
Abstract
The inherited retinal diseases (IRDs) have traditionally been described phenotypically with the description evolving to incorporate more sophisticated structural and functional assessments. In the last 25 years there has been considerable advances in the understanding of underlying genetic aetiologies. The role of the ophthalmologist is now to work in a multi-disciplinary team to identify the disease-causing genotype, which might be amenable to gene-directed intervention. Visual electrophysiology is an important tool to assist the ophthalmologist in guiding the clinical geneticist to reach a final molecular diagnosis. This review outlines the physiological basis for the ISCEV standard electrophysiology tests, the role of electrophysiology in localising the functional deficit, correlation with structural findings to guide diagnosis and finally management of IRDs in the era of genomics with emphasis on the outer retina.
Similar content being viewed by others
Introduction
The electroretinogram (ERG) has been a useful functional investigation used to interrogate different components of the afferent visual pathway for over a 100 years. With recent advances in retinal imaging, and the genomic revolution, the question arises, where does the ERG fit in the current diagnostic paradigm? In this review we will demonstrate the continued importance that the ERG plays in understanding the structure–function correlation in the diagnosis, natural history and monitoring treatment for inherited retinal disorders in the genomics era, with particular emphasis on the outer retina.
Retinal structure and function: the electroretinogram
In understanding the origins and cellular contributions of the retina that is recorded by the standardised ERG, the clinician will be assisted in determining the site of dysfunction in the visual pathway. Interpreting the ERG also requires an understanding of the retinal circuitry and the relative concentration and distribution of the individual elements. Cone photoreceptors comprise 5% of the total photoreceptors in the retina (4.6 million from a total 92 million) with 90% of the cones located in the retinal periphery [1]. The cones are crowded into the rod-free foveola at very high density facilitating our high visual acuity [2, 3]. Spatial acuity is low in the periphery due to pooling by peripheral retinal ganglion cells from photopic signals across extensive retinal areas [4].
The electrical potentials of the visual system are altered by stimulation with flashes of light or pattern variation, producing a mass response from the retina. Traces recorded from different electrodiagnostic procedures represent pooled cellular responses from different levels within the retina (Fig. 1). These traces are helpful in determining the objective function of the visual system, particularly when clinical phenotype does not correlate with multimodal imaging (MMI) or subjective function. These non-invasive procedures have been standardised by the International Society for Clinical Electrophysiology of Vision (ISCEV) so that electrophysiology results can be meaningfully interpreted and compared worldwide [5, 6].

This diagram represents the high level retinal circulatory assessed by the common electrophysiology tests. The electro-oculogram (EOG) represents the electrical response from the outer retina (photoreceptor–RPE complex). The electroretinogram (ERG) measures the electrical response from the photoreceptor and inner retina. The visual evoked potential (VEP) represents the response from ganglion cells to the occipital cortex. NFL nerve fibre layer, GCL ganglion cell layer, IPL inner plexiform layer, INL inner nuclear layer, OPL outer plexiform layer, ONL outer nuclear layer, RPE retinal pigment epithelium, PR photoreceptor.
Full-field ERG
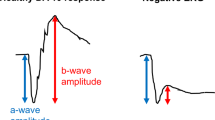
Full-field ERG testing can help differentiate different types of photoreceptor dystrophies (rod–cone or cone–rod) and direct clinical geneticists in their investigations for a specific genetic diagnosis. The ISCEV standard full-field ERG (ffERG) gives a mass response of the retina under both scotopic and photopic conditions. The waveform recorded varies depending on the stimulation and recording conditions with the result being the sum of overlapping retinal pathway contributions [7,8,9] and is able to distinguish between generalised inner and outer dysfunction. The classic waveform (Fig. 2A) arising from bright-flash stimulation under dark- or light-adapted conditions is characterised by an electronegative a-wave representing hyperpolarisation of the photoreceptors’ inner segments and the b-wave arising from the inner retina, predominantly the bipolar cells with a contribution from Muller cells. The oscillatory potentials are thought to be generated by post-receptor dopamine pathways with a presumed significant contribution from amacrine cells [10] (Fig. 2).

A Typical ISCEV standard wave forms. The scotopic standard tests are dim flash (DA 0.01), standard flash (DA 3.0), strong flash (DA 12.0) and oscillatory potentials (OP). The photopic standard tests are 30 Hz flicker (LA 30 Hz) and single flash (LA 3.0). B The selected ISCEV standard tests are sensitive or specific for the localisation of the rod or cone system defect, which provides guidance for further investigation. The weak flash (DA 0.01) ERG arises in the inner retinal rod bipolar cells and this standardised test is the only one that selectively monitors rod system function, making it rod sensitive. The standard flash (DA 3.0 and DA 10.0 or DA 12.0) ERG helps localise the dysfuction to either the rod photoreceptor (a- and b-wave reduction) or the inner retina (normal a-wave with reduced b-wave response). The DA 3.0 and DA 10.0 ERGs represent the response from both rod and cone system, however the rod system dictates the response in a healthy retina. The 30 Hz flash stimulus generates a post-receptoral response from the cone, making it a sensitive response of the cone system. The single flash (LA 3.0) ERG consists of an a-wave, representing the cone and off-bipolar cell response, and a b-wave, representing the response of both on- and off-bipolar cells. This makes the LA 3.0 ERG useful in specific localisation within the cone system [6]. C The ISCEV standard tests have been further developed to assist in identifying the origin of retinal dysfunction. The components of each wave form under different background lighting and stimulus further refine localisation. DA dark adapted, LA light adapted, ISCEV international society for clinical electrophysiology of vision.
Patients need “dark adaptation” of at least 20 min and a dim light stimulus below cone threshold, to test the scotopic system, as rods are 1000 times more sensitive to light than cones. The response of the very dim light (DA 0.01) has an almost undetectable a-wave but a prominent b-wave, which is the response of the rod bipolar cells collectively. This dim flash tests the sensitivity of the scotopic system (Fig. 2B) and anatomically, an abnormal response to this weak flash stimulus is either due to rod pathway dysfunction or that of the rod bipolar cell. Increasing the intensity of the light stimulus to DA 3.0 and DA 10.0, incorporates a response that includes both rod and cone systems. Under scotopic conditions the rod system dominates the final waveform with the a-wave representing rod photoreceptors [11], the a-wave slope phototransduction kinetics [12, 13] and the b-wave the rod bipolar cells [11] (Fig. 2C). The ISCEV standard strong flash [5] is DA 10.0 cd s m−2, the standard allows for some variability in the clinical case examples presented in this paper reports responses from the strong flash stimulus of DA 12.0 cd s m−2, which is minimally different in log sensitivity terms and is interchangeable. Figures in this review use traces representing the DA 12.0 response.
