STAT3, thereby blocking HSP110 effect in STAT3 phosphorylation. These molecules significantly reduced cancer cell proliferation both in vitro and in vivo. STAT3 is active in ~70% of human cancers and STAT3 activation is associated with adverse clinical outcomes, particularly in CRC [33, 34]. As a consequence, the development STAT3 inhibitors remains an active area of research. However, no inhibitors have yet been approved in cancer treatment. One of the main drawbacks is that STAT3 is an essential transcriptional factor, required for many cellular functions. In consequence, its inhibition may lead to adverse effects [35, 36]. Although more studies are needed, the use of molecules as the foldamers described here, targeting instead of STAT3, its chaperone HSP110 might be a way to circumvent this problem.
The HSP110-targeting compounds selected here, inhibited HSP110 chaperone function. HSP110 has essentially two chaperone functions. A direct anti-aggregation function and an indirect chaperone function, through its role as a nuclear exchanging factor for HSP70 proteins. Therefore, HSP110 is part of a chaperone network that, as mentioned above, is essential for cancer cells’ survival (but not for normal cells). That explains the effect observed after in vivo systemic administration of the foldamer 33, which induced tumor cell death without affecting normal cell viability, such as the epithelial cells from the intestinal crypts.
We and others have found that HSP110 could be secreted and was abundant in tumor microenvironment [5, 37]. This extracellular HSP110 had an effect in macrophage polarization: it favored the development of pro-inflammatory macrophages, thereby facilitating tumor progression [5]. In this work, we demonstrated in our in vivo experiments that foldamer 33 was most probably also able to block this extracellular effect of HSP110, since this chemical HSP110-inhibitor induced intra-tumor infiltration of macrophages expressing anti-tumor (M1-like) markers (Suppl. Fig. 8). Whether this increase in cytotoxic macrophages is a consequence of extracellular HSP110 inactivation by the foldamer needs to be explored in depth.
In conclusion, in this study we successfully identified at least one potential lead inhibitor (foldamer 33) targeting the nucleotide-binding domain of HSP110. In our in vitro and in vivo models, the foldamer blocked cancer cell proliferation and induced apoptosis. Lack of weight loss and absence of cell death in intestinal crypts of the treated animals suggests that foldamer 33 does not cause adverse effects and thus warrant further development. ADMET predictions indicated that compound 33 followed the Veber rule [38] and had a 55% estimated bioavailability, suggesting that it could be a good candidate for oral administration [39]. Advantageously, this compound presented low probability to cross blood brain barrier and to be a P-glycoprotein substrate [40]. Further, our proposed lead compound was also negative for mutagenicity risks, as predicted by Ames test.
Materials and methods
NBD-HSP110 crystallization and structure determination
Human Hsp105alpha (UNIPROT ID: Q92598-1) was studied. Truncated HSP110 forms carrying either the nucleotide-binding domain (NBD-HSP110, amino acids 1–384) or the peptide binding domain (PBD-HSP110, amino acids 385–858) were generated by gene synthesis performed by Geneart©. We set-up their heterologous expression in Escherichia coli. HSP110 domains were produced and purified using a nickel-ion affinity resin and diluted at 4 mg/mL in Tris 50 mM, NaCl 300 mM, and pH 8. NBD-HSP110 crystallization was performed using the hanging drop vapor diffusion method. 30–40 µm crystals appeared after 24 h in different conditions the best being in 30% (w/v) PEG4000, Tris 100 mM, pH 8.5, and MgCl2 0.2 M. Data collection was performed at 100 K on the ID23-1 beamline [41] at the ESRF (Grenoble, France). X-ray diffraction data were integrated, scaled and merged using the XDS package suite [42]. The structure of a monomer of the Sse1p NBD (PDBID: 2QXL) was used as a search model for molecular replacement with PHASER [43]. The structure was refined with the use ARP/wARP, REFMAC, the PHENIX package suite and COOT [44,45,46,47]. Final model was validated using Polygon/Molprobity from the PHENIX package suite [45, 48]. Data reduction and refinement statistics are shown in Supplementary Table 1. The 2.0 Å of the NBD-HSP110 was deposited in the Protein Data Bank (PDBID: 6GFA). All structural figures were generated using Pymol [49].
