
Abstract
Peroxisomal disorders are a clinically and genetically heterogeneous group of diseases caused by defects in peroxisomal biogenesis or function, usually impairing several metabolic pathways. Peroxisomal disorders are rare; however, the incidence may be underestimated due to the broad spectrum of clinical presentations. The inclusion of X-linked adrenoleukodystrophy to the Recommended Uniform Screening Panel for newborn screening programs in the United States may increase detection of this and other peroxisomal disorders. The current diagnostic approach relies heavily on biochemical genetic tests measuring peroxisomal metabolites, including very long–chain and branched-chain fatty acids in plasma and plasmalogens in red blood cells. Molecular testing can confirm biochemical findings and identify the specific genetic defect, usually utilizing a multiple-gene panel or exome/genome approach. When next-generation sequencing is used as a first-tier test, evaluation of peroxisome metabolism is often necessary to assess the significance of unknown variants and establish the extent of peroxisome dysfunction. This document provides a resource for laboratories developing and implementing clinical biochemical genetic testing for peroxisomal disorders, emphasizing technical considerations for sample collection, test performance, and result interpretation. Additionally, considerations on confirmatory molecular testing are discussed.
Similar content being viewed by others
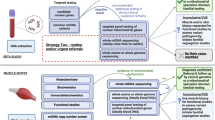

Background
Peroxisome structure and function
Peroxisomes are ubiquitous single membrane-bound organelles responsible for several anabolic and catabolic reactions.1 The most well-characterized peroxisomal pathways are those used in clinical evaluation, including β-oxidation of very long–chain fatty acids (VLCFAs), α-oxidation of methyl-branched phytanic acid, and the synthesis of mature bile acids (BAs) and plasmalogens1 (Fig. 1 Supplemental).
VLCFAs, defined as having a carbon chain with 22 carbons or more, are “tethered” to the peroxisome membrane by ACBD52 and transported into the peroxisome by the integral peroxisome membrane protein ABCD1.3 Once in the peroxisome, fatty acid shortening occurs by a mechanism similar to mitochondrial β-oxidation although producing H2O2, which is further catabolized into water and oxygen by peroxisomal catalase.4 Phytanic acid and C27-bile acid intermediates, dihydroxycholestanoic acid (DHCA) and trihydroxycholestanoic acid (THCA), require auxiliary modifications before they can undergo β-oxidation.1,5 These substrates enter the peroxisome by ABCD3.6 Phytanoyl-CoA requires α-oxidation by phytanoyl-CoA hydroxylase (PhyH);7 the resulting pristanoyl-CoA, as well as DHCA and THCA-CoA derivatives, require racemization from R to S forms by α-methylacyl-CoA racemase (AMACR).1,5,8 The first step in peroxisomal β-oxidation utilizes an oxidase: acyl-CoA oxidase (ACOX) 19 for straight-chain fatty acids, ACOX25,10 for bile acids, and ACOX2 and ACOX3 for pristanic acid. The next steps, hydration and dehydrogenation, are accomplished by the bifunctional enzyme (DBP),11 followed by thiolysis (Sterol Carrier Protein X [SCPx]12 or ACAA1.5) After several β-oxidation cycles, fatty acids shortened to approximately 16 carbons exit the peroxisome as carnitine derivatives. The C27-bile acid intermediates, DHCA and THCA, and the docosahexaenoic acid (DHA) precursor (C24:6n-3) undergo only one cycle of β-oxidation, which results in the production of the C24 mature bile acids, 3α,7α,12α-trihydroxy-5β-cholanoic acid (CA) and 3α,7α-dihydroxy-5β-cholanoic acid (CDCA)5 and DHA (C22:6n-3).13 Subsequently, CA and CDCA are conjugated to glycine or taurine by the bile acid-CoA:amino acid N-acyltransferase (BAAT).14
An additional oxidative pathway occurring in peroxisomes is the oxidation of L-pipecolic acid, an intermediate of lysine catabolism, to Δ1-piperidine-6-carboxylate (P6C).15 The main pathway for lysine degradation occurs in liver mitochondria with conversion to saccharopine, and then to α-aminoadipic semialdehyde (AASA).16 The two pathways converge on AASA, since the P6C ring spontaneously opens forming AASA. AASA is a substrate for the enzyme AASA dehydrogenase, encoded by ALDH7A1 and found both in the cytosol and within mitochondria. Pathogenic variants inALDH7A1 cause pyridoxine-dependent epilepsy (PDE; OMIM 266100),17,18 an infantile epilepsy unresponsive to standard anticonvulsants.
Peroxisomes are also responsible for the initial steps of plasmalogen biosynthesis. Plasmalogens are a subset of glycerophospholipids distinguished by the presence of a vinyl-ether bond at the sn-1 position and enriched polyunsaturated fatty acids at the sn-2 position. They are abundant in cellular and subcellular membranes of many tissues. Synthesis of plasmalogens begins with two enzymes associated with the peroxisomal luminal membrane, dihydroxyacetonephosphate acyltransferase (DHAPAT)19 and alkyl-dihydroxyacetonephosphate synthase (AGPS).20 In forming a vinyl-ether bond, AGPS utilizes fatty alcohols made by fatty acyl-CoA reductase (FAR1)21 from fatty acyl-CoAs specifically of chain-lengths C16:0, C18:0, and C18:1. FAR1 is a membrane protein located on the cytoplasmic surface of the peroxisome membrane; it displays feedback inhibition to cellular plasmalogen levels and thus regulates this pathway. The 1-alkyl-DHAP product, generated from the reaction catalyzed by AGPS, is reduced to alkyl-glycerophosphate by an acyl/alkyl-DHP reductase. Further modifications occur in the endoplasmic reticulum to generate a mature vinyl-ether glycerophospholipid (plasmalogen).
Lastly, glyoxylate detoxification to glycine by the enzyme alanine-glyoxylate aminotransferase (AGT) takes place predominantly in liver peroxisomes. Deficiencies in AGT cause the kidney condition primary hyperoxaluria type 1 (OMIM 259900).22
Peroxisome biogenesis
Peroxisomes are versatile compartments that can increase their capacity in response to the metabolic needs of the cell. Peroxisome biogenesis refers to the formation of new peroxisome membranes, division of existing peroxisomes, and enzyme import into the peroxisome matrix.1,23 These processes require the coordinated action of 16 PEX proteins, or peroxins, encoded by their cognate PEX genes (Table 1 Supplemental). Peroxisomal enzymes are synthesized on cytosolic ribosomes and contain one of two peroxisome-targeting sequences (PTS). The majority of enzymes contain the targeting sequence PTS1, recognized by PEX5 receptor.24,25 A few enzymes, notably ACAA1, PhyH,7 and AGPS,20 contain a PTS2 sequence recognized by PEX7 receptor.26 PEX5 and PEX7 receptors bind to their respective PTS1 or PTS2 targeted enzymes in the cytosol; in addition, PEX7 binds the PEX5 long isoform (PEX5L),25 which is critical for PEX7-mediated import of PTS2-tagged proteins.
The receptor–enzyme complexes dock at the peroxisome membranes using PEX1327,28 and PEX1429 (importomer complex) and release their cargo enzymes into the matrix. Three integral membrane proteins PEX2,30 PEX10,31 and PEX1232 participate in monoubiquitination of PEX5, which allows PEX5 (presumably with PEX7) to be recycled into the cytosol by PEX1–PEX6–PEX2633,34,35,36 (exportomer complex) for another round of import. PEX3, PEX16,37 and PEX1938 are required to form new peroxisome membranes from the endoplasmic reticulum and from mitochondrial membranes.39 PEX11 (there are 3 isoforms, PEX11 alpha, beta,40 and gamma) is required for fission of existing peroxisomes, a process requiring mitochondrial fission proteins, MFF, FIS1, and DLP141 (DNM1L).
Clinical description of peroxisomal disorders
Peroxisomal disorders are a heterogeneous group of inherited diseases due to defects in peroxisomal biogenesis (PBD, Table 1 Supplemental) or single peroxisomal proteins (SPPD; Table 2 Supplemental). PBDs are divided into two groups: Zellweger spectrum disorders (ZSD) and rhizomelic chondrodysplasia punctata (RCDP). ZSDs are a highly heterogeneous group of disorders due to pathogenic variants in one of several PEX genes (Table 1 Supplemental). Core features of ZSDs include liver dysfunction, neurological abnormalities including developmental delays, adrenocortical dysfunction, and vision and hearing impairment.42,43 RCDP is characterized by proximal shortening of the long bones, a distinctive facial appearance, cataracts, congenital contractures, and intellectual disability. Punctate calcification within the epiphyses of long bones (chondrodysplasia punctata) is apparent on radiographs, particularly in infancy.44
X-linked adrenoleukodystrophy (X-ALD), caused by pathogenic variants in ATP-binding cassette transporter type D1 (ABCD1),3 typically presents with one of three phenotypes:45 (1) childhood cerebral form with progressive neurological impairment (cognition, behavior, vision, hearing, motor function) and adrenal insufficiency; (2) late-adolescent or early-adulthood adrenomyeloneuropathy (AMN) with progressive paraparesis and adrenocortical dysfunction; (3) isolated adrenal insufficiency at any age. Approximately 20–50% of adult females with an ABCD1 variant may develop symptoms of AMN. Frequently, X-ALD is misdiagnosed as attention deficit–hyperactivity disorder (ADHD), multiple sclerosis, or idiopathic adrenal insufficiency. Other SPPDs are less common than X-ALD (Table 2 Supplemental). While some features are common to patients with RCDP, ZSDs, or X-ALD, others are not. For example, ulcerating oral gangrene is typically observed in patients with catalase deficiency. Accumulation of calcium oxalate in the kidney progressing to renal failure is common in patients with hyperoxalurias, but also described in ZSD patients. Retinitis pigmentosa, peripheral neuropathy, cerebellar ataxia, and elevated protein levels in the cerebrospinal fluid are seen in Refsum disease (neurological and visual abnormalities are also seen in ZSD patients). Adult-onset neurodegeneration is present in AMACR deficiency. Elevated serum BA concentrations, itching, and fat malabsorption are seen in familial hypercholanemia. Encephalopathy, developmental delay, and hypotonia are present in patients with encephalopathy due to defective mitochondrial peroxisomal fission 1 (EMPF1).
