
Abstract
Chimeric antigen receptor (CAR)-engineered T-cell (CAR-T) therapy has demonstrated impressive therapeutic efficacy against hematological malignancies, but multiple challenges have hindered its application, particularly for the eradication of solid tumors. Innate killer cells (IKCs), particularly NK cells, NKT cells, and γδ T cells, employ specific antigen-independent innate tumor recognition and cytotoxic mechanisms that simultaneously display high antitumor efficacy and prevent tumor escape caused by antigen loss or modulation. IKCs are associated with a low risk of developing GVHD, thus offering new opportunities for allogeneic “off-the-shelf” cellular therapeutic products. The unique innate features, wide tumor recognition range, and potent antitumor functions of IKCs make them potentially excellent candidates for cancer immunotherapy, particularly serving as platforms for CAR development. In this review, we first provide a brief summary of the challenges hampering CAR-T-cell therapy applications and then discuss the latest CAR-NK-cell research, covering the advantages, applications, and clinical translation of CAR- and NK-cell receptor (NKR)-engineered IKCs. Advances in synthetic biology and the development of novel genetic engineering techniques, such as gene-editing and cellular reprogramming, will enable the further optimization of IKC-based anticancer therapies.
Similar content being viewed by others
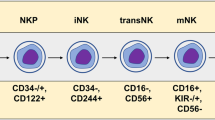

Introduction
Cancer immunotherapy is becoming an increasing promising modality. Adoptive cell immunotherapy involves the transfer of ex vivo-expanded and activated immune effector cells, including tumor-infiltrating lymphocytes (TILs), cytotoxic T cells (CTLs), natural killer (NK) cells, and natural killer T (NKT) cells, to a recipient for the eradication of tumor cells [1, 2]. With recent advances in immunobiology, synthetic biology, and bioengineering, the genetic manipulation of immune cells has become an innovative solution for the design of effective cancer immunotherapies [3,4,5]. In particular, the adoptive transfer of chimeric antigen receptor (CAR)-engineered T (CAR-T) cells has shown impressive therapeutic efficacy in patients with relapsed and refractory B-cell lymphoma and leukemia, demonstrating high remission rates, and great promise in the treatment of other hematological malignancies [6, 7]. However, the success of CAR-T-cell therapy is still limited to patients with hematological malignancies, remaining ineffective for those with solid tumors. The major obstacles interfering with the clinical efficacy of CAR-T-cell therapy include cytokine release syndrome (CRS), neurotoxicity, graft-versus-host disease (GVHD), “on-target, off-tumor” toxicities, poor homing, and trafficking into solid tumor sites, and the immunosuppressive qualities of the tumor microenvironment (TME). A wide range of gene modification strategies have been exploited to improve the safety and efficacy and broaden the application of CAR-T-cell therapy to other disease settings, particularly solid tumors [8, 9]. The use of CAR-T cells in combination with other therapeutic approaches also promises to enhance the antitumor efficacy and may be used to overcome resistance to treatment. In addition to T cells, the CAR construct can be engineered into other immune effector cells. Innate killer cells (IKCs), such as NK cells, NKT cells, and γδ T cells, have unique biological features, including a wide range of tumor recognition mechanisms, potent antitumor capacity, and potentially improved safety in clinical settings [10, 11]. In this review, we first provide a brief summary of the challenges in CAR-T-cell therapy application and then offer an overview of the current research involving IKCs, discussing the advantages and clinical translation of CAR- and NK-cell receptor (NKR)-engineered IKCs for use in cancer immunotherapy.
Challenges in CAR-T cell research
CARs are genetically engineered receptors that recognize specific surface antigens and subsequently activate downstream signaling pathways to elicit the T-cell-mediated killing of target cells. The CAR construct structure usually comprises a single-chain variable fragment (scFv) extracellular tumor antigen recognition domain, a hinge region, and transmembrane and intracellular signaling domains. The intracellular domain usually contains a CD3ζ signaling domain and a CD28 and/or 4-1BB costimulatory domain, which are required to activate T cells, resulting in proliferation, cytokine secretion, and/or T-cell-mediated cytotoxicity. To date, CAR-T-cell therapy has been used with great success in the treatment of B-cell hematological cancers, particularly relapsed or refractory B-cell acute lymphoblastic leukemia (ALL), with a complete remission (CR) rate of 80–90% and long-lasting CR in the majority of ALL patients [12, 13]. CAR-T-cell therapy targeting the B-cell maturation antigen (BCMA) in patients with multiple myeloma has also showed promising clinical responses and a good safety record [14, 15]. However, there are still multiple hurdles to overcome before widespread application of CAR-T therapy can become the norm. The most serious life-threatening toxicity associated with CAR-T-cell therapy is CRS triggered by high levels of proinflammatory cytokines (such as tumor necrosis factor (TNF)-α, interleukin (IL-1)β, IL-6, and granulocyte-macrophage colony-stimulating factor (GM-CSF)) secreted by infused CAR-T cells and other activated immune cells [16]. CRS may lead to fever, nausea, fatigue, vascular leakage, multiple organ failure, and even death. Elevated levels of cytokines and T-cell-mediated inflammation can cause neurotoxicity, which has been observed in several clinical trials [17]. Since the majority of tumor antigens are tumor-associated antigens (TAAs) expressed on both tumor and healthy tissues, CAR-T cells often attack and destroy normal cells, defined as “on-target off-tumor” toxicity. Transfusion of allogeneic CAR-T cells can thus lead to the development of GVHD. The diverse antigen heterogeneity of solid tumors hampers the recognition of tumor cells by T cells and impairs the efficacy of CAR-T therapy. Antigen downregulation and loss are two common mechanisms employed by tumors to escape T-cell immunosurveillance, leading to CAR-T-cell therapeutic resistance [18].
The major obstacles limiting CAR-T therapeutic efficacy against solid tumors include the immunosuppressive microenvironment and the poor capacity of CAR-T cells to migrate to solid tumor sites. Aberrant vasculature hinders T-cell trafficking from blood vessels; low levels of chemokines in the immediate tumor environment and poor chemokine receptor expression on CAR-T cells impede CAR-T-cell migration. Furthermore, the existence of the extracellular matrix (ECM) barrier around solid tumors and the thick collagen fiber network surrounding some tumor islets impede CAR-T cells from entering tumor sites [19]. More importantly, the potency of CAR-T cells that are able to infiltrate into tumor beds is attenuated by the immunosuppressive TME, interfering with CAR-T-cell effector functions, rendering them dysfunctional, exhausted, and unable to kill tumor cells [20]. High frequencies of regulatory T cells (Tregs), myeloid-derived suppressor cells (MDSCs), tumor-associated macrophages (TAMs), neutrophils, and multiple immunosuppressive factors (particularly transforming growth factor (TGF)-β, IL-10, IL-4, prostaglandin E2 (PGE2), indoleamine 2,3-dioxygenase (IDO), and adenosine) impair the activation, survival, proliferation, and effector function of CAR-T cells through a variety of mechanisms [21, 22]. Blocking inhibitory factors in the TME, for example, by using an IDO inhibitor or adenosine receptor antagonist or using a dominant-negative (DN) TGF-β receptor, has been shown to restore CAR-T-cell activity [23,24,25]. Tumor cells, stromal cells, and certain immunosuppressive cells, which express high levels of immune checkpoint molecules such as programmed-death ligand 1 (PD-L1), induce the dysfunction of CAR-T cells, thereby reducing their antitumor activity [26]. Using combination therapy with checkpoint inhibitors (e.g., anti-PD-1 or anti-PD-L1 antibodies), engineering CAR-T cells to secrete antagonistic anti-PD-1 or anti-PD-L1 antibodies, or disrupting the PD-1 pathway (e.g., by transducing a DN PD-1 receptor or PD-1 switch receptor) have enhanced the antitumor efficacy of CAR-T cells and prolonged their survival in several preclinical models [27,28,29,30]. With the advent of novel synthetic biology and genome-editing techniques, researchers are increasingly focusing on the development of innovative strategies to overcome the hurdles encountered in clinical practice through the optimization of engineered CAR design to unleash the full antitumor potential of CAR-T cells while limiting potential side effects [31,32,33,34,35].
CAR- and NKR-engineered innate killer cells for use in cancer immunotherapy
Innate lymphocytes constitute a heterogeneous group of cells that includes innate lymphoid cells (ILCs, comprising NK, ILC1, ILC2, and ILC3 cells) and innate-like T lymphocytes (such as NKT cells, γδ T cells, and mucosal-associated invariant T cells (MAITs)) [11, 36, 37]. Among these innate lymphocytes, NK cells, NKT cells, and γδ T cells have a direct cytolytic function and display potent antitumor efficacy with features that differ from those of conventional αβ T cells. They usually have specific cytolytic mechanisms and unique tissue distribution and safety profiles. Most innate lymphocytes express innate recognition receptors, such as NKG2D, thus exerting cytotoxicity independent of T-cell receptor (TCR) recognition. In addition, IKCs have a distinct cytokine profile, indicating that these cells are less likely to induce CRS. High antitumor potency of IKCs has been observed in both preclinical and clinical investigations [36, 38, 39]. Owing to the unique biological features and potent antitumor capacity of innate lymphocytes, as well as the shortcomings of CAR-T-cell therapy in clinical application, IKCs (particularly NK cells, NKT cells, and γδ T cells) have recently gained much attention as alternative candidates for CAR-mediated therapy [40]. For instance, IKCs have been engineered to express NK-cell receptors, giving rise to NKR-engineered IKCs with innate recognition features. CAR- and NKR-engineered IKCs can overcome multiple hurdles that prevent the application of conventional CAR-T cells in the clinic. For instance, innate lymphocytes can kill target cells in an antigen- and major histocompatibility complex (MHC)-independent manner, thus avoiding tumor escape due to antigen loss. Importantly, compared to conventional CAR-T cells, IKCs are associated with a low risk of GVHD, making them suitable for use as allogeneic ‘off-the-shelf’ cellular therapeutic agents. Preclinical studies and clinical trials have shown promising results when CAR-engineered NK cells and CAR-engineered γδ T cells have been used for the treatment of both hematological cancers and solid tumors [41].
CAR- and NKR-engineered NK cells for use in cancer immunotherapy
Advantages of CAR- and NKR-modified NK cells
NK cells are important innate cytotoxic effectors specializing in the first line of defense against invading pathogens and tumors without requiring prior sensitization or MHC compatibility. NK cells are currently regarded as ILC members and are also referred to as cytotoxic ILCs [42]. As professional IKCs, NK cells have characteristics distinct from conventional T cells. For instance, they do not express a TCR or CD3, instead recognizing malignant cells and pathogens using NKRs independent of MHC restriction. NK-cell function is tightly regulated by multiple signals that originate from an array of germline-encoded activating and inhibitory receptors, which recognize specific ligands present on the surface of target cells [43]. In addition, NK cells can kill target cells via antibody-dependent cellular cytotoxicity (ADCC), which is triggered by CD16 (FcγRIII)-mediated recognition of antibody (Ab)-coated target cells.
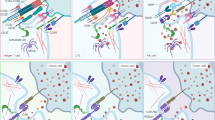
NK cells may be the most promising candidate for CAR- or NKR-based therapies because of their superior safety profile and efficacy [44, 45,]. Similar to CAR-T cells, CAR-engineered NK cells target TAAs expressed on tumor cells via their extracellular scFv fragment. NKR-modified NK cells are engineered to overexpress NK-cell receptors not TAA-targeting scFvs to target cancer cells expressing NK-cell receptor ligands. The ligands critical for the activation of NKRs, such as NKG2D, NKp30, and DNAX accessory molecule 1 (DNAM-1), are widely expressed on most solid tumors and hematological malignancies but are not present on normal cells. Thus, these ‘activating’ NKR-engineered NK cells can target a wide spectrum of tumor types as “universal NKR” effectors. In contrast, ‘inhibitory’ NKR-modified NK cells can target tumors that express high levels inhibitory ligands such as PD-L1. For instance, the modification of NK cells with a truncated DN inhibitory receptor (e.g., DN PD-1) lacking its intracellular signaling domains can render NK cells resistant to checkpoint receptor-induced cell exhaustion. Furthermore, the PD-1 switch receptor contains an extracellular PD-1 domain and intracellular costimulatory domains; therefore, engineered NK cells can switch PD-L1-mediated inhibitory signals into activating signals, reversing TME-mediated immunosuppression. The various features of CAR- and NKR-engineered NK cells are shown in Fig. 1.