The photopic system is tested following light adaptation for 10 min at a background luminance of 30 cd m−2. The rod system is suppressed by this luminance and by the 30 Hz flicker stimulus as rods only respond up to 20 Hz stimulus [7, 14]. The 30 Hz flicker (LA 30 Hz) is the most sensitive measure of cone system function, however it does not distinguish between cone photoreceptor and inner retinal layer connection dysfunction (Fig. 2B). The single flash (LA 3.0) stimulus is used to elicit specific components of the photopic system with the single flash (LA 3.0) a-wave reflecting the response of the hyperpolarising off-bipolar cells and cone photoreceptors [15], whilst the b-wave has contributions from both on- and off-bipolar cells [6, 7, 16, 17] (Fig. 2C). The standard flash (3.0 cd s m−2) ERG is specific for the location in the cone system.
The ffERG is a measure of the entire retinal function, which means that loss of function from focal lesions, including the macula, is averaged with the remainder of the retinal function and the focal functional significance is often not demonstrated. To address this issue ISCEV standard topographical retinal testing has been developed. The available tests are the pattern ERG (PERG) [18] and the multifocal ERG (mfERG) [19] (Fig. 3). Assessing macular function is important for diagnostic and functional assessments. The PERG and mfERG achieve this in different ways. Optimal refraction is required for both tests as well as fixation. The PERG is performed with undilated pupils but the mfERG protocol requires pupil dilation and supplementary lenses. Pattern ERG tests average macular function to a set stimulus field size, whereas the mfERG response demonstrates a topographic local macular response to a local luminance change in the hexagon display.

The pattern and multifocal ERG have been developed to assess localised macular function. A High contrast chequerboard reversal image of a pattern ERG (PERG) stimulus seen by the patient. Testing is performed with best corrected visual acuity and the patient is required to fixate on the centre of the screen. B The PERG waveform to a 15° field is an indirect measure of macular function. The PERG arises largely in the ganglion cells, driven by the macular photoreceptors. The positive P50 represents the response of the ganglion cells at the inner retina and is usually between 2.0 and 8.0 µV. The negative N95 reflects ganglion cell action potentials at the optic nerve head. C The PERG to the 30° field enables assessment of both the macular and para-macular function. The P50 amplitude typically doubles from the 15° stimulus. D Hexagonal stimulus pattern scaled in size to produce mfERG responses of almost equal amplitudes across the central retina. Stable fixation is essential, which requires moderate visual acuity for accurate topographical analysis. E Multifocal traces array for each spatial hexagon. It represents a map of macular function. F Three-dimensional density map shows the overall signal strength per unit area of retina. This assists in assessing the quality of fixation by observing the blind spot location.
Pattern ERG (PERG)
The PERG waveform is the average of several hundred alternating stimulus responses and forms a discrete response with a positive and negative component (Fig. 3B). The positive deflection of the waveform is at a latency of approximately 50 ms and is referred to as P50. The larger negative component, N95, has a latency of approximately 95 ms (Fig. 3B). In contrast to the ffERG, the PERG stimulates a smaller area of retina and the resultant response has a lower amplitude. Averaging is needed to improve the signal-to-noise ratio with sometimes more than 100 trials per average is required. The N95 component of the human PERG is a contrast-related component generated in relation to the retinal ganglion cells [20, 21]. The PERG P50 component has its partial origins in the inner retina, but depends on the functional integrity of the macular photoreceptors and is used as an objective measure of preganglionic macular function [22]. Good, steady fixation is required by the patient, as with the mfERG. Doubling the stimulus size from 15° to 30° can assess the paramacular region [22, 23]. The larger field helps distinguish between localised central, predominantly paracentral, and widespread macular dysfunction. This provides an alternative to a multifocal ERG when precise steady fixation is not possible [22].
Multifocal ERG (mfERG)
The mfERG provides a topographic measure of retinal electrophysiological activity by stimulating localised areas of the central 45° or 60° field. The stimulus is an array of hexagonal elements. With this technique, many local ERG responses, typically 61 or 103, are recorded from the cone-driven retina under light-adapted conditions. The size of the hexagons of the ISCEV mfERG standard are scaled to gain comparable response amplitudes resulting in smaller hexagons for the central most stimuli size (Fig. 3D) [19]. Each element follows the same pseudo-random sequence of illumination. Cross-correlation of the continuous ERG signal with the sequence of on- and off-phases of each element enables calculation of an ERG signal at each location. The mfERG waveform is biphasic with an initial negative N1, a response from cones and their bipolar cells, and a later positive P2 component from the cone bipolar cells (Fig. 3E) [24, 25]. Good fixation is required to ensure topographic mapping (Fig. 3F) and a visual acuity of better than 6/60 is required. The mfERG is useful in identifying topographical location of retinal pathology up to 50° centred on the fovea.
Electro-oculogram (EOG)
The Electro-oculogram (EOG) assesses generalised retinal pigment epithelium (RPE) function as there is a potential difference between the basal and apical surface of the RPE of about 60 mV. The RPE itself is not a light receptor, however activation of the photoreceptors leads to a change in the ionic gradients across the RPE. The EOG is recorded during 15 min of dark adaptation followed by a further 15 min of light adaptation. The recordings are made with the eye (a dipole) making horizontal movements (saccades) from left to right. After about 7–10 min the dark trough (DT) (the lowest resting potential) is recorded. Similarly after about 7–10 min of light adaptation, the resting potential reaches its highest, known as the light peak (LP). This rise is due to the change in potassium currents of the photoreceptor–RPE matrix [26]. The ratio of LP/DT, termed the Arden ratio, provides a measure of function of this photoreceptor–RPE complex: normal is greater than 1.7 [26] (Fig. 4A). Clinically, if the photoreceptors are dysfunctional then the EOG will also be affected [26]. There are a small group of disorders with a normal ffERG and an abnormal EOG. The most important is Best vitelliform macular dystrophy (Fig. 4B–D).

A Changes in the ocular standing potential are recorded to an alternating horizontal stimulus, at each time point, following dark adaptation. The normal response results in a dark trough (DT) [down arrowhead] and a light peak (LP) (up arrowhead). The ratio of the LP/DT also termed the Arden ratio and is normally ≥1.7. B EOG response recorded for a patient with Best disease. LP/DT = 1.0. C Colour fundus photo of a patient with Best disease showing the pseudohypopyon stage. D Optical coherence tomography (OCT) through the Best disease lesion above the pseudohypopyon level demonstrating disorganisation of the ellipsoid zone with hyperreflective deposits and an optical empty subretinal space.