pSTAT3 level in synchronized cells
Cells were plated at a density of 2.5 × 106 cell in T75 cm2 flask and incubated with 2 mM thymidine during 20 h, washed twice with PBS and grown 9 h in complete medium. Thereafter, cells were treated with 2 mM thymidine for 20 h. After synchronization, cells (3.0 × 105 cells/well) were treated with 10 µM of the indicated molecules or DMSO during 48 h, harvested and lysed using Cell signaling 10× lysis buffer (Cell Signaling Technology), Tris 20 mM (pH 7.5), NaCl 150 mM, Na2EDTA 1 mM, EGTA 1 mM, Triton 1%, sodium pyrophosphate 2.5 mM, beta-glycerophosphate 1 mM, Na3VO4 1 mM, and leupeptin 1 µg/mL supplemented with Roche complete protease inhibitor cocktail (Roche), PMSF (Sigma-Aldrich), and phosphatase inhibitor cocktail 2 and 3 (Sigma-Aldrich). Cellular extracts were run on western blot to assess pSTAT3 and cyclin E level.
Protein–protein interactions assays
Cells were lysed with co-immunoprecipitation buffer (0.5% (w/v) Triton X-100, 0.5% (w/v) deoxycholate, 0.05% (w/v) SDS, Tris 10 mM pH 8, NaCl 50 mM, EDTA 10 mM, Na3VO4 1 mM, pyrophosphate 30 mM, glycerophosphate 10 mM, and 0.01% (w/v) NaN3). After centrifugation, 1 mg of protein lysate was incubated with 3 µg of STAT3 antibody (dilution: 1:1000, Cell Signaling Technology, 9139S) and then incubated with 30 µL of protein A agarose beads (Merck Millipore) during 25 min at 4 °C. After extensive washing in co-IP buffer, extracts were boiled in 2× Laemmli buffer.
Alphascreen
Assay was performed according to the manufacturer’s recommendations using increasing concentration of non-biotinylated HSP110 versus 50 nM of GST-Stat-3 (abcam) and 50 nM recombinant HSP110 both coated to alphascreen beads (i.e., GST-bead for Stat-3 and biotin bead for biotinylated HSP110). All experiments were performed in duplicate in Tris 50 mM, NaCl 150 mM, pH 7.5. Fluorescence was detected at 570 nm using EnVision (Perkin Elmer).
Biolayer interferometry
For in vitro interaction assays, we used Biolayer interferometry technology (Octet Red96, Forté-Bio). Both HSP110 and NBD-HSP110 were produced using the same protocol. Proteins were biotinylated with NHS-PEG4-biotin reagent (Thermo Scientific 21330) at a ratio of protein:biotin of 1:3 and 1:2, respectively. Excess biotin was then removed (using ZebaTM Spin Desalting Columns, 7 K MWCO, 0.5 mL, Thermo Scientific B2162579). Super Streptavidin biosensors (Forté-Bio) were hydrated for 15 min at RT in running buffer (Tris 50 mM pH 8, NaCl 300 mM, 0.01% (w/v) Tween-20, and 0.1% (w/v) BSA) prior kinetic experiment. Experiment was performed in black 96-well plates with 200 µL volume/well, under constant shaking (1000 rpm) and RT temperature. Baseline was measured in running buffer (120 s), then pins were moved out in 15 µg/mL of HSP110 or NDB-HSP110 (600 s). Sensors tethered with proteins were washed in running buffer (60 s), then a second baseline was measured in running buffer (120 s). Association step was performed for 60 s in wells containing several compound concentrations diluted in running buffer with 1% DMSO. Dissociation was evaluated in running buffer (60 s). The Kd values (1:1 binding model) were calculated by the software provided by the manufacturer using double reference subtraction of sensors loaded with a control protein (here hemoglobin was used, 15 µg/mL).
Proximity ligation assay (PLA)
Cells were seeded on coverslips in 12-well plates during 48 h with DMSO (Sigma-Aldrich) or indicated molecules (10 µM, DMSO < 0.001%). After 48 h of culture, cells were fixed using 4% PBS-PFA, permeabilized using 100% chilled methanol and incubated at 4 °C with HSP110 (1:1000, Abcam, EPR4576) or STAT3 (1:1000, Cell Signaling Technology, 124H6) antibodies. Duolink® experiments were performed according to the manufacturer’s protocol (Sigma-Aldrich). Microscopy images were obtained by an Axio Imager 2 (Carl Zeiss Microscopy GmbH) and images were acquired using AxioCam MRm CCD camera (Carl Zeiss GmbH, ×63 oil objective). The Spot detector plugin of ICY software was used and statistical analysis was performed with GraphPad Prism (Unpaired Man and Whitney, at least 100 cells counted for each condition).