Incidence
Collectively, peroxisomal disorders have an estimated combined incidence of 1:5,000 individuals.1 The most common SPPD is X-ALD3,45 with an estimated incidence of 1:17,000.1 The most common PBDs are ZSDs,43 with an estimated frequency of 1:50,000 in North America.23 Although ZSDs are pan-ethnic, the incidence varies by region, ranging from 1:12,000 in the French Canadian region of Quebec46 to 1:500,000 in Japan.47
Mode of inheritance
Most PBDs and SPPDs are autosomal recessive (AR) conditions. The most notable exception to this is X-ALD, which is X-linked; hemizygous males will develop a clinical phenotype and heterozygous females may develop symptoms over time. Other exceptions include autosomal dominant (AD) de novo variants inDLP1 in patients with EMPF141 and a recently described autosomal dominant form of ZSD due to allelic imbalance for the PEX6-p.Arg860Trp allele (NM_000287.4:c.2578C>T).48
Peroxisomal disorders treatment
Several symptomatic or supportive interventions are available to treat peroxisomal disorders, such as feeding tubes to ensure adequate nutrition, eye glasses and hearing aids, antiepileptic medications, and treatment for nephrolithiasis.42,43,49 Adrenal hormone replacement for those with adrenal insufficiency is lifesaving. Specific interventions are available for selected diagnoses. Prevention of oxalate calculi with urine alkalinization and hydration preserves renal function in patients with hyperoxaluria type I;49 clinical trials using RNA-mediated drugs are ongoing (Alnylam Pharmaceuticals 2016, NCT02706886). In patients with Refsum disease, dietary intake of phytanic acid is restricted to reduce accumulation, and fasting is avoided to prevent its mobilization from stored fat.50 Cholic acid therapy has been used in specific disorders of bile acid synthesis, such as AMACR deficiency, to reduce bile precursor accumulation and normalize liver function.49 Cholic acid therapy reduces the production of bile acid intermediates in patients with ZSD; however, its use may be limited due to possible hepatotoxicity.51,52
Patients with X-ALD and cerebral involvement can benefit from hematopoietic stem cell transplantation (HSCT) if performed prior to significant involvement.53,54 HSCT is associated with up to a 20% mortality rate due to transplant-related complications.53,54 Early results of the STARBEAM study suggest that transplantation with autologous hematopoietic stem cells transfected with elivaldogene tavalentivec (Lenti-D) lentiviral vector is as effective as conventional allogeneic transplantation.55 Currently, there is no targeted therapy for AMN.
Newborn screening for peroxisomal disorders
In February 2016, the US Secretary of Health and Human Services approved the inclusion of X-ALD in the Recommended Uniform Screening Panel (RUSP) for state newborn screening (NBS) programs.56 Attempts to screen for this condition by analyzing VLCFAs in dried blood spots (DBS) were carried out in the late 1980s and early 1990s; however, cumbersome extraction procedures, long run times, high false positive rates, and the discovery of endogenous VLCFAs in filter paper used for NBS made analysis of VLCFAs in DBS unreliable.57 For these reasons, other lipid fractions were evaluated and the lysophosphatidylcholine (LPC) species were found not only to be elevated in DBS of patients with X-ALD, but could be measured accurately and efficiently using standard NBS technology.58 Current NBS protocols utilize measurement of hexacosanoyl-lysophatidylcholine (C26:0 LPC); other markers, such as the ratio of C26:0 LPC to C20:0 LPC, and hexacosanoylcarnitine (C26-carnitine) may also be useful. New York State began screening for X-ALD at the end of 2013; its experience, together with data on the efficacy of early treatment, was instrumental in obtaining RUSP approval.56 To date, several methods are available to measure C20:0-C26:0 LPCs, either alone or in combination with other analytes,59,60,61,62,63,64,65 and additional states have enacted X-ALD screening (for a list of states, refer to www.babysfirsttest.org). Guidelines on follow-up evaluation of a positive screen are available.66,67 Although NBS was designed to identify newborns with X-ALD, LPCs may detect other conditions, such as ZSDs, and ACOX1 and DBP deficiencies. In addition, some neonates with Aicardi Goutières syndrome have elevated levels of C26:0 LPC.68
Methods
The laboratory technical standard was informed by a review of the literature, including current guidelines, and expert opinion. Resources consulted included PubMed (search terms: peroxisomes; peroxisome biogenesis; peroxisomal disorders [AND newborn screening]; very long–chain fatty acids [AND liquid chromatography OR gas chromatography]; plasmalogen [AND liquid chromatography OR gas chromatography]; pipecolic acid [AND liquid chromatography OR gas chromatography]; bile acid synthesis [AND peroxisomal disorders]; X-linked adrenoleukodystrophy; Zellweger spectrum disorder; rhizomelic chondrodysplasia), the American College of Medical Genetics and Genomics (ACMG) Standards and Guidelines for Clinical Genetics Laboratories, Clinical and Laboratory Standards Institute (CLSI) guidelines, CLIA regulations, and the Centers for Disease Control and Prevention Morbidity and Mortality Weekly Report (MMWR) on Good Laboratory Practices for Biochemical Genetics Testing and Newborn Screening for Inherited Metabolic Disorders. When the literature provided conflicting evidence about a topic or when there was insufficient evidence, the authors used expert opinion to inform the recommendations. Expert opinion included the coauthors of the document, members of the ACMG Laboratory Quality Assurance Committee, as well as the experts consulted outside of the Committee but acknowledged in this document. The standard Human Genome Variation Society (HGVS) nomenclature69 was used to describe the sequence variants cited in this document.
Any conflicts of interest for workgroup members or consultants are listed. A draft was delivered to the ACMG Board of Directors for review and member comment. The draft document was posted on the ACMG website and an email link was sent inviting ACMG members to provide comment. The authors assessed all comments. When appropriate, additional evidence was included to address member comments and the draft was amended. Both member comments and author responses were reviewed by a representative of the ACMG Laboratory Quality Assurance Committee and by the ACMG Board of Directors. The ACMG Board of Directors approved the final document before submission for publication.
Preanalytical requirements
Specimen requirements
Laboratory testing for peroxisomal disorders includes different classes of metabolites that accumulate in different metabolic pathways. Hence, a variety of assays are utilized, each being routinely performed in a certain specimen type depending on analyte abundance, ease of collection, or other analytical and clinical considerations. In general, the laboratory should evaluate sample requirements and factors influencing its collection, handling, shipping, and storage at the time of method validation. Sample requirements must be made available to ordering physicians.