Visual electrophysiology in the clinic
The care of medical retina patients (e.g. diabetic retinopathy, age-related macular degeneration, autoimmune retinopathy), inherited retinal diseases (IRDs) and diagnostic dilemmas increasingly require objective functional assessments. Questions of when to refer and what tests to choose are now common clinical questions. Visual electrophysiology referrals are recommended to assist in establishing a clinical diagnosis and in determining the level of visual function [6]. Further roles arise from these investigations in adding the establishment of a genetic diagnosis and monitoring the natural history [27].
The paediatric patient also requires objective functional vision assessment; particularly when there are concerns regarding visual development or a nonvisual infant, a family history of poor vision, or systemic or metabolic disease.
Younger children are not capable of undertaking a full-length ISCEV protocol, however a meaningful electrophysiological examination that is comparable to the ISCEV standard. These modifications include shorter dark adaptation, modified protocols and the use of less invasive skin electrodes [5]. The recordings from skin electrodes have smaller amplitudes but similar waveforms to those of gold foil electrodes. Many paediatric electrophysiology testing centres are able to study young children without sedation or general anaesthesia, using swaddling, patience and teamwork.
For both adults and children, the specific electrophysiology tests performed will be directed by the clinical presentation, incorporating a combination of history, examination and ocular investigations. The clinical history pertinent to IRDs include symptoms of the following: rod dysfunction (nyctalopia and/or peripheral vision issues), cone dysfunction (photophobia, reduced distance and near visual acuity and/or dyschromatopsia), age at onset and progression of symptoms [27,28,29]. IRDs can also occur as part of a multi-system condition and systemic medical problems need to be specifically inquired about and evaluated, particularly hearing, renal dysfunction, neurological dysfunction, skeletal anomalies and metabolic disturbance. The effects of medication also need to be evaluated [30].
The clinical question being asked determines the initial visual electrophysiology tests to be performed. When differentiating whether a patient has poor cone function, or no cone function the ffERG is helpful (Fig. 5A–F). If determining whether a patient has a retinopathy or a maculopathy the addition of the PERG ± mfERG to the ffERG is required (Fig. 6A, B). Poor central vision requires a pattern visual evoked potential (VEP) to exclude an optic neuropathy, which may be performed simultaneously with a PERG to determine whether there is a macular dysfunction and a ffERG to confirm whether there is generalised retinal dysfunction. Nyctalopia requires a ffERG to identify rod dysfunction, determine whether the nyctalopia is likely to be progressive or stationary and a PERG to determine residual macular function (Fig. 7).

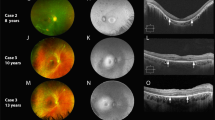
A congenital stationary cone disorder from a cone or cone–rod dystrophy. Achromatopsia should be considered if the LA 30 Hz flicker response is severely attenuated or absent. (Top panel ISCEV ffERG) both LA 30 Hz and LA 3.0 significantly attenuated. (A–C) Patient with Achromatopsia: A colour image, B fundus autofluorescence (FAF) image and C optical coherence tomography (OCT). (Middle panel ISCEV ffERG) LA 30 Hz reduced but identifiable and reduced LA 3.0 with variable reduction in DA 0.01, DA 3.0 and DA 12.0). (D–F) Patient with cone dystrophy: D colour image, E FAF and F OCT. (Bottom panel) normal ISCEV standard ffERG (G–I) Normal patient. G colour image H FAF I OCT. DA dark adapted, LA light adapted ISCEV International Society for Clinical Electrophsyiology in Vision ffERG ful-field Electroretinogram.

Poor central vision may arise due to isolated macular dysfunction or generalised retinal dysfunction. In this case a normal ffERG excludes generalised retinal dysfunction. The abnormal PERG localises the pathology to the macular region. A multifocal ERG further refines the topographic defect. Electrophysiology can further assist in Stargardt disease by grouping the phenotype according to the criteria described by Lois et al. [74]. A Top panel demonstrates a normal ffERG and a reduced PERG indicating macular dysfunction. B The mfERG further refines the topographic localisation of the macular abnormality. Multimodal imaging (MMI) in the patient with Stargardt disease. C Wide field imaging of the patient’s right retina. D Fundus autofluorescence (FAF) imaging of the patient’s right eye. E Macular OCT confirming loss of outer retina and normal macular architecture.

Nyctalopia is a common clinical presentation. Differentiating a progressive disorder (e.g. rod–cone dystrophy – retinitis pigmentosa or enhanced S-cone syndrome) from a stationary disorder (congenital stationary night blindness) is an important indication for a ffERG. In this patient, A the ffERG demonstrates an almost undetectable DA 0.01 indicating rod system abnormality. The reduced a-wave in the DA 3.0 and DA 12.0 further localises the defect to the rod photoreceptor. The reduced 30 Hz flicker and LA 3.0 a-wave indicates cone photoreceptor dysfunction as well, consistent with a diagnosis of rod–cone dystrophy. The pERG is reduced indicating associated macular dysfunction. B mfERG demonstrating reduced parafoveal responses, with moderate preservation of the foveal response. C Ultra-wide field image, left eye, demonstrating retinal pigment epithelial pigment migration resulting in the typical bone-spicule pattern. There is also evidence of outer retinal atrophy peripheral to the vascular arcades. D Ultra-wide field fundus autofluorescence, left eye demonstrating hyper-autofluorescence ring at the posterior pole (red arrow) with patchy and blotchy hypo-autofluorescence in the retinal periphery. E Macular ocular coherence tomography (OCT) demonstrating outer retinal layer disruption of the foveal ellipsoid zone (EZ). The EZ is preserved in the central macular (between the blue bars), which corresponds to the segment within the ring of hyper-autofluorescence (D) (scale bar 200 μm).
In rod–cone dystrophies (retinitis pigmentosa, RP), secondary cone degeneration follows rod degeneration centripetally and is slowest at the macular where cone density is greatest. This correlates with the clinical finding of central visual acuity preservation until advanced stages. Measuring the rate of cone degeneration with the ffERG provides one measure of RP progression. The “fused” flicker amplitude (FFAmp) is a non-ISCEV standard waveform that enables assessment of residual cone function, described by Berson et al. [31]. The electrophysiology system records at the frequency of 30 ± 1 Hz and improves the signal-to-noise ratio by computer averaging 1200 trials over 10 min enabling amplitudes as low as 0.05 μV to be recorded with Burian Allen electrode. The more common clinically used electrodes (gold foil and DTL) may only record to 1 μV [31]. The importance of recording FFAmp is that useful vision is retained even when the cone function drops below 10 μV and is maintained until 0.05 μV. Below this critical level, virtual blindness ensues. The rate of decline in both 30 Hz flicker amplitude and FFAmp provide a guide to predict maintenance of useful vision [31, 32].