Through their extracellular scFv domain, CAR- and NKR-engineered NK cells. CAR-engineered NK cells specifically target TAAs expressed on tumor cells. They exert cytolytic activity against tumor cells in both a CAR-dependent and CAR-independent (through NKR recognition of NKR ligands and through CD16-mediated ADCC effect) manner. NKR-engineered NK cells can target a wide spectrum of tumor types with high expression of NKR ligands through NKRs. Activated receptor-modified NKR-NK cells (such as NKG2D-, NKp30- or DNAM-1-engineered NK cells) exhibit direct cytotoxicity against tumors with high expression of NKR ligands. Inhibitory NKR-modified NK cells can target tumors that express high levels of corresponding inhibitory ligands such as PD-L1. Modification with a truncated DN inhibitory receptor (e.g., DN PD-1), lacking its intracellular signaling domains, can resist checkpoint receptor-induced cell exhaustion. Inhibitory switch receptors containing an extracellular domain of inhibitory receptors (e.g., PD-1) and intracellular activating costimulatory domains (e.g., CD28) in engineered NK cells enable checkpoint receptor-mediated inhibitory signals to be switched to activating signals, thus reversing TME-mediated immunosuppression. The potential of inhibitory switch receptors, including PD-1, TIGIT, and NKG2A, used alone or in combination with other strategies for the treatment of solid tumors is under investigation
CAR- or NKR-engineered NK cells present several advantages over CAR-T cells [46, 47]. For example, allogeneic NK cells can easily cross the human leukocyte antigen barrier without inducing GVHD. Moreover, the triggering of NK-cell alloreactivity, referred to as the graft-versus-tumor (GVT) effect, can eliminate leukemia and induce remission in acute myeloid leukemia. In addition, NK cells can inhibit alloreactive T-cell-mediated GVHD by direct lysis of activated alloreactive T cells or the elimination of host antigen-presenting cells (APCs) [48, 49]. The shorter lifespan and a cytokine profile that is different than that of T cells render NK cells much safer and more applicable to cancer immunotherapy. Activated NK cells mainly produce interferon (IFN)-γ and GM-CSF but seldom secrete high levels of proinflammatory cytokines [50, 51], such as TNF-α, IL-6, or IL-1β, which are the major inducers of CRS during CAR-T-cell therapy. Thus, both CAR-NK and NKR-NK cells present a low risk of inducing severe adverse effects, including CRS, and are therefore much safer than CAR-T cells for use in the clinic. Preclinical studies and clinical trials have demonstrated the safety and feasibility of CAR- and NKR-engineered NK cells in the treatment of both hematological malignancies and solid tumors [52,53,54]. CAR-NK cells exert cytotoxicity via several distinct mechanisms. In addition to killing tumor cells by recognizing tumor antigens through the scFv portion of the CAR structure, CAR-NK cells have the intrinsic ability to recognize and target tumor cells through their natural activating receptors (including NKG2D, NKp46, NKp44, NKp30, and DNAM-1). These activating NK-cell receptors recognize stress-induced ligands on the surface of tumors but not on normal cells. The dominant activating signals arising from specific receptor/ligand engagement override any tumor-mediated inhibitory signals, leading to NK-cell activation and potent tumor elimination through the release of perforin and granzyme or via death-receptor pathways. NK cells can also kill tumor cells through CD16-mediated ADCC. Therefore, CAR-engineered NK cells can eradicate tumor cells by employing CAR-dependent and CAR-independent mechanisms. In addition, NK cells are able to induce tumor cell lysis in tumors on the basis of the “missing self” hypothesis, whereby cells undergo a loss in MHC expression. Antigenic downregulation represents the most common mechanism employed by tumors to evade T-cell recognition. Therefore, CAR- and NKR-engineered NK cells have the ability to efficiently eradicate tumor cells even when MHC expression is downregulated or TAAs are lost/mutated in tumors, which typically leads to CAR-T-cell therapy resistance. Importantly, this effect may potentially reduce the risk of relapse due to CAR target-specific tumor antigen loss. Finally, CAR- and NKR-engineered NK cells can be generated from a variety of sources, including autologous or allogeneic peripheral blood, umbilical cord blood (UCB), hematopoietic stem/progenitor cells, induced pluripotent stem cells (iPSCs), human embryonic stem cells, memory-like NK cells, and the NK-92 cell line [55]. Of these cell lines, iPSCs and UCB have been recently proven to be the most promising sources of CAR- and NKR-NK cells. The safety and efficacy of anti-CD19 CAR-engineered UCB-NK cells in the first-in-human phase I/II clinical trial treating patients with relapsed/refractory CD19+ non-Hodgkin’s lymphoma (NHL) or chronic lymphocytic leukemia (CLL) were recently revealed (NCT03056339). The highly encouraging results showed that 7/11 enrolled patients achieved CR with no evidence of neurotoxicity, CRS, or GVHD [56]. Induced PSCs can be reprogrammed and undergo unlimited ex vivo expansion without losing pluripotency [57], thereby serving as ideal sources of readily expanded “off-the-shelf” CAR- and NKR-engineered NK cells suitable for clinical-scale cancer therapy [58, 59]. Induced PSC-derived anti-mesothelin CAR-NK cells have demonstrated enhanced antitumor activity, prolonged survival, and reduced toxicity compared to conventional anti-mesothelin CAR-T cells in a mouse xenograft ovarian cancer model [60]. The safety and validity of UCB- and iPSC-derived NK cells are currently under investigation in several clinical trials (NCT03056339, NCT03579927, and NCT04245722).
Genetic engineering strategies based on NK-cell activation mechanisms
The majority of early CAR constructs used in NK-cell engineering were based on the design strategy originally developed for producing CAR-T cells. In these early constructs, the scFv extracellular domain, transmembrane region, and intracellular signaling domains (identical to those in CAR-T cells) were simply “transplanted” into NK cells. Despite the efficacy of some of these engineered CAR-T NK cells, they may not represent an optimized strategy for the design of therapeutic NK cells.
Receptor recognition and intracellular signal transduction are crucial for the formation of immunological synapses and subsequent immune cell activation. These processes also determine the cell state, including exhaustion, memory, and effector function of immune cells, including CAR- and NKR-engineered effector cells [61, 62]. NK-cell-specific signaling pathways are closely related to but distinct from those in T cells [63]. Although signal domains such as CD3ζ and 4-1BB are shared by T and NK cells, some costimulatory domains (e.g., CD28) and the CD8α transmembrane domain are absent from NK cells. To enhance the activation and cytolytic capacities of NK cells, it is therefore particularly important to design optimized CAR or NKR constructs based on NK-cell-specific signal activation features [55]. The ability of activating NK-cell receptors to transduce activating signals depends on their intracellular adaptor molecules. DNAX-activating protein 10 (DAP10) and DAP12 are NK-cell-specific adaptor molecules containing immunoreceptor tyrosine-based activation motifs (ITAMs). DAP10, as the adapter protein of the NKG2D receptor, can promote and stabilize the membrane expression of NKG2D. The phosphorylation of DAP10 recruits additional downstream signaling molecules and, ultimately, triggers the activation and cytotoxicity of NK cells [64]. DAP12 is an adapter for NKp44, NKG2C, and activating killer immunoglobulin receptors (KIRs). 2B4 is a member of the signaling lymphocytic activation molecule (SLAM) receptor family. Upon engagement with its ligand, CD48, 2B4 recruits the adaptor molecule SLAM-associated protein through its immunoreceptor tyrosine-based switch motif (ITSM), mediating signal transduction [65, 66]. Considering this evidence, researchers have increasingly designed CAR or NKR constructs containing NK-cell-specific costimulatory domains, including DAP10, DAP12, and 2B4, with or without the conventional CD3ζ domain. An NKG2D-engineered NKR construct containing the NKG2D ectodomain and intracellular DAP10 and CD3ζ costimulatory domains has been shown to considerably enhance the activation and antitumor capacity of primary NK cells in an osteosarcoma xenograft mouse model [67]. However, another study testing anti-CD19 CAR constructs containing the intracellular CD3ζ portion or DAP10 showed that the anti-CD19-ζ CAR triggered higher NK-cell-mediated cytotoxicity than the anti-CD19-DAP10 CAR [68]. It is likely that the optimization of CAR- and NKR-mediated NK-cell activation may require the use of different combinations of extracellular, transmembrane, and intracellular signaling domains. Other researchers demonstrated that DAP12 was superior to CD3ζ as an intracellular domain for NKG2D-engineered primary NK cells. In this study, anti-prostate stem cell Ag (PSCA) CAR-NK cells containing DAP12 exhibited enhanced cytotoxicity, cytokine secretion, and more efficient tumor eradication in a mouse model [69, 70]. In addition, anti-CD5 CAR-NK cells engineered with a construct containing the 2B4 intracellular domain were reported to have superior antitumor capacity compared to NK cells engineered using a 4-1BB-containing construct [71]. In a recent study comparing the contribution of intracellular costimulatory domains of CD28, DNAM-1, and 2B4 to CAR-NK-cell activation, it was reported that anti-glypican-3 (GPC3) CAR-NK-92 cells engineered with a construct containing NK-cell-associated DNAM-1 and/or 2B4 costimulatory domains displayed enhanced proliferative capacity, persistence, and cytotoxicity against hepatocellular carcinoma (HCC) cells compared with NK-92 cells transduced with the CD28-containing CAR construct. CAR-NK-92 cells containing both the DNAM-1 and 2B4 costimulatory domains exhibited the highest cytolytic activity [72]. An optimized NK-specific CAR construct strategy was adopted by Kaufman and colleagues, who screened 9 different CAR constructs containing various combinations of activating NK receptor transmembrane domains (NKG2D, NKp44, NKp46, and CD16) and intracellular costimulatory domains (DAP10, DAP12, 2B4, 4-1BB, and CD3ζ) in NK-92 cells. These researchers found that the optimal construct comprised an NKG2D transmembrane domain, a 2B4 costimulatory element, and a CD3ζ signaling domain (NKG2D-2B4ζ); this construct exhibited antitumor potency via the effective stimulation of antigen-specific NK-cell activating signaling pathways, including the phospholipase C (PLC)-γ, Syk-vav1-Erk, and nuclear factor-k-gene binding (NF-kB) pathways. NKG2D-2B4ζ-engineered iPSC-derived anti-mesothelin NK cells displayed greater antitumor activity than traditional CAR-T construct-engineered iPSC-derived CAR-NK cells. NK-cell-specific iPSC-CAR-NK cells also exhibited effective tumor growth inhibition and prolonged survival and inducing less side effects in an ovarian cancer xenograft model [60]. Notably, a comparison of the 2B4 and 4-1BB intracellular domains in engineered NK-92 cells revealed that the 2B4-containing CAR construct induced more-rapid proliferation, increased cytokine production, and degranulation. However, the incorporation of DAP10, DAP12, or 4-1BB into the NKG2D-2B4ζ CAR construct did not enhance CAR-mediated cytotoxicity. Collectively, these findings imply that more detailed studies are needed to design and optimize NK-cell-specific CAR or NKR structures for use in NK-cell-mediated therapy.
Clinical translation of CAR- and NKR-engineered NK cells
Mounting evidence from preclinical studies has revealed the therapeutic efficacy (including the effective control of tumor growth and associated prolonged patient survival) of CAR-NK cells in cancer therapy, particularly in the treatment of hematological malignancies. Encouragingly, CAR-NK therapy has also demonstrated promising results in solid tumor models, including glioblastoma, ovarian cancer, hepatocellular carcinoma, and prostate cancer [73,74,75,76]. To date, numerous clinical trials have evaluated the efficacy of CAR-NK cells as anticancer agents, some of which have shown great promise. These trials have included clinical evaluations of CAR-NK cells targeting common TAAs such as CD19, CD22, CD7, CD33, and BCMA in the context of hematological malignancy and human epidermal growth factor receptor (HER)-2, PSMA, mesothelin, Mucin-1 (MUC1), and roundabout guidance receptor 1 (ROBO1) in the treatment of solid tumors (Table 1). In addition, NKG2D-engineered NKR-NK cells targeting NKG2D ligands are being tested in the clinic for their potency against metastatic solid tumors, as well as relapsed/refractory AML and myelodysplastic syndromes (MDS) (Table 1). Although most of the clinical trials are in their infancy, CAR- and NKR-NK cells have been proven to be generally safe and well tolerated (NCT02944162, NCT03415100, and NCT03056339) [77]. No serious toxic effects were observed in the first-in-human phase I and II clinical trials of an allogeneic UCB-derived anti-CD19 CAR-NK-cell therapy used for patients with relapsed/refractory NHL or CLL (NCT03056339). Notably, the expansion of adoptive CAR-NK cells was seen as early as 3 days post infusion and persisted at low levels for at least 12 months [56]. The first off-the-shelf, iPSC-derived CAR-NK-cell therapy is currently being evaluated in a phase I clinical trial sponsored by Fate Therapeutics. iPSC-derived NK cells (named FT596) were generated using a construct containing a CD19-targeting CAR, a high-affinity uncleavable CD16 (hnCD16) Fc receptor, and an IL-15/IL-15R fusion. Several doses of FT596 as a monotherapy and in combination with an anti-CD20 monoclonal antibody (mAb) were given to patients with relapsed/refractory B-cell lymphoma and CLL (NCT04245722). Even low doses of FT596 were able to induce rapid clinical responses, as evidenced by a patient showing partial response and over 50% reduction in tumor size shortly after a second cycle of FT596 monotherapy [78]. Another small-cohort phase I clinical study provided the first clinical results relating to the application of NKG2D-engineered primary NK cells for the treatment of patients with solid tumors (NCT03415100). Three patients with metastatic colorectal cancer received a local infusion of NKG2D-engineered CAR-NK cells (engineered using NKG2D-DAP12 mRNA transient transduction). Treatment with low doses of an intraperitoneal infusion of NKR-NK cells caused a marked reduction in the volume of ascites and the number of ascites-associated tumors in two patients. Moreover, rapid tumor regression in the liver region was observed in the third patient who received therapy with ultrasound-guided percutaneous injection, followed by intraperitoneal infusion of NKR-NK cells [69]. No serious adverse effects were observed in any of the three patients. Even with a small-cohort, the results of NKR-NK-cell therapy of solid tumors are encouraging and provide evidence supporting CAR and NKR-NK-cell use in cancer immunotherapy.
Challenges of administering CAR- and NKR-NK-cell therapy and new genetic manipulation strategies aimed at improving the efficacy of NK-cell cancer immunotherapy
Intratumoral persistence hinders the activation and efficacy of CAR- and NKR-NK cells
Although recent clinical trials involving CAR- and NKR-NK cells in cancer therapy have shown good levels of safety and a certain level of promise, multiple factors still hinder their clinical application. One of the major limitations of the successful clinical application of CAR- and NKR-NK cells is their low persistence in vivo. The administration of exogenous cytokines, such as rhIL-15, can enhance the in vivo proliferation and survival of NK cells, with several related phase I/II clinical studies currently underway to verify this outcome. However, rhIL-15 has a short serum half-life and can cause undesirable toxicity, including CRS, neurotoxicity, and even autoimmune diseases [79]. Based on the characteristics of IL-15 trans-presentation [80, 81], the IL-15 superagonist N-803, an IL-15-IL-15Rα complex fused to the IgG1 Fc region (IL15N72D:IL15RαSu/IgG1 Fc complex), was developed as a therapeutic drug to mimic the physiologic trans-presentation of IL-15 and extend its in vivo half-life. The capacity of N-803 to improve the proliferation and in vivo survival of both NK and CD8+ T cells, as well as its immunogenicity and pharmacokinetics, has shown great promise in both hematologic malignancy and solid tumor models [82,83,84] Several clinical trials are underway to evaluate its safety and effectiveness [85, 86]. Although the preliminary results demonstrated that N-803 was highly effective without causing serious toxicity, there is still concern regarding the induction of hyperresponsiveness’ and the functional impairment of NK cells due to repeated N-803 treatment [79]. Another strategy involves engineering NK cells with the IL-15 gene, which can render NK cells capable of endogenous IL-15 expression, thus avoiding the potential side effects associated with cytokine administration. IL-15 gene-modified NK cells have shown greater persistence and antitumor activity with little induced toxicity in several different tumor models [87, 88]. Expression of IL-15 in membrane-bound form (mbIL-15) in NK cells also resulted in significant cell expansion and survival independent of exogenous IL-2 and enhanced antitumor capacity [89]. Incorporating the IL15 gene into UCB-derived anti-CD19 CAR-NK cells led to their long-term persistence (at least 12 months in vivo) and potent antitumor activity without increasing systemic levels of IL-15 or inducing IL-15-mediated toxicity during the treatment of patients with relapsed/refractory NHL or CLL [56]. Cytokine-inducible Src-homology-2-containing protein (CIS), encoded by the CISH gene, is a member of the suppressor-of-cytokine-signaling (SOCS) family and is the key negative regulator of IL-15 signaling [90]. A recent study reported a promising strategy combining CISH deletion using clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated endonuclease (Cas9) gene-editing with IL-15 engineering to better improve the in vivo persistence of UCB-derived anti-CD19 CAR-NK cells. The coupling of CISH deletion and constitutive IL-15 expression doubled the in vivo persistence of CAR-NK cells, compared the that of IL-15-engineered CAR-NK cells alone, leading to enhanced antitumor capacity in lymphoma xenografts [91]. Importantly, no serious off-target toxicity or uncontrolled expansion of CAR-NK cells was detected. Similarly, CISH deletion in human iPSC-derived NK cells also prolonged in vivo persistence and enhanced the tumor eradication capacity of these engineered NK cells [92].