Quantitative analysis of very long–chain fatty acids (VLCFAs) and branched-chain fatty acids (BCFAs; e.g., phytanic and pristanic acids) can be performed in plasma or serum70 separated from venous blood (2–5 mL) ideally within 1 hour from collection to avoid hemolysis; sodium or lithium heparin (green top) or EDTA (lavender top) tubes can also be used.71 Metabolite analysis, immunochemical or enzymatic assays can be performed on primary fibroblasts derived from a 2–3 mm skin biopsy under sterile conditions and cultured according to standard protocols. Free and conjugated bile acids (BA) are usually quantified in serum samples; EDTA plasma and serum (red top) contain similar BA concentrations.72
Although plasmalogens can be detected in plasma and serum, these specimens are less sensitive for distinguishing peroxisomal disease states from controls, due to their lower plasmalogen concentrations compared with erythrocytes and possibly due to the influence of diet on plasma/serum levels.70,73 In general, erythrocytes are separated by centrifugation from 1–5 mL of EDTA anticoagulated blood, washed twice in phosphate-buffered saline to remove residual plasma and buffy coat, flushed with nitrogen, and stored frozen until processing.74
Pipecolic acid accumulates in several body fluids. For ease of collection, clinical testing is usually carried out in plasma (sodium or lithium heparin, EDTA, or ACD),75 serum,76 or urine. While pipecolic acid can be measured in cerebral spinal fluid (CSF), normal levels in this specimen type are very low, approximately tenfold less than plasma; thus, detection requires very sensitive methods.77 Pipecolic acid is not consistently elevated in the urine of patients with PBDs,76 so plasma is preferred with one exception: in neonates, plasma pipecolic acid in affected individuals may be within normal limits, thus urine is recommended (unpublished data from the Peroxisomal Diseases Laboratory, Kennedy Krieger Institute). Pipecolic acid urinary excretion is measured using random urine, and is normalized to creatinine. Urine should be collected in a clean container without preservatives.75,76
Molecular genetic studies are typically performed using DNA isolated from leukocytes, usually using 1–2 mL EDTA whole blood. Some laboratories offer testing of RNA by complementary DNA (cDNA) sequencing. Other tissue sources, including cultured chorionic villus sample (CVS) or amniocytes for preimplantation/prenatal diagnosis, can be used if necessary. When no pathogenic variant(s) have been previously identified, biochemical testing in CVS or amniocytes can be used in at-risk pregnancies. When testing CVS or amniocytes, maternal cell contamination must be excluded.78
Sample handling, shipping, and storage
Plasma, urine, and CSF should be immediately frozen upon collection, and shipped on dry ice. Stability under different conditions varies among metabolites, and laboratories should establish proper handling, shipping, and storage conditions. VLCFAs and BCFAs are stable up to 15 days at room temperature (18–25 °C) or refrigerated (2–8 °C), and at least 92 days at ≤−20 °C (unpublished data from the Biochemical Genetics Laboratory at Mayo Clinic). Plasmalogens are stable in whole blood at room temperature (18–25 °C) or refrigerated (2–8 °C) for up to 72 hours. However, once erythrocytes have been isolated and washed, they should be stored under nitrogen at ≤−65 °C, where plasmalogens are stable for up to 12 months (unpublished data from the Peroxisomal Diseases Laboratory, Kennedy Krieger Institute). Pipecolic acid and the metabolites P6C and AASA are stable in plasma or urine up to 5 hours at room temperature (18–25 °C) and up to 24 hours refrigerated (2–8 °C); once samples are frozen, they are stable at least 40 days at ≤−20 °C and up to 1 year at ≤−65 °C.75,79 BAs are stable in EDTA plasma up to 24 hours at room temperature (18–25 °C), at least for several weeks once samples are frozen (≤−20 °C).72 Cultured primary fibroblasts are shipped at room temperature (18–25 °C) in T-25 flasks filled with culture media. Whole blood for DNA extraction should be shipped at room temperature if delivery is within 24 hours or refrigerated if it takes longer (2–8 °C).
Preanalytical variables
In general, fasting or preprandial blood specimens are recommended for metabolite testing; the effect on metabolite concentrations following feeding has not been evaluated for all analytes and may differ among analytes. VLCFAs and BCFAs levels have been shown to increase slightly after a meal.80 BA concentrations also increase after a meal; the increase is modest in healthy individuals, but significant in patients with various liver conditions.81 Other factors affect VLCFA and/or BCFA concentrations leading to an abnormal result: a ketogenic diet,82 high peanut butter consumption,83 severe liver disease, and hyperlipidemia. Hemolysis increases VLCFAs through release from erythrocyte membranes. Hence, plasma/serum should be separated within 24 hours from collection,74 ideally within the first hour, and conditions compromising blood integrity should be avoided. On the other hand, pipecolic acid levels in plasma/serum are not affected by hemolysis or hyperlipidemia,75 and there is no evidence of significant changes with postprandial collection.15
Hemolyzed blood samples are not optimal for plasmalogen analysis, as loss of erythrocytes will artificially lower absolute levels of plasmalogens measured as C16:0 and C18:0 dimethyl acetals (DMAs). Specimens demonstrating moderate to severe hemolysis should be rejected; erythrocytes collected from slightly hemolyzed blood may be used if plasmalogen content is evaluated as a ratio to C16 and C18 fatty acids. Testing performed in blood collected following a blood transfusion could result in erroneously normal or partially reduced plasmalogen levels, as transfused erythrocytes can survive for up to 120 days.74 Results obtained in this scenario requires clinical correlation and reanalysis at least 4 weeks post-transfusion should be considered.
Method validation
Protocols should be established by each laboratory to determine and periodically verify performance characteristics of methods used in peroxisomal disorder testing, in accordance with CLIA and the College of American Pathologists (CAP) regulations (see CLSI document C2484). The laboratory should implement procedures to address values outside of their established criteria for an assay’s performance.
Gas chromatography–mass spectrometry (GC-MS) and tandem mass spectrometry (MS/MS), with or without liquid chromatography (LC), are routinely used to quantify VLCFAs/BCFAs, plasmalogens, pipecolic acid, and BAs. Quantitation with these methods requires a calibration curve with purified synthetic standards, and isotope-labeled internal standards. With few exceptions (e.g., the metabolites P6C and AASA), high-grade standards and internal standards are commercially available from several sources; however, standards may not be available for all components of a multianalyte panel. Reagent performance should be verified by the laboratory prior to clinical use.
As for any analytical assay, quality control (QC) samples should be included with each batch of patient samples processed. Ideally, QC samples should be of the appropriate sample matrix and include two levels of quality control, typically normal and abnormal. Target ranges for QC samples should be established by analyzing replicates (at least 15–20) over several runs, possibly by multiple operators. Plasma or serum specimens from several normal individuals can be pooled and used as normal QC for metabolites assayed in plasma, serum, or CSF. Similarly, urine from a normal individual is used for metabolites assayed in urine. Often, normal plasma/serum or urine samples spiked with standards are used as abnormal QC, also referred to as “high control,” while the normal control can be also referred to as “low control.” Ideally, the abnormal QC is around 75% of the highest calibrator used, which usually corresponds to the upper limit of quantification, and within the range of values observed in patients. A readily available source of erythrocytes with normal plasmalogen levels can be obtained from expired blood purchased from the American Red Cross. Canine blood, which has low erythrocyte plasmalogen levels relative to human blood, is a potential source for an abnormal control and may be obtained from veterinary clinics.
Proficiency testing
Laboratories should participate in an ongoing proficiency testing (PT) program, at least semiannually, as required by regulating agencies. PT outcome should be carefully reviewed to monitor performance and identify results requiring investigation; any remedial action should be carefully documented. The European Research Network for Evaluation and Improvement of Screening, Diagnosis and Treatment of Inherited Disorders of Metabolism (ERNDIM) provides a commercially available, external proficiency testing service for the quantitative analysis of VLCFAs (C:22, C24:0, C26:0), phytanic and pristanic acids in serum, and L-pipecolic acid in serum and urine. Scheduled interlaboratory comparison (alternative PT) can be used when external proficiency testing is unavailable.
Reference intervals
Laboratory-specific reference intervals should be established for all analytes and periodically verified following guidelines (e.g., CLSI document EP28-A3c85). Literature-based ranges should be verified using specific methods/platforms employed by the laboratory to validate the test. Anonymized samples from the general population are acceptable once relevant conditions affecting test results have been excluded; for some analytes, reference intervals are age-specific. If possible, the range of values observed in affected individuals should also be determined. Since there are often significant differences between sample types, reference intervals should be established or verified for each specimen type used for clinical testing.