Inherited retinal disorders (IRD) with pathognomonic ERGs with phenotype–genotype correlation
Investigations with an ERG establishes whether a patient has a retinal or macular dysfunction and what anatomical level it is. In conjunction with the patient’s symptoms and MMI, this dysfunction can be classified as a probable dystrophy, facilitating a more directed clinical genetic workup within a genetic multi-disciplinary team (MDT).
Several IRDs have specific visual electrophysiology changes that are pathognomonic and correlate with genotype [33]. These include KCNV2 retinopathy, congenital stationery night blindness, enhanced S-cone syndrome and bradyopsia.
Cone dystrophy with supernormal rod ERG
KCNV2-associated retinopathy (cone dystrophy with supernormal rod responses) first described by Gouras et al. [34] is an autosomal recessive (AR) cone–rod dystrophy caused by mutation in the KCNV2 gene and has characteristic ERG findings. This gene encodes Kv8.2, a voltage-gated potassium channel, which sets the vertebrate photoreceptor resting potential and voltage-gated response [35].
Affected individuals usually present in the first two decades of life with poor visual acuity, abnormal colour vision and variable night vision difficulties [36]. Most patients are myopic and nystagmus is common. The fundus appearance can appear normal in the early stages, but later there may be macular pigmentary disturbance or macular atrophy (Fig. 8B–D).

KCNV2 retinopathy (cone dystrophy with supranormal rod response) is an inherited retinal disease with diagnostic electrophysiology findings. A Compares a KCNV2 ffERG with normal ISCEV standard ffERG. The specific characteristics are a delayed and supra-normal b-wave DA 0.01 (red arrow). The strong flash (DA 10.0 or DA 12.0) has a normal a-wave slope and normal amplitude with a broadened flat trough with a “squared” or “rhomboid,” shape followed by a steeply rising high amplitude b-wave (*) best seen in the DA 12.0. Photopic LA 30 Hz flicker and single flash (LA 3.0) responses are significantly reduced. KCNV2 clinical images: B wide-field image with macular atrophy, C wide-field fundus autofluorescence with mild bulls-eye pattern of alternating hyper- and hypo-autofluorescence. D OCT with disruption of the ellipsoid zone at the fovea.
The ERG (Fig. 8A) shows a markedly delayed rod ERG (DA 0.01). The arrow highlights the marked delay in b-wave implicit time compared to the normal control (same time scale) red dotted line indicates mean normal implicit time. The strong flash (DA 10.0) reveals a normal a-wave slope and normal or near normal amplitude with a broadened flat trough with a “squared” or “rhomboid” shape [37], with a steeply rising high amplitude b-wave (* in Fig. 8) best seen in the dark-adapted (DA) maximal (DA 12) recording. Photopic flicker and single flash ERGs are significantly reduced [36, 37]. Additional non-ISCEV standard luminance response series in the dark-adapted phase are useful in the diagnosis of this condition [38]. The KCNV2 transcript has been demonstrated in the inner segments of both cone and rod photoreceptors, which might explain the typical ERG findings in keeping with dysfunction occurring after phototransduction but before generation of b-wave [39].
Enhanced S-cone syndrome (ESCS)
Enhanced S-cone syndrome first described by Marmor et al. [40] is an AR disorder characterised by early onset night blindness, maculopathy with variable loss of visual acuity and increased sensitivity to blue light caused by mutations in NR2E3 gene [41]. NR2E3, a nuclear receptor specifically expressed in photoreceptors, has a key role in rod and cone photoreceptor development and maintenance by repressing cone-specific genes and activating several rod-specific genes. Mutations lead to excess S-cone proliferation at the expense of other cones/rods (abnormal cell fate determination) [42]. Mutations in NR2E3 gene have also been identified in Goldmann-Favre syndrome [43, 44].
Patients with ESCS present with nyctalopia with or without reduced VA in early childhood usually in the first decade of life [41, 45]. Posterior segment examination reveals optically empty vitreous, typical nummular pigmentary changes at the level of RPE along or outside the vascular arcades, macular schisis which is more obvious on spectral domain optical coherence tomography (SD-OCT) and sometimes whitish retinal deposits or hyperpigmented lesions (Fig. 9D). Thickening of the outer nuclear layer (ONL) on SD-OCT is another common structural finding [46, 47]. Fundus autofluorescence (FAF) shows partial or complete loss of autofluorescence (AF) outside the arcade presumably due to photoreceptor loss (Fig. 9C) [48].

Enhanced S-cone syndrome is an inherited retinal disease with diagnostic electrophysiology findings. A Full-field electroretinogram (ffERG) of a patient with ESCS compared to normal ffERG ISCEV standard below. The top panel ffERG demonstrated an essentially undetectable DA 0.01, the ISCEV standard responses under both scotopic (DA 3.0) and photopic (LA 3.0) conditions are of similar waveform, being simplified and delayed. The LA 30 Hz flicker ERG is delayed and lower amplitude than the single flash cone photopic a-wave (LA 3.0). B Ultra-wide field image of the left retina demonstrating retinal vascular arcade and peripheral pigmentary changes associated with retinal atrophy. C Ultra-wide field autofluorescence image of the same left retina demonstrates hypo-autofluorescence at the corresponding areas of pigmentary retinal change. Areas of hyper-autofluorescence inferior to the inferior retinal arcade. D Ocular coherence tomography (OCT) demonstrating retinal schisis (scale bar 200 μm).
Patients with ESCS show characteristic full-field ERG waveform abnormalities. The dark-adapted (DA) 0.01 response (rod specific) is bilaterally undetectable. The DA 3.0 and light-adapted (LA) 3.0 responses have the same simplified waveform and are markedly delayed. The 30 Hz flicker electroretinography is both delayed and of lower amplitude than the LA 3.0 response a-wave (Fig. 9A). S-cone-specific testing provides further confirmation of the diagnosis. The S-cone ERG is elicited using short-wavelength (blue) flashes on a long-wavelength background to saturate the rod system and to reduce contributions from the L/M-cone systems. The S-cones are isolated using a 5-millisecond duration short-wavelength (blue) stimulus (445 nm, 80 cd m−2) superimposed on a long-wavelength background (orange, 620 nm, background luminance 560 cd m−2) to saturate the rod system and to reduce contributions from the L/M-cone systems [49, 50]. These are typically larger than normal and resemble the waveform of LA 3.0 ERG due to the predominant contribution of S-cones to both these responses. PERG abnormalities vary from being reduced and/or delayed or completely unrecordable [41].
Bradyopsia
Bradyopsia is a rare stationary form of retinal dysfunction due to biallelic mutations in the gene RGS9 (regulator of G-protein signalling 9) or R9AP (regulator of G-protein signal 9-anchoring protein). It was first described by Kooijman et al. [51], and the term bradyopsia (“slow vision”) coined by Nishiguchi et al. [52]. RGS9 facilitates the hydrolysis of GTP (α-transducin bound) to GDP and ultimately opening of cGMP-gated cation channels in darkness [52]. Mutations in these genes impair the retina’s ability to rapidly adjust to and from bright light by slowing guanosine triphosphate (GTP) hydrolysis [53].