Overcoming challenges in NK-cell homing
Similar to that of CAR-T-cell application, one challenge of CAR- and NKR-NK-cell application to the eradication of solid tumors is the poor ability of these cells to translocate to or infiltrate the tumor site. The efficacy of CAR- and NKR-NK-cell therapy largely relies on the efficient infiltration of NK cells into the tumor sites [93]. To improve the trafficking of NK cells to tumor beds, many researchers have modified CAR- and NKR-NK cells with chemokine receptors to accelerate NK-cell migration into tumor regions expressing the corresponding chemokines. The chemokine CXCL12/SDF-1α, which is mainly expressed in bone marrow stromal cells, is also highly expressed in glioblastoma. Therefore, its receptor, CXCR4, has been chosen as a candidate for incorporation into CAR constructs to promote CAR-NK-cell homing to the bone marrow or permit their entry into glioblastoma tumor sites. CXCR4-engineered anti-epidermal growth factor receptor (EGFR)vIII CAR-NK cells have demonstrated improved trafficking into glioblastoma sites, which secrete the corresponding chemokine CXCL12/SDF-1α, resulting in complete tumor remission and prolonged survival, in contrast to treatment with anti-EGFRvIII CAR-NK cells [94]. Primary anti-CD19 CAR-NK cells overexpressing CXCR4 displayed markedly higher migration ability in the presence of recombinant SDF-1 or bone marrow stromal cells than anti-CD19 CAR-NK cells [95]. Expression of CXCR1 significantly improved the antitumor effectiveness of NKG2D-engineered NK cells by promoting NK-cell migration and infiltration into tumor tissues in subcutaneous and intraperitoneal ovarian cancer xenograft models [96]. Engineering primary NK cells with CXCR2 significantly enhanced the migration of NK cells to renal cell carcinoma sites [97]. CAR-T cells equipped with other chemokine receptors, such as CXCR2, CXCR5, CCR2b, and CCR4, have demonstrated improved migration to tumor sites and consequently more potent eradication of solid tumors, including hepatocellular carcinoma (HCC), breast cancer, non-small cell lung carcinoma (NSCLC), colorectal cancer, ovarian cancer, pancreatic cancer, and glioma [98,99,100,101], some of which are currently being evaluated in clinical trials (NCT04153799, NCT01740557). Similar strategies are likely suitable for use in CAR- or NKR-NK-cell-mediated therapy of solid tumors.
Overcoming the immunosuppressive TME
The TME is a major obstacle to successful CAR- and NKR-NK-cell therapy. Immunosuppressive cells (e.g., Tregs, MDSCs, and TAMs), immunosuppressive soluble factors (TGF-β, IDO, IL-10, and IL-4) or metabolites, hypoxia, the ECM, and other suppressive molecules (e.g., ligands of checkpoint receptors) expressed on tumor cells or immunosuppressive cells found at tumor sites form a complex network that hampers the proliferation, survival, and activity of NK cells, leading to their dysfunction and eventual exhaustion [102,103,104,105,106]. For example, TGF-β secreted by Tregs, MDSCs, TAMs, and tumor cells directly inhibits NK-cell activity by downregulating the expression of the activating receptors NKG2D and NKp30 and impairing NK-cell-mediated IFN-γ secretion, thus attenuating the antitumor efficacy of tumor-infiltrating NK cells. Numerous gene modification approaches are being developed to overcome the immunosuppressive nature of the TME and to augment the cytolytic activity of CAR- and NKR-NK cells against solid tumors. The possibility of genetically engineering CAR- and NKR-NK cells by specifically targeting inhibitory components in the TME to directly block the sources of immunosuppression or reverse inhibitory signals has been investigated. Primary UCB-derived NK cells engineered with DN TGF-β receptor II (DNRII) lacking a cytoplasmic domain displayed an activating receptor phenotype (expression of NKG2D and DNAM-1) and retained cytotoxicity against glioblastoma tumor cells even in the presence of TGF-β [107]. Similarly, DNRII-transduced NK-92 cells displayed enhanced cytolytic activity against lung carcinoma compared to unmodified NK-92 cells in a xenograft mouse model [108]. Another strategy to reverse TGF-β-mediated inhibitory signaling by employing a TGF-β receptor II-based switch receptor was investigated. NK-92 cells genetically modified with a switch chimeric receptor (consisting of the TGF-β type II receptor extracellular and transmembrane domains and the intracellular domain of NKG2D), effectively resisted TGF-β-induced suppressive signaling, leading to enhanced IFN-γ secretion and cytotoxicity and ultimately the suppression of tumor growth in a hepatocellular carcinoma xenograft model [109]. UCB-NK cells modified with TGF-β receptor II-based switch receptor (containing a truncated TGFβRII fused to an intracellular DAP12 or a synthetic Notch-like receptor (SynNotch) domain) displayed higher cytotoxicity against neuroblastoma in the presence of TGF-β, as well as improved progression-free survival and tumor eradication, in a neuroblastoma xenograft model [110]. This switch receptor-based strategy used to combat the antagonizing effects of TGFβ-mediated suppression has shown great promise, particularly in the treatment of TGFβ-secreting solid cancers. A similar switch receptor strategy has been adopted in designing CAR constructs by combining the IL-4 receptor ectodomain with the endodomain of the IL-7 receptor (4/7 inverted cytokine receptor (ICR)) to convert IL-4-mediated suppressive signals transmitted by IL-7into proliferative signals. The strategy has been tested by incorporating the 4/7 ICR into anti-PSCA CAR-T cells to treat PSCA+ pancreatic cancer, which is associated with an IL-4-rich TME [111].
Interrupting the inhibition of NK cells mediated by inhibitory checkpoints in the TME is critical to the antitumor function of NK cells because it ends NK-cell exhaustion. In addition to combination therapy with checkpoint inhibitors, designing switch receptors to reverse the negative signals transmitted by checkpoint receptors, including PD-1 and TIGIT, has also been investigated, with promising results for both CAR-T and CAR-NK cells have been realized. A chimeric PD-1-NKG2D-41BB receptor consisting of the extracellular domain of PD-1, the transmembrane and cytoplasmic domains of NKG2D, and the cytoplasmic domain of 4-1BB was recently reported to switch the negative PD-1 signal into an activating signal in NK cells. Chimeric receptor-engineered NK-92 cells exhibited significant tumor growth inhibition by reversing the immunosuppressive effects of PD-1 in a lung cancer xenograft model [112]. The clinical investigation of PD-1-NKG2D-41BB receptor-engineered NK-92 cells for the treatment of advanced NSCLC is currently in a phase I clinical trial (NCT03656705). PD-1-CD28 switch receptor-modified anti-CD19 CAR-T cells have been evaluated for use in salvage therapy for patients with relapsed/refractory diffuse large B-cell lymphoma (DLBCL) and PD-L1+ large B-cell lymphoma (in a phase Ib study), eliciting both potent and durable anticancer responses [113, 114]. PD-1-CD28 switch receptor-engineered CAR-T cells have demonstrated superior eradication of advanced solid tumors (e.g., prostate cancer and mesothelioma) compared to CAR-T-cell therapy alone or anti-PD-1 Abs [30]. Similarly, T-cell immunoreceptor with Ig and ITIM domains (TIGIT)-CD28 switch receptor-modified T cells have demonstrated superior antitumor function in a melanoma xenograft model [115]. Similar strategies are worthy of investigation in CAR-NK cells, particularly for the treatment of solid tumors. With the rapid development of gene-editing technologies, the deletion of inhibitory receptor-encoding genes or the reprogramming of NKR expression patterns is currently employed to limit immunosuppression and augment NK-cell antitumor activity [116,117,118,119]. Deletion of PD-1 in primary NK cells using CRISPR/Cas9 technology significantly improved NK-cell-mediated cytolytic activity and cytokine production [120]. The depletion of immunosuppressive cells populating the TME is critical to restoring NK-cell function. It has been reported that MDSCs and Tregs in the TME express high levels of NKG2D ligands (NKG2DLs). NKG2D-engineered NK cells not only eradicate NKG2DL+ cancer cells but also directly eliminate NKG2DL+ MDSCs, thus abrogating the suppressive effects exerted by MDSCs in the TME [121]. Interestingly, it has been demonstrated that NKG2D-engineered NK cells can secrete chemokines in response to MDSCs in the TME and thus enhance the infiltration and antitumor efficacy of infused CAR-T cells. NK-92 cells engineered to express high-affinity CD16, endoplasmic reticulum (ER)-retained IL-2, and a PD-L1-specific CAR (PD-L1 t-haNK cells, NantKwest Inc.) exhibited high levels of cytotoxicity against PD-L1+ solid tumors and inhibited the growth of breast, lung, and bladder cancers in xenograft models [122]. Notably, these PD-L1 t-haNK cells preferentially killed suppressive MDSCs. The safety and preliminary efficacy of PD-L1 t-haNK therapy in patients with locally advanced or metastatic solid cancers is being evaluated in a phase I clinical trial (NCT04050709). The evaluation of PD-L1 t-haNK cells in combination with N-803 and conventional chemotherapy in patients with locally advanced or metastatic pancreatic cancer is ongoing in several phase II studies, and positive interim data on survival rates has been reported (NCT04390399). PD-L1 t-haNK cells in combination with N-803, conventional chemotherapy and anti-PD-1 or PD-L1 Abs are currently being evaluated in the treatment of patients with recurrent/metastatic gastric or head and neck cancer and other solid tumors (NCT04847466 and NCT03228667).
Strategies to overcome tumor escape: bispecific NKR- or CAR-NK cells
To enhance tumor specificity and overcome tumor escape due to antigen loss and tumor heterogeneity of solid tumors, multiple bispecific modifying strategies have been developed for CAR-T therapy [123,124,125]. For example, bispecific CARs consisting of two extracellular scFv domains that recognize two different TAAs synergistically (“tandem” CARs) or constructs containing two CARs, each targeting a different TAA (Dual CARs), have been designed. CAR constructs containing two CARs, each comprising different intracellular activation domains targeting different TAAs (split CARs; e.g., one CAR containing a CD3ζ signaling domain and another CAR containing CD28/4-1BB costimulatory domains), whereby CAR-T-cell activation occurs only when both TAAs are recognized simultaneously, have also been developed. For instance, in a synNotch system, T-cell activation is triggered only when a synthetic Notch receptor recognizing one TAA induces the expression of a CAR construct specific for a second TAA. Thus, CAR-T-cell activation requires the simultaneous binding of two TAAs. To date, CD19/CD20- or CD19/CD22-targeting bispecific CAR-T cells have exhibited significant clinical efficacy in patients with relapsed or refractory B-cell malignancies [126,127,128]. Recently, a bispecific CAR strategy was designed to target two TAAs and simultaneously block TME-mediated checkpoint inhibition. A CAR construct targeting c-Met and PD-L1 was developed, and the resulting anti-c-Met/PD-L1-bispecific CAR-T cells displayed high cytolytic activity against c-Met and PD-L1 double-positive HCC cells and exhibited marked in vivo antitumor efficacy, including significant tumor growth inhibition and prolonged survival, compared with monovalent c-Met CAR-T cells or PD-L1 CAR-T cells in xenograft HCC models [129]. In another study, bispecific Trop2/PD-L1 CAR-T cells significantly reduced the growth of gastric cancer overexpressing Trop2 and PD-L1 in a xenograft model [130]. Bispecific CAR-modifying strategies can effectively improve antitumor efficacy and overcome tumor heterogeneity, antigen escape, and TME-mediated immunosuppression exhibited by solid tumors. These results indicate that similar techniques can be readily applied to NK-cell therapy. An early phase I clinical trial of anti-CD19/CD22-bispecific CAR-NK cells is recruiting patients for treatment of relapsed or refractory B-cell lymphoma (NCT03824964). A novel bispecific chimeric PD-1-DAP10/NKG2D receptor comprising a PD-1 extracellular domain fused with transmembrane and intracellular domains, as well as full-length NKG2D, can simultaneously target tumor cells expressing high levels of PD-L1 and NKG2D ligands (MICA/B and ULBPs) in the TME. Bispecific PD-1-DAP10/NKG2D-engineered NK-92 cells exhibited potential antitumor efficacy against gastric cancer in a xenograft model [131]. Other bispecific NK cells containing both a switch receptor (e.g., NKG2A, TIGIT, or Tim-3) and a CAR or NKR are worthy of further investigation. These bispecific engineering strategies not only can be used to target tumor cells but also to block inhibitory signals emitted from checkpoint molecules simultaneously, thus preventing or reversing immune exhaustion. Rational engineering strategies need to be adopted in the design of bispecific NKR- or CAR-NK cells to see improve the therapeutic efficacy of NK-cell therapy, particularly for solid tumors.
Genetic manipulation of NKT cells for use in cancer immunotherapy
The biological characteristics of NKT cells and their roles in antitumor immunity
NKT cells share features with both T cells and NK cells. In contrast to traditional αβ T cells, NKT cells recognize CD1d-presented glycolipids, glycosphingolipids, or lipid antigens and respond rapidly to secrete a wide variety of cytokines. The secreted cytokines can further regulate both innate and adaptive immune cells. Therefore, NKT cells serve as bridges linking innate and adaptive immunity. Depending on receptor usage, NKT cells can be categorized into invariant NKT (iNKT) cells (also known as type I NKT cells) and diverse or variant NKT cells (type II NKT cells). Invariant NKT cells express a semi-invariant TCR (Vα24–Jα18 in humans, Vα14-Jα18 in mice) paired with a limited repertoire of Vβ chains, while diverse NKT cells express diverse αβ TCRs. Following antigen recognition, iNKT cells activate rapidly and secrete large amounts of both Th1 (IFN-γ and TNF-α) and Th2 (IL-4, IL-10, and IL-13) cytokines, which can further regulate the activation and function of other immune cells. Invariant NKT cells can eliminate CD1d-expressing tumor cells by direct cytolysis through the secretion of perforin and granzyme B or via the Fas-Fas ligand (FasL) pathway. Invariant NKT cells can also promote the antitumor effect of other innate and adaptive immune cells through the rapid release of Th1 cytokines and chemokines [132]. For instance, activated iNKT cells can directly activate NK cells, CD8+ T cells, and dendritic cells (DCs) through IFN-γ secretion. They can also facilitate the activation of DCs through CD1d-TCR/lipid antigen and CD40/CD40L interactions and promote DC maturation via the secretion of IL-12. Mature DCs can in turn induce the activation of CD8+ T cells, CD4+ T cells, NK cells, and NKT cells, thereby further enhancing both innate and adaptive antitumor immune responses [133]. Intriguingly, iNKT cells can also regulate and alter the function of immunosuppressive cells in the TME. For instance, iNKT cells were reported to kill CD1d+ TAMs in neuroblastoma in an IL-15- and CD1d-dependent manner [134]. The activation of iNKT cells was also shown to significantly increase the frequency of inducible nitric oxide synthase (iNOS)+ CD206− M1 macrophages while reducing the number of iNOS-CD206+ M2 macrophages in the spleen and at the tumor site, enhancing the antitumor immune response [135]. Moreover, iNKT cells are able to reverse the suppressive effect of MDSCs and promote the maturation of immunosuppressive immature MDSCs into APCs that can prime antitumor CD8+ T cells and NK cells [136]. Finally, iNKT cells have been shown to abolish the suppressive role of neutrophils in the context of melanoma by simultaneously inhibiting neutrophil-mediated IL-10 production and enhancing IL-12 secretion, thus facilitating the proliferation and activity of antigen-specific CD8+ T cells [137].
