
Abstract
Spatial patterns of genetic variation can help understand how environmental factors either permit or restrict gene flow and create opportunities for regional adaptations. Organisms from harsh environments such as the Brazilian semiarid Caatinga biome may reveal how severe climate conditions may affect patterns of genetic variation. Herein we combine information from mitochondrial DNA with physical and environmental features to study the association between different aspects of the Caatinga landscape and spatial genetic variation in the whiptail lizard Ameivula ocellifera. We investigated which of the climatic, environmental, geographical and/or historical components best predict: (1) the spatial distribution of genetic diversity, and (2) the genetic differentiation among populations. We found that genetic variation in A. ocellifera has been influenced mainly by temperature variability, which modulates connectivity among populations. Past climate conditions were important for shaping current genetic diversity, suggesting a time lag in genetic responses. Population structure in A. ocellifera was best explained by both isolation by distance and isolation by resistance (main rivers). Our findings indicate that both physical and climatic features are important for explaining the observed patterns of genetic variation across the xeric Caatinga biome.
Similar content being viewed by others


Introduction
Genetic diversity and structure are usually not evenly distributed across the landscape, and many factors may influence this spatial heterogeneity (e.g., Funk et al. 2005; Lawson 2013; Ortego et al. 2012; Pease et al. 2009; Pérez-Espona et al. 2008). Processes that reduce or increase the exchange of individuals and genes among populations may influence the spatial patterns of genetic variation (Anderson et al. 2010; Sork and Waits 2010; Storfer et al. 2006; Zeller et al. 2012). The landscape itself and environmental features, for instance, may influence the connectivity among populations. In addition, given habitat preferences and the ecological niche of each organism, dispersal matrices may show different levels of permeability (e.g., Funk et al. 2005; Lawson 2013; Ortego et al. 2012; Pease et al. 2009; Pérez-Espona et al. 2008). Recent human-induced shifts in landscape such as habitat fragmentation and large roads may also impact gene flow (e.g., Pérez-Espona et al. 2008; Zellmer and Knowles 2009). Thus, investigating how landscape and environmental features shape genetic variation is an important step to understanding species distribution ranges, evolutionary trajectories, speciation, and may ultimately help improve biodiversity management and conservation (Sork and Waits 2010; Storfer et al. 2006; Zeller et al. 2012).
One of the most well-documented patterns of genetic differentiation is isolation by distance (hereafter IBD). IBD occurs when gene flow is reduced among populations located at greater distances from each other (Jenkins et al. 2010; Wright 1943) and has been commonly detected among wide range species (see review in Jenkins et al. 2010). However, the landscape is often complex and many abiotic or physical features can influence spatial patterns of genetic variation more than distance. Therefore, incorporating landscape complexity may help generate more realistic models to understand gene flow among populations, spatial patterns of genetic variation, and local adaptation (Anderson et al. 2010; Sork and Waits 2010; Storfer et al. 2006; Zeller et al. 2012).
Physical features of the landscape can act as barriers, affecting the spatial connectivity and rates of gene flow among populations. Rivers and mountains have been frequently found to reduce gene flow (e.g., Funk et al. 2005; Lawson 2013), and may ultimately facilitate allopatric speciation (e.g., Soltis et al. 2006). How rivers resist dispersal varies depending on their width and depth, as well as the ability of the particular organism to cross them. Some physical aspects vary less abruptly in the landscape and yet may strongly affect patterns of dispersal and gene flow. For example, elevation and slope gradients have been associated with increased genetic differentiation among populations (e.g., Funk et al. 2005; Wang 2009), even in species with high potential for gene flow (Bulgarella et al., 2012; Ortego et al. 2012; Pérez-Espona et al. 2008). In this case, energetic costs of moving up steep slopes, natural selection against nonlocal genotypes, and/or asynchrony in reproductive phenology may have generated patterns of reduced gene flow along these gradients (Bulgarella et al. 2012; Funk et al. 2005; Ortego et al. 2012; Pérez-Espona et al. 2008).
Present and past environmental conditions may also influence the distribution of genetic variability (Anderson et al. 2010). For instance, historically stable areas may sustain more genetic diversity than unstable areas (Carnaval et al. 2009). In addition, populations experiencing different environmental conditions have shown genetic differentiation, even in species that have high vagility (e.g., Pease et al. 2009) and relatively small distribution ranges (e.g., Ortego et al. 2012). Understanding how suitable climates are spatially and temporally distributed can be useful for identifying corridors for gene flow as well as for detecting populations in areas featuring low habitat suitability or isolated by unsuitable patches (Lawson 2013; Ortego et al. 2012; Pease et al. 2009). Past events such as the historical fluctuations of a species’ range may explain its genetic differentiation (Knowles and Alvarado-Serrano 2010). Hence, understanding the spatial and temporal history of the colonization process can also help create a more accurate picture of how genetic variation is distributed across species ranges (Sork and Waits 2010).
Examining genetic variation in organisms inhabiting harsh environments such as deserts or arid regions can reveal the specific requirements for populations to persist under challenging conditions (Wang 2009). The Caatinga is an arid biome in north-eastern Brazil that represents the largest, most isolated and species-rich nucleus of Seasonally Dry Tropical Forests, characterized by semiarid vegetation, high temperatures, and severe droughts (for a review on Caatinga environment, see Werneck 2011; Werneck et al. 2011). Intuitively, water availability appears to be a limiting factor for survival under hot and arid conditions (Hawkins et al. 2003). Considering that water and temperature availability can maintain high genetic diversity by increasing the rate of biotic interactions (Moya-Laraño 2010), the Caatinga biota offers an excellent opportunity to study the contribution of severe climatic conditions in defining patterns of genetic variation.
Herein, we evaluate the relative importance of different mechanisms such as IBD, physical barriers, environmental conditions, current and past climate, and colonization events in shaping the spatial genetic variation in whiptail lizard Ameivula ocellifera (Spix 1825), an abundant and widespread heliothermic species from Caatinga, preferentially occupying open and sandy areas. This species has a large effective population size, is genetically structured as a single population and, apparently, both males and females migrate (Oliveira 2014; Oliveira et al. 2015). We combined information from genetic data, ecological niche modeling, and phylogeographic history of A. ocellifera to test the association between landscape and environmental features and the spatial distribution of genetic variation. We tested five plausible hypotheses that may explain genetic diversity in A. ocellifera: (i) climatic suitability, (ii) climatic stability, (iii) water and energy availability, (iv) environmental heterogeneity, and (v) colonization history. We also tested six potential drivers of genetic differentiation in A. ocellifera: (i) geographical distance, (ii) connectivity through differences in the current climatic suitability, (iii) connectivity through differences in the past climatic suitability, (iv) resistance through differences in terrain slope, (v) resistance through differences in terrain roughness, and (vi) resistance of rivers. Our findings highlight how physical barriers, climate, and geography interact to affect the accumulation of genetic diversity and differentiation across the xeric Caatinga biome.