Patients with this condition have photophobia, mildly reduced visual acuity, difficulty tracking moving objects, trichromatic colour discrimination and delayed light/dark adaptation [52, 54,55,56]. Vision may be better under reduced luminance conditions and improve with pinhole [55]. Fundus examination and macular OCT are normal.
The ISCEV standard ERGs will often misdiagnose bradyopsia. The PERGs are usually undetectable. The DA 0.01 ERG is normal and DA 3.0 ERGs are often normal to only mildly reduced in amplitude (Fig. 10). The photopic ERGs (30 Hz and LA 3.0) are undetectable or profoundly subnormal. The ISCEV DA red flash ERG extended protocol presents a red flash under scotopic conditions [57]. The resultant waveform normally has two distinct positive peaks, the first positive component is the cone-mediated x-wave with a peak at 30–50 ms; the second is a rod-mediated b-wave with a peak time of approximately 100 ms. The combination of a preserved DA red flash ERG x-wave and undetectable or severely abnormal standard LA ERGs is pathognomonic for bradyopsia [33, 53, 57].

Bradyopsia is an inherited retinal disease with diagnostic electrophysiology findings. A Compares a bradyopsia ffERG (top panel) with an ISCEV standard ffERG (lower panel). The ISCEV standard ERGs show normal dark-adapted responses, an undetectable LA 30 Hz flicker ERG and a severely reduced LA 3.0 response, which frequently suggests incomplete achromatopsia. Using the ISCEV extended protocol (shaded column), dark-adapted dim red flash ERG (DA red), a normal early cone response (x-wave) excludes achromatopsia. The diagnosis of bradyopsia requires the testing of cone function under dark adaptation. Adapted from Audo et al. [71].
Electrodiagnostic role in genetic diagnosis
IRDs caused by monogenic gene mutations are the commonest cause of blindness in young and working age individuals in many countries causing a huge economic burden [58, 59]. In the 1990s the primary goal of the ophthalmologists with visual electrophysiology information and Clinical Geneticists was to give the IRD a name, try to establish an inheritance pattern and estimate recurrence risk for other family members. Molecular diagnosis was a challenge since the genetic testing was either single gene based or a chip approach searching for a limited number of known gene mutations.
Next-generation sequencing technology has enabled vastly improved mutation detection rates. The advances have also highlighted variants of uncertain significance (VUS) that are simultaneously identified [27]. The American College of Medical Genetics and Genomics (ACMG) provides a systematic methodology to classify these variants [60].
Advances in understanding the cell biology of the visual system, gene replacement approaches, and CRISPR-based genome editing have led to successful use of genetic therapies for IRDs [61] and multiple clinical trials are now underway across a spectrum of conditions. In the genomics era, the multi-disciplinary team now consists of input from ophthalmologists with visual electrophysiology information, clinical geneticists and genomic laboratory scientists, and the clinical goal is strongly focussed on identifying the disease-causing genotype and evaluating for therapy suitability.
Electrophysiology contributing to MDT genetic review
Pathogenic variants in more than 250 genes can give rise to IRDs with multiple modes of inheritance [62]. However, there exists a considerable genetic and phenotypic heterogeneity [63]. Due to this, prioritisation in examining the genes known to be associated with a certain IRD is challenging.
A genetic MDT approach where the ophthalmologist liaises with the clinical genetics team is extremely important in the diagnosis and appropriate management of IRDs. Visual electrophysiology has two broad roles in assisting genetic diagnosis. Firstly, the electrophysiology provides functional results that assist in localisation of the defect to a particularly retinal cell type and thus a diagnostic group. This information guides the clinical geneticist in selecting gene panels that encompass the most common genetic causes for the group. Secondly, structure–function outcomes obtained with visual electrophysiology assists in interpreting molecular genetic results. This commonly involves confirming that the identified molecular genetic change is consistent with the patient phenotype. Importantly the electrophysiology assists in interpreting variants of uncertain significance (VUS).
Electrodiagnostic testing as a guide for genetic panel screening
Multigene panel testing is a well-established approach to dissect genetically heterogenous diseases and has been a validated method for clinical diagnosis [64,65,66]. Application of a focussed approach to gene panel testing can increase the chance of the correct molecular diagnosis and reduce the presence of confounding variants of uncertain significance (VUSs). Electrodiagnostic tests can help guide this testing. As an example, an abnormal PERG ± mfERG with normal ffERG will help direct the genetic panel testing to a maculopathy as against a generalised retinopathy. These of course, need to be considered in the context of clinical presentation, family history and MMI.
A “bulls-eye” maculopathy is a relatively common clinical presentation and is frequently seen in cone/cone–rod dystrophies. However, as with the example in Fig. 11, other IRDs can have the phenotype of a bulls-eye maculopathy, but functional studies demonstrate a different diagnosis. The ffERG of Fig. 11 is consistent with achromatopsia and this information would lead to an expansion of the gene panel being evaluated so that a CNGB3 mutation may be correctly identified.

Expanding phenotypes for known IRD genes are now being revealed with increasing genetic testing. A 60-year-old patient had been diagnosed with a cone dystrophy based on the bulls-eye maculopathy clinical appearance. A Widefield fundus autofluorescence, and B macular OCT. Genetic testing had been initiated based on these clinical findings. C Electrophysiology was undertaken in parallel. The scotopic ERG (DA 0.01, DA 3.0 and DA 12.0) were within normal limits. The photopic ERG showed an undetectable LA 30 Hz response and significantly reduced LA 3.0. These ffERG results were suggestive of achromatopsia. This information led to including the achromatopsia genes in the initial gene group examined bioinformatically from the whole exome genetic sequencing. Compound heterozygous pathogenic mutations were identified in CNGB3, which account for a small fraction of the later onset progressive form of cone photoreceptor disorders. The ffERG was critical in providing the evidence to support the expanded phenotype for this gene [75].