Current NKT cell research and the development of CAR-engineered NKT cells for cancer immunotherapy
The unique biological features of iNKT cells, such as their cytolytic and regulatory capacities, make them ideal effector cells for use in cancer immunotherapy. Several approaches have been employed to modulate iNKT cells for use in cancer immunotherapy, including the administration of α-GalCer, a potent stimulator of iNKT cells isolated from a marine sponge (used either alone or in combination with IL-12), α-GalCer-loaded DCs, irradiated tumor cells, or adoptive transfer of ex vivo expanded and activated iNKT cells [138]. Preclinical studies of iNKT cell treatment of melanoma, colon cancer, liver metastasis of colorectal cancer, multiple myeloma, B-cell lymphoma, ovarian carcinoma, breast cancer, and renal carcinoma have shown promising results, as exemplified by the significant inhibition of tumor growth and metastasis and prolonged survival [132, 138]. Many clinical trials are underway to evaluate the safety, feasibility, and validity of iNKT cell-mediated cancer immunotherapy. To date, the majority of therapeutic iNKT regimens have been well tolerated without severe off-target toxicity. However, administration of α-GalCer alone failed to achieve a clinical response, mainly due to limited NKT cell numbers in circulation and the induction of NKT cell anergy. Administration of autologous α-GalCer-pulsed APCs and adoptive transfer of ex vivo expanded and activated iNKT cells has shown some promise. The evidence supporting these strategies is indicated by increased in vivo iNKT expansion with either elevated IFN-γ levels or increased numbers of T and NK cells in patients with advanced and recurrent NSCLC, head and neck squamous cell carcinoma, advanced melanoma, and other solid tumors [139, 140]. A recently completed phase II clinical trial involving the intravenous administration of α-GalCer-pulsed APCs to patients with advanced or recurrent NSCLC has reported encouraging results, including prolonged overall survival [141].
Preclinical studies and several clinical trials involving CAR-iNKT effector cells are underway and have produced encouraging results (Table 2). Anti-disialoganglioside GD2-CAR-iNKT cells have been developed and evaluated in the treatment of neuroblastoma in a NOD/SCID/IL2Rγ-null (hu-NSG) mouse model. GD2-CAR modification significantly enhanced the cytolysis of GD2-positive neuroblastoma and CD1d-positive M2 macrophages. Interestingly, 4-1BB endodomain-containing GD2-CAR promotes the Th1-like polarization of iNKT cells. Importantly, these GD2-CAR-iNKT cells exhibited improved in vivo persistence and trafficking to tumor areas, effectively inhibited tumor growth, and prolonged survival without inducing GVHD [142]. IL-15 expression in GD2-CAR-iNKT cells further enhanced their long-term persistence and infiltration to tumor sites and reduced PD-1 expression on their cell surface, leading to more efficient tumor eradication compared with conventional GD2-CAR iNKT cells in a neuroblastoma xenograft model [143]. Currently, GD2-specific CAR- and IL-15-expressing autologous iNKT cells are under investigation in a phase I study for the treatment of children with relapsed or resistant neuroblastoma (NCT03294954). The initial results indicated their safety with no dose-limiting toxicity. More importantly, the in vivo expansion of these CAR-iNKT cells and their localization to tumor sites were observed, with one patient achieving an objective response leading to the regression of bone metastatic lesions [144]. Another research group developed CD19-targeting CAR-iNKT cells that demonstrated more effective cytotoxicity against CD1d-expressing lymphomas in both a CD1d- and CAR-dependent manner than conventional anti-CD19-CAR-T cells [145]. Enhanced in vivo antitumor effects, particularly the eradication of intracranial and relapsed lymphoma, as well as long-term survival, were observed in tumor-bearing NSG mice. A transcriptome analysis revealed that anti-CD19 CAR-iNKT cells expressed higher levels of numerous chemokine receptors than conventional anti-CD19 CAR-T cells [145]. A phase I clinical study of allogeneic anti-CD19 CAR iNKT cells used for the treatment of patients with relapsed or refractory B-cell malignancies is currently at the recruitment stage (NCT03774654). A central memory-like CD62L+ iNKT subpopulation was transduced with an anti-CD19 CAR, resulting in anti-CD19 CAR-iNKT cells with high proliferative potential and prolonged in vivo persistence. The CD19-specific CD62L+ CAR-iNKT subsets also demonstrated superior therapeutic efficacy in lymphoma models, signaling promising prospects for CAR-engineered memory iNKT subpopulations in cancer immunotherapy [146].
Collectively, CAR-engineered iNKT cells have several unique features compared with conventional CAR-T cells. For instance, they are able to eradicate tumor cells in both a CAR- and CD1d-dependent manner. This synergistic cytotoxicity may not only potentially limit the likelihood of tumor immune escape but also eliminate suppressive cells (e.g., by killing CD1d+ TAMs) residing within the TME. Compared to CAR-T cells, CAR-iNKT cells express high levels of chemokine receptors, endowing these cells with a superior ability to localize to tumor sites. Since allogeneic iNKT cells do not mediate GVHD (and have even been shown to inhibit GVHD) while maintaining GVT activity [147], they are considered suitable for universal (“off-the-shelf”) clinical application. Despite the efficacy and safety of CAR-iNKT cells, there are still challenges associated with their translation to the clinic. One of the major limitations of CAR-iNKT cell therapy is the extremely low numbers of iNKT cells in the human body. Ex vivo expansion approaches involve initial activation with α-GalCer, followed by further activation and expansion with cytokines, anti-CD3/CD28 mAb stimulation, and/or the use of CD1d-expressing APCs in combination with α-GalCer [145]. Even after following these steps, producing large quantities of clinical-grade CAR-iNKT cells remains a challenge. Hematopoietic stem cells (HSCs) and iPSCs can be reprogrammed and differentiated into iNKT cells [148,149,150]. The safety, feasibility, and antitumor potential of HSC-derived iNKT cell therapy are being evaluated in preclinical studies [148, 151, 152]. HSCs, particularly iPSCs, may serve as ideal sources of easily expanded “off-the-shelf” CAR-iNKT cells suitable for clinical cancer therapy.
Genetic manipulation of γδ T cells for use in cancer immunotherapy
The biological characteristics of γδ T cells and their roles in antitumor immunity
Gamma delta T cells are a small subpopulation of T cells that express a γδ TCR. They account for only 1–5% of CD3+ T cells in peripheral blood but are more abundant at the mucosal barrier, including in the gut mucosa, skin, and reproductive tracts [153]. In contrast to αβ T cells, γδ T cells recognize nonpeptidic phosphoantigens (pAgs) or phosphorylated metabolites. Upon detecting invading pathogens, inflammation, or malignant transformation, γδ T cells expand rapidly and exert protective immune responses generally independent of antigen processing and MHC restriction [154]. Thus, γδ T cells are defined as innate immune cells, serving as the first line of defense against foreign infections and malignancy. Despite this label, γδ T cells have characteristics of both innate and adaptive immune cells and act as bridges between these two types of immune responses. Upon activation, γδ T cells exhibit cytotoxicity against tumors and infected cells and secrete multiple cytokines, such as IFN-γ, TNF-α, and IL-17. Gamma delta T cells express NKRs, including NKG2D, NKp30, and/or NKp44, thus synergistically recognizing tumors via both TCRs and NKRs. Their cytotoxic mechanisms include the release of perforin and granzymes, the expression of FasL and TNF-related apoptosis-inducing ligand (TRAIL), and the secretion of IFN-γ and TNF-α. Gamma delta T cells can also influence the antitumor efficacy of other immune cells (e.g., DCs, NK, and CD8+ T cells) by producing IFN-γ or expressing costimulatory molecules and can act as APCs to present antigens and augment the cytolytic capacity of CD8+ T cells to kill tumor cells [155, 156].
Human γδ T cells can be classified into different subpopulations based on their usage of TCRγ or TCRδ chains. Most of the studies have focused on the Vδ1 and Vδ2 subsets. The Vδ2 chain mainly pairs with the Vγ9 chain to form Vγ9Vδ2 T cells, which account for ~50–95% of peripheral γδ T cells. Vγ9Vδ2 T cells have potent antitumor capacity, recognize TAAs, such as tumor-derived isoprene pyrophosphate (IPP), via their butyrophilin 3A1 (BTN3A1, CD277)- and BTN2A1-dependent TCRs [157,158,159], and detect stress proteins through NKG2D and DNAM-1 receptors. They also express CD16, which can promote ADCC-mediated cytotoxicity. Vγ9Vδ2 T cells exhibit high levels of cytolytic capacity for the recognition of unique innate-like nonpeptidic antigens presented by BTN3A1 and BTN2A1, not classical MHC molecules, which makes them attractive candidates for cancer immunotherapy [160, 161]. Vδ1 T cells predominantly reside in solid tissues, such as the skin, intestine, liver, spleen, and various tumors. Mucosal tissues are particularly enriched with Vδ1 T cells, which compose approximately 20-50% of all intraepithelial lymphocytes (IELs) in the dermis and intestinal epithelia. Vδ1 T cells also recognize and kill tumor cells in both a TCR- and NKR-dependent manner. The TCR of Vδ1 T cells is not activated by pAgs, the ligand for which remains to be identified. It has been demonstrated that MHC class I-related chain A (MICA) can be recognized by both NKG2D and the Vδ1 TCR in a sequentially recognized and temporally organized manner [162]. Several reports have shown that Vδ1 T cells recognize glycolipids (e.g., α-GalCer) presented by CD1d or CD1c [163, 164]. However, the precise recognition mechanism employed by the Vδ1 TCR needs to be further clarified. Similar to Vγ9Vδ2 T cells, Vδ1 T cells also recognize tumors through NKG2D, binding to its ligands, MICA/B and ULBPs, on stressed or tumor cells. After activation, particularly upon in vitro stimulation with a strong TCR agonist and cytokines (IL-2 and IL-15), Vδ1 T cells express NKp30, NKp44 and NKp46, which are not expressed by Vγ9Vδ2 T cells, and thus exert antitumor activity combined with IFN-γ secretion [165,166,167]. In addition, it has been demonstrated that Vδ1 T cells readily infiltrate tumor tissues, possibly due to the higher expression of chemokine receptors compared to Vδ2 T cells [168]. Moreover, Vδ1 T cells are resistant to activation-induced cell death (AICD) and T-cell exhaustion upon continuous stimulation, whereas Vδ2 T cells are susceptible to T-cell exhaustion and AICD [168]. These unique features render Vδ1 T cells promising effector cells suitable for cancer immunotherapy.
Current γδ T-cell research and the genetic engineering of γδ T cells for use in cancer immunotherapy
The unique biological features of γδ T cells, such as the effective and extensive recognition of tumor cells, potent cytolytic capacity, rapid secretion of abundant cytokines, no MHC restriction, and low risk of inducing GVHD, make γδ T cells excellent candidates for use in cancer therapy. Cancer therapies relying on γδ T cells have been widely investigated, and multiple clinical studies have been performed to evaluate their safety and efficacy in the treatment of cancer patients with both hematological malignancies and solid tumors.
Multiple clinical trials involving the use of Vγ9Vδ2 T cells in the treatment of hematological malignancies (NCT02656147 and NCT03862833) or solid tumors, including HCC, renal carcinoma, mammary carcinoma, lung cancer, head and neck cancer, neuroblastoma, and prostate cancer, have been performed or are ongoing [161, 169, 170]. Most initial therapies rely on the adoptive transfer of ex vivo-expanded Vγ9Vδ2 T cells and/or their in vivo stimulation with synthetic pAgs. The most widely used stimulants of Vγ9Vδ2 T cells include zoledronic acid (ZOL, a type of aminobiphosphonate) and a synthetic pAg bromohydrin pyrophosphate (BrHPP) in combination with IL-2. A recent clinical trial revealed that the adoptive transfer of autologous or allogenic Vγ9Vδ2 T cells is safe and feasible, with only low levels of side effects being observed [171,172,173,174,175]. Promising clinical responses have been obtained in the majority of the studies conducted to date. For instance, six of ten patients with metastatic renal cell carcinoma displayed stable disease after receiving repeated infusions of autologous Vγ9Vδ2 T cells [171]. A patient with stage IV cholangiocarcinoma and recurrent mediastinal lymph node metastasis receiving repeated allogenic Vγ9Vδ2 T cells responded well to treatment with minimal side effects [176]. Peritoneal lymph node metastasis was gradually eliminated. Interestingly, γδ T-cell therapy greatly improved the patients’ overall immune status, as evidenced by the increased proportions of functional CD4+ and CD8+ T cells and a decrease in the percentages of exhausted CD4+ and CD8+ T cells [176]. In vivo stimulation of Vγ9Vδ2 T cells with pAgs or ZOL has also exhibited potential, but the clinical response rates observed have been lower than those achieved with adoptive transfer [177, 178]. In summary, Vγ9Vδ2 T-cell-based clinical trials have shown some promising clinical responses; however, the complete response rates are still relatively low, and improved overall survival has not been reached, particularly in the treatment of solid tumors (such as HCC, lung cancer, and renal cell carcinoma).
Although Vδ1 T cells constitute only a minor proportion of peripheral blood cells, their unique recognition and activation features, long-term in vivo survival, and resistance to AICD and exhaustion render them very attractive candidates for adoptive cancer therapy. A clinical-grade protocol for the selective expansion and differentiation of highly cytotoxic Vδ1+ cells, refined as Delta One T (DOT) cells, was established [179]. Highly cytotoxic DOT cells are Vδ1-enriched (>60%) γδ T cells produced through a two-step method. The protocol is designed to selectively expand and differentiate DOT cells with anti-CD3 mAb and various cytokines (IL-4, IL-15, IFN-γ, and IL-21) for over 3 weeks, achieving 2000-fold expansion. Expanded DOT cells display enhanced expression of NKRs, particularly NKp30 and NKp44, and upregulated expression of NKG2D and DNAM-1, contributing to their potent cytolytic activity. Hence, expanded DOT cells can recognize a broad range of tumor signals through their NKRs and TCR, thus endowing them with cytotoxicity against a broad range of tumors and potentially the ability to overcome tumor-mediated evasion mechanisms, such as antigen loss. Indeed, in a xenograft model of CLL and acute myeloid leukemia (AML), the adoptive transfer of in vitro-expanded DOT cells exhibited significant tumor growth inhibition, prevented tumor metastasis, and extended overall survival [179, 180]. Therefore, Vδ1 T cells, particularly DOT cells, may represent promising candidates for use in adoptive cell immunotherapy.