Anti-aggregation activity of HSP110
HSP110 anti-aggregation function was evaluated as previously described with minor modifications [26]. Human HSP110 (1.5 µM) was incubated with firefly luciferase (Sigma-Aldrich, L9506) in reaction buffer (HEPES 25 mM pH 7.6, MgCl2 5 mM, DTT 2 mM, ATP 2 mM) and heated in water bath for 30 min at 42 °C. Compounds were added in reaction buffer to achieve the indicated concentrations, with a held final concentration of 1% DMSO. The heated reaction was diluted 10-fold into buffer containing 60% of rabbit reticulocyte lysate (TnT® T7 Quick Master Mix, Promega) and incubated for 2 h RT. For luciferase activity measurement, the solution was diluted fivefold in HEPES (25 mM at pH 7.6), then 5 µL were added to 50 µL of luciferase substrate (Luciferase Assay System, Promega) prior bioluminescence reading (Envision Perkin Elmer).
In silico studies
Docking of derivatives into HSP110 crystal structure was carried out with the GOLD program. Molecular dynamics simulations were carried out using NAMD 2.12 with the all-atom CHARMM 36 forcefield for protein and CGENFF.
Cell line culture
CRC cell lines were purchased from the American Type Culture Collection (ATCC). CT26 and HCT116 cells were cultured in Roswell Park Memorial Institute 1640 medium. SW480 cells were engineering to express a control (SHC016-1EA) or a hsph1 targeting shRNA (TRCN0000275617) using Sigma-Aldrich Mission pLKO.1 hPGK-Puro-CMV-tGFP plasmids and were in cultured in high-glucose DMEM containing puromycin (ThermoFisher Scientific, 2.5 μg/mL), 10% SBF, penicillin 100 U/mL, streptomycin 100 μg/mL, and amphotericin B 0.25 μg/mL. Cells were maintained at 37 °C in 5% CO2.
Compounds
Foldamers were dissolved in DMSO (ACS-Sigma-Aldrich) and stored at −20 °C. To achieve indicated concentrations, compounds were diluted in culture medium (for cell assays) or reaction buffer (for in vitro assays). Controls with or without DMSO (maximal 0.1% for cells and 0.5% for in vitro) were carried out in each assay. The physicochemical properties and predictions of compound 33 were calculated by the web tool Swiss ADME. In vitro Ames test was performed using the QSAR ToolBox 4.1.
Cell proliferation and viability
Proliferation was determined by staining the cells with Cell Trace Violet (Invitrogen, C34557), according to the manufacturer’s procedure. HCT116 and CT-26 were plated in 24-well plates at 4 × 104 cells/well and SW480 at 6 × 104 cells/well. Cell divisions were determined during 96 h using LSRII cytometer and number of cell generations was estimated by ModFit software. To quantify the effect of HSP110 on cell proliferation rate (x), the following equation was applied:
\(x = {\sum} {A - {\sum} B }\) where A is the percentage of cells SW480shRNAHSP110 or HCT116siRNAHSP110 increased in early generations and B is the percentage of cells SW480shRNAControl or HCT116siRNAControl present in generations identified in A. Dead cells were excluded from analysis by AnnexinV-FITC labelling (BD Pharmigen, 556419) and 7-AAD (eBioscience, 00-6993-50). Cell viability quantification was performed with FlowJo software.
Real-time proliferation
Real-time proliferation was monitored using xCELLigence RTCA DP (ACEA Bioscience), which measures electrical impedance of attached cells on E-Plate view 16 PET (300600890). HCT116 and SW480 (shRNA engineered) were seeded at 2.5 × 103 cells/well and CT26 at 6 × 103 cells/well. Cells were maintained at 37 °C in 5% CO2 and impedance was measured every 10 min for the first 6 h and every 15 min during the subsequent 166 h.