Patients with retinitis punctata albescens (RPA) and fundus albipunctatus (FA) present with nyctalopia and similar fundal phenotype with whitish, glistening small deposits across the outer retina (white fleck syndrome) (Fig. 12). Distinguishing the two diagnosis functionally with electrophysiology again guides genomic diagnosis. RPA is a subtype of RP with a similar progressive time course, and in most cases is caused by pathogenic variants in RLBP1 gene [67, 68]. Electrophysiology on these patients demonstrates a rod–cone dysfunction (Fig. 12, left ffERG panel). In contrast, FA is a recessively inherited, usually stationary disorder [69] and is most commonly associated with RDH5 gene mutations [70]. Bright-flash dark-adapted DA 12.0 ERG of patients with FA have subnormal a-wave and reduced b-wave amplitude commonly resulting in an electronegative ERG appearance (Fig. 12, right ffERG panel). Repeating the ffERG following an extended period of dark adaptation (prolonged dark adaption), the DA 0.01 and DA 10.0 ERGs wave form returns to normal (red box Fig. 12) [71]. Prolonged dark adaption is performed by patching and occluding one eye overnight and then performing standard ISCEV dark-adapted ffERG with the patch taken off after the 20 min of further dark adaption. This enables the unpatched (control eye) and the prolonged dark adaption to be compared. Demonstration of this electrophysiology pattern helps the ophthalmologist give prognosis to the patient and help guide clinical geneticists with their genetic panel screen.

Nyctalopia associated with white or yellow retinal flecks is a relatively common presentation. Retinitis punctata albescens (RPA) [left column] and fundus albipunctatus (FA) [right column] have this presentation. The ffERG can assist in differentiating these two conditions by adding extended dark adaption to the ISCEV standard ffERG. The standard ISCEV scotopic ERG (DA 0.01.DA 3.0 and DA 12.0) are significantly attenuated amplitudes in each condition. Prolonged dark adaption performed by occlusion overnight results in recovery of the scotopic ERG amplitudes in FA (red box) but not in RPA. Electrophysiology assists in distinguishing FA usually associated with RDH5 mutations from RPA usually associated with RLBP1 mutations.
Electrodiagnostic testing role in assessing molecular genetic results including variants of uncertain significance (VUS)
Genetic testing to identify the cause of disease has become increasingly important as more clinical trials for IRD focus on patient populations with specific genotypes [72], for example, studies recruiting USH2A, RPGR, CHM, ABCA4, MERTK, CNGA3, CNGB3, PDE6B or RLBP1‐ affected patients (https://clinicaltrials.gov/). In addition, IRD patients due to RPE65 mutations would be eligible for the first Food and Drug Administration (FDA)‐approved gene therapy, voretigene neparvovec (Luxturna) [73]; Patients without a genetic diagnosis are not eligible for gene‐specific treatments.
IRD genetic testing is the same as other panel-based genomic testing strategies in that candidates with VUS are often identified with insufficient evidence to deem as disease causing, or in genes which have not been described in the literature matching the particular phenotypic retinal presentations. Incorporating an ocular genetics MDT approach can improve diagnostic rates due to clarification of ocular phenotype review of clinical features in family members, and segregation studies of candidate genetic variants. Visual electrophysiology contributes important objective functional phenotypic information to the MDT review process.
Conclusions
Visual electrophysiology plays an important role in the management of IRDs across all stages of their management from diagnosis, determination of retinal and visual function, guidance for genomic testing, interpretation of genomic results and monitoring visual function over time as part of natural history evaluation.
Methodology
We used the following databases and search terms to research this review: MEDLINE/PubMed: Inherited Retinal Degeneration; Electrophysiology; ERG; Pathognomonic ERG; Enhanced S Cone Syndrome; KCNV2 retinopathy; Bradyopsia; ISCEV standard ERG; full-field electroretinogram; pattern electroretinogram; multifocal electroretinogram; ABCA4 associated retinal degeneration; Genetic basis of Inherited Retinal disease; Molecular genetics in inherited retinal disorders; Genomic applications in diagnosis of retinal dystrophies; gene therapy for retinal dystrophies. Non-English papers were excluded.
References
Lamb TD. Why rods and cones? Eye. 2016;30:179–85.
Cornish EE, Hendrickson AE, Provis JM. Distribution of short-wavelength-sensitive cones in human fetal and postnatal retina: early development of spatial order and density profiles. Vis Res. 2004;44:2019–26.
Provis JM, Penfold PL, Cornish EE, Sandercoe TM, Madigan MC. Anatomy and development of the macula: specialisation and the vulnerability to macular degeneration. Clin Exp Optom. 2005;88:269–81.
Anderson S, Mullen K, Hess R. Human peripheral spatial resolution for achromatic and chromatic stimuli: limits imposed by optical and retinal factors. J Physiol. 1991;442:47–64.
McCulloch DL, Marmor MF, Brigell MG, Hamilton R, Holder GE, Tzekov R, et al. ISCEV standard for full-field clinical electroretinography (2015 update). Doc Ophthalmol. 2015;130:1–12.
Robson AG, Nilsson J, Li S, Jalali S, Fulton AB, Tormene AP, et al. ISCEV guide to visual electrodiagnostic procedures. Doc Ophthalmol. 2018;136:1–26.
Bush RA, Sieving PA. Inner retinal contributions to the primate photopic fast flicker electroretinogram. J Opt Soc Am A Opt Image Sci Vis. 1996;13:557–65.
Robson JG, Saszik SM, Ahmed J, Frishman LJ. Rod and cone contributions to the a-wave of the electroretinogram of the macaque. J Physiol (Lond). 2003;547(Pt 2):509–30.
Robson J, Frishman L. The rod-driven a-wave of the dark-adapted mammalian electroretinogram. Prog Retin Eye Res. 2014;39:1–22.
Wachtmeister L. Oscillatory potentials in the retina: what do they reveal. Prog Retinal Eye Res. 1998;17:485–521.
Robson J, Frishman L. Dissecting the dark-adapted electroretinogram. Doc Ophthalmol. 1999;95:187–215.
Lamb T, Pugh EJ. A quantitative account of the activation steps involved in phototransduction in amphibian photoreceptors. J Physiol. 1992;449:719–58.
Lamb TD, Pugh EN Jr. Phototransduction, dark adaptation, and rhodopsin regeneration the proctor lecture. Invest Ophthalmol Vis Sci. 2006;47:5137–52.
Dodt E, Wadensten L. The use of flicker electroretinography in the human eye; observations on some normal and pathological retinae. Acta Ophthalmol (Copenh). 1954;32:165–80.
van Hateren JH, Lamb TD. The photocurrent response of human cones is fast and monophasic. BMC Neurosci. 2006;7:34.
Sieving PA, Murayama K, Naarendorp F. Push–pull model of the primate photopic electroretinogram: a role for hyperpolarizing neurons in shaping the b-wave. Vis Neurosci. 1994;11:519–32.
Friedburg C, Allen CP, Mason PJ, Lamb TD. Contribution of cone photoreceptors and post-receptoral mechanisms to the human photopic electroretinogram. J Physiol (Lond). 2004;556(Pt 3):819–34.
Bach M, Brigell M, Hawlina M, Holder G, Johnson M, McCulloch D, et al. ISCEV standard for clinical pattern electroretinography (PERG) – 2012 update. Doc Ophthalmol. 2013;126:1–7.