Although promising, immunotherapy based on the adoptive transfer of natural γδ T cells has yet to demonstrate impressive clinical efficacy. Novel technologies and strategies are needed to overcome both the intrinsic functional constraints of γδ T cells and the suppression of the TME. Table 3 summarizes the CAR- and NKR-engineered technologies employed to date in γδ T-cell-based cancer immunotherapy. CAR- and NKR-modified γδ T cells have several advantages over conventional CAR-αβ T cells. They can exert antitumor effects more efficiently by recognizing tumor antigens both through CAR specificity and through endogenous γδ TCRs. Activated γδ T cells can act as APCs to prime activated CD8+ T cells, thus enhancing their antitumor capacity. Vδ1 T cells display high levels of tropism toward tumor tissues and resistance to AICD and exhaustion, which are advantageous traits for the treatment of solid tumors. Furthermore, Vδ1 T cells do not rely on MHC restriction and are associated with a low risk of GVHD induction, rendering them suitable candidates for the generation of allogeneic “off-the-shelf” products for use in cancer therapy. CD19-targeting polyclonal CAR-γδ T cells have been shown to significantly enhance the cytolytic activity of γδ T cells against CD19+ tumor cells in vitro and in vivo, pointing to a synergistic stimulatory effect between γδ TCRs and CARs [181]. Another study showed that CD19 CAR-transduced highly expanded γδ T cells readily eliminated bone marrow leukemic cells in an NSG model. Importantly, CD19 CAR-γδ T cells primed with ZOL showed the ability to kill CD19 antigen-negative leukemia cells [182]. A phase I clinical study is currently underway to evaluate the safety and efficacy of anti-CD19 CAR-γδ T cells for the treatment of lymphoma and leukemia (NCT02656147). Other clinical trials have been established to assess CAR-engineered γδ T cells targeting hematologic malignancies, namely, anti-CD7 CAR-γδ T cells and anti-CD20 CAR-γδ T cells (NCT04702841 and NCT04735471). A phase I trial has recently been initiated to test the efficacy of ADI-001, the first-in-human allogeneic CAR-γδ T-cell therapy targeting CD20 for the treatment of B-cell NHL (NCT04735471); the preliminary safety and tolerability data are expected by the end of 2021. Regarding solid tumors, GD2-targeted CAR-γδ T cells have been developed and have shown promising results. Expanded and GD2-CAR-engineered γδ T cells have not only demonstrated enhanced GD2-specific killing of GD2-expressing tumors but have also retained their capability to migrate toward tumor sites and cross-present antigens to effector αβ T cells [183, 184]. These results highlight the potential advantages of CAR-γδ T cells over conventional CAR-T cells, especially in application to solid tumors. NKR-modified γδ T cells have also shown promising results. NKG2D-modified Vγ9Vδ2 T cells transduced by mRNA electroporation displayed substantially augmented cytotoxicity against multiple solid tumors. Repeated transfusion of NKG2D-engineered Vγ9Vδ2 T cells resulted in tumor regression and prolonged survival in xenograft models of ovarian cancer, colorectal cancer, and hepatocarcinoma [185]. A phase I clinical trial of NKG2D-engineered CAR-γδ T cells is currently being undertaken for the treatment of relapsed or refractory solid tumors, including triple-negative breast cancer, colorectal cancer, nasopharyngeal carcinoma, sarcoma, gastric cancer, and prostate cancer (NCT04107142).
A novel gene manipulation strategy is based on γδ T cells as sources of high-affinity Vγ9Vδ2 TCRs for the transduction of αβ T cells, forming novel T cells engineered with defined γδ TCRs (TEGs). The engineered Vγ9Vδ2 TCR reprograms both CD4+ and CD8+ T cells and redirects αβ T cells to selectively target a broad panel of cancer cells while ignoring normal cells [186, 187]. Owing to the broad reactivity of the Vγ9Vδ2 TCR, TEGs can target a wide range of tumor cells while retaining features of conventional αβ T cells (high proliferative capacity and long-term memory), thus overcoming the limitations of γδ T cells (low persistence and impaired activation in the TME) [188]. Indeed, TEGs displayed potent cytolytic capacity against both hematological malignancies and solid tumors, including leukemia stem cells [189, 190]. To date, TEGs have been generated under GMP conditions and are being evaluated in a phase I clinical trial treating patients with relapsed and refractory AML and multiple myeloma (NTR6541) [191, 192].
Mounting evidence has demonstrated the promising prospects of γδ T-cell-based cancer therapy, particularly as a safe and effective platform for allogeneic “off-the-shelf” cell products. However, to date, the current clinical applications of CAR- and NKR-γδ T cells have not yet demonstrated superior therapeutic efficacy over conventional CAR-T cells, particularly for solid tumors. A deeper understanding of the recognition mechanisms, the intracellular signaling pathways implicated, and the molecular interactions relevant to CARs and NKRs will provide insight for the optimization of CAR γδ T cells. Taking advantage of the novel gene manipulation techniques available for the rational modification of γδ T cells to optimize their proliferation, survival, and persistence can further improve the validity of genetically engineered γδ T cells in clinical therapy. Combination with other therapeutic approaches, such as conventional CAR-αβ T cells or immune checkpoint blockade, may be required to achieve more significant clinical efficacy. More advanced-phase clinical trials are needed to determine the efficacy of genetically engineered γδ T-cell-based immunotherapy.
Conclusions and future perspectives
The development and characteristics of CAR- and NKR-engineered IKCs are summarized in Fig. 2. IKCs have some unique advantages over conventional αβ T cells when used as candidates for CAR cancer immunotherapy (Table 4): (1) they respond rapidly in response to cellular stress or malignant transformation, (2) they employ multiple antigen recognition mechanisms and exert CAR-dependent and CAR-independent cytolytic effects (e.g., NKR and CD1d), thus leading to more effective antitumor efficacy. Their NKRs recognize a broad spectrum of antigens, endowing IKCs with the ability to kill various cancer cells even following antigen loss or MHC downregulation, thus preventing tumor escape, (3) Upon activation, IKCs can enhance the antitumor activities of other effector cells by producing cytokines or acting as APCs, (4) IKCs do not induce CRS, GVHD, or other types of autoreactivity and have demonstrated an excellent safety record to date, and (5) IKCs can be generated from many sources, including autologous or allogeneic cells, UCB, hematopoietic stem/progenitor cells, iPSCs, and human embryonic stem cells. They are therefore suitable for the development of allogeneic “off-the-shelf” products from healthy donors, reducing manufacturing time and cost. Intriguingly, certain IKCs (e.g., Vδ1 cells and NKT cells) display high levels of tropism to tumor tissues and are resistant to AICD and exhaustion, while others (NK and NKT cells) have the ability to kill immunosuppressive cells (e.g., MDSCs and TAMs) in the TME, thereby helping to control solid tumors.

CAR- and NKR-engineered innate killer cells. IKCs can be engineered to express CAR and NKR, giving rise to CAR- or NKR-engineered IKCs with more powerful tumor targeting and efficient cytolytic capacity to kill tumor cells. CAR- or NKR-engineered NK cells exert direct cytolysis against tumor cells in both a CAR- and NKR-dependent manner and through antibody-dependent cellular cytotoxicity (ADCC). NKT cell activation depends on the recognition of glycosphingolipids, glycolipids, or lipid antigens (e.g., α-GalCer). CAR-engineered NKT cells exhibit a more effective antitumor immune response and are able to eradicate tumor cells in both a CAR- and CD1d-dependent manner. γδ T cells recognize phosphoantigens (pAgs). They can be engineered to include a CAR or NKR, thus driving direct antitumor responses through CAR or NKR recognition and by ADCC
However, there are still challenges facing the translation of these IKCs into the clinic. One of these challenges is the low frequency of IKCs in the human body. Large-scale ex vivo expansion is required prior to infusion or gene modification. Numerous research teams have tried to overcome this hurdle by using various amplification techniques and have been able to obtain enough clinical-grade expanded effector cells (NK, NKT, and γδ T cells) for use in therapy [179, 193, 194]. Induced PSCs can be reprogrammed to directionally differentiate into different effector cells and represent ideal sources of easily expanded clinical-grade standardized “off-the-shelf” CAR- or NKR-engineered IKCs suitable for cancer therapy [59, 195]. Another problem is the efficiency of IKC gene engineering. Some IKCs are difficult to transduce or transfect, particularly peripheral blood-derived NK cells and γδ T cells. Various strategies, including changing the virus pseudotype (e.g., BaEV) or stimulation with cytokines and/or some compounds, have been developed to enhance viral transduction of IKCs [55, 196]. Several recent studies have reported that the transduction efficiency can be improved to achieve ~30–70% NK cells, 40–60% γδ T cells, and 60–80% NKT cells [145, 182, 197]. Precisely optimizing CAR or NKR structures, including the extracellular recognition, transmembrane, and intracellular signaling domains, is critical for fine-tuning CAR activity [198, 199]. It is important to design and optimize these engineering strategies, tailoring them to specific cell activation features and diseases [200]. For example, constructing NK-specific CARs or NKRs to generate optimized CAR- or NKR-NK cells depends on the features of specific intracellular signal pathway features. Due to the heterogeneity of IKCs, distinct subsets may have different functions. For instance, some subpopulations, such as IL-17-producing γδ T cells, might play an immunosuppressive or protumor role. It is worth exploring strategies to selectively expand and modify particular IKC subsets for the treatment of corresponding tumor types. The longer lifespan of memory-like NK cells may render them ideal sources for NK-cell therapy [201,202,203]. Cytomegalovirus (CMV)-induced adaptive memory NK cells not only survive longer in vivo but also have the ability to resist the suppressive effects of Tregs and MDSCs [204, 205]. Cytokine (IL-12, IL-15, and IL-18)-induced memory-like NK cells have exhibited potent antitumor responses and have been shown to induce CR in 50% of relapsed/refractory AML patients with poor prognosis [206]. Furthermore, when modified with an anti-CD19 CAR, cytokine-induced memory-like NK cells displayed enhanced responses against NK-resistant leukemia and lymphoma [207]. The use of CAR- or NKR-engineered memory-like NK cells or a specific NK-cell subset would likely lead to considerable benefits. The major issues facing CAR-T-cell therapy, particularly in the treatment of solid tumors, include tumor heterogeneity, poor trafficking and infiltration into the tumor sites, low in vivo survival and persistence, and the immunosuppressive TME. The same obstacles must also be overcome by CAR- or NKR-engineered IKCs. Researchers are addressing these current obstacles with a wide range of engineering strategies to improve the safety, efficacy, and applicability of gene-modified cell therapies. Recent advances in synthetic biology and novel genetic engineering approaches (such as gene-editing and cellular reprogramming) have provided new opportunities for the genetic modification and manipulation of immune cells [208, 209]. These strategies will be employed to optimize the antitumor efficacy of IKCs to minimize their intrinsic drawbacks and overcome extrinsic challenges presented by the TME, thus maximizing the efficacy of future cancer therapies.
Despite showing some promise, immunotherapy involving genetically engineered IKCs is still in its infancy. A more comprehensive understanding of the biological characteristics of these intriguing cells will help to provide new strategies and improve their antitumor efficacy. The optimization of engineering strategies due to advances in synthetic biology and precise gene-editing techniques will endow these currently used drugs with the ability to target tumors more precisely and effectively, as well as widen their applications. A more in-depth investigation of the clinical therapeutic efficacy of engineered IKCs in cancer therapy is necessary. In conclusion, IKCs and adaptive CTLs each have their own unique characteristics and complementary features, the rational combination of which, together with other therapeutic approaches, will certainly lead to significantly improved clinical efficacy.
References
Leon E, Ranganathan R, Savoldo B. Adoptive T cell therapy: boosting the immune system to fight cancer. Semin Immunol. 2020;49:101437.
Varade J, Magadan S, Gonzalez-Fernandez A. Human immunology and immunotherapy: main achievements and challenges. Cell Mol Immunol. 2021;18:805–28.
Weber EW, Maus MV, Mackall CL. The emerging landscape of immune cell therapies. Cell. 2020;181:46–62.
Brenner MJ, Cho JH, Wong NML, Wong WW. Synthetic biology: immunotherapy by design. Annu Rev Biomed Eng. 2018;20:95–118.
Wu MR, Jusiak B, Lu TK. Engineering advanced cancer therapies with synthetic biology. Nat Rev Cancer. 2019;19:187–95.
El-Khazragy N, Ghozy S, Emad P, Mourad M, Razza D, Farouk YK, et al. Chimeric antigen receptor T cells immunotherapy: challenges and opportunities in hematological malignancies. Immunotherapy. 2020;12:1341–57.
Wei J, Guo Y, Wang Y, Wu Z, Bo J, Zhang B, et al. Clinical development of CAR T cell therapy in China: 2020 update. Cell Mol Immunol. 2021;18:792–804.
MacKay M, Afshinnekoo E, Rub J, Hassan C, Khunte M, Baskaran N, et al. The therapeutic landscape for cells engineered with chimeric antigen receptors. Nat Biotechnol. 2020;38:233–44.
Arndt C, Fasslrinner F, Loureiro LR, Koristka S, Feldmann A, Bachmann M, et al. Adaptor CAR platforms-next generation of T cell-based cancer immunotherapy. Cancers. 2020;12:1302.
Krabbendam L, Bernink JH, Spits H. Innate lymphoid cells: from helper to killer. Curr Opin Immunol. 2021;68:28–33.
Chen Y, Tian Z. Innate lymphocytes: pathogenesis and therapeutic targets of liver diseases and cancer. Cell Mol Immunol. 2021;18:57–72.
Wei J, Han X, Bo J, Han W. Target selection for CAR-T therapy. J Hematol Oncol. 2019;12:62.
Fleischer LC, Spencer HT, Raikar SS. Targeting T cell malignancies using CAR-based immunotherapy: challenges and potential solutions. J Hematol Oncol. 2019;12:141.
Wu C, Zhang L, Brockman QR, Zhan F, Chen L. Chimeric antigen receptor T cell therapies for multiple myeloma. J Hematol Oncol. 2019;12:120.
Yu B, Jiang T, Liu D. BCMA-targeted immunotherapy for multiple myeloma. J Hematol Oncol. 2020;13:125.
Siegler EL, Kenderian SS. Neurotoxicity and cytokine release syndrome after chimeric antigen receptor T cell therapy: insights into mechanisms and novel therapies. Front Immunol. 2020;11:1973.
Bonifant CL, Jackson HJ, Brentjens RJ, Curran KJ. Toxicity and management in CAR T-cell therapy. Mol Ther Oncolytics. 2016;3:16011.
Shalabi H, Kraft IL, Wang HW, Yuan CM, Yates B, Delbrook C, et al. Sequential loss of tumor surface antigens following chimeric antigen receptor T-cell therapies in diffuse large B-cell lymphoma. Haematologica. 2018;103:e215–8.
Donnadieu E, Dupre L, Pinho LG, Cotta-de-Almeida V. Surmounting the obstacles that impede effective CAR T cell trafficking to solid tumors. J Leukoc Biol. 2020;108:1067–79.