Testing for peroxisomal disorders
Analysis of very long–chain fatty acids and branched-chain fatty acids in plasma
Traditionally, the measurement of very long–chain fatty acids (VLCFAs) and branched-chain fatty acids (BCFAs) in plasma/serum was carried out by GC-MS.74 This method quantifies fatty acids containing hydrocarbon chains with 8 to 30 carbons, but only a subset is used for diagnosing peroxisomal disorders. The first step of sample preparation consists of hydrolysis of fatty acids bound to complex molecules, using an acid hydrolysis followed by basic hydrolysis and reacidification. After extraction with hexane, the fatty acids are derivatized using pentafluorobenzyl bromide (PFBBr).74 Capillary GC-MS using negative chemical ionization with ammonia as the reagent gas and selected negative-ion monitoring allows separation and detection of the corresponding PFB-esters. Quantitation is achieved using stable isotope-labeled internal standards.74 VLCFA/BCFA quantitation by GC-MS requires a lengthy and laborious sample preparation (approximately 8 hours), and run time of approximately 30 minutes per sample. Several methods using positive-ion electrospray ionization tandem mass spectrometry (ESI-MS/MS)86,87,88 or negative-ion electrospray ionization mass spectrometry (ESI-MS)89 have been developed to simplify sample preparation and reduce run time. The ESI-MS/MS method originally developed by Johnson,86 detecting fatty acids as dimethylaminoethyl ester derivatives, offered a rapid sample preparation (approximately 2 hours) and a short run time (3.5 minutes/sample). However, BCFAs cannot be distinguished from their straight-chain isomers with this method. Method limitations were resolved by the inclusion of a second derivatization step to trimethylaminoethyl ester derivative, and the use of liquid chromatography;87 however, these changes resulted in sample preparation and run time comparable with GC-MS methods. The method developed by Al-Dirbashi et al. utilizes ultraperformance liquid chromatography (UPLC) and derivatization with 4-[2-(N,N-dimethylamino)ethylaminosulfonyl]-7-(2-aminoethylamino)-2,1,3-benzoxadiazole (DAABD-AE) to rapidly detect (5 minutes/sample) plasma BCFAs/VLCFAs after a relatively simple sample preparation (approximately 3 hours).88 Overall, VLCFA/ BCFA concentrations are comparable among methods.88
VLCFAs/BCFAs analysis is a particularly useful biochemical test for the diagnosis of peroxisomal disorders, since abnormalities in these metabolites are noted in the majority of patients. Elevations in hexacosanoic acid (C26:0) and the C26:0 to docosanoic acid (C22:0) ratio are typical of X-ALD patients. Some cases, especially adults, might also show an elevation in tetracosanoic acid (C24:0). The test is very sensitive in males, with almost all X-ALD or AMN patients presenting an abnormal profile. However, 15–20% of female heterozygotes have a normal profile, whether or not they are symptomatic.90,91 Recently, C26:0 LPC in DBS has been shown to be a more sensitive marker to identify X-ALD heterozygotes compared with C26:0-carnitine in plasma, being abnormal in all X-ALD heterozygotes, including those with normal plasma results.92 The degree of the VLCFA abnormality does not correlate with age of onset or severity of phenotype,93 and cannot be used as a predictor of clinical outcome. Abnormal results for this test are not specific to X-ALD/AMN; peroxisomal biogenesis disorders, ACOX1 deficiency, and DBP deficiency have a similar VLCFA profile but different clinical features and other biochemical abnormalities. Elevated phytanic acid accompanies the abnormal VLCA profile in ZSD and some DBP deficiencies. As a consequence of impaired α-oxidation, isolated elevations in phytanic acid are seen in Refsum disease, RCDP types 1 and 5; however, the VLCFA profile is normal.7,50,94 AMACR deficiency is characterized by elevation of BCFAs, in particular pristanic acid, and by an increased pristanic to phytanic acid ratio;8 similar abnormalities can be seen in SCPx deficiency.12 Since VLCFA levels do not change over time, age is not a significant factor in result interpretation.90 In contrast, BCFAs are low at birth, and increase with dietary intake of phytanic acid; hence, affected newborns may not show BCFA abnormalities. As noted previously, nonfasting and hemolyzed specimens and samples from individuals on a ketogenic diet can produce abnormal results; the pattern of abnormalities in different VLCFA and BCFA species can assist in differentiation from peroxisomal disorders; in particular, the C24/C22 ratio is less influenced by diet. A normal result in plasma does not exclude a peroxisomal disorder, as false negative results have been reported.95,96
Analysis of plasmalogens in red blood cells
Historically, plasmalogen deficiency was identified by measuring plasmalogens as DMAs in erythrocytes.73 Several analytical methods for measuring plasmalogen levels have been published and include gas–liquid chromatography (GC), GC-MS, and liquid chromatography/mass spectrometry (LC-MS) techniques.70,73,97,98,99,100 GC and GC-MS methodologies require solvent extraction of lipids and conversion of fatty acids to fatty acid methyl esters (FAME) and plasmalogens to DMA derivatives by acid methanolysis.74 DMA derivatives are separated by GC using a nonpolar capillary column and plasmalogen levels may be expressed as a ratio of C16:0 DMA and C18:0 DMA compared with their corresponding FAME or quantified using a nonendogenous free fatty acid of similar chain length (e.g., C19:0 free fatty acid). Ratio analysis may have greater sensitivity, as samples from individuals with RCDP have a higher level of long-chain alcohols, which are subsequently converted to C16 fatty acid.101 When GC alone is used, appropriate standards must be analyzed concurrently to confirm relative retention times for DMAs and FAMEs. GC-MS methods use selected-ion monitoring (SIM) mass spectrometry to increase assay specificity by utilizing mass assignments, m/z 255 for C16:0 DMA andm/z 283 for C18:0 DMA, in addition to retention time for accurate identification. More recently, LC-MS methods measuring plasmalogens have been published.99,100 The advantages of these techniques include reduced sample processing time and more accurate quantification using synthetic plasmalogens as internal standards, although these internal standards also could be used in SIM-GC-MS analysis. The disadvantage is that not all plasmalogens are measured, only the major species of the ethanolamine and choline plasmalogens. One source of variability in analyte measurement is the GC injection port. Recovery may be reduced as a result of inadequate passage of DMAs and FAMEs through the injection port due to interaction with septal particles produced during the injection process. Thus, for GC and GC-MS analyses, a specialized septum such as the Merlin microseal septum, which is designed to eliminate septal coring, may be required. Likewise, for LC-MS assays, recovery may be influenced by carryover, as plasmalogens may adhere to LC polyetheretherketone (PEEK) tubing.
Conditions affecting plasmalogen synthesis, including PBDs and isolated deficiencies of DHAPAT, AGPS, or FAR1, can lead to reduced plasmalogen levels and ultimately result in an abnormal phenotype. Plasmalogen levels are typically low at birth, gradually increasing to plateau by 6 months of age.102 Because unaffected neonates can have levels that overlap those in older children confirmed to have a PBD, each laboratory, based on their analytical method of choice, must establish age-specific reference intervals. Still, plasmalogen levels in PBD patients may normalize with age,103 decreasing the utility of this assay for the investigation PBDs in older children. In RCDP, the extent of plasmalogen deficiency correlates with phenotypic severity.104 While severely affected individuals have a significant reduction in red blood cell (RBC) plasmalogen content, those with less severe phenotypes show a more modest reduction. In very mild cases, the RBC plasmalogen level may be near normal. While RCDP is often the primary clinical indication for measuring plasmalogen levels, this assay also is performed as part of an overall investigation for inborn errors of metabolism affecting peroxisomal function or assembly. Secondary plasmalogen deficiency has been observed in a number of common conditions such as respiratory disease, inflammatory conditions, Parkinson disease, Down syndrome, and Alzheimer disease.105
Analysis of pipecolic acid in urine and plasma
Amino acid analysis performed with traditional methods, like ion exchange chromatography, can detect L-pipecolic acid when present in large amounts;106 but several more sensitive methods have been developed using mass spectrometry combined with gas or liquid chromatography.74,76,77,107,108,109 Pipecolic acid can be measured by GC-MS in negative74,77 or positive107 mode following derivatization of the amine group with methyl chloroformate, acidic ethyl acetate extraction, and further derivatization with pentafluorobenzyl bromide. Some LC-MS/MS methods do not require sample derivatization, thus requiring smaller sample volumes and less processing time.108,109 A deuterated stable isotope, like 2PA-d11 or 2PA-d9, is utilized as internal standard. Quantitation can be performed by stable isotope dilution or using a calibration curve. 2H5-phenylalanine has also been used as internal standard.108,109 A chiral column can specifically discriminate L-pipecolic acid from the D-enantiomer.108 However, in both affected and normal individuals, the D-enantiomer represents only 2% of pipecolic acid.110
Interestingly, elevated pipecolic acid was the first biochemical abnormality described in ZSD patients.111 Patients with single peroxisomal protein defects, like ACOX1 deficiency or DBP deficiency, display pipecolic acid within the normal range.23,94,112 Pipecolic acid accumulation is not exclusive to peroxisomal biogenesis disorders. In addition to lysine, patients with hyperlysinuria type I accumulate pipecolic acid in plasma and urine.113 Pipecolic acid accumulates and is considered a secondary biomarker in patients with PDE,114 although normal levels have been described.115 In PDE, the specific metabolites P6C and AASA are markedly increased,17,116 and pipecolic acid is routinely assessed as part of multianalyte panels that also include AASA and P6C.75,117 Elevated pipecolic acid has also been described in a patient with pathogenic variants in NADK2,118 a gene encoding a mitochondrial enzyme implicated in several pathways, including lysine degradation.118 Elevated pipecolic acid is also seen in chronic liver dysfunction119 and has been reported in an individual without any clinical features.120,121
Other biochemical tests