Materials and methods
Study species, sample collection, and sequencing
In a previous study, Oliveira et al. (2015) analyzed genetic diversity and structure, phylogeographic history, and diversification of a widespread Caatinga whiptail lizard based on large geographical sampling for multiple loci. They inferred two well-delimited lineages: one is distributed from north to southeastern Caatinga, occupying a large part of this biome (Northeast lineage, that is, Ameivula ocellifera), and the other from the southwest of the Caatinga (Espinhaço Mountain Range) and adjacent areas of Cerrado biome (Southwest lineage, that is, Ameivula xacriaba). Oliveira et al. (2015) findings suggest that A. xacriaba is more widespread in the Cerrado biome. Hence, other populations formerly designated as A. ocellifera from central Brazil (Arias et al. 2014) are most likely A. xacriaba and/or other still undescribed taxa. The Northeast lineage originated from the Southwest lineage and expanded continuously, leaving a genetic signature that was concordant with a founder effect (Oliveira et al. 2015). Here, we use the genus Ameivula instead of Cnemidophorus due to the congruence among recent phylogenomic analyses (Tucker et al. 2016) and taxonomic revisions using morphological data from previous analyses (Harvey et al. 2012).
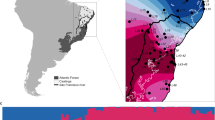
Based on the results of Oliveira et al. (2015) and the description of the type locality of A. ocellifera, we infer that the distribution of A. ocellifera corresponds to the distribution of the Northeast lineage found by Oliveira et al. (2015). From fieldwork and collection loans, we obtained 336 tissue samples from 46 localities distributed throughout the range of Ameivula ocellifera (Fig. 1 and Supplementary Table S1; see also Oliveira et al. (2015)). We sampled an average of seven individuals per locality (range: 4-16; see Table 1). We extracted DNA from liver or muscle tissue using Qiagen DNeasy kits. Following the protocols adopted by Oliveira et al. (2015), we amplified a fragment of the mitochondrial gene 12S ribosomal RNA (12S; 394 bp) via polymerase chain reaction (PCR) using GoTaq Green MasterMix (Promega Corporation) and primers 12Sa (5′-AAACTGGGATTAGATACCCCACTAT-3′) and 12Sb (5′-GAGGGTGACGGGCGGTGTGT-3′). We cleaned PCR products with 2 µl of ExoSap (USB Corporation) and sequenced products using 1 µl of each primer, 2 µl of DTCS (Beckman-Coulter), and 4 µl of ultrapure water. All sequences were generated using Sanger sequencing, aligned with the Clustal algorithm (Sievers et al. 2011), and checked by eye using Geneious 6.1 (Biomatters). We removed gaps using Gblocks (Talavera and Castresana 2007), which reduces the need for manually editing multiple alignments and makes the automation of phylogenetic analysis of large data sets feasible. In addition, this program also facilitates the reproduction of alignments and subsequent phylogenetic analysis by other researchers. All sequences obtained from Oliveira et al. (2015) and in this study are available in GenBank (accession numbers are shown in Supplementary Table S1).

Map with localities sampled for genetic data of the whiptail lizard Ameivula ocellifera along the Caatinga biome (light gray). Haplotypes genealogy from Maximum Likelihood analysis of the 12S gene tree performed in the software Haploviewer. Each haplotype is represented by a circle whose size is proportional to its frequency (indicated in legend). The localities where each haplotype (coded as a number) occurs are available in Table 1
We provide one caveat about the use of only a single mitochondrial marker here. The addition of more loci would be desirable if they reinforced the patterns we found and helped identify the influence of additional predictors that could not be detected through matrilineal heritage alone. However, the four nuclear genes analyzed by Oliveira et al. (2015) all showed low nucleotide diversity and genotypic diversity. Therefore, our present mitochondrial sampling allowed us to expand our geographical coverage, which was important for addressing our specific questions. In addition, mitochondrial DNA has commonly been used in landscape genetic studies and is considered well suited for handeling questions related to historical change over large spatial scales (Anderson et al. 2010).
Genetic diversity and differentiation
To access genetic diversity, we calculated haplotype number (h), haplotype diversity (Hd), and nucleotide diversity (π) for each locality using DnaSP 5.10 (Librado and Rozas 2009). Because there is no deep population structure (Oliveira et al. 2015), each locality was considered as a population, which is a simple way to investigate the spatial distribution of genetic variation. In addition, considering each locality as a population allowed us to model climatic and geographical features throughout the species geographical coverage, which was crucial for our specific questions. Although localities have different number of samples, genetic diversity (i.e., π) was independent of sample size (r = 0.026; P = 0.862). To assess genetic differentiation, we calculated genetic distances among 46 sampling locations with analysis of molecular variance (AMOVA) and tested their significance using 10,000 permutations in ARLEQUIN 3.5 (Excoffier and Lischer 2010). This analysis resulted in a matrix of pairwise F ST-values (Supplementary Table S2). Because AMOVA accounts for sampling bias, it is possible to obtain negative values of F ST that normally are interpreted as zeros (Bird et al. 2011). We detected 20 negative values in our F ST matrix and replaced them with zeros to perform the analyses of genetic differentiation.
We also estimated the genealogical relationships among haplotypes to visualize the genetic diversity and structure in A. ocellifera. First, we used a Maximum Likelihood (ML) approach in PHYML 3.1 (Guindon et al. 2010), using default options and the best-fit model for 12S (i.e., HKY) inferred using the Bayesian Information Criterion (BIC) in jModeltest (Posada 2008). We then used the ML tree to estimate a haplotype network in Haploviewer (Salzburger et al. 2011). To better illustrate the spatial distribution of genetic variation, we generated maps by interpolating both genetic diversity and differentiation across the study region. Interpolation provides a way to predict values and corresponding levels of uncertainty for the variable of interest between points where observations have been made. We used π values from sampling localities and average pairwise genetic distances (F ST) of each locality to generate interpolated genetic variation maps. Using the R package geoR (Ribeiro Jr and Diggle 2001), we derived the final raster maps through the interpolation of ML values by Gaussian process regression (kriging) for non-sampled localities.