Tumor growth analysis in vivo
CT26 (1 × 106) or HCT116 cells (10 × 106) were injected s.c. into the right flank of BALB/c or NOD/SCID female mice, respectively (Charles River Laboratories). Mice were intraperitoneally treated with compound 33 at 5 mg/kg or a control vehicle (BALB/c n = 10/group, NOD SCID n = 6/group) twice a week. Mice were euthanized when tumors reached 2000 mm3 and tumor IHC staining was performed.
Soft agar colony formation
Colorectal cancer SW480 cells, depleted or not for HSP110, were cultured, in the presence of absence of molecule 33 (20 μM) in a six-well dish (10,000 cells/well) in 0.45% agarose in growth media, layered on top of 0.75% agarose growth media. Colonies were counted under a light microscope 2–3 weeks post plating. For each experiment, cells were seeded in triplicate and three fields per well were quantified.
References
Factsheet of WHO 2012 on cancer available here: http://www.cancerresearchuk.org/sites/default/files/cs_report_world.pdf
Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 blockade in tumors with mismatch-repair deficiency. New Engl J Med. 2015;372:2509–20.
Chung KY, Gore I, Fong L, Venook A, Beck SB, Dorazio P, et al. Phase II study of the anti-cytotoxic T-lymphocyte-associated antigen 4 monoclonal antibody, tremelimumab, in patients with refractory metastatic colorectal cancer. J Clin Oncol. 2010;28:3485–90.
Champiat S, Dercle L, Ammari S, Massard C, Hollebecque A, Postel-Vinay S, et al. Hyperprogressive disease is a new pattern of progression in cancer patients treated by anti-PD-1/PD-L1. Clin Cancer Res. 2017;23:1920–8.
Berthenet K, Boudesco C, Collura A, Svrcek M, Richaud S, Hammann A, et al. Extracellular HSP110 skews macrophage polarization in colorectal cancer. Oncoimmunology. 2016;5:e1170264.
Berthenet K, Bokhari A, Lagrange A, Marcion G, Boudesco C, Causse S, et al. HSP110 promotes colorectal cancer growth through STAT3 activation. Oncogene. 2017;36:2328–36.
Guttmann DM, Koumenis C. The heat shock proteins as targets for radiosensitization and chemosensitization in cancer. Cancer Biol Ther. 2011;12:1023–31.
Calderwood SK, Khaleque MA, Sawyer DB, Ciocca DR. Heat shock proteins in cancer: chaperones of tumorigenesis. Trends Biochem Sci. 2006;31:164–72.
Schmitt E, Maingret L, Puig PE, Rerole AL, Ghiringhelli F, Hammann A, et al. Heat shock protein 70 neutralization exerts potent antitumor effects in animal models of colon cancer and melanoma. Cancer Res. 2006;66:4191–7.
Bonniaud P, Burgy O, Garrido C. Heat shock protein-90 toward theranostics: a breath of fresh air in idiopathic pulmonary fibrosis. Eur Respir J. 2018;51:1702612.
Collura A, Lagrange A, Svrcek M, Marisa L, Buhard O, Guilloux A, et al. Patients with colorectal tumors with microsatellite instability and large deletions in HSP110 T17 have improved response to 5-fluorouracil-based chemotherapy. Gastroenterology. 2014;146:401–11 e401.
Rampelt H, Kirstein-Miles J, Nillegoda NB, Chi K, Scholz SR, Morimoto RI, et al. Metazoan Hsp70 machines use Hsp110 to power protein disaggregation. EMBO J. 2012;31:4221–35.
Mattoo RU, Sharma SK, Priya S, Finka A, Goloubinoff P. Hsp110 is a bona fide chaperone using ATP to unfold stable misfolded polypeptides and reciprocally collaborate with Hsp70 to solubilize protein aggregates. J Biol Chem. 2013;288:21399–411.
Slaby O, Sobkova K, Svoboda M, Garajova I, Fabian P, Hrstka R, et al. Significant overexpression of Hsp110 gene during colorectal cancer progression. Oncol Rep. 2009;21:1235–41.
Zappasodi R, Ruggiero G, Guarnotta C, Tortoreto M, Tringali C, Cavane A, et al. HSPH1 inhibition downregulates Bcl-6 and c-Myc and hampers the growth of human aggressive B-cell non-Hodgkin lymphoma. Blood. 2015;125:1768–71.
Hwang TS, Han HS, Choi HK, Lee YJ, Kim YJ, Han MY, et al. Differential, stage-dependent expression of Hsp70, Hsp110 and Bcl-2 in colorectal cancer. J Gastroenterol Hepatol. 2003;18:690–700.