Zhao Y, Shao Q, Peng G. Exhaustion and senescence: two crucial dysfunctional states of T cells in the tumor microenvironment. Cell Mol Immunol. 2020;17:27–35.
Cheng H, Ma K, Zhang L, Li G. The tumor microenvironment shapes the molecular characteristics of exhausted CD8(+) T cells. Cancer Lett. 2021;506:55–66.
Pan X, Zheng L. Epigenetics in modulating immune functions of stromal and immune cells in the tumor microenvironment. Cell Mol Immunol. 2020;17:940–53.
Ninomiya S, Narala N, Huye L, Yagyu S, Savoldo B, Dotti G, et al. Tumor indoleamine 2,3-dioxygenase (IDO) inhibits CD19-CAR T cells and is downregulated by lymphodepleting drugs. Blood. 2015;125:3905–16.
Beavis PA, Henderson MA, Giuffrida L, Mills JK, Sek K, Cross RS, et al. Targeting the adenosine 2A receptor enhances chimeric antigen receptor T cell efficacy. J Clin Investig. 2017;127:929–41.
Kloss CC, Lee J, Zhang A, Chen F, Melenhorst JJ, Lacey SF, et al. Dominant-negative TGF-beta receptor enhances PSMA-targeted human CAR T cell proliferation and augments prostate cancer eradication. Mol Ther. 2018;26:1855–66.
Zebley CC, Gottschalk S, Youngblood B. Rewriting history: epigenetic reprogramming of CD8(+) T cell differentiation to enhance immunotherapy. Trends Immunol. 2020;41:665–75.
Rafiq S, Yeku OO, Jackson HJ, Purdon TJ, van Leeuwen DG, Drakes DJ, et al. Targeted delivery of a PD-1-blocking scFv by CAR-T cells enhances anti-tumor efficacy in vivo. Nat Biotechnol. 2018;36:847–56.
Li S, Siriwon N, Zhang X, Yang S, Jin T, He F, et al. Enhanced cancer immunotherapy by chimeric antigen receptor-modified T cells engineered to secrete checkpoint inhibitors. Clin Cancer Res. 2017;23:6982–92.
Cherkassky L, Morello A, Villena-Vargas J, Feng Y, Dimitrov DS, Jones DR, et al. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J Clin Investig. 2016;126:3130–44.
Liu X, Ranganathan R, Jiang S, Fang C, Sun J, Kim S, et al. A chimeric switch-receptor targeting PD1 augments the efficacy of second-generation CAR T cells in advanced solid tumors. Cancer Res. 2016;76:1578–90.
Hyrenius-Wittsten A, Roybal KT. Paving new roads for CARs. Trends Cancer. 2019;5:583–92.
Brandt LJB, Barnkob MB, Michaels YS, Heiselberg J, Barington T. Emerging approaches for regulation and control of CAR T cells: a mini review. Front Immunol. 2020;11:326.
Arcangeli S, Mestermann K, Weber J, Bonini C, Casucci M, Hudecek M. Overcoming key challenges in cancer immunotherapy with engineered T cells. Curr Opin Oncol. 2020;32:398–407.
Liu G, Rui W, Zhao X, Lin X. Enhancing CAR-T cell efficacy in solid tumors by targeting the tumor microenvironment. Cell Mol Immunol. 2021;18:1085–95.
Chen F, Fraietta JA, June CH, Xu Z, Joseph Melenhorst J, Lacey SF. Engineered T cell therapies from a drug development viewpoint. Engineering. 2019;5:140–9.
Ruf B, Heinrich B, Greten TF. Immunobiology and immunotherapy of HCC: spotlight on innate and innate-like immune cells. Cell Mol Immunol. 2021;18:112–27.
Kansler ER, Li MO. Innate lymphocytes-lineage, localization and timing of differentiation. Cell Mol Immunol. 2019;16:627–33.
Rameshbabu S, Labadie BW, Argulian A, Patnaik A. Targeting innate immunity in cancer therapy. Vaccines. 2021;9:138.
de Araújo ND, Gama FM, de Souza Barros M, Ribeiro T, Alves FS, Xabregas LA, et al. Translating unconventional T cells and their roles in leukemia antitumor immunity. J Immunol Res. 2021;2021:6633824.
Cortes-Selva D, Dasgupta B, Singh S, Grewal IS. Innate and innate-like cells: the future of chimeric antigen receptor (CAR) cell therapy. Trends Pharmacol Sci. 2021;42:45–59.
Kabelitz D, Serrano R, Kouakanou L, Peters C, Kalyan S. Cancer immunotherapy with gammadelta T cells: many paths ahead of us. Cell Mol Immunol. 2020;17:925–39.
Vivier E, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, et al. Innate lymphoid cells: 10 years on. Cell. 2018;174:1054–66.
Sivori S, Vacca P, Del Zotto G, Munari E, Mingari MC, Moretta L. Human NK cells: surface receptors, inhibitory checkpoints, and translational applications. Cell Mol Immunol. 2019;16:430–41.
Zhang C, Hu Y, Shi C. Targeting natural killer cells for tumor immunotherapy. Front Immunol. 2020;11:60.
Myers JA, Miller JS. Exploring the NK cell platform for cancer immunotherapy. Nat Rev Clin Oncol. 2021;18:85–100.
Daher M, Rezvani K. Outlook for new CAR-based therapies with a focus on CAR NK cells: what lies beyond CAR-engineered T cells in the race against cancer. Cancer Discov. 2020;11:45–58.
Yilmaz A, Cui H, Caligiuri MA, Yu J. Chimeric antigen receptor-engineered natural killer cells for cancer immunotherapy. J Hematol Oncol. 2020;13:168.
Olson JA, Leveson-Gower DB, Gill S, Baker J, Beilhack A, Negrin RS. NK cells mediate reduction of GVHD by inhibiting activated, alloreactive T cells while retaining GVT effects. Blood. 2010;115:4293–301.
Sheng L, Mu Q, Wu X, Yang S, Zhu H, Wang J, et al. Cytotoxicity of donor natural killer cells to allo-reactive T cells are related with acute graft-vs.-host-disease following allogeneic stem cell transplantation. Front Immunol. 2020;11:1534.
Klingemann H. Are natural killer cells superior CAR drivers? Oncoimmunology. 2014;3:e28147.
Rezvani K, Rouce R, Liu E, Shpall E. Engineering natural killer cells for cancer immunotherapy. Mol Ther. 2017;25:1769–81.
Wang W, Jiang J, Wu C. CAR-NK for tumor immunotherapy: clinical transformation and future prospects. Cancer Lett. 2020;472:175–80.
Hu Y, Tian ZG, Zhang C. Chimeric antigen receptor (CAR)-transduced natural killer cells in tumor immunotherapy. Acta Pharmacol Sin. 2018;39:167–76.
Hu Y, Tian ZG, Zhang C. Natural killer cell-based immunotherapy for cancer: advances and prospects. Engineering. 2019;5:106–14.
Gong Y, Klein Wolterink RGJ, Wang J, Bos GMJ, Germeraad WTV. Chimeric antigen receptor natural killer (CAR-NK) cell design and engineering for cancer therapy. J Hematol Oncol. 2021;14:73.
Liu E, Marin D, Banerjee P, Macapinlac HA, Thompson P, Basar R, et al. Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors. N Engl J Med. 2020;382:545–53.
Valamehr B, Robinson M, Abujarour R, Rezner B, Vranceanu F, Le T, et al. Platform for induction and maintenance of transgene-free hiPSCs resembling ground state pluripotent stem cells. Stem Cell Rep. 2014;2:366–81.
Saetersmoen ML, Hammer Q, Valamehr B, Kaufman DS, Malmberg KJ. Off-the-shelf cell therapy with induced pluripotent stem cell-derived natural killer cells. Semin Immunopathol. 2019;41:59–68.
Shankar K, Capitini CM, Saha K. Genome engineering of induced pluripotent stem cells to manufacture natural killer cell therapies. Stem Cell Res Ther. 2020;11:234.
Li Y, Hermanson DL, Moriarity BS, Kaufman DS. Human iPSC-derived natural killer cells engineered with chimeric antigen receptors enhance anti-tumor activity. Cell Stem Cell. 2018;23:181–92.e185.
Wu L, Wei Q, Brzostek J, Gascoigne NRJ. Signaling from T cell receptors (TCRs) and chimeric antigen receptors (CARs) on T cells. Cell Mol Immunol. 2020;17:600–12.
Liu D, Badeti S, Dotti G, Jiang JG, Wang H, Dermody J, et al. The role of immunological synapse in predicting the efficacy of chimeric antigen receptor (CAR) immunotherapy. Cell Commun Signal. 2020;18:134.
Zenere G, Olwenyi OA, Byrareddy SN, Braun SE. Optimizing intracellular signaling domains for CAR NK cells in HIV immunotherapy: a comprehensive review. Drug Discov Today. 2019;24:983–91.
Upshaw JL, Arneson LN, Schoon RA, Dick CJ, Billadeau DD, Leibson PJ. NKG2D-mediated signaling requires a DAP10-bound Grb2-Vav1 intermediate and phosphatidylinositol-3-kinase in human natural killer cells. Nat Immunol. 2006;7:524–32.
Chen S, Dong Z. NK cell recognition of hematopoietic cells by SLAM-SAP families. Cell Mol Immunol. 2019;16:452–9.
Dong Z, Davidson D, Pérez-Quintero LA, Kurosaki T, Swat W, Veillette A. The adaptor SAP controls NK cell activation by regulating the enzymes Vav-1 and SHIP-1 and by enhancing conjugates with target cells. Immunity. 2012;36:974–85.
Chang YH, Connolly J, Shimasaki N, Mimura K, Kono K, Campana D. A chimeric receptor with NKG2D specificity enhances natural killer cell activation and killing of tumor cells. Cancer Res. 2013;73:1777–86.
Imai C, Iwamoto S, Campana D. Genetic modification of primary natural killer cells overcomes inhibitory signals and induces specific killing of leukemic cells. Blood. 2005;106:376–83.
Xiao L, Cen D, Gan H, Sun Y, Huang N, Xiong H, et al. Adoptive transfer of NKG2D CAR mRNA-engineered natural killer cells in colorectal cancer patients. Mol Ther. 2019;27:1114–25.
Töpfer K, Cartellieri M, Michen S, Wiedemuth R, Müller N, Lindemann D, et al. DAP12-based activating chimeric antigen receptor for NK cell tumor immunotherapy. J Immunol. 2015;194:3201–12.
Xu Y, Liu Q, Zhong M, Wang Z, Chen Z, Zhang Y, et al. 2B4 costimulatory domain enhancing cytotoxic ability of anti-CD5 chimeric antigen receptor engineered natural killer cells against T cell malignancies. J Hematol. Oncol. 2019;12:49.
Huang Y, Zeng J, Liu T, Xu Q, Song X, Zeng J. DNAM1 and 2B4 costimulatory domains enhance the cytotoxicity of Anti-GPC3 chimeric antigen receptor-modified natural killer cells against hepatocellular cancer cells in vitro. Cancer Manag Res. 2020;12:3247–55.
Burger MC, Zhang C, Harter PN, Romanski A, Strassheimer F, Senft C, et al. CAR-engineered NK cells for the treatment of glioblastoma: turning innate effectors into precision tools for cancer immunotherapy. Front Immunol. 2019;10:2683.
Cao B, Liu M, Wang L, Liang B, Feng Y, Chen X, et al. Use of chimeric antigen receptor NK-92 cells to target mesothelin in ovarian cancer. Biochem Biophys Res Commun. 2020;524:96–102.
Mantovani S, Oliviero B, Varchetta S, Mele D, Mondelli MU. Natural killer cell responses in hepatocellular carcinoma: implications for novel immunotherapeutic approaches. Cancers. 2020;12:926.
Montagner IM, Penna A, Fracasso G, Carpanese D, Pietà AD, Barbieri V, et al. Anti-PSMA CAR-engineered NK-92 cells: an off-the-shelf cell therapy for prostate cancer. Cells. 2020;9,:1382.
Tang X, Yang L, Li Z, Nalin AP, Dai H, Xu T, et al. First-in-man clinical trial of CAR NK-92 cells: safety test of CD33-CAR NK-92 cells in patients with relapsed and refractory acute myeloid leukemia. Am J Cancer Res. 2018;8:1083–9.
Bachanova V, Cayci Z, Lewis D, Maakaron JE, Janakiram M, Bartz A, et al. Initial clinical activity of FT596, a first-in-class, multi-antigen targeted, off-the-shelf, iPSC-derived CD19 CAR NK cell therapy in relapsed/refractory B-Cell lymphoma. Blood. 2020;136:8.
Fiore PF, Di Matteo S, Tumino N, Mariotti FR, Pietra G, Ottonello S, et al. Interleukin-15 and cancer: some solved and many unsolved questions. J Immunother Cancer. 2020;8:e001428.
Rautela J, Huntington ND. IL-15 signaling in NK cell cancer immunotherapy. Curr Opin Immunol. 2017;44:1–6.
Tamzalit F, Barbieux I, Plet A, Heim J, Nedellec S, Morisseau S, et al. IL-15.IL-15Ralpha complex shedding following trans-presentation is essential for the survival of IL-15 responding NK and T cells. Proc Natl Acad Sci USA. 2014;111:8565–70.
Desbois M, Béal C, Charrier M, Besse B, Meurice G, Cagnard N, et al. IL-15 superagonist RLI has potent immunostimulatory properties on NK cells: implications for antimetastatic treatment. J Immunother Cancer. 2020;8:e000632.
Desbois M, Le Vu P, Coutzac C, Marcheteau E, Béal C, Terme M, et al. IL-15 trans-signaling with the superagonist RLI promotes effector/memory CD8+ T cell responses and enhances antitumor activity of PD-1 antagonists. J Immunol. 2016;197:168–78.
Liu B, Jones M, Kong L, Noel T, Jeng EK, Shi S, et al. Evaluation of the biological activities of the IL-15 superagonist complex, ALT-803, following intravenous versus subcutaneous administration in murine models. Cytokine. 2018;107:105–12.
Romee R, Cooley S, Berrien-Elliott MM, Westervelt P, Verneris MR, Wagner JE, et al. First-in-human phase 1 clinical study of the IL-15 superagonist complex ALT-803 to treat relapse after transplantation. Blood. 2018;131:2515–27.
Margolin K, Morishima C, Velcheti V, Miller JS, Lee SM, Silk AW, et al. Phase I trial of ALT-803, a novel recombinant IL15 complex, in patients with advanced solid tumors. Clin Cancer Res. 2018;24:5552–61.
Jiang W, Zhang C, Tian Z, Zhang J. hIL-15-gene modified human natural killer cells (NKL-IL15) exhibit anti-human leukemia functions. J Cancer Res Clin Oncol. 2018;144:1279–88.
Jiang W, Zhang C, Tian Z, Zhang J. hIL-15 gene-modified human natural killer cells (NKL-IL15) augments the anti-human hepatocellular carcinoma effect in vivo. Immunobiology. 2014;219:547–53.