Hood D, Bach M, Brigell M, Keating D, Kondo M, Lyons J. et al. ISCEV standard for clinical multifocal electroretinography (2011 edition). Doc Ophthalmol. 2012;124:1–13.
Holder GE. The pattern electroretinogram in anterior visual pathway dysfunction and its relationship to the pattern visual evoked potential: a personal clinical review of 743 eyes. Eye. 1997;11(Pt 6):924–34.
Holder GE. Pattern electroretinography (PERG) and an integrated approach to visual pathway diagnosis. Prog Retinal Eye Res. 2001;20:531–61.
Lenassi E, Robson AG, Hawlina M, Holder GE. The value of two-field pattern electroretinogram in routine clinical electrophysiologic practice. Retina. 2012;32:588–99.
Bach M, Holder GE. Check size tuning of the pattern electroretingoram: a reappraisal. Doc Ophthalmol. 1996;92:193–202.
Hood DC. Assessing retinal function with the multifocal technique. Prog Retinal Eye Res. 2000;19:607–46.
Hood DC, Frishman LJ, Saszik S, Viswanathan S. Retinal origins of the primate multifocal ERG: implications for the human response. Invest Ophthalmol Vis Sci. 2002;43:1673–85.
Constable PA, Bach M, Frishman LJ, Jeffrey BG, Robson AG. ISCEV standard for clinical electro-oculography (2017 update). Doc Ophthalmol. 2017;134:1–9.
Grigg J, Jamieson R, Chen F, Simunovic M, Huq A, Vincent A, et al. Guidelines for the assessment and management of patients with inherited retinal diseases (IRD) 2020. RANZCO; 2020. p. 1–14.
Gill JS, Georgiou M, Kalitzeos A, Moore AT, Michaelides M. Progressive cone and cone–rod dystrophies: clinical features, molecular genetics and prospects for therapy. Br J Ophthalmol. 2019;24:24.
Kumaran N, Moore AT, Weleber RG, Michaelides M. Leber congenital amaurosis/early-onset severe retinal dystrophy: clinical features, molecular genetics and therapeutic interventions. Br J Ophthalmol. 2017;101:1147–54.
Verbakel SK, van Huet RAC, Boon CJF, den Hollander AI, Collin RWJ, Klaver CCW, et al. Non-syndromic retinitis pigmentosa. Prog Retinal Eye Res. 2018;66:157–86.
Berson EL. Long-term visual prognoses in patients with retinitis pigmentosa: the Ludwig von Sallmann lecture. Exp Eye Res. 2007;85:7–14.
Arsiwalla TA, Cornish EE, Nguyen PV, Korsakova M, Ali H, Saakova N, et al. Assessing residual cone function in retinitis pigmentosa patients. Transl Vis Sci Technol. 2020;9:29.
Vincent A, Robson AG, Holder GE. Pathognomonic (diagnostic) ERGs. A review and update. Retina. 2013;33:5–12.
Gouras P, Eggers HM, MacKay CJ. Cone dystrophy, nyctalopia, and supernormal rod responses. A new retinal degeneration. Arch Ophthalmol. 1983;101:718–24.
Gayet-Primo J, Yaeger DB, Khanjian RA, Puthussery T. Heteromeric KV2/KV8.2 channels mediate delayed rectifier potassium currents in primate photoreceptors. J Neurosci. 2018;38:3414–27.
Robson AG, Webster AR, Michaelides M, Downes SM, Cowing JA, Hunt DM, et al. “Cone dystrophy with supernormal rod electroretinogram”: a comprehensive genotype/phenotype study including fundus autofluorescence and extensive electrophysiology. Retina. 2010;30:51–62.
Grigg JR, Holder GE, Billson FA, Korsakova M, Jamieson RV. The importance of electrophysiology in revealing a complete homozygous deletion of KCNV2. J AAPOS. 2013;17:641–3.
McCulloch DL, Kondo M, Hamilton R, Lachapelle P, Messias AMV, Robson AG, et al. ISCEV extended protocol for the stimulus–response series for light-adapted full-field ERG. Doc Ophthalmol. 2019;138:205–15.
Wu H, Cowing JA, Michaelides M, Wilkie SE, Jeffery G, Jenkins SA, et al. Mutations in the gene KCNV2 encoding a voltage-gated potassium channel subunit cause “cone dystrophy with supernormal rod electroretinogram” in humans. Am J Hum Genet. 2006;79:574–9.
Marmor MF, Jacobson SG, Foerster MH, Kellner U, Weleber RG. Diagnostic clinical findings of a new syndrome with night blindness, maculopathy, and enhanced S cone sensitivity. Am J Ophthalmol. 1990;110:124–34.
Audo I, Michaelides M, Robson AG, Hawlina M, Vaclavik V, Sandbach JM, et al. Phenotypic variation in enhanced S-cone syndrome. Invest Ophthalmol Vis Sci. 2008;49:2082–93.
Milam AH, Rose L, Cideciyan AV, Barakat MR, Tang WX, Gupta N, et al. The nuclear receptor NR2E3 plays a role in human retinal photoreceptor differentiation and degeneration. Proc Natl Acad Sci USA. 2002;99:473–8.
Schorderet DF, Escher P. NR2E3 mutations in enhanced S-cone sensitivity syndrome (ESCS), Goldmann-Favre syndrome (GFS), clumped pigmentary retinal degeneration (CPRD), and retinitis pigmentosa (RP). Hum Mutat. 2009;30:1475–85.
Sharon D, Sandberg MA, Caruso RC, Berson EL, Dryja TP. Shared mutations in NR2E3 in enhanced S-cone syndrome, Goldmann-Favre syndrome, and many cases of clumped pigmentary retinal degeneration. Arch Ophthalmol. 2003;121:1316–23.
Hull S, Arno G, Sergouniotis PI, Tiffin P, Borman AD, Chandra A, et al. Clinical and molecular characterization of enhanced S-cone syndrome in children. JAMA Ophthalmol. 2014;132:1341–9.
Jacobson SG, Sumaroka A, Aleman TS, Cideciyan AV, Schwartz SB, Roman AJ, et al. Nuclear receptor NR2E3 gene mutations distort human retinal laminar architecture and cause an unusual degeneration. Hum Mol Genet. 2004;13:1893–902.
Sustar M, Perovsek D, Cima I, Stirn-Kranjc B, Hawlina M, Brecelj J. Electroretinography and optical coherence tomography reveal abnormal post-photoreceptoral activity and altered retinal lamination in patients with enhanced S-cone syndrome. Doc Ophthalmol. 2015;130:165–77.
Tsang SH, Sharma T. Enhanced S-cone syndrome (Goldmann-Favre Syndrome). Adv Exp Med Biol. 2018;1085:153–6.