Bile acids (BAs) are hydroxylated steroids synthetized from cholesterol in the liver and conjugated to the amino acids taurine or glycine. BAs facilitate the absorption of fat and fat-soluble vitamins in the small intestine; after being metabolized to unconjugated and dehydroxylated species by the gut bacteria, they are reabsorbed and recycled by the liver. Serum concentrations are directly affected by liver function and represent a sensitive marker for common liver conditions, including intrahepatic cholestasis of pregnancy. The final step of BA synthesis takes place in the peroxisomes, where the C27-bile acid intermediates DHCA and THCA are converted to the primary C24-bile acids. In PBD patients, the C27-bile acid intermediates accumulate and cause progressive liver disease.122 Testing for these intermediates specifically aids in the diagnosis of peroxisomal disorders.123,124,125 The quantitative analysis of serum BAs by GC-MS is sensitive, accurate, and allows for good discrimination between species.123 However, since GC-MS methods require a large sample volume and cleavage of conjugated bile acids followed by derivatization, they have been largely replaced by LC-MS/MS methods in the clinical laboratory.72,124,125 BA abnormalities can be severe in ZSDs, and disease severity correlates with the impairment in BA synthesis; mildly affected individuals can have normal BA levels.126
Metabolite levels can also be assessed in cultured skin fibroblasts, although these tests are seldom performed using this sample type, since it requires an invasive skin biopsy and time to establish primary fibroblast cultures. In a minority of individuals (<5%) suspected of having a peroxisomal disorder, measurement of peroxisomal metabolites in fibroblasts could be useful (Steven Steinberg, personal communication); several individuals with abnormalities in fibroblast VLCFAs and BCFAs suggestive of a peroxisomal disorder, with normal plasma values, have been reported.95,96 Assays have been described that measure peroxisomal enzyme activities in patient fibroblasts, usually using radioisotope-labeled substrates.127 Only phytanic acid oxidase and pristanic acid oxidase are routinely offered clinically. Fibroblasts can be used to assess the cellular distribution of the enzyme catalase (catalase solubility), which is normally localized to peroxisomes; catalase localization to the cytosol is a sensitive marker of peroxisomal dysfunction.127
Molecular testing
Molecular testing can be used to confirm (e.g., X-ALD) or identify (e.g., ZSD) the specific gene defect in individuals with highly suggestive clinical and biochemical findings. In some individuals, molecular testing may be used to clarify equivocal laboratory findings in the presence of a nonspecific clinical presentation. Carrier testing for at-risk relatives by molecular testing is the best approach, since carrier testing by biochemical methods is not possible for most peroxisomal disorders, and has only about 80% sensitivity for X-ALD heterozygotes. The classification of variants can be challenging and may require further research-based studies for clarification, but should be made using the ACMG variant classification guidelines.128 In families where a peroxisomal defect has been confirmed biochemically (e.g., abnormal VLCFA results in cultured skin fibroblasts from the index case), but molecular testing has identified variants of uncertain clinical significance, predictive prenatal testing using biochemical methods is best, if available. When two definitive pathogenic variants are identified in trans in a proband, predictive testing, including preimplantation genetic diagnosis, can be reliably offered to families. For confirmatory testing, test options range from a single-gene test to a multigene panel to exome or genome sequencing. The best approach varies depending on the clinical presentation and certainty of the biochemical profile. Testing ABCD1 in males with a biochemical and clinical diagnosis of X-ALD should be adequate, but males with a mild or dominant presentation of a ZSD may overlap129 (PEX6-p.Arg860Trp; NM_000287.4:c.2578C>T) and thus require follow-up of a negative ABCD1 test with a ZSD gene panel. Likewise, individuals with a suspected diagnosis of acatalasemia or primary hyperoxaluria type 1 would be candidates for single-gene testing. For individuals with a suspected diagnosis of ZSD, but who have undergone plasma VLCFA testing only, a broader panel that includes PEX genes and genes associated with SPPD should be considered. Similarly, a patient with a suspected diagnosis of RCDP who has deficient erythrocyte plasmalogens and phytanic acid oxidation (elevated plasma phytanic acid) could have molecular testing limited to PEX7, but if there is uncertainty then a broader RCDP multigene panel is recommended. For individuals with a mild or equivocal biochemical profile, a comprehensive multigene panel that includes most PEX genes and genes associated with SPPDs would be ideal. Exome or genome sequencing may be considered for patients with negative single-gene and multigene panel tests or when the most appropriate single or multigene test cannot be determined. The majority of pathogenic variants associated with peroxisomal disorders involve single-nucleotide substitutions or small insertion/deletions that can be detected by sequencing (either Sanger or next-generation sequencing platforms); however, in some instances, exon-level deletions or duplications are present, and require alternative techniques for detection. Although some laboratories may be able to detect multiexon deletions or duplications using next-generation sequencing platforms, an orthogonal method such as multiplex ligation-dependent probe amplification (MLPA) will be usually needed to confirm a single exon copy-number alteration. About 5–6% of ABCD1 pathogenic variants are large deletions that can be detected by Southern blot or MLPA; in these cases, ABCD1 sequencing results are negative in a female heterozygote, but would fail to amplify in a male hemizygote. In both cases, further testing to detect large deletions is required. Moreover, when sequencing ABCD1 it is important to be aware of pseudogene regions, homologous to exons 7–10, duplicated on chromosomes 2, 10, 16, and 22.130 Both Sanger and next-generation sequencing platforms necessitate careful validation to be certain that these pseudogene regions are avoided.
For autosomal recessive disorders, parental testing is useful to be able to offer carrier testing to family members in each lineage. If a pathogenic variant is detected in apparent homozygosity, parental targeted testing will confirm that one pathogenic variant was inherited from each parent. If both parents are not carriers, the second variant may be a large deletion; alternatively, the variant could be present in two copies, but of the same parental chromosome (uniparental isodisomy has been reported in association with ZSD131 and RCDP132); apparent homozygosity could also be caused by allele dropout due to technical reasons.
References
Waterham HR, Ferdinandusse S, Wanders RJ. Human disorders of peroxisome metabolism and biogenesis. Biochim Biophys Acta. 2016;1863:922–933.
Ferdinandusse S, Falkenberg KD, Koster J, et al. ACBD5 deficiency causes a defect in peroxisomal very long-chain fatty acid metabolism. J Med Genet. 2017;54:330–337.
Mosser J, Douar AM, Sarde CO, et al. Putative X-linked adrenoleukodystrophy gene shares unexpected homology with ABC transporters. Nature. 1993;361:726–730.
Wen JK, Osumi T, Hashimoto T, Ogata M. Molecular analysis of human acatalasemia. Identification of a splicing mutation. J Mol Biol. 1990;211:383–393.
Vaz FM, Ferdinandusse S. Bile acid analysis in human disorders of bile acid biosynthesis. Mol Aspects Med. 2017;56:10–24.
Ferdinandusse S, Jimenez-Sanchez G, Koster J, et al. A novel bile acid biosynthesis defect due to a deficiency of peroxisomal ABCD3. Hum Mol Genet. 2015;24:361–370.
Mihalik SJ, Morrell JC, Kim D, Sacksteder KA, Watkins PA, Gould SJ. Identification of PAHX, a Refsum disease gene. Nat Genet. 1997;17:185–189.
Ferdinandusse S, Denis S, Clayton PT, et al. Mutations in the gene encoding peroxisomal alpha-methylacyl-CoA racemase cause adult-onset sensory motor neuropathy. Nat Genet. 2000;24:188–191.
Fournier B, Saudubray JM, Benichou B, et al. Large deletion of the peroxisomal acyl-CoA oxidase gene in pseudoneonatal adrenoleukodystrophy. J Clin Invest. 1994;94:526–531.
Ferdinandusse S, Denis S, van Roermund CWT, et al. A novel case of ACOX2 deficiency leads to recognition of a third human peroxisomal acyl-CoA oxidase. Biochim Biophys Acta. 2018;1864:952–958.
Ferdinandusse S, van Grunsven EG, Oostheim W, et al. Reinvestigation of peroxisomal 3-ketoacyl-CoA thiolase deficiency: identification of the true defect at the level of d-bifunctional protein. Am J Hum Genet. 2002;70:1589–1593.
Ferdinandusse S, Kostopoulos P, Denis S, et al. Mutations in the gene encoding peroxisomal sterol carrier protein X (SCPx) cause leukencephalopathy with dystonia and motor neuropathy. Am J Hum Genet. 2006;78:1046–1052.
Su HM, Moser AB, Moser HW, Watkins PA. Peroxisomal straight-chain Acyl-CoA oxidase and D-bifunctional protein are essential for the retroconversion step in docosahexaenoic acid synthesis. J Biol Chem. 2001;276:38115–38120.
Carlton VE, Harris BZ, Puffenberger EG, et al. Complex inheritance of familial hypercholanemia with associated mutations in TJP2 and BAAT. Nat Genet. 2003;34:91–96.
Fujita T, Hada T, Higashino K. Origin of D- and L-pipecolic acid in human physiological fluids: a study of the catabolic mechanism to pipecolic acid using the lysine loading test. Clin Chim Acta. 1999;287:145–156.
Sacksteder KA, Biery BJ, Morrell JC, et al. Identification of the alpha-aminoadipic semialdehyde synthase gene, which is defective in familial hyperlysinemia. Am J Hum Genet. 2000;66:1736–1743.
Plecko B, Paul K, Paschke E, et al. Biochemical and molecular characterization of 18 patients with pyridoxine-dependent epilepsy and mutations of the antiquitin (ALDH7A1) gene. Hum Mutat. 2007;28:19–26.
Mills PB, Struys E, Jakobs C, et al. Mutations in antiquitin in individuals with pyridoxine-dependent seizures. Nat Med. 2006;12:307–309.
Ofman R, Hettema EH, Hogenhout EM, Caruso U, Muijsers AO, Wanders RJ. Acyl-CoA:dihydroxyacetonephosphate acyltransferase: cloning of the human cDNA and resolution of the molecular basis in rhizomelic chondrodysplasia punctata type 2. Hum Mol Genet. 1998;7:847–853.
de Vet EC, Ijlst L, Oostheim W, Wanders RJ, van den Bosch H. Alkyl-dihydroxyacetonephosphate synthase. Fate in peroxisome biogenesis disorders and identification of the point mutation underlying a single enzyme deficiency. J Biol Chem. 1998;273:10296–10301.
Buchert R, Tawamie H, Smith C, et al. A peroxisomal disorder of severe intellectual disability, epilepsy, and cataracts due to fatty acyl-CoA reductase 1 deficiency. Am J Hum Genet. 2014;95:602–610.