Dorard C, de Thonel A, Collura A, Marisa L, Svrcek M, Lagrange A, et al. Expression of a mutant HSP110 sensitizes colorectal cancer cells to chemotherapy and improves disease prognosis. Nat Med. 2011;17:1283–9.
Yu N, Kakunda M, Pham V, Lill JR, Du P, Wongchenko M, et al. HSP105 recruits protein phosphatase 2A to dephosphorylate beta-catenin. Mol Cell Biol. 2015;35:1390–1400.
Flaherty KM, DeLuca-Flaherty C, McKay DB. Three-dimensional structure of the ATPase fragment of a 70K heat-shock cognate protein. Nature. 1990;346:623–8.
Liu Q, Hendrickson WA. Insights into Hsp70 chaperone activity from a crystal structure of the yeast Hsp110 Sse1. Cell. 2007;131:106–20.
Santos JS, Voisin-Chiret AS, Burzicki G, Sebaoun L, Sebban M, Lohier JF, et al. Structural characterizations of oligopyridyl foldamers, alpha-helix mimetics. J Chem Inf Model. 2012;52:429–39.
Raviol H, Sadlish H, Rodriguez F, Mayer MP, Bukau B. Chaperone network in the yeast cytosol: Hsp110 is revealed as an Hsp70 nucleotide exchange factor. EMBO J. 2006;25:2510–8.
Joly AL, Deepti A, Seignez A, Goloudina A, Hebrard S, Schmitt E, et al. The HSP90 inhibitor, 17AAG, protects the intestinal stem cell niche and inhibits graft versus host disease development. Oncogene. 2016;35:2948.
Dragovic Z, Broadley SA, Shomura Y, Bracher A, Hartl FU. Molecular chaperones of the Hsp110 family act as nucleotide exchange factors of Hsp70s. EMBO J. 2006;25:2519–28.
Raviol H, Bukau B, Mayer MP. Human and yeast Hsp110 chaperones exhibit functional differences. FEBS Lett. 2006;580:168–74.
Oh HJ, Easton D, Murawski M, Kaneko Y, Subjeck JR. The chaperoning activity ofhsp110. Identification of functional domains by use of targeted deletions. J Biol Chem. 1999;274:15712–8.
Yamagishi N, Ishihara K, Hatayama T. Hsp105alpha suppresses Hsc70 chaperone activity by inhibiting Hsc70 ATPase activity. J Biol Chem. 2004;279:41727–33.
Andreasson C, Fiaux J, Rampelt H, Mayer MP, Bukau B. Hsp110 is a nucleotide-activated exchange factor for Hsp70. J Biol Chem. 2008;283:8877–84.
Buhard O, Lagrange A, Guilloux A, Colas C, Chouchene M, Wanherdrick K, et al. HSP110 T17 simplifies and improves the microsatellite instability testing in patients with colorectal cancer. J Med Genet. 2016;53:377–84.
Kimura A, Ogata K, Altan B, Yokobori T, Ide M, Mochiki E, et al. Nuclear heat shock protein 110 expression is associated with poor prognosis and chemotherapy resistance in gastric cancer. Oncotarget. 2016;7:18415–23.
Kimura A, Ogata K, Altan B, Yokobori T, Mochiki E, Yanai M, et al. Nuclear heat shock protein 110 expression is associated with poor prognosis and hyperthermo-chemotherapy resistance in gastric cancer patients with peritoneal metastasis. World J Gastroenterol. 2017;23:7541–50.
Scott DE, Bayly AR, Abell C, Skidmore J. Small molecules, big targets: drug discovery faces the protein-protein interaction challenge. Nat Rev Drug Discov. 2016;15:533–50.
Morikawa T, Baba Y, Yamauchi M, Kuchiba A, Nosho K, Shima K, et al. STAT3 expression, molecular features, inflammation patterns, and prognosis in a database of 724 colorectal cancers. Clin Cancer Res. 2011;17:1452–62.
Huang W, Dong Z, Chen Y, Wang F, Wang CJ, Peng H, et al. Small-molecule inhibitors targeting the DNA-binding domain of STAT3 suppress tumor growth, metastasis and STAT3 target gene expression in vivo. Oncogene. 2016;35:802.