Ecological niche modeling
We used ecological niche modeling (ENM) to estimate the geographic distribution of climatically suitable regions for A. ocellifera. ENM results were used to analyze whether current or past climatic conditions were responsible for patterns of genetic diversity and differentiation in A. ocellifera (see below). We used all 46 localities from the present study plus additional 45 confirmed records (museums and literature) as species occurrence data set (Supplementary Table S3). We first modeled current climatically suitable regions for A. ocellifera and then projected the model into three past climatic scenarios: mid-Holocene (6 thousand years before present; 6 kyr), last glacial maximum (LGM, 21 kyr), and last interglacial (LIG, ~ 130 kyr). Present climatic variables were downloaded from the WorldClim database (see http://www.worldclim.org/ for variable descriptions) interpolated to 2.5 arc-min resolution (Hijmans et al. 2005). We obtained past climate data for the mid-Holocene and LGM from ECHAM3 atmospheric General Circulation Model (GCM; DKRZ, 1992) available at the Palaeoclimatic Modeling Intercomparison Project webpage (PMIP; http://pmip.lsce.ipsl.fr/) and for the LIG from Otto-Bliesner et al. (2006). To avoid over-prediction and low specificity, we cropped the bioclimatic layers to span from latitudes 0 to −20 and longitudes −50 to −34. This background encompassed the current extent of Caatinga and adjacent areas. To avoid model overparameterization, we removed strongly correlated variables (r > 0.85) based on their presumed biological relevance for A. ocellifera. We built our models using 11 out of 19 original environmental variables (see Results). We used values of permutation importance (i.e., the loss of model predictive power when each variable is excluded) to determine variables importance.
We implemented ENM using the maximum entropy algorithm (Phillips and Dudik 2008) and the package dismo (Hijmans et al. 2015) in the R platform (R Development Core Team 2017). First, we trained the model under current climatic scenarios based on 75% of randomly selected presence records and used remaining 25% to test the model in 20 bootstrap repetitions, using Maxent default parameters (Phillips and Dudik 2008). We evaluated model performance using the area under the curve (AUC) for the test data. AUC statistics assess the sensitivity (absence of omission error) and the specificity (absence of commission error) of a model (Fielding and Bell 1997). AUC value of 0.50 indicates model performance that was no different than null expectations (random prediction), while higher AUC values indicate better models, with maximum prediction being 1 (Hanley and Mcneil 1982).
Effect of historic and environmental factors on genetic diversity
We used π values to represent genetic diversity. We tested five plausible hypotheses that may explain genetic diversity in A. ocellifera: (i) climatic suitability, (ii) climatic stability, (iii) water and energy availability, (iv) environmental heterogeneity, and (v) colonization. These hypotheses are not mutually exclusive and two or more hypotheses combined may better explain the genetic pattern. Explanatory variables related to each hypothesis are described below in each subtopic (see also Table 2). Using the R package raster (Hijmans and van Etten 2014) and several databases (described below), we extracted environmental data for all 46 localities with genetic information. All environmental values are available at Supplementary Table S4.
Climatic suitability hypothesis
Considering that climatic suitability increases the probability of a population to persist in the environment, this hypothesis predicts higher genetic variability in areas with high habitat suitability. Past environmental conditions may also have had an important influence on the current genetic variability (Anderson et al. 2010). Thus, this hypothesis also predicts higher genetic diversity in areas with high habitat suitability in the past. Because the LGM experienced the most extreme climatic conditions, we only used LGM values to represent past climates. We implemented two alternative approaches to test this hypothesis. Firstly, we assessed the current and LGM climatic suitability for A. ocellifera by extracting values from ENM results. Secondly, we used the best predictors in the ENM [isothermality (BIO3); temperature seasonality (BIO4); and minimum temperature of coldest month (BIO6)] as a simplified measure of A. ocellifera climatic suitability because together they explain 73% of the model (see Results). A possible advantage is that this approach does not consider less important predictors that could mask the influence of the three main predictors. In addition, these three predictors had positive and significant model coefficients (Supplementary Table S5). BIO3 indicates how large the daily temperature oscillation is in comparison to the annual oscillation, whereas BIO4 shows annual range in temperature. From WorldClim and ECHAM3 atmospheric GCM databases, we extracted current and LGM values of BIO3, BIO4, and BIO6.
Climatic stability hypothesis
Population size and persistence should be higher in areas that experienced little climatic variation over time (e.g., Carnaval et al. 2009). Therefore, this hypothesis predicts that genetic diversity should be positively associated with stability. To obtain a stability map that represents regions where suitable climates for A. ocellifera have persisted since the LIG (i.e., refugia), we overlapped the presence/absence projections of each climatic scenario (current, mid-Holocene, LGM, and LIG) generated in ENM. We then obtained a map with stability values ranging from 0 (unsuitable climate in all climate projections) to 4 (suitable climate in all climate projections).
Water and energy availability hypothesis
Temperature and/or water availability can accelerate evolutionary rates (Allen et al. 2002) and maintain high genetic diversity by increasing the rate of biotic interactions (Moya-Laraño 2010). This hypothesis then predicts that genetic variability should increase with water and energy availability. Four variables represented this hypothesis: annual mean temperature (BIO1) and annual precipitation (BIO12) were used as a direct measure of energy and water availability, respectively; actual evapotranspiration (AET) and net primary productivity (NPP) were used as measures of the water-energy balance. We obtained values for BIO1 and BIO12 from WorldClim. AET and NPP values were obtained from the FAO GeoNetwork (http://www.fao.org/geonetwork/srv/en/main.home) and National Aeronautics and Space Administration (NASA; http://daac.ornl.gov/), respectively.
Environmental heterogeneity hypothesis
This hypothesis predicts that genetic diversity should be higher in areas with high environmental heterogeneity, due to local adaptations that lead to small-scale genetic differentiation (e.g., Garant et al. 2005). We considered topographic complexity as a surrogate for environmental heterogeneity. We used altitude data at a 30″ spatial resolution (~ 1 km) obtained from NASA webpage (www2.jpl.nasa.gov/srtm/) to build a topographic complexity index at 2.5 arc-min resolution (~ 5 km). Our index consists of the standard deviation calculated for the 25 cells (30″ resolution) contained in each 2.5 arc-min cell. Therefore, our index reflects the variance in topographic relief within each cell of our grid.