Nishiyama K, Funai T, Katafuchi R, Hattori F, Onoyama K, Ichiyama A. Primary hyperoxaluria type I due to a point mutation of T to C in the coding region of the serine:pyruvate aminotransferase gene. Biochem Biophys Res Commun. 1991;176:1093–1099.
Steinberg SJ, Dodt G, Raymond GV, Braverman NE, Moser AB, Moser HW. Peroxisome biogenesis disorders. Biochim Biophys Acta. 2006;1763:1733–1748.
Dodt G, Braverman N, Wong C, et al. Mutations in the PTS1 receptor gene, PXR1, define complementation group 2 of the peroxisome biogenesis disorders. Nat Genet. 1995;9:115–125.
Baroy T, Koster J, Stromme P, et al. A novel type of rhizomelic chondrodysplasia punctata, RCDP5, is caused by loss of the PEX5 long isoform. Hum Mol Genet. 2015;24:5845–5854.
Braverman N, Steel G, Obie C, et al. Human PEX7 encodes the peroxisomal PTS2 receptor and is responsible for rhizomelic chondrodysplasia punctata. Nat Genet. 1997;15:369–376.
Shimozawa N, Suzuki Y, Zhang Z, et al. Nonsense and temperature-sensitive mutations in PEX13 are the cause of complementation group H of peroxisome biogenesis disorders. Hum Mol Genet. 1999;8:1077–1083.
Liu Y, Bjorkman J, Urquhart A, Wanders RJ, Crane DI, Gould SJ. PEX13 is mutated in complementation group 13 of the peroxisome-biogenesis disorders. Am J Hum Genet. 1999;65:621–634.
Shimozawa N, Tsukamoto T, Nagase T, et al. Identification of a new complementation group of the peroxisome biogenesis disorders and PEX14 as the mutated gene. Hum Mutat. 2004;23:552–558.
Shimozawa N, Tsukamoto T, Suzuki Y, et al. A human gene responsible for Zellweger syndrome that affects peroxisome assembly. Science. 1992;255:1132–1134.
Warren DS, Morrell JC, Moser HW, Valle D, Gould SJ. Identification of PEX10, the gene defective in complementation group 7 of the peroxisome-biogenesis disorders. Am J Hum Genet. 1998;63:347–359.
Chang CC, Lee WH, Moser H, Valle D, Gould SJ. Isolation of the human PEX12 gene, mutated in group 3 of the peroxisome biogenesis disorders. Nat Genet. 1997;15:385–388.
Reuber BE, Germain-Lee E, Collins CS, et al. Mutations in PEX1 are the most common cause of peroxisome biogenesis disorders. Nat Genet. 1997;17:445–448.
Fukuda S, Shimozawa N, Suzuki Y, et al. Human peroxisome assembly factor-2 (PAF-2): a gene responsible for group C peroxisome biogenesis disorder in humans. Am J Hum Genet. 1996;59:1210–1220.
Yahraus T, Braverman N, Dodt G, et al. The peroxisome biogenesis disorder group 4 gene, PXAAA1, encodes a cytoplasmic ATPase required for stability of the PTS1 receptor. EMBO J. 1996;15:2914–2923.
Matsumoto N, Tamura S, Furuki S, et al. Mutations in novel peroxin gene PEX26 that cause peroxisome-biogenesis disorders of complementation group 8 provide a genotype-phenotype correlation. Am J Hum Genet. 2003;73:233–246.
Honsho M, Tamura S, Shimozawa N, Suzuki Y, Kondo N, Fujiki Y. Mutation in PEX16 is causal in the peroxisome-deficient Zellweger syndrome of complementation group D. Am J Hum Genet. 1998;63:1622–1630.
Matsuzono Y, Kinoshita N, Tamura S, et al. Human PEX19: cDNA cloning by functional complementation, mutation analysis in a patient with Zellweger syndrome, and potential role in peroxisomal membrane assembly. Proc Natl Acad Sci USA. 1999;96:2116–2121.
Sugiura A, Mattie S, Prudent J, McBride HM. Newly born peroxisomes are a hybrid of mitochondrial and ER-derived pre-peroxisomes. Nature. 2017;542:251–254.
Ebberink MS, Koster J, Visser G, et al. A novel defect of peroxisome division due to a homozygous non-sense mutation in the PEX11beta gene. J Med Genet. 2012;49:307–313.
Waterham HR, Koster J, van Roermund CW, Mooyer PA, Wanders RJ, Leonard JV. A lethal defect of mitochondrial and peroxisomal fission. N Engl J Med. 2007;356:1736–1741.
Steinberg SJ, Raymond GV. Braverman NE, Moser AB. Zellweger spectrum disorder. In: Adam MP, Ardinger HH, Pagon RA et al., editors. GeneReviews. Seattle, WA: University of Washington; 1993–2017.
Braverman NE, Raymond GV, Rizzo WB, et al. Peroxisome biogenesis disorders in the Zellweger spectrum: an overview of current diagnosis, clinical manifestations, and treatment guidelines. Mol Genet Metab. 2016;117:313–321.
Wanders RJ, Waterham HR. Peroxisomal disorders I: biochemistry and genetics of peroxisome biogenesis disorders. Clin Genet. 2005;67:107–133.
Moser HW, Mahmood A, Raymond GV. X-linked adrenoleukodystrophy. Nat Clin Pract Neurol. 2007;3:140–151.
Levesque S, Morin C, Guay SP, et al. A founder mutation in the PEX6 gene is responsible for increased incidence of Zellweger syndrome in a French Canadian population. BMC Med Genet. 2012;15:13:72.
Klouwer FC, Berendse K, Ferdinandusse S, Wanders RJ, Engelen M, Poll-The BT. Zellweger spectrum disorders: clinical overview and management approach. Orphanet J Rare Dis. 2015;10:151.
Falkenberg KD, Braverman NE, Moser AB, et al. Allelic expression imbalance promoting a mutant PEX6 allele causes Zellweger spectrum disorder. Am J Hum Genet. 2017;101:965–976.
Wanders RJA. Peroxisomal disorders: improved laboratory diagnosis, new defects and the complicated route to treatment. Mol Cell Probes. 2018;40:60–69.
Wanders RJA, Waterham HR. Leroy BP. Refsum disease. In: Adam MP, Ardinger HH, Pagon RA et al., editors. GeneReviews. Seattle, WA: University of Washington; 1993.
Berendse K, Klouwer FC, Koot BG, et al. Cholic acid therapy in Zellweger spectrum disorders. J Inherit Metab Dis. 2016;39:859–868.
Klouwer FCC, Koot BGP, Berendse K, et al. The cholic acid extension study in Zellweger spectrum disorders: results and implications for therapy. 2019;42:303–312.
Miller WP, Rothman SM, Nascene D, et al. Outcomes after allogeneic hematopoietic cell transplantation for childhood cerebral adrenoleukodystrophy: the largest single-institution cohort report. Blood. 2011;118:1971–1978.
Kuhl JS, Suarez F, Gillett GT, et al. Long-term outcomes of allogeneic haematopoietic stem cell transplantation for adult cerebral X-linked adrenoleukodystrophy. Brain. 2017;140:953–966.
Eichler F, Duncan C, Musolino PL, et al. Hematopoietic stem-cell gene therapy for cerebral adrenoleukodystrophy. N Engl J Med. 2017;377:1630–1638.
Kemper AR, Brosco J, Comeau AM, et al. Newborn screening for X-linked adrenoleukodystrophy: evidence summary and advisory committee recommendation. Genet Med. 2017;19:121–126.
Jakobs C, van den Heuvel CM, Stellaard F, Largilliere C, Skovby F, Christensen E. Diagnosis of Zellweger syndrome by analysis of very long-chain fatty acids in stored blood spots collected at neonatal screening. J Inherit Metab Dis. 1993;16:63–66.
Hubbard WC, Moser AB, Tortorelli S, Liu A, Jones D, Moser H. Combined liquid chromatography-tandem mass spectrometry as an analytical method for high throughput screening for X-linked adrenoleukodystrophy and other peroxisomal disorders: preliminary findings. Mol Genet Metab. 2006;89:185–187.
Hubbard WC, Moser AB, Liu AC, et al. Newborn screening for X-linked adrenoleukodystrophy (X-ALD): validation of a combined liquid chromatography-tandem mass spectrometric (LC-MS/MS) method. Mol Genet Metab. 2009;97:212–220.
Haynes CA, De Jesus VR. Improved analysis of C26:0-lysophosphatidylcholine in dried-blood spots via negative ion mode HPLC-ESI-MS/MS for X-linked adrenoleukodystrophy newborn screening. Clin Chim Acta. 2012;413:1217–1221.
Sandlers Y, Moser AB, Hubbard WC, Kratz LE, Jones RO, Raymond GV. Combined extraction of acyl carnitines and 26:0 lysophosphatidylcholine from dried blood spots: prospective newborn screening for X-linked adrenoleukodystrophy. Mol Genet Metab. 2012;105:416–420.
Turgeon CT, Moser AB, Morkrid L, et al. Streamlined determination of lysophosphatidylcholines in dried blood spots for newborn screening of X-linked adrenoleukodystrophy. Mol Genet Metab. 2015;114:46–50.