Welte T, Zhang SS, Wang T, Zhang Z, Hesslein DG, Yin Z, et al. STAT3 deletion during hematopoiesis causes Crohn’s disease-like pathogenesis and lethality: a critical role of STAT3 in innate immunity. Proc Natl Acad Sci USA. 2003;100:1879–84.
Mantel C, Messina-Graham S, Moh A, Cooper S, Hangoc G, Fu XY, et al. Mouse hematopoietic cell-targeted STAT3 deletion: stem/progenitor cell defects, mitochondrial dysfunction, ROS overproduction, and a rapid aging-like phenotype. Blood. 2012;120:2589–99.
Shen Y, Guo D, Weng L, Wang S, Ma Z, Yang Y, et al. Tumor-derived exosomes educate dendritic cells to promote tumor metastasis via HSP72/HSP105-TLR2/TLR4 pathway. Oncoimmunology. 2017;6:e1362527.
Veber DF, Johnson SR, Cheng HY, Smith BR, Ward KW, Kopple KD. Molecular properties that influence the oral bioavailability of drug candidates. J Med Chem. 2002;45:2615–23.
Martin YC. A bioavailability score. J Med Chem. 2005;48:3164–70.
Daina A, Zoete V. A boiled-egg to predict gastrointestinal absorption and brain penetration of small molecules. ChemMedChem. 2016;11:1117–21.
Nurizzo D, Mairs T, Guijarro M, Rey V, Meyer J, Fajardo P, et al. The ID23-1 structural biology beamline at the ESRF. J Synchrotron Radiat. 2006;13(Pt 3):227–38.
Kabsch W. Xds. Acta Crystallogr Sect D, Biol Crystallogr. 2010;66(Pt 2):125–32.
McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40(Pt 4):658–74.
Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr Sect D, Biol Crystallogr. 2004;60(Pt 12 Pt 1):2126–32.
Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr Sect D, Biol Crystallogr. 2010;66(Pt 2):213–21.
Perrakis A, Harkiolaki M, Wilson KS, Lamzin VS. ARP/wARP and molecular replacement. Acta Crystallogr Sect D, Biol Crystallogr. 2001;57(Pt 10):1445–50.
Murshudov GN, Skubak P, Lebedev AA, Pannu NS, Steiner RA, Nicholls RA, et al. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr Sect D, Biol Crystallogr. 2011;67(Pt 4):355–67.
Chen VB, Arendall WB 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr Sect D, Biol Crystallogr. 2010;66(Pt 1):12–21.
DeLano WL. The PyMOL Molecular Graphics System, Version 2.0. Schrödinger, LLC. http://www.pymol.org.
Ashkenazy H, Abadi S, Martz E, Chay O, Mayrose I, Pupko T, et al. ConSurf 2016: an improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res. 2016;44(W1):W344–350.
Acknowledgements
The authors thank R. Seigneuric for helpful discussions and acknowledge ESRF for access to beamlines via its in-house research program. We thank also I. Gregoire for English revision of the manuscript. This work has been supported by the “Fondation de France” (grant n° Engt: 00057934), the “Labex” LipSTIC, “la ligue national contre le cancer”, the “initiative d’excellence ISITE”, the Burgundy Council, the FEDER and MATWIN. The authors thank Cellimap and the cytometry platform. Part of this work was performed using computing resources of CRIANN (Normandy, France).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by S. Fulda
Rights and permissions
About this article

Cite this article
Gozzi, G.J., Gonzalez, D., Boudesco, C. et al. Selecting the first chemical molecule inhibitor of HSP110 for colorectal cancer therapy. Cell Death Differ 27, 117–129 (2020). https://doi.org/10.1038/s41418-019-0343-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41418-019-0343-4
This article is cited by
-
Inhibition of Hsp110-STAT3 interaction in endothelial cells alleviates vascular remodeling in hypoxic pulmonary arterial Hypertension model
Respiratory Research (2023)
-
A first-in-class inhibitor of Hsp110 molecular chaperones of pathogenic fungi
Nature Communications (2023)
-
Purification and biochemical characterization of Msi3, an essential Hsp110 molecular chaperone in Candida albicans
Cell Stress and Chaperones (2021)
-
Lactobacillus stress protein GroEL prevents colonic inflammation
Journal of Gastroenterology (2021)
-
Heat-shock proteins: chaperoning DNA repair
Oncogene (2020)