Colonization hypothesis
Areas closer to the center of diffusion presumably had more time to accumulate genetic diversity due to an earlier colonization. Therefore, this hypothesis predicts that genetic diversity should be negatively related to geographic distance from the center of diffusion. A previous study defined the center of diffusion for A. ocellifera using multilocus phylogeographic reconstructions (Oliveira et al. 2015). We overlapped the center of diffusion for five genes (12S, ATPSB, NKTR, R35, and RP40) and calculated its centroid. The geographic coordinates of this centroid were used as an estimate of the center of diffusion. We calculated Euclidean distance from each locality to the center of diffusion using R package ecodist (Goslee and Urban 2007).
Statistical analyses: genetic diversity
To analyze genetic diversity patterns under the above hypotheses, we initially used simple or multiple linear regression (see Table 2) between π (response) and predictors related to each hypothesis. We used the variance inflation factor (VIF) as a measure of the degree of collinearity among multiple predictors (see Dormann et al. 2013). Collinearity is common in ecological data and can artificially inflate predictor regression coefficients, leading to wrong identification of relevant predictors in a statistical model (Dormann et al. 2013). Therefore, we removed predictors with high VIF values and only retained those with VIF < 3 (Groß 2003), which represent the best model (see Table 3). Spatial structure (autocorrelation) can inflate Type I error rates due to non-independence of the data. We examined correlograms of Moran’s I coefficient for ten geographic distance classes (Legendre 1993). Because autocorrelation was observed in model residuals (see Results; and Supplementary Figs. S1 and S2), we performed univariate and multivariate simultaneous autoregression (SAR), using the Akaike’s Information Criterion (AIC) to select the best model. We then compared models by calculating the difference between the AIC of each model and the minimum AIC found for the set of models compared (∆AIC, Burnham and Anderson 2002; Diniz-Filho et al. 2008). Lower ∆AIC scores indicate closer fit to the best model and models with ∆AIC > 2 were considered statistically different (Burnham and Anderson 2002). We built SAR models and performed model selection with SAM 4.0—Spatial Analysis in Macroecology (Rangel et al. 2010).
Effect of isolation by distance vs. isolation by resistance on genetic differentiation
We considered a priori six potential drivers of genetic differentiation in A. ocellifera: (i) geographical distance, (ii) connectivity through differences in the current climatic suitability (CSCURRENT), (iii) connectivity through differences in the LGM climatic suitability (CSLGM), (iv) resistance through differences in terrain slope, (v) resistance through differences in terrain roughness, and (vi) resistance of rivers. The first driver (geographical distance) represents the predicted pattern of IBD, whereas the others are based on assumptions regarding the permeability of landscape features to dispersal and represent the probable isolation by resistance pattern. These hypotheses are not mutually exclusive and, in combination, may better explain the genetic pattern. Predictors related to each hypothesis are described below in each subtopic (see also Table 2).
Isolation by distance
This hypothesis predicts that genetic differentiation increases with geographical distance simply due to the restricted gene flow between populations located far from each other. We used the geographical coordinates of each locality to calculate the geographical distances among all 46 localities (more details in the section “Statistical analyses: genetic differentiation” below).
Isolation by resistance
This hypothesis predicts that landscape features can restrict gene flow among populations due to variations in habitat suitability and the presence of barriers. We used circuit theory to calculate the environmental cost of all possible routes connecting pairs of populations and identify the corridor with the least environmental resistance using CIRCUITSCAPE 3.5.8 (McRae and Beier 2007). We applied this method for slope, roughness, and rivers, as well as CSCURRENT and CSLGM raster grids obtained from ENM analyses. Following Wilson et al. (2007), we derived roughness from altitude using the R package raster (Hijmans and van Etten 2014). We calculated roughness for each grid cell as the difference between the maximum and minimum altitude value of the eight surrounding grid cells. Roughness is an indication of how surface complexity changes over the study area. We used weighting coefficients for the nearest elevation values (eight grid cells) to derive a slope surface (Horn 1981). These coefficients are proportional to the reciprocal of the square of the distance from the central grid cell. From the slope, it is possible to identify flat areas that could facilitate dispersal. Our river raster comprises the main perennial rivers in the Caatinga. Because CIRCUITSCAPE interprets values of 0 as hard barriers, we changed all zeros to 0.0001, following Lawson (2013). Rasters of main rivers, slope, and roughness are available in Supplementary Fig. S3. We used CIRCUITSCAPE to generate three matrices of connectivity distances (CSCURRENT, CSLGM, and rivers) and two matrices of resistance distances (slope and roughness). All matrices are available in Supplementary Tables S6–S10.
Statistical analyses: genetic differentiation
We used Generalized Dissimilarity Modeling (GDM) to evaluate the contribution of environment and space (i.e., the six predictors described above) in explaining genetic differentiation in A. ocellifera. GDM is a matrix regression technique that can fit nonlinear relationships of environmental variables to biological variation through I-spline basis functions (Ferrier et al. 2007). GDM accommodates two types of nonlinearity: (i) variation in the rate of genetic differentiation (non-stationarity) at different positions along a given gradient, and (ii) the curvilinear relationship between genetic differentiation and environmental/geographical distances (Fitzpatrick and Keller 2015). When plotted, the maximum height of each I-spline represents the total amount of genetic differentiation associated with that predictor and the slope shows the rate of change in genetic differentiation along the environmental/geographical gradient. Thus, the splines provide insight into the total magnitude of genetic differentiation as a function of each gradient and where along each gradient those changes are most pronounced.