Tortorelli S, Turgeon CT, Gavrilov DK, et al. Simultaneous testing for 6 lysosomal storage disorders and X-adrenoleukodystrophy in dried blood spots by tandem mass spectrometry. Clin Chem. 2016;62:1248–1254.
Haynes CA, De Jesus VR. Simultaneous quantitation of hexacosanoyl lysophosphatidylcholine, amino acids, acylcarnitines, and succinylacetone during FIA-ESI-MS/MS analysis of dried blood spot extracts for newborn screening. Clin Biochem. 2016;49:161–165.
Hong X, Kumar AB, Ronald Scott C, Gelb MH. Multiplex tandem mass spectrometry assay for newborn screening of X-linked adrenoleukodystrophy, biotinidase deficiency, and galactosemia with flexibility to assay other enzyme assays and biomarkers. Mol Genet Metab. 2018;124:101–108.
Vogel BH, Bradley SE, Adams DJ, et al. Newborn screening for X-linked adrenoleukodystrophy in New York State: diagnostic protocol, surveillance protocol and treatment guidelines. Mol Genet Metab. 2015;114:599–603.
Regelmann MO, Kamboj MK, Miller BS, et al. Adrenoleukodystrophy: guidance for adrenal surveillance in males identified by newborn screen. J Clin Endocrinol Metab. 2018;103:4324–4331.
Armangue T, Orsini JJ, Takanohashi A, et al. Neonatal detection of Aicardi Goutieres syndrome by increased C26:0 lysophosphatidylcholine and interferon signature on newborn screening blood spots. Mol Genet Metab. 2017;122:134–139.
den Dunnen JT, Dalgleish R, Maglott DR, et al. HGVS recommendations for the description of sequence variants: 2016 update. Hum Mutat. 2016;37:564–569.
Takemoto Y, Suzuki Y, Horibe R, Shimozawa N, Wanders RJ, Kondo N. Gas chromatography/mass spectrometry analysis of very long chain fatty acids, docosahexaenoic acid, phytanic acid and plasmalogen for the screening of peroxisomal disorders. Brain Dev. 2003;25:481–487.
Hall NA, Lynes GW, Hjelm NM. Ratios for very-long-chain fatty acids in plasma of subjects with peroxisomal disorders, as determined by HPLC and validated by gas chromatography-mass spectrometry. Clin Chem. 1988;34:1041–1045.
Scherer M, Gnewuch C, Schmitz G, Liebisch G. Rapid quantification of bile acids and their conjugates in serum by liquid chromatography-tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2009;877:3920–3925.
Bjorkhem I, Sisfontes L, Bostrom B, Kase BF, Blomstrand R. Simple diagnosis of the Zellweger syndrome by gas-liquid chromatography of dimethylacetals. J Lipid Res. 1986;27:786–791.
Steinberg S, Jones R, Tiffany C, Moser A. Investigational methods for peroxisomal disorders. selected-ion monitoring. Curr Protoc Hum Genet. 2008; Chapter 17:Unit 17.6.
Yuzyuk T, Liu A, Thomas A, et al. A novel method for simultaneous quantification of alpha-aminoadipic semialdehyde/piperideine-6-carboxylate and pipecolic acid in plasma and urine. J Chromatogr B Analyt Technol Biomed Life Sci. 2016;1017–8:145–152.
van den Berg GA, Breukelman H, Elzinga H, Trijbels JM, Monnens LA, Muskiet FA. Determination of pipecolic acid in urine and plasma by isotope dilution mass fragmentography. Clin Chim Acta. 1986;159:229–237.
Kok RM, Kaster L, de Jong AP, Poll-The B, Saudubray JM, Jakobs C. Stable isotope dilution analysis of pipecolic acid in cerebrospinal fluid, plasma, urine and amniotic fluid using electron capture negative ion mass fragmentography. Clin Chim Acta. 1987;168:143–152.
Steinberg S, Katsanis S, Moser A, Cutting G. Biochemical analysis of cultured chorionic villi for the prenatal diagnosis of peroxisomal disorders: biochemical thresholds and molecular sensitivity for maternal cell contamination detection. J Med Genet. 2005;42:38–44.
Struys EA, Bok LA, Emal D, Houterman S, Willemsen MA, Jakobs C. The measurement of urinary Delta(1)-piperideine-6-carboxylate, the alter ego of alpha-aminoadipic semialdehyde, in Antiquitin deficiency. J Inherit Metab Dis. 2012;35:909–916.
Moser HW, Moser AB, Frayer KK, et al. Adrenoleukodystrophy: increased plasma content of saturated very long chain fatty acids. Neurology. 1981;31:1241–1249.
Heikkinen J. Effect of a standard test meal on serum bile acid levels in healthy nonpregnant and pregnant women and in patients with intrahepatic cholestasis of pregnancy. Ann Clin Res. 1983;15:183–188.
Stradomska TJ, Bachanski M, Pawlowska J, Syczewska M, Stolarczyk A, Tylki-Szymanska A. The impact of a ketogenic diet and liver dysfunction on serum very long-chain fatty acids levels. Lipids. 2013;48:405–409.
Lam C, Wong D, Cederbaum S, Lim B, Qu Y. Peanut consumption increases levels of plasma very long chain fatty acids in humans. Mol Genet Metab. 2012;107:620–622.
CLSI. Statistical quality control for quantitative measurement procedures: principles and definitions. 4th Ed. CLSI guideline C24. Wayne, PA: Clinical and Laboratory Standards Institute; 2016.
CLSI. Defining, establishing, and verifying reference intervals in the clinical laboratory; approved guideline. 3rd Ed. CLSI document EP28-A3c. Wayne, PA: Clinical and Laboratory Standards Institute; 2008.
Johnson DW. A rapid screening procedure for the diagnosis of peroxisomal disorders: quantification of very long-chain fatty acids, as dimethylaminoethyl esters, in plasma and blood spots, by electrospray tandem mass spectrometry. J Inherit Metab Dis. 2000;23:475–486.
Johnson DW, Trinh MU, Oe T. Measurement of plasma pristanic, phytanic and very long chain fatty acids by liquid chromatography-electrospray tandem mass spectrometry for the diagnosis of peroxisomal disorders. J Chromatogr B Analyt Technol Biomed Life Sci. 2003;798:159–162.
Al-Dirbashi OY, Santa T, Rashed MS, et al. Rapid UPLC-MS/MS method for routine analysis of plasma pristanic, phytanic, and very long chain fatty acid markers of peroxisomal disorders. J Lipid Res. 2008;49:1855–1862.
Valianpour F, Selhorst JJ, van Lint LE, van Gennip AH, Wanders RJ, Kemp S. Analysis of very long-chain fatty acids using electrospray ionization mass spectrometry. Mol Genet Metab. 2003;79:189–196.
Moser AB, Kreiter N, Bezman L, et al. Plasma very long chain fatty acids in 3,000 peroxisome disease patients and 29,000 controls. Ann Neurol. 1999;45:100–110.
Engelen M, Barbier M, Dijkstra IM, et al. X-linked adrenoleukodystrophy in women: a cross-sectional cohort study. Brain. 2014;137:693–706.
Huffnagel IC, van de Beek MC, Showers AL, et al. Comparison of C26:0-carnitine and C26:0-lysophosphatidylcholine as diagnostic markers in dried blood spots from newborns and patients with adrenoleukodystrophy. Mol Genet Metab. 2017;122:209–215.
Tran C, Patel J, Stacy H, et al. Long-term outcome of patients with X-linked adrenoleukodystrophy: a retrospective cohort study. Eur J Paediatr Neurol. 2017;21:600–609.
Hoefler G, Hoefler S, Watkins PA, et al. Biochemical abnormalities in rhizomelic chondrodysplasia punctata. J Pediatr. 1988;112:726–733.
Rosewich H, Waterham HR, Wanders RJ, et al. Pitfall in metabolic screening in a patient with fatal peroxisomal beta-oxidation defect. Neuropediatrics. 2006;37:95–98.
Lusebrink N, Porto L, Waterham HR, et al. Absence of biochemical evidence at an early age delays diagnosis in a patient with a clinically severe peroxisomal biogenesis disorder. Eur J Paediatr Neurol. 2016;20:331–335.
Moser AB, Jones DS, Raymond GV, Moser HW. Plasma and red blood cell fatty acids in peroxisomal disorders. Neurochem Res. 1999;24:187–197.
Caruso U. Simple analysis of plasmalogens in erythrocytes using gas chromatography/mass spectrometry with selected-ion monitoring acquisition. Rapid Commun Mass Spectrom. 1996;10:1283–1285.
Zemski Berry KA, Murphy RC. Electrospray ionization tandem mass spectrometry of glycerophosphoethanolamine plasmalogen phospholipids. J Am Soc Mass Spectrom. 2004;15:1499–1508.
Hui SP, Chiba H, Kurosawa T. Liquid chromatography-mass spectrometric determination of plasmalogens in human plasma. Anal Bioanal Chem. 2011;400:1923–1931.
Rizzo WB, Craft DA, Judd LL, Moser HW, Moser AB. Fatty alcohol accumulation in the autosomal recessive form of rhizomelic chondrodysplasia punctata. Biochem Med Metab Biol. 1993;50:93–102.