Arden G, Wolf J, Berninger T, Hogg CR, Tzekov R, Holder GE. S-cone ERGs elicited by a simple technique in normals and in tritanopes. Vis Res. 1999;39:641–50.
Perlman I, Kondo M, Chelva E, Robson AG, Holder GE. ISCEV extended protocol for the S-cone ERG. Doc Ophthalmol. 2020;140:95–101.
Kooijman AC, Houtman A, Damhof A, van Engelen JP. Prolonged electro-retinal response suppression (PERRS) in patients with stationary subnormal visual acuity and photophobia. Doc Ophthalmol. 1991;78:245–54.
Nishiguchi KM, Sandberg MA, Kooijman AC, Martemyanov KA, Pott JW, Hagstrom SA, et al. Defects in RGS9 or its anchor protein R9AP in patients with slow photoreceptor deactivation. Nature. 2004;427:75–78.
Khan AO. The clinical presentation of bradyopsia in children. J AAPOS. 2017;21:507–9.e1.
Cheng JY, Luu CD, Yong VH, Mathur R, Aung T, Vithana EN. Bradyopsia in an Asian man. Arch Ophthalmol. 2007;125:1138–40.
Hartong DT, Pott JW, Kooijman AC. Six patients with bradyopsia (slow vision): clinical features and course of the disease. Ophthalmology. 2007;114:2323–31.
Michaelides M, Li Z, Rana NA, Richardson EC, Hykin PG, Moore AT. et al. Novel mutations and electrophysiologic findings in RGS9- and R9AP-associated retinal dysfunction (bradyopsia). Ophthalmology. 2010;117:120–7.e1.
Thompson DA, Fujinami K, Perlman I, Hamilton R, Robson AG. ISCEV extended protocol for the dark-adapted red flash ERG. Doc Ophthalmol. 2018;136:191–7.
Liew G, Michaelides M, Bunce C. A comparison of the causes of blindness certifications in England and Wales in working age adults (16–64 years), 1999–2000 with 2009–10. BMJ open. 2014;4:e004015.
Solebo AL, Teoh L, Rahi J. Epidemiology of blindness in children. Arch Dis Child. 2017;102:853–7.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24.
Russell S, Bennett J, Wellman JA, Chung DC, Yu ZF, Tillman A, et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. [Erratum appears in Lancet. 2017;390:848]. Lancet. 2017;390:849–60.
Farrar GJ, Carrigan M, Dockery A, Millington-Ward S, Palfi A, Chadderton N, et al. Toward an elucidation of the molecular genetics of inherited retinal degenerations. Hum Mol Genet. 2017;26(R1):R2–11.
Nash B, Wright D, Grigg J, Bennetts B, Jamieson R. Retinal dystrophies, genomic applications in diagnosis and prospects for therapy. Transl Pediatrics. 2015;4:139–63.
Gargis AS, Kalman L, Berry MW, Bick DP, Dimmock DP, Hambuch T, et al. Assuring the quality of next-generation sequencing in clinical laboratory practice. Nat Biotechnol. 2012;30:1033–6.
Rehm HL, Bale SJ, Bayrak-Toydemir P, Berg JS, Brown KK, Deignan JL, et al. ACMG clinical laboratory standards for next-generation sequencing. Genet Med. 2013;15:733–47.
Cottrell CE, Al-Kateb H, Bredemeyer AJ, Duncavage EJ, Spencer DH, Abel HJ, et al. Validation of a next-generation sequencing assay for clinical molecular oncology. J Mol Diagn. 2014;16:89–105.
Hipp S, Zobor G, Glockle N, Mohr J, Kohl S, Zrenner E, et al. Phenotype variations of retinal dystrophies caused by mutations in the RLBP1 gene. Acta Ophthalmol. 2015;93:e281–6.
Torres-Costa S, Ferreira CS, Grangeia A, Santos-Silva R, Brandao E, Estrela-Silva S, et al. A novel homozygous frameshift variant in the cellular retinaldehyde-binding protein 1 (RLBP1) gene causes retinitis punctata albescens. Eur J Ophthalmol 2020. https://doi.org/10.1177/1120672120919064. [Epub ahead of print].
Sergouniotis PI, Sohn EH, Li Z, McBain VA, Wright GA, Moore AT, et al. Phenotypic variability in RDH5 retinopathy (fundus albipunctatus). Ophthalmology. 2011;118:1661–70.
Skorczyk-Werner A, Pawlowski P, Michalczuk M, Warowicka A, Wawrocka A, Wicher K, et al. Fundus albipunctatus: review of the literature and report of a novel RDH5 gene mutation affecting the invariant tyrosine (p.Tyr175Phe). J Appl Genet. 2015;56:317–27.
Audo I, Holder GE, Moore AT. Inherited stationary disorders of the retina. In: Puech B, De Laey J-J, Holder GE, editors. Inherited chorioretinal dystrophies: a textbook and atlas. Springer-Verlag Berlin and Heidelberg GmbH & Co.; 2014. p. 80–84.
Wan A, Place E, Pierce EA, Comander J. Characterizing variants of unknown significance in rhodopsin: a functional genomics approach. Hum Mutat. 2019;40:1127–44.
Russell S, Bennett J, Wellman JA, Chung DC, Yu Z-F, Tillman A, et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet. 2017;390:849–60.
Lois N, Holder GE, Fitzke FW, Plant C, Bird AC. Intrafamilial variation of phenotype in Stargardt macular dystrophy-fundus flavimaculatus. Invest Ophthalmol Vis Sci. 1999;40:2668–75.
Thiadens AA, Roosing S, Collin RW, van Moll-Ramirez N, van Lith-Verhoeven JJ, van Schooneveld MJ. et al. Comprehensive analysis of the achromatopsia genes CNGA3 and CNGB3 in progressive cone dystrophy. Ophthalmology. 2010;117:825–30.e1.
Author information
Authors and Affiliations
Contributions
J.G., E.C., A.V. and R.J. conceived and designed the work. J.G., E.C., A.V. and R.J. drafted and revised the manuscript. J.G., E.C., A.V. and R.J. approved the final version. J.G., E.C., A.V. and R.J. agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Cornish, E.E., Vaze, A., Jamieson, R.V. et al. The electroretinogram in the genomics era: outer retinal disorders. Eye 35, 2406–2418 (2021). https://doi.org/10.1038/s41433-021-01659-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41433-021-01659-y
This article is cited by
-
Group comparisons of the individual electroretinogram time trajectories for the ascending limb of the b-wave using a raw and registered time series
BMC Research Notes (2023)
-
Visual electrophysiology and “the potential of the potentials”
Eye (2023)
-
Early recognition of CLN3 disease facilitated by visual electrophysiology and multimodal imaging
Documenta Ophthalmologica (2023)