Imamura M, Shook D, Kamiya T, Shimasaki N, Chai SM, Coustan-Smith E, et al. Autonomous growth and increased cytotoxicity of natural killer cells expressing membrane-bound interleukin-15. Blood. 2014;124:1081–8.
Delconte RB, Kolesnik TB, Dagley LF, Rautela J, Shi W, Putz EM, et al. CIS is a potent checkpoint in NK cell-mediated tumor immunity. Nat Immunol. 2016;17:816–24.
Daher M, Basar R, Gokdemir E, Baran N, Uprety N, Nunez Cortes AK, et al. Targeting a cytokine checkpoint enhances the fitness of armored cord blood CAR-NK cells. Blood. 2021;137:624–36.
Zhu H, Blum RH, Bernareggi D, Ask EH, Wu Z, Hoel HJ, et al. Metabolic reprograming via deletion of CISH in human iPSC-Derived NK cells promotes in vivo persistence and enhances anti-tumor activity. Cell Stem Cell. 2020;27:224–37.e226.
Habif G, Crinier A, Andre P, Vivier E, Narni-Mancinelli E. Targeting natural killer cells in solid tumors. Cell Mol Immunol. 2019;16:415–22.
Müller N, Michen S, Tietze S, Töpfer K, Schulte A, Lamszus K, et al. Engineering NK cells modified with an EGFRvIII-specific chimeric antigen receptor to overexpress CXCR4 improves immunotherapy of CXCL12/SDF-1alpha-secreting glioblastoma. J Immunother. 2015;38:197–210.
Jamali A, Hadjati J, Madjd Z, Mirzaei HR, Thalheimer FB, Agarwal S, et al. Highly efficient generation of transgenically augmented CAR NK cells overexpressing CXCR4. Front Immunol. 2020;11:2028.
Ng YY, Tay JCK, Wang S. CXCR1 expression to improve anti-cancer efficacy of intravenously injected CAR-NK cells in mice with peritoneal xenografts. Mol Ther Oncolytics. 2020;16:75–85.
Kremer V, Ligtenberg MA, Zendehdel R, Seitz C, Duivenvoorden A, Wennerberg E, et al. Genetic engineering of human NK cells to express CXCR2 improves migration to renal cell carcinoma. J Immunother Cancer. 2017;5:73.
Jin L, Tao H, Karachi A, Long Y, Hou AY, Na M, et al. CXCR1- or CXCR2-modified CAR T cells co-opt IL-8 for maximal antitumor efficacy in solid tumors. Nat Commun. 2019;10:4016.
Liu G, Rui W, Zheng H, Huang D, Yu F, Zhang Y, et al. CXCR2-modified CAR-T cells have enhanced trafficking ability that improves treatment of hepatocellular carcinoma. Eur J Immunol. 2020;50:712–24.
Whilding LM, Halim L, Draper B, Parente-Pereira AC, Zabinski T, Davies DM, et al. CAR T-cells targeting the integrin alphavbeta6 and Co-expressing the chemokine receptor CXCR2 demonstrate enhanced homing and efficacy against several solid malignancies. Cancers. 2019;11:674.
Wang Y, Wang J, Yang X, Yang J, Lu P, Zhao L, et al. Chemokine receptor CCR2b enhanced anti-tumor function of chimeric antigen receptor T cells targeting mesothelin in a non-small-cell lung carcinoma model. Front Immunol. 2021;12:628906.
Riggan L, Shah S, O’Sullivan TE. Arrested development: suppression of NK cell function in the tumor microenvironment. Clin Transl Immunol. 2021;10:e1238.
Cozar B, Greppi M, Carpentier S, Narni-Mancinelli E, Chiossone L, Vivier E, et al. Tumor-Infiltrating natural killer cells. Cancer Discov. 2020;11:34–44.
Huang Q, Huang M, Meng F, Sun R. Activated pancreatic stellate cells inhibit NK cell function in the human pancreatic cancer microenvironment. Cell Mol Immunol. 2019;16:87–9.
Nakamura K, Smyth MJ. Myeloid immunosuppression and immune checkpoints in the tumor microenvironment. Cell Mol Immunol. 2020;17:1–12.
Zhang Y, Zhang Z. The history and advances in cancer immunotherapy: understanding the characteristics of tumor-infiltrating immune cells and their therapeutic implications. Cell Mol Immunol. 2020;17:807–21.
Yvon ES, Burga R, Powell A, Cruz CR, Fernandes R, Barese C, et al. Cord blood natural killer cells expressing a dominant negative TGF-beta receptor: implications for adoptive immunotherapy for glioblastoma. Cytotherapy. 2017;19:408–18.
Yang B, Liu H, Shi W, Wang Z, Sun S, Zhang G, et al. Blocking transforming growth factor-beta signaling pathway augments antitumor effect of adoptive NK-92 cell therapy. Int Immunopharmacol. 2013;17:198–204.
Wang Z, Guo L, Song Y, Zhang Y, Lin D, Hu B, et al. Augmented anti-tumor activity of NK-92 cells expressing chimeric receptors of TGF-betaR II and NKG2D. Cancer Immunol Immunother. 2017;66:537–48.
Burga RA, Yvon E, Chorvinsky E, Fernandes R, Cruz C, Bollard CM. Engineering the TGFbeta receptor to enhance the therapeutic potential of natural killer cells as an immunotherapy for neuroblastoma. Clin Cancer Res. 2019;25:4400–12.
Mohammed S, Sukumaran S, Bajgain P, Watanabe N, Heslop HE, Rooney CM, et al. Improving chimeric antigen receptor-modified T cell function by reversing the immunosuppressive tumor microenvironment of pancreatic cancer. Mol Ther. 2017;25:249–58.
Lu C, Guo C, Chen H, Zhang H, Zhi L, Lv T, et al. A novel chimeric PD1-NKG2D-41BB receptor enhances antitumor activity of NK92 cells against human lung cancer H1299 cells by triggering pyroptosis. Mol Immunol. 2020;122:200–6.
Liang Y, Liu H, Lu Z, Lei W, Zhang C, Li P, et al. CD19 CAR-T expressing PD-1/CD28 chimeric switch receptor as a salvage therapy for DLBCL patients treated with different CD19-directed CAR T-cell therapies. J Hematol Oncol. 2021;14:26.
Liu H, Lei W, Zhang C, Yang C, Wei J, Guo Q, et al. CD19-specific CAR T cells that express a PD-1/CD28 chimeric switch-receptor are effective in patients with PD-L1-positive B-cell lymphoma. Clin Cancer Res. 2021;27:473–84.
Hoogi S, Eisenberg V, Mayer S, Shamul A, Barliya T, Cohen CJ. A TIGIT-based chimeric co-stimulatory switch receptor improves T-cell anti-tumor function. J Immunother Cancer. 2019;7:243.
Huang RS, Lai MC, Shih HA, Lin S. A robust platform for expansion and genome editing of primary human natural killer cells. J Exp Med. 2021;218:e20201529.
Huang RS, Shih HA, Lai MC, Chang YJ, Lin S. Enhanced NK-92 cytotoxicity by CRISPR genome engineering using Cas9 ribonucleoproteins. Front Immunol. 2020;11:1008.
Afolabi LO, Adeshakin AO, Sani MM, Bi J, Wan X. Genetic reprogramming for NK cell cancer immunotherapy with CRISPR/Cas9. Immunology. 2019;158:63–9.
Zhang Q, Bi J, Zheng X, Chen Y, Wang H, Wu W, et al. Blockade of the checkpoint receptor TIGIT prevents NK cell exhaustion and elicits potent anti-tumor immunity. Nat Immunol. 2018;19:723–32.
Pomeroy EJ, Hunzeker JT, Kluesner MG, Lahr WS, Smeester BA, Crosby MR, et al. A genetically engineered primary human natural killer cell platform for cancer immunotherapy. Mol Ther. 2020;28:52–63.
Parihar R, Rivas C, Huynh M, Omer B, Lapteva N, Metelitsa LS, et al. NK cells expressing a chimeric activating receptor eliminate MDSCs and rescue impaired CAR-T cell activity against solid tumors. Cancer Immunol Res. 2019;7:363–75.
Fabian KP, Padget MR, Donahue RN, Solocinski K, Robbins Y, Allen CT, et al. PD-L1 targeting high-affinity NK (t-haNK) cells induce direct antitumor effects and target suppressive MDSC populations. J Immunother. Cancer. 2020;8:e000450.
Rafiq S, Hackett CS, Brentjens RJ. Engineering strategies to overcome the current roadblocks in CAR T cell therapy. Nat Rev Clin Oncol 2020;17:147–67.
Chen N, Li X, Chintala NK, Tano ZE, Adusumilli PS. Driving CARs on the uneven road of antigen heterogeneity in solid tumors. Curr Opin Immunol. 2018;51:103–10.
Ramakrishna S, Barsan V, Mackall C. Prospects and challenges for use of CAR T cell therapies in solid tumors. Expert Opin Biol Ther. 2020;20:503–16.
Shah NN, Zhu F, Schneider D, Taylor C, Krueger W, Worden A, et al. Results of a phase I study of bispecific anti-CD19, anti-CD20 chimeric antigen receptor (CAR) modified T cells for relapsed, refractory, non-Hodgkin lymphoma. J Clin Oncol. 2019;37:2510.
Mahadeo KM, Khazal SJ, Abdel-Azim H, Fitzgerald JC, Taraseviciute A, Bollard CM, et al. Management guidelines for paediatric patients receiving chimeric antigen receptor T cell therapy. Nat Rev Clin Oncol. 2019;16:45–63.
Dai H, Wu Z, Jia H, Tong C, Guo Y, Ti D, et al. Bispecific CAR-T cells targeting both CD19 and CD22 for therapy of adults with relapsed or refractory B cell acute lymphoblastic leukemia. J Hematol Oncol. 2020;13:30.
Jiang W, Li T, Guo J, Wang J, Jia L, Shi X, et al. Bispecific c-Met/PD-L1 CAR-T Cells have enhanced therapeutic effects on hepatocellular carcinoma. Front Oncol. 2021;11:546586.
Zhao W, Jia L, Zhang M, Huang X, Qian P, Tang Q, et al. The killing effect of novel bi-specific Trop2/PD-L1 CAR-T cell targeted gastric cancer. Am J Cancer Res. 2019;9:1846–56.
Li M, Zhi L, Yin M, Guo C, Zhang H, Lu C, et al. A novel bispecific chimeric PD1-DAP10/NKG2D receptor augments NK92-cell therapy efficacy for human gastric cancer SGC-7901 cell. Biochem Biophys Res Commun. 2020;523:745–52.
Nair S, Dhodapkar MV. Natural killer T cells in cancer immunotherapy. Front Immunol. 2017;8:1178.
Keller CW, Freigang S, Lunemann JD. Reciprocal crosstalk between dendritic cells and natural killer T cells: mechanisms and therapeutic potential. Front Immunol. 2017;8:570.
Song L, Asgharzadeh S, Salo J, Engell K, Wu HW, Sposto R, et al. Valpha24-invariant NKT cells mediate antitumor activity via killing of tumor-associated macrophages. J Clin Investig. 2009;119:1524–36.
Paul S, Chhatar S, Mishra A, Lal G. Natural killer T cell activation increases iNOS(+)CD206(-) M1 macrophage and controls the growth of solid tumor. J Immunother Cancer. 2019;7:208.
Ko HJ, Lee JM, Kim YJ, Kim YS, Lee KA, Kang CY. Immunosuppressive myeloid-derived suppressor cells can be converted into immunogenic APCs with the help of activated NKT cells: an alternative cell-based antitumor vaccine. J Immunol. 2009;182:1818–28.
De Santo C, Arscott R, Booth S, Karydis I, Jones M, Asher R, et al. Invariant NKT cells modulate the suppressive activity of IL-10-secreting neutrophils differentiated with serum amyloid A. Nat Immunol. 2010;11:1039–46.
Bedard M, Salio M, Cerundolo V. Harnessing the power of invariant natural killer T cells in cancer immunotherapy. Front Immunol. 2017;8:1829.
Exley MA, Friedlander P, Alatrakchi N, Vriend L, Yue S, Sasada T, et al. Adoptive transfer of invariant NKT cells as immunotherapy for advanced melanoma: a phase I clinical trial. Clin Cancer Res. 2017;23:3510–9.
Yamasaki K, Horiguchi S, Kurosaki M, Kunii N, Nagato K, Hanaoka H, et al. Induction of NKT cell-specific immune responses in cancer tissues after NKT cell-targeted adoptive immunotherapy. Clin Immunol. 2011;138:255–65.
Toyoda T, Kamata T, Tanaka K, Ihara F, Takami M, Suzuki H, et al. Phase II study of alpha-galactosylceramide-pulsed antigen-presenting cells in patients with advanced or recurrent non-small cell lung cancer. J Immunother Cancer. 2020;8:e000316.
Heczey A, Liu D, Tian G, Courtney AN, Wei J, Marinova E, et al. Invariant NKT cells with chimeric antigen receptor provide a novel platform for safe and effective cancer immunotherapy. Blood. 2014;124:2824–33.
Xu X, Huang W, Heczey A, Liu D, Guo L, Wood M, et al. NKT cells coexpressing a GD2-specific chimeric antigen receptor and IL15 show enhanced in vivo persistence and antitumor activity against neuroblastoma. Clin Cancer Res. 2019;25:7126–38.
Heczey A, Courtney AN, Montalbano A, Robinson S, Liu K, Li M, et al. Anti-GD2 CAR-NKT cells in patients with relapsed or refractory neuroblastoma: an interim analysis. Nat Med. 2020;26:1686–90.
Rotolo A, Caputo VS, Holubova M, Baxan N, Dubois O, Chaudhry MS, et al. Enhanced anti-lymphoma activity of CAR19-iNKT cells underpinned by dual CD19 and CD1d targeting. Cancer Cell. 2018;34:596–610. e511
Tian G, Courtney AN, Jena B, Heczey A, Liu D, Marinova E, et al. CD62L+ NKT cells have prolonged persistence and antitumor activity in vivo. J Clin Investig. 2016;126:2341–55.
Pillai AB, George TI, Dutt S, Teo P, Strober S. Host NKT cells can prevent graft-versus-host disease and permit graft antitumor activity after bone marrow transplantation. J Immunol. 2007;178:6242–51.
Zhu Y, Smith DJ, Zhou Y, Li YR, Yu J, Lee D, et al. Development of hematopoietic stem cell-engineered invariant natural killer T cell therapy for cancer. Cell Stem Cell. 2019;25:542–57.e549.
Murray MP, Kronenberg M. Engineered stem cells provide cancer-killing iNKT cells. Cell Stem Cell. 2019;25:454–5.
Smith DJ, Liu S, Ji S, Li B, McLaughlin J, Cheng D, et al. Genetic engineering of hematopoietic stem cells to generate invariant natural killer T cells. Proc Natl Acad Sci. USA. 2015;112:1523–8.