Labadaridis I, Moraitou M, Theodoraki M, Triantafyllidis G, Sarafidou J, Michelakakis H. Plasmalogen levels in full-term neonates. Acta Paediatr. 2009;98:640–642.
Wanders RJ, Purvis YR, Heymans HS, et al. Age-related differences in plasmalogen content of erythrocytes from patients with the cerebro-hepato-renal (Zellweger) syndrome: implications for postnatal detection of the disease. J Inherit Metab Dis. 1986;9:335–342.
Braverman N, Chen L, Lin P, et al. Mutation analysis of PEX7 in 60 probands with rhizomelic chondrodysplasia punctata and functional correlations of genotype with phenotype. Hum Mutat. 2002;20:284–297.
Braverman NE, Moser AB. Functions of plasmalogen lipids in health and disease. Biochim Biophys Acta. 2012;1822:1442–1452.
Hutzler J, Dancis J. The determination of pipecolic acid: method and results of hospital survey. Clin Chim Acta. 1983;128:75–82.
Zee T, Stellaard F, Jakobs C. Analysis of pipecolic acid in biological fluids using capillary gas chromatography with electron-capture detection and [2H11]pipecolic acid as internal standard. J Chromatogr. 1992;574:335–339.
Rashed MS, Al-Ahaidib LY, Aboul-Enein HY, Al-Amoudi M, Jacob M. Determination of L-pipecolic acid in plasma using chiral liquid chromatography-electrospray tandem mass spectrometry. Clin Chem. 2001;47:2124–2130.
Semeraro M, Muraca M, Catesini G, et al. Determination of plasma pipecolic acid by an easy and rapid liquid chromatography-tandem mass spectrometry method. Clin Chim Acta. 2015;440:108–112.
Armstrong DW, Zukowski J, Ercal N, Gasper M. Stereochemistry of pipecolic acid found in the urine and plasma of subjects with peroxisomal deficiencies. J Pharm Biomed Anal. 1993;11:881–886.
Danks DM, Tippett P, Adams C, Campbell P. Cerebro-hepato-renal syndrome of Zellweger. A report of eight cases with comments upon the incidence, the liver lesion, and a fault in pipecolic acid metabolism. J Pediatr. 1975;86:382–387.
Peduto A, Baumgartner MR, Verhoeven NM, et al. Hyperpipecolic acidaemia: a diagnostic tool for peroxisomal disorders. Mol Genet Metab. 2004;82:224–230.
Tondo M, Calpena E, Arriola G, et al. Clinical, biochemical, molecular and therapeutic aspects of 2 new cases of 2-aminoadipic semialdehyde synthase deficiency. Mol Genet Metab. 2013;110:231–236.
Willemsen MA, Mavinkurve-Groothuis AM, Wevers RA, Rotteveel JJ, Jakobs C. Pipecolic acid: a diagnostic marker in pyridoxine-dependent epilepsy. Ann Neurol. 2005;58:653.
Mercimek-Mahmutoglu S, Donner EJ, Siriwardena K. Normal plasma pipecolic acid level in pyridoxine dependent epilepsy due to ALDH7A1 mutations. Mol Genet Metab. 2013;110:197.
Yuzyuk T, Thomas A, Viau K, et al. Effect of dietary lysine restriction and arginine supplementation in two patients with pyridoxine-dependent epilepsy. Mol Genet Metab. 2016;118:167–172.
Sadilkova K, Gospe SM Jr., Hahn SH. Simultaneous determination of alpha-aminoadipic semialdehyde, piperideine-6-carboxylate and pipecolic acid by LC-MS/MS for pyridoxine-dependent seizures and folinic acid-responsive seizures. J Neurosci Methods. 2009;184:136–141.
Tort F, Ugarteburu O, Torres MA, et al. Lysine restriction and pyridoxal phosphate administration in a NADK2 patient. Pediatrics. 2016;138:e20154534.
Matsuda Y, Fujita T, Hada T, Higashino K. Comparative study on the correlation of plasma gamma-aminobutyric acid and pipecolic acid with liver function in patients with liver cirrhosis. Hepatol Res. 2000;18:132–140.
Vallat C, Denis S, Bellet H, Jakobs C, Wanders RJ, Mion H. Major hyperpipecolataemia in a normal adult. J Inherit Metab Dis. 1996;19:624–626.
Baas JC, van de Laar R, Dorland L, et al. Plasma pipecolic acid is frequently elevated in non-peroxisomal disease. J Inherit Metab Dis. 2002;25:699–701.
Wanders RJ, Ferdinandusse S. Peroxisomes, peroxisomal diseases, and the hepatotoxicity induced by peroxisomal metabolites. Curr Drug Metab. 2012;13:1401–1411.
Courillon F, Gerhardt MF, Myara A, Rocchiccioli F, Trivin F. The optimized use of gas chromatography-mass spectrometry and high performance liquid chromatography to analyse the serum bile acids of patients with metabolic cholestasis and peroxisomal disorders. Eur J Clin Chem Clin Biochem. 1997;35:919–922.
Bootsma AH, Overmars H, van Rooij A, et al. Rapid analysis of conjugated bile acids in plasma using electrospray tandem mass spectrometry: application for selective screening of peroxisomal disorders. J Inherit Metab Dis. 1999;22:307–310.
Johnson DW, ten Brink HJ, Schuit RC, Jakobs C. Rapid and quantitative analysis of unconjugated C(27) bile acids in plasma and blood samples by tandem mass spectrometry. J Lipid Res. 2001;42:9–16.
Berendse K, Engelen M, Ferdinandusse S, et al. Zellweger spectrum disorders: clinical manifestations in patients surviving into adulthood. J Inherit Metab Dis. 2016;39:93–106.
Ferdinandusse S, Ebberink MS, Vaz FM, Waterham HR, Wanders RJ. The important role of biochemical and functional studies in the diagnostics of peroxisomal disorders. J Inherit Metab Dis. 2016;39:531–543.
Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424.
Tran C, Hewson S, Steinberg SJ, Mercimek-Mahmutoglu S. Late-onset Zellweger spectrum disorder caused by PEX6 mutations mimicking X-linked adrenoleukodystrophy. Pediatr Neurol. 2014;51:262–265.
Eichler EE, Budarf ML, Rocchi M, et al. Interchromosomal duplications of the adrenoleukodystrophy locus: a phenomenon of pericentromeric plasticity. Hum Mol Genet. 1997;6:991–1002.
Turner CL, Bunyan DJ, Thomas NS, et al. Zellweger syndrome resulting from maternal isodisomy of chromosome 1. Am J Med Genet A. 2007;143A:2172–2177.
Nimmo G, Monsonego S, Descartes M, Franklin J, Steinberg S, Braverman N. Rhizomelic chrondrodysplasia punctata type 2 resulting from paternal isodisomy of chromosome 1. Am J Med Genet A. 2010;152A:1812–1817.
Acknowledgements
The authors gratefully acknowledge Ann Moser for her significant contribution to the section “Analysis of plasmalogens in red blood cells” and for carefully reviewing the final manuscript, and the members of the ACMG Biochemical Genetics Subcommittee (Laboratory Quality Assurance Committee) for their valuable input and editorial assistance.
Author information
Authors and Affiliations
Consortia
Ethics declarations
Disclosure
The following authors direct laboratories that perform laboratory testing for the diagnosis and management of peroxisomal disorders as a fee for service: I.D.B., S.T., S.S., L.K., and K.C.-O. N.B. is a member of the medical and scientific advisory board for the Global Foundation for Peroxisome Disorders.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Disclaimer:
This technical standard is designed primarily as an educational resource for clinical laboratory geneticists to help them provide quality clinical laboratory genetic services. Adherence to this standard is voluntary and does not necessarily assure a successful medical outcome. This standard should not be considered inclusive of all proper procedures and tests or exclusive of other procedures and tests that are reasonably directed to obtaining the same results. In determining the propriety of any specific procedure or test, the clinical laboratory geneticist should apply his or her own professional judgment to the specific circumstances presented by the individual patient or specimen.
Clinical laboratory geneticists are encouraged to document in the patient’s record the rationale for the use of a particular procedure or test, whether or not it is in conformance with this standard. They also are advised to take notice of the date any particular standard was adopted, and to consider other relevant medical and scientific information that becomes available after that date. It also would be prudent to consider whether intellectual property interests may restrict the performance of certain tests and other procedures.
The Board of Directors of the American College of Medical Genetics and Genomics approved this technical standard on 15 October 2019.
Correspondence: ACMG (documents@acmg.net)
Supplementary information
Rights and permissions
About this article

Cite this article
De Biase, I., Tortorelli, S., Kratz, L. et al. Laboratory diagnosis of disorders of peroxisomal biogenesis and function: a technical standard of the American College of Medical Genetics and Genomics (ACMG). Genet Med 22, 686–697 (2020). https://doi.org/10.1038/s41436-019-0713-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-019-0713-9
Keywords
This article is cited by
-
A unique Levey–Jennings control chart used for internal quality control in human papillomavirus detection
Virology Journal (2022)
-
An integrated multiomic approach as an excellent tool for the diagnosis of metabolic diseases: our first 3720 patients
European Journal of Human Genetics (2022)
-
A novel use for Levey-Jennings charts in prenatal molecular diagnosis
BMC Medical Genomics (2020)