We first fit a model using the matrix of genetic distances (F ST) as the response and geographic distance (geographical coordinates as input) plus five matrices of resistance and connectivity distances as predictors. We plotted the I-splines to assess how magnitudes and rates of genetic differentiation varied along and between gradients. We also obtained predictor importance by summing its I-spline coefficients (using the default of three I-spline functions). Next, we selected the best predictors using a stepwise matrix permutation and backward elimination approach (see Ferrier et al. 2007; Fitzpatrick et al. 2011; Jones et al. 2016). At each step, the least important predictor, as determined by summing the I-spline coefficients, was removed and a second GDM model was then fit using the reduced set of n-1 predictors and the unique contribution of each predictor to total explained deviance was calculated. We ran 500 random permutations and excluded the predictor with the least significant contribution to explained deviance at each step. We repeated this procedure until all variables retained in the final model made significant unique contributions to explained deviance (P ≤ 0.05). These analyses were performed using the R package gdm (Manion et al. 2017). To evaluate the unique contributions of environmental and geographic distances to genetic differentiation in A. ocellifera, we partitioned the explained deviance resulting from GDM.
Results
Genetic diversity and differentiation
The 336 sequences of the 12S gene preserved ~ 95% (375 bp) of its original size after gap exclusion and corresponded to 88 distinct haplotypes (Fig. 1). The number of haplotypes per locality varied from one to eight (Table 1). H2 was the most widespread haplotype, occurring in 17 different localities in north-northeastern Caatinga. Nucleotide diversity per locality ranged from 0 to 0.01067 (Table 1). Genetic diversity in A. ocellifera was not uniformly distributed across the species range (Fig. 2a) and the highest diversity was concentrated in two patches in central Caatinga.

Spatial distribution of the genetic variation in Ameivula ocellifera represented by interpolated π values a and interpolated pairwise genetic distances b
Most pairwise F ST values ranged from 0 to 1 (except by some few negative values) and 919 of the 1035 pairwise comparisons were significant (Supplementary Table S2). Localities with highest mean genetic distance in A. ocellifera were concentrated in two patches in central-southern Caatinga and along the eastern coastline (Fig. 2b). The very small genetic distances in north-eastern Caatinga (see Fig. 2b) corresponded to the distribution of the most widespread haplotype H2 (see Fig. 1 and Table 1).
Ecological niche modeling
The climatic predictors included in the final habitat suitability model and their values of percent contribution and permutation importance values are shown in Supplementary Table S11. The average training AUC for the replicate runs was 0.925 (SD = 0.010; n = 20 replicate model runs), indicating a high-performance model (Fielding and Bell 1997). The three most important predictors, measured as the percent drop in test AUC when the variable is excluded (permutation importance), were temperature seasonality (BIO4, 39.7%), isothermality (BIO3, 18.5%), and minimum temperature of coldest month (BIO6, 14.9%).
The distribution of climatically suitable regions for A. ocellifera suggests a decrease in habitat suitability over the species range from current to LIG period (Supplementary Fig. S4). The current and mid-Holocene predictions did not differ substantially and suggest numerous suitable habitats in the Caatinga biome and adjacent coastline areas, whereas the largest patches of unsuitable habitats are concentrated in the extreme southwest and northwest of Caatinga. The LGM predictions showed a range contraction of suitable areas for A. ocellifera, which are restricted to a narrow corridor that extends from center to northeast of the Caatinga region. The LIG model suggests low suitability areas for A. ocellifera in most part of Caatinga biome. In this period, favorable climates were distributed in the central-western portion of the Caatinga and outside of its current geographic boundaries.
Effect of historic and environmental factors on genetic diversity
Among five hypotheses tested to explain genetic diversity (Table 3), two were significant: climatic suitability, and water and energy availability. Genetic diversity was positively associated with climatic suitability in both LGM (BIO3LGM + BIO4LGM; AIC = −427.465, ∆AIC = 0, P < 0.001) and current conditions (BIO3CURRENT + BIO4CURRENT; AIC = −427.161, ∆AIC = 3.3, P < 0.001). In addition, genetic diversity was negatively associated with annual precipitation (BIO12; AIC = −414.834, ∆AIC = 12.6, P = 0.036). Because the five hypotheses are not mutually exclusive, we tested all combinations of explanatory variables. The model that mixed variables from both current and LGM climate suitability (BIO3LGM + BIO4CURRENT) explained genetic diversity (AIC = −429.369, ∆AIC = 0.1, P < 0.001) as well as the LGM climate suitability model (BIO3LGM + BIO4LGM; AIC = −427.465, ∆AIC = 0, P < 0.001). Altogether, these two models show that factors related to temperature variability play an important role in driving genetic diversity patterns in A. ocellifera. Areas with higher temperature variability (i.e., more suitable climate) have higher genetic diversity (Supplementary Table S12). Past climate conditions were important for shaping genetic diversity since both models included LGM conditions. Our results support the hypothesis of climatic suitability as the best explanation for genetic diversity in A. ocellifera.
Effect of isolation by distance vs. isolation by resistance on genetic differentiation
The full GDM model of genetic differentiation across the study region (in which all six variables were entered simultaneously) explained 13.5% of the total observed variation. However, three predictors (i.e., CSLGM, resistance through differences in terrain slope and roughness) were not important in determining patterns of genetic differentiation in A. ocellifera, showing I-spline values equal zero. Thus, simpler GDM models performed equally well. Three predictors had unique contributions explaining the total deviance (Fig. 3): connectivity through differences in the CSCURRENT (I-spline value = 0.34), resistance of main rivers (1.15), and geographical distance (0.94); but only the last two made significant contributions (P ≤ 0.05). Therefore, the best model that describes genetic differentiation in A. ocellifera includes resistance of rivers and geographical distance, which explains 13.4% of the total observed variation. The variance partitioning analysis indicated that resistance of rivers and geographical distance accounted for 83.5% and 16.5% of the explained variation, respectively.

Generalized dissimilarity model-fitted I-splines (partial regression fits, a–c panels) for variables associated with genetic differentiation in Ameivula ocellifera. The maximum height reached by each curve indicates the total amount of genetic differentiation associated with that variable, holding all other variables constant. The shape of each function provides an indication of how the rate of genetic differentiation varies along the gradient. The final panel d illustrates the relationship between observed genetic dissimilarity for each site pair in the data set and the linear predictor of the GDM (predicted ecological distance between site pairs)
Discussion
Effects of historic and environmental factors on genetic diversity
Our results showed that the spatial distribution of genetic diversity in A. ocellifera is mostly explained by present and past distributions of suitable climates (i.e., climatic suitability hypothesis). Multiple factors can explain higher genetic diversity in areas with high suitability. First, suitable areas may maximize population fitness, and hence promote accumulation of genetic variability (Nagaraju et al. 2013). Second, suitable areas may shelter large population sizes and retain higher genetic diversity (Gugger et al. 2013). Third, suitable areas may also regularly receive immigrants from neighboring unsuitable areas, increasing genetic diversity. These assumptions are consistent with the expectation that genetic diversity should be higher in populations that occur in suitable areas when compared to unsuitable areas. Hence, populations with repeated local extinctions contain lower genetic diversity (Berendonk et al. 2009). Our data and analyses cannot differentiate among these different mechanisms. Future studies exploring population parameters such as growth rates, migration rates and effective population sizes in regions within high climatic suitability vs. low climatic suitability would help clarify the relative contribution of these different mechanisms.