Yamada D, Iyoda T, Vizcardo R, Shimizu K, Sato Y, Endo TA, et al. Efficient regeneration of human Valpha24(+) invariant natural killer T cells and their anti-tumor activity in vivo. Stem Cells. 2016;34:2852–60.
Sun W, Wang Y, East JE, Kimball AS, Tkaczuk K, Kesmodel S, et al. Invariant natural killer T cells generated from human adult hematopoietic stem-progenitor cells are poly-functional. Cytokine. 2015;72:48–57.
Chien YH, Meyer C, Bonneville M. gammadelta T cells: first line of defense and beyond. Annu Rev Immunol. 2014;32:121–55.
Deseke M, Prinz I. Ligand recognition by the gammadelta TCR and discrimination between homeostasis and stress conditions. Cell Mol Immunol. 2020;17:914–24.
Khan MW, Curbishley SM, Chen HC, Thomas AD, Pircher H, Mavilio D, et al. Expanded human blood-derived gammadelta T cells display potent antigen-presentation functions. Front Immunol. 2014;5:344.
Himoudi N, Morgenstern DA, Yan M, Vernay B, Saraiva L, Wu Y, et al. Human gammadelta T lymphocytes are licensed for professional antigen presentation by interaction with opsonized target cells. J Immunol. 2012;188:1708–16.
Yang Y, Li L, Yuan L, Zhou X, Duan J, Xiao H, et al. A structural change in butyrophilin upon phosphoantigen binding underlies phosphoantigen-mediated Vgamma9Vdelta2 T cell activation. Immunity. 2019;50:1043–53.e1045.
Karunakaran MM, Willcox CR, Salim M, Paletta D, Fichtner AS, Noll A, et al. Butyrophilin-2A1 directly binds germline-encoded regions of the Vgamma9Vdelta2 TCR and is essential for phosphoantigen sensing. Immunity. 2020;52:487–98.e486.
Rigau M, Ostrouska S, Fulford TS, Johnson DN, Woods K, Ruan Z, et al. Butyrophilin 2A1 is essential for phosphoantigen reactivity by gammadelta T cells. Science. 2020;367:eaay5516.
Raverdeau M, Cunningham SP, Harmon C, Lynch L. Gammadelta T cells in cancer: a small population of lymphocytes with big implications. Clin Transl Immunol. 2019;8:e01080.
Yazdanifar M, Barbarito G, Bertaina A, Airoldi I. Gammadelta T cells: the ideal tool for cancer immunotherapy. Cells. 2020;9:1305.
Xu B, Pizarro JC, Holmes MA, McBeth C, Groh V, Spies T, et al. Crystal structure of a gammadelta T-cell receptor specific for the human MHC class I homolog MICA. Proc Natl Acad Sci USA. 2011;108:2414–9.
Spada FM, Grant EP, Peters PJ, Sugita M, Melián A, Leslie DS, et al. Self-recognition of CD1 by gamma/delta T cells: implications for innate immunity. J Exp Med. 2000;191:937–48.
Uldrich AP, Le Nours J, Pellicci DG, Gherardin NA, McPherson KG, Lim RT, et al. CD1d-lipid antigen recognition by the gammadelta TCR. Nat Immunol. 2013;14:1137–45.
Silva-Santos B, Strid J. Working in “NK Mode”: natural killer group 2 Member D and natural cytotoxicity receptors in stress-surveillance by gammadelta T cells. Front Immunol. 2018;9:851.
Hudspeth K, Silva-Santos B, Mavilio D. Natural cytotoxicity receptors: broader expression patterns and functions in innate and adaptive immune cells. Front Immunol. 2013;4:69.
Mikulak J, Oriolo F, Bruni E, Roberto A, Colombo FS, Villa A, et al. NKp46-expressing human gut-resident intraepithelial Vdelta1 T cell subpopulation exhibits high antitumor activity against colorectal cancer. Jci Insight 2019;4:e125884.
Siegers GM, Lamb LS Jr. Cytotoxic and regulatory properties of circulating Vdelta1+ gammadelta T cells: a new player on the cell therapy field? Mol Ther. 2014;22:1416–22.
Liu Y, Zhang C. The role of human gammadelta T cells in anti-tumor immunity and their potential for cancer immunotherapy. Cells. 2020;9:1206.
Li Y, Li G, Zhang J, Wu X, Chen X. The dual roles of human gammadelta T cells: anti-tumor or tumor-promoting. Front Immunol. 2020;11:619954.
Bennouna J, Bompas E, Neidhardt EM, Rolland F, Philip I, Galéa C, et al. Phase-I study of Innacell gammadelta, an autologous cell-therapy product highly enriched in gamma9delta2 T lymphocytes, in combination with IL-2, in patients with metastatic renal cell carcinoma. Cancer Immunol Immunother. 2008;57:1599–609.
Sakamoto M, Nakajima J, Murakawa T, Fukami T, Yoshida Y, Murayama T, et al. Adoptive immunotherapy for advanced non-small cell lung cancer using zoledronate-expanded gammadeltaTcells: a phase I clinical study. J Immunother. 2011;34:202–11.
Nicol AJ, Tokuyama H, Mattarollo SR, Hagi T, Suzuki K, Yokokawa K, et al. Clinical evaluation of autologous gamma delta T cell-based immunotherapy for metastatic solid tumours. Br J Cancer. 2011;105:778–86.
Kobayashi H, Tanaka Y, Yagi J, Minato N, Tanabe K. Phase I/II study of adoptive transfer of gammadelta T cells in combination with zoledronic acid and IL-2 to patients with advanced renal cell carcinoma. Cancer Immunol Immunother. 2011;60:1075–84.
Kakimi K, Matsushita H, Masuzawa K, Karasaki T, Kobayashi Y, Nagaoka K, et al. Adoptive transfer of zoledronate-expanded autologous Vgamma9Vdelta2 T-cells in patients with treatment-refractory non-small-cell lung cancer: a multicenter, open-label, single-arm, phase 2 study. J Immunother Cancer. 2020;8:e001185.
Alnaggar M, Xu Y, Li J, He J, Chen J, Li M, et al. Allogenic Vgamma9Vdelta2 T cell as new potential immunotherapy drug for solid tumor: a case study for cholangiocarcinoma. J Immunother Cancer. 2019;7:36.
Lang JM, Kaikobad MR, Wallace M, Staab MJ, Horvath DL, Wilding G, et al. Pilot trial of interleukin-2 and zoledronic acid to augment gammadelta T cells as treatment for patients with refractory renal cell carcinoma. Cancer Immunol Immunother. 2011;60:1447–60.
Pressey JG, Adams J, Harkins L, Kelly D, You Z, Lamb LS Jr. In vivo expansion and activation of gammadelta T cells as immunotherapy for refractory neuroblastoma: a phase 1 study. Medicine. 2016;95:e4909.
Almeida AR, Correia DV, Fernandes-Platzgummer A, da Silva CL, da Silva MG, Anjos DR, et al. Delta one T cells for immunotherapy of chronic lymphocytic leukemia: clinical-grade expansion/differentiation and preclinical proof of concept. Clin Cancer Res. 2016;22:5795–804.
Di Lorenzo B, Simões AE, Caiado F, Tieppo P, Correia DV, Carvalho T, et al. Broad cytotoxic targeting of acute myeloid leukemia by polyclonal delta one T cells. Cancer Immunol Res. 2019;7:552–8.
Deniger DC, Switzer K, Mi T, Maiti S, Hurton L, Singh H, et al. Bispecific T-cells expressing polyclonal repertoire of endogenous gammadelta T-cell receptors and introduced CD19-specific chimeric antigen receptor. Mol Ther. 2013;21:638–47.
Rozenbaum M, Meir A, Aharony Y, Itzhaki O, Schachter J, Bank I, et al. Gamma-delta CAR-T cells show CAR-directed and independent activity against leukemia. Front Immunol. 2020;11:1347.
Capsomidis A, Benthall G, Van Acker HH, Fisher J, Kramer AM, Abeln Z, et al. Chimeric antigen receptor-engineered human gamma delta T cells: enhanced cytotoxicity with retention of cross presentation. Mol Ther. 2018;26:354–65.
Fisher J, Abramowski P, Wisidagamage Don ND, Flutter B, Capsomidis A, Cheung GW, et al. avoidance of on-target off-tumor activation using a co-stimulation-only chimeric antigen receptor. Mol Ther. 2017;25:1234–47.
Ang WX, Ng YY, Xiao L, Chen C, Li Z, Chi Z, et al. Electroporation of NKG2D RNA CAR improves Vgamma9Vdelta2 T cell responses against human solid tumor xenografts. Mol Ther Oncolytics. 2020;17:421–30.
Marcu-Malina V, Heijhuurs S, van Buuren M, Hartkamp L, Strand S, Sebestyen Z, et al. Redirecting alphabeta T cells against cancer cells by transfer of a broadly tumor-reactive gammadeltaT-cell receptor. Blood. 2011;118:50–9.
Johanna I, Straetemans T, Heijhuurs S, Aarts-Riemens T, Norell H, Bongiovanni L, et al. Evaluating in vivo efficacy—toxicity profile of TEG001 in humanized mice xenografts against primary human AML disease and healthy hematopoietic cells. J Immunother Cancer. 2019;7:69.
Sebestyen Z, Prinz I, Dechanet-Merville J, Silva-Santos B, Kuball J. Translating gammadelta (gammadelta) T cells and their receptors into cancer cell therapies. Nat Rev Drug Discov. 2020;19:169–84.
Braham MVJ, Minnema MC, Aarts T, Sebestyen Z, Straetemans T, Vyborova A, et al. Cellular immunotherapy on primary multiple myeloma expanded in a 3D bone marrow niche model. Oncoimmunology. 2018;7:e1434465.
Johanna I, Hernández-López P, Heijhuurs S, Bongiovanni L, de Bruin A, Beringer D, et al. TEG011 persistence averts extramedullary tumor growth without exerting off-target toxicity against healthy tissues in a humanized HLA-A*24:02 transgenic mice. J Leukoc Biol. 2020;107:1069–79.
Straetemans T, Kierkels G, Doorn R, Jansen K, Heijhuurs S, Dos Santos JM, et al. GMP-grade manufacturing of T cells engineered to express a defined gammadeltaTCR. Front Immunol. 2018;9:1062.
Straetemans T, Gründer C, Heijhuurs S, Hol S, Slaper-Cortenbach I, Bönig H, et al. Untouched GMP-ready purified engineered immune cells to treat cancer. Clin Cancer Res. 2015;21:3957–68.
Kundu S, Gurney M, O’Dwyer M. Generating natural killer cells for adoptive transfer: expanding horizons. Cytotherapy. 2021;23:559–66.
Yu H, Chen W, Li C, Lin D, Liu J, Yang Z, et al. Large scale ex vivo expansion of clinical-grade effector cells for adoptive immunotherapy. Exp Ther Med. 2017;14:5678–86.
Zhu H, Kaufman DS. An improved method to produce clinical-scale natural killer cells from human pluripotent stem cells. Methods Mol Biol. 2019;2048:107–19.
Schmidt P, Raftery MJ, Pecher G. Engineering NK cells for CAR therapy-recent advances in gene transfer methodology. Front Immunol. 2020;11:611163.
Müller S, Bexte T, Gebel V, Kalensee F, Stolzenberg E, Hartmann J, et al. High cytotoxic efficiency of lentivirally and alpharetrovirally engineered CD19-specific chimeric antigen receptor natural killer cells against acute lymphoblastic leukemia. Front Immunol. 2019;10:3123.
Roselli E, Frieling JS, Thorner K, Ramello MC, Lynch CC, Abate-Daga D. CAR-T engineering: optimizing signal transduction and effector mechanisms. BioDrugs. 2019;33:647–59.
Wan Z, Shao X, Ji X, Dong L, Wei J, Xiong Z, et al. Transmembrane domain-mediated Lck association underlies bystander and costimulatory ICOS signaling. Cell Mol Immunol. 2020;17:143–52.
Etxeberria I, Olivera I, Bolaños E, Cirella A, Teijeira Á, Berraondo P, et al. Engineering bionic T cells: signal 1, signal 2, signal 3, reprogramming and the removal of inhibitory mechanisms. Cell Mol Immunol. 2020;17:576–86.
Wang X, Peng H, Tian Z. Innate lymphoid cell memory. Cell Mol Immunol. 2019;16:423–9.
Chen Y, Tian Z, Peng H. Immunological memory: ILC1s come into view. Cell Mol Immunol. 2019;16:895–6.
Peng H, Tian Z. Natural killer cell memory: progress and implications. Front Immunol. 2017;8:1143.
Sarhan D, Cichocki F, Zhang B, Yingst A, Spellman SR, Cooley S, et al. Adaptive NK cells with low TIGIT expression are inherently resistant to myeloid-derived suppressor cells. Cancer Res. 2016;76:5696–706.
Sarhan D, Hippen KL, Lemire A, Hying S, Luo X, Lenvik T, et al. Adaptive NK cells resist regulatory T-cell suppression driven by IL37. Cancer Immunol Res. 2018;6:766–75.
Romee R, Rosario M, Berrien-Elliott MM, Wagner JA, Jewell BA, Schappe T, et al. Cytokine-induced memory-like natural killer cells exhibit enhanced responses against myeloid leukemia. Sci Transl Med. 2016;8:357ra123.
Gang M, Marin ND, Wong P, Neal CC, Marsala L, Foster M, et al. CAR-modified memory-like NK cells exhibit potent responses to NK-resistant lymphomas. Blood. 2020;136:2308–18.
Roybal KT, Lim WA. Synthetic immunology: hacking immune cells to expand their therapeutic capabilities. Annu Rev Immunol. 2017;35:229–53.
Kim S, Shah SB, Graney PL, Singh A. Multiscale engineering of immune cells and lymphoid organs. Nat Rev Mater. 2019;4:355–78.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (81788101) and the CAMS Innovation Fund for Medical Sciences (CIFMS 2019-I2M-5-073).
Author information
Authors and Affiliations
Contributions
ZT and CZ conceptualized this review. CZ wrote the paper. YH and WX engaged in discussion and offered suggestions for the paper. ZT provided guidance on the outline of this review and revised the paper. All authors approved the final submitted version of the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
About this article

Cite this article
Zhang, C., Hu, Y., Xiao, W. et al. Chimeric antigen receptor- and natural killer cell receptor-engineered innate killer cells in cancer immunotherapy. Cell Mol Immunol 18, 2083–2100 (2021). https://doi.org/10.1038/s41423-021-00732-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41423-021-00732-6
Keywords
This article is cited by
-
Chimeric antigen receptor-based natural killer cell immunotherapy in cancer: from bench to bedside
Cell Death & Disease (2024)
-
Understanding the Interplay of CAR-NK Cells and Triple-Negative Breast Cancer: Insights from Computational Modeling
Bulletin of Mathematical Biology (2024)
-
Exploiting innate immunity for cancer immunotherapy
Molecular Cancer (2023)
-
Exploring the promising potential of induced pluripotent stem cells in cancer research and therapy
Molecular Cancer (2023)
-
Remote control of cellular immunotherapy
Nature Reviews Bioengineering (2023)