Our results do suggest that genetic diversity is related to past climate suitability. Many studies have shown that genetic structure and diversity are explained by past environmental conditions (e.g., Anderson et al. 2010; Gugger et al. 2013; Ortego et al. 2012). This is probably related to a delayed genetic response when changes in landscape features occur so rapidly that a considerable lag will exist between causal events and biological response (Anderson et al. 2010). The temporal lag in genetic response is greater for organisms with large effective population sizes (Excoffier 2004) and long generation times (Ortego et al. 2012). Obviously, the temporal lag is also dependent on the time scale considered and the genetic markers used (Anderson et al. 2010), even for makers like mitochondrial DNA, with high rates of substitution and sorting (Wan et al. 2004). Considering that A. ocellifera has a large effective population size (Oliveira et al. 2015), the temporal lag in the genetic responses is even more plausible.
We found that temperature oscillation was the most important predictor of genetic variation. Localities with higher temperature variability tend to present higher genetic diversity. Temperature variability is correlated with A. ocellifera climatic suitability, and we interpret this as evidence for the climatic suitability hypothesis as described above. However, we cannot rule out the possibility that genetic diversity is directly affected by temperature variation. Many studies suggest that higher temperatures may lead to faster mutation rates (Allen et al. 2002; Rohde 1992), but it is still not clear how temperature oscillation per se could increase genetic diversity.
Isolation by distance vs. isolation by resistance
Physical aspects of the landscape can potentially influence the spatial connectivity and rates of gene flow among populations. However, landscape features associated to surface complexity (i.e., slope and roughness) had no effect on genetic differentiation among A. ocellifera populations. On the other hand, the main Caatinga rivers explained around 83.5% of the genetic differentiation. Rivers are important phylogeographic breaks in several lizard species (e.g., Jackson and Austin 2010; Pellegrino et al. 2005; Torres-Pérez et al. 2007). The longest perennial river in Caatinga, the São Francisco River (SFR), is a barrier for many species or lineages, including rodents (Nascimento et al. 2013, 2011) and lizards (Passoni et al. 2008; Siedchlag et al. 2010; Werneck et al. 2012, 2015). Conversely, some widespread Caatinga lineages do not show genetic differentiation on either side of the SFR (e.g., Machado et al. 2014; Recoder et al. 2014; São-Pedro et al., unpublished). Ameivula ocellifera also does not show deep genetic structure associated with the SFR (Oliveira et al. 2015). However, we identified that the network of Caatinga rivers played an important role in the genetic differentiation at finer level, suggesting they provide enough resistance to reduce gene flow among populations.
As expected, our results show that gene flow in A. ocellifera is influenced by IBD, given that genetic distance increases with geographic distance. IBD is especially common among ectotherms, suggesting a metabolic basis underlying gene flow (Jenkins et al. 2010). Ectotherms must modulate activity and location based on external temperature; hence, they are necessarily constrained to disperse within strict temperature limits and temporal windows of opportunity (Ghalambor et al. 2006; Janzen 1967). In our case, temperature oscillation can be considered an indirect measure of habitat quality for A. ocellifera. Some evidence suggests that variation in habitat quality drives differential dispersal and, therefore, population differentiation (Garant et al. 2005). Although our results also support IBD, landscape features such as rivers added unique contributions to the observed genetic pattern. Our results show that gene flow across Caatinga biome is influenced by combination of IBD and resistance imposed by rivers.
Our results constitute a first approach to understanding drivers of genetic variation in Caatinga organisms at a landscape level, based on a sampling effort covering the entire range of a species. Overall, this study suggests that landscape and environmental features can shape patterns of genetic variation in species that inhabit arid regions under challenging conditions. This study also illustrates the benefits of taking into account spatial and temporal information when interpreting patterns of genetic variation.
Data archiving
DNA sequences are publicly available at GenBank. Most sequences were deposited by Oliveira et al. (2015) and some by this study (MF817564-MF817614; see Supplementary Table S1). Voucher number and geographic data of individuals (Supplementary Table S1), GPS coordinates used in ENM (Supplementary Table S3), climate and environmental values for each locality (Supplementary Table S4), and pairwise matrices (Supplementary Tables S2 and S6 to S10) are available from Dryad (https://doi.org/10.5061/dryad.8v5p3).
References
Allen AP, Brown JH, Gillooly JF (2002) Global biodiversity, biochemical kinetics, and the energetic-equivalence rule. Science 297(5586):1545–1548
Anderson CD, Epperson BK, Fortin MJ, Holderegger R, James P, Rosenberg MS et al. (2010) Considering spatial and temporal scale in landscape-genetic studies of gene flow. Mol Ecol 19(17):3565–3575
Arias F, Carvalho CM, Zaher H, Rodrigues MT (2014) A new species of Ameivula (Squamata, Teiidae) from southern Espinhaço mountain range, Brazil. Copeia 2014(1):95–105
Berendonk T, Spitze K, Kerfoot W (2009) Ephemeral metapopulations show high genetic diversity at regional scales. Ecology 90(10):2670–2675
Bird CE, Karl SA, Mouse PE, Toonen RJ (2011). Detecting and measuring genetic differentiation. In: Held C, Koenemann S, Schubart CD (eds) Phylogeography and population genetics in Crustacea, CRC Press: Boca Raton, FL, USA. pp 31–55.
Bulgarella M, Peters JL, Kopuchian C, Valqui T, Wilson RE, McCracken KG (2012) Multilocus coalescent analysis of haemoglobin differentiation between low- and high-altitude populations of crested ducks (Lophonetta specularioides). Mol Ecol 21(2):350–368
Burnham KP, Anderson DR (2002) Model selection and mutlimodel inference: A pratical information-theoretic approach, 2nd edn. Springer Science & Business Media: Berlin, Germany
Carnaval AC, Hickerson MJ, Haddad CFB, Rodrigues MT, Moritz C (2009) Stability predicts genetic diversity in the Brazilian Atlantic Forest hotspot. Science 323(5915):785–789
Diniz-Filho JAF, Rangel TFLVB, Bini LM (2008) Model selection and information theory in geographical ecology. Global Ecol Biogeogr 17(4):479–488
Dormann CF, Elith J, Bacher S, Buchmann C, Carl G, Carré G et al. (2013) Collinearity: a review of methods to deal with it and a simulation study evaluating their performance. Ecography 36(1):027–046
Excoffier L (2004) Patterns of DNA sequence diversity and genetic structure after a range expansion: lessons from the infinite-island model. Mol Ecol 13(4):853–864
Excoffier L, Lischer HEL (2010) Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 10(3):564–567
Ferrier S, Manion G, Elith J, Richardson K (2007) Using generalized dissimilarity modelling to analyse and predict patterns of beta diversity in regional biodiversity assessment. Divers Distrib 13(3):252–264
Fielding AH, Bell JF (1997) A review of methods for the assessment of prediction errors in conservation presence/absence models. Environ Conserv 24:38–49
Fitzpatrick MC, Keller SR (2015) Ecological genomics meets community-level modelling of biodiversity: mapping the genomic landscape of current and future environmental adaptation. Ecol Lett 18(1):1–16
Fitzpatrick MC, Sanders NJ, Ferrier S, Longino JT, Weiser MD, Dunn R (2011) Forecasting the future of biodiversity: a test of single- and multi-species models for ants in North America. Ecography 34(5):836–847
Funk WC, Blouin MS, Corn PS, Maxell BA, Pilliod DS, Amish S et al. (2005) Population structure of Columbia spotted frogs (Rana luteiventris) is strongly affected by the landscape. Mol Ecol 14(2):483–496
Garant D, Kruuk LE, Wilkin TA, McCleery RH, Sheldon BC (2005) Evolution driven by differential dispersal within a wild bird population. Nature 433(7021):60–65
Ghalambor CK, Huey RB, Martin PR, Tewksbury JJ, Wang G (2006) Are mountain passes higher in the tropics? Janzen’s hypothesis revisited. Integr Comp Biol 46(1):5–17
Goslee SC, Urban DL (2007) The ecodist package for dissimilarity-based analysis of ecological data. J Stat Softw 22(7):1–19
Groß J (2003) Variance inflation factors. R news 3(1):13–15
Gugger PF, Ikegami M, Sork VL (2013) Influence of late Quaternary climate change on present patterns of genetic variation in valley oak, Quercus lobata Née. Mol Ecol 22(13):3598–3612
Guindon S, Dufayard J-F, Lefort V, Anisimova M, Hordijk W, Gascuel O (2010) New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol 59(3):307–321
Hanley JA, Mcneil BJ (1982) The meaning and use of the area under a receiver operating characteristic (Roc) curve. Radiology 143(1):29–36
Harvey MB, Ugueto GN, Gutberlet Jr. RL (2012) Review of teiid morphology with a revised taxonomy and phylogeny of the Teiidae (Lepidosauria: Squamata). Zootaxa 3459:1–156
Hawkins BA, Field R, Cornell HV, Currie DJ, Guegan JF, Kaufman DM et al. (2003) Energy, water, and broad-scale geographic patterns of species richness. Ecology 84(12):3105–3117
Hijmans RJ, Cameron SE, Parra JL, Jones PG, Jarvis A (2005) Very high resolution interpolated climate surfaces for global land areas. Int J Climatol 25:1965–1978
Hijmans RJ, Phillips S, Leathwick J, Elith J (2015) dismo: Species distribution modeling. The R Foundation for Statistical Computing, Vienna, R package version 1.0-12. https://cran.r-project.org/
Hijmans RJ, van Etten J (2014) raster: Geographic data analysis and modeling. The R Foundation for Statistical Computing, Vienna, R package version 2.2-31. https://cran.r-project.org/
Horn BK (1981) Hill shading and the reflectance map. Proc IEEE 69(1):14–47
Jackson ND, Austin CC (2010) The combined effects of rivers and refugia generate extreme cryptic fragmentation within the common ground skink (Scincella lateralis). Evolution 64(2):409–428
Janzen DH (1967) Why mountain passes are higher in the tropics. Am Nat 101(919):233–249
Jenkins DG, Carey M, Czerniewska J, Fletcher J, Hether T, Jones A et al. (2010) A meta-analysis of isolation by distance: relic or reference standard for landscape genetics? Ecography 33(2):315–320
Jones MM, Gibson N, Yates C, Ferrier S, Mokany K, Williams KJ et al. (2016) Underestimated effects of climate on plant species turnover in the Southwest Australian Floristic Region. J Biogeogr 43(2):289–300
Knowles L, Alvarado-Serrano DF (2010) Exploring the population genetic consequences of the colonization process with spatio-temporally explicit models: insights from coupled ecological, demographic and genetic models in montane grasshoppers. Mol Ecol 19(17):3727–3745
Lawson LP (2013) Diversification in a biodiversity hot spot: landscape correlates of phylogeographic patterns in the African spotted reed frog. Mol Ecol 22(7):1947–1960
Legendre P (1993) Spatial autocorrelation: trouble or new paradigm? Ecology 74(6):1659–1673
Librado P, Rozas J (2009) DnaSPv5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25(11):1451–1452
Machado T, Silva VX, Silva MJdJ (2014) Phylogenetic relationships within Bothrops neuwiedi group (Serpentes, Squamata): geographically highly-structured lineages, evidence of introgressive hybridization and Neogene/Quaternary diversification. Mol Phylogen Evol 71(0):1–14
Manion G, Lisk M, Ferrier S, Nieto-Lugilde D, Mokany K, Fitzpatrick MC (2017) gdm: Generalized Dissimilarity Modeling. The R Foundation for Statistical Computing, Vienna, R package version 1.3.4. https://cran.r-project.org/
McRae BH, Beier P (2007) Circuit theory predicts gene flow in plant and animal populations. Proc Natl Acad Sci 104(50):19885–19890
Moya-Laraño J (2010) Can temperature and water availability contribute to the maintenance of latitudinal diversity by increasing the rate of biotic interactions? Open Ecol J 3:1–13
Nagaraju SK, Gudasalamani R, Barve N, Ghazoul J, Narayanagowda GK, Ramanan US (2013) Do ecological niche model predictions reflect the adaptive landscape of species?: a test using Myristica malabarica Lam., an endemic tree in the Western Ghats, India. PLoS ONE 8(11):e82066
Nascimento FF, Lazar A, Menezes AN, Durans AdM, Moreira JC, Salazar-Bravo J et al. (2013) The role of historical barriers in the diversification processes in open vegetation formations during the Miocene/Pliocene using an ancient rodent lineage as a model. PLoS ONE 8(4):e61924
Nascimento FF, Pereira LG, Geise L, Bezerra AMR, D’Andrea PS, Bonvicino CR (2011) Colonization pprocess of the Brazilian common vesper mouse, Calomys expulsus (Cricetidae, Sigmodontinae): a biogeographic hypothesis. J Hered 102:260–268
Oliveira EF (2014) Filogeografia de Cnemidophorus ocellifer (Squamata: Teiidae) na Caatinga. PhD Thesis, Universidade Federal do Rio Grande do Norte, Natal, RN, Brazil
Oliveira EF, Gehara M, São-Pedro VA, Chen X, Myers EA, Burbrink FT et al. (2015) Speciation with gene flow in whiptail lizards from a Neotropical xeric biome. Mol Ecol 24(23):5957–5975
Ortego J, Riordan EC, Gugger PF, Sork VL (2012) Influence of environmental heterogeneity on genetic diversity and structure in an endemic southern Californian oak. Mol Ecol 21(13):3210–3223
Otto-Bliesner BL, Marsha SJ, Overpeck JT, Miller GH, Hu AX, Mem CLIP (2006) Simulating arctic climate warmth and icefield retreat in the last interglaciation. Science 311(5768):1751–1753
Passoni J, Benozzati M, Rodrigues M (2008) Phylogeny, species limits, and biogeography of the Brazilian lizards of the genus Eurolophosaurus (Squamata: Tropiduridae) as inferred from mitochondrial DNA sequences. Mol Phylogen Evol 46(2):403–414
Pease KM, Freedman AH, Pollinger JP, McCormack JE, Buermann W, Rodzen J et al. (2009) Landscape genetics of California mule deer (Odocoileus hemionus): the roles of ecological and historical factors in generating differentiation. Mol Ecol 18(9):1848–1862
Pellegrino K, Rodrigues MT, Waite AN, Morando M, Yassuda YY, Sites JW (2005) Phylogeography and species limits in the Gymnodactylus darwinii complex (Gekkonidae, Squamata): genetic structure coincides with river systems in the Brazilian Atlantic Forest. Biol J Linn Soc 85(1):13–26
Pérez-Espona S, Pérez-Barbería F, McLeod J, Jiggins C, Gordon I, Pemberton J (2008) Landscape features affect gene flow of Scottish Highland red deer (Cervus elaphus). Mol Ecol 17(4):981–996
Phillips SJ, Dudik M (2008) Modeling of species distributions with Maxent: new extensions and a comprehensive evaluation. Ecography 31(2):161–175
Posada D (2008) jModelTest: phylogenetic model averaging. Mol Biol Evol 25(7):1253–1256
R Development Core Team (2017) R: Language and Enviroment for Statistical Computing. R Foundation for Statistical Computing, Vienna
Rangel TF, Diniz JAF, Bini LM (2010) SAM: a comprehensive application for Spatial Analysis in Macroecology. Ecography 33(1):46–50
Recoder RS, Werneck FdP, Teixeira M, Colli GR, Sites JW, Rodrigues MT (2014) Geographic variation and systematic review of the lizard genus Vanzosaura (Squamata, Gymnophthalmidae), with the description of a new species. Zool J Linn Soc 171(1):206–225
Ribeiro Jr PJ, Diggle PJ (2001) geoR: A package for geostatistical analysis. R News 1(2):14–18
Rohde K (1992) Latitudinal gradients in species diversity: the search for the primary cause. Oikos 65(3):514–527.
Salzburger W, Ewing GB, Von Haeseler A (2011) The performance of phylogenetic algorithms in estimating haplotype genealogies with migration. Mol Ecol 20(9):1952–1963
Siedchlag AC, Benozzati ML, Passoni JC, Rodrigues MT (2010) Genetic structure, phylogeny, and biogeography of Brazilian eyelid-less lizards of genera Calyptommatus and Nothobachia (Squamata, Gymnophthalmidae) as inferred from mitochondrial DNA sequences. Mol Phylogen Evol 56(2):622–630
Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W et al. (2011) Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 7(1):539
Soltis DE, Morris AB, McLachlan JS, Manos PS, Soltis PS (2006) Comparative phylogeography of unglaciated eastern North America. Mol Ecol 15(14):4261–4293
Sork VL, Waits L (2010) Contributions of landscape genetics–approaches, insights, and future potential. Mol Ecol 19(17):3489–3495
Spix JB (1825) Animalia Nova, sive, species novae Lacertarum quas in itinere per Brasiliam annis MDCCCXVII-MDCCCXX jussu et auspiciis Maximiliani Josephi I. Bavariae regis suscepto / collegit et descripsit Dr. JB de Spix. Typis Franc., Seraph, Hubschmanni: Monachii
Storfer A, Murphy M, Evans J, Goldberg C, Robinson S, Spear S et al. (2006) Putting the ‘landscape’ in landscape genetics. Heredity 98(3):128–142
Talavera G, Castresana J (2007) Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst Biol 56(4):564–577
Torres-Pérez F, Lamborot M, Boric-Bargetto D, Hernández CE, Ortiz JC, Palma RE (2007) Phylogeography of a mountain lizard species: an ancient fragmentation process mediated by riverine barriers in the Liolaemus monticola complex (Sauria: Liolaemidae). J Zool Syst Evol Res 45(1):72–81
Tucker DB, Colli GR, Giugliano LG, Hedges SB, Hendry CR, Lemmon EM et al. (2016) Methodological congruence in phylogenomic analyses with morphological support for teiid lizards (Sauria: Teiidae). Mol Phylogen Evol 103:75–84
Wan QH, Wu H, Fujihara T, Fang SG (2004) Which genetic marker for which conservation genetics issue? Electrophoresis 25(14):2165–2176
Wang IJ (2009) Fine‐scale population structure in a desert amphibian: landscape genetics of the black toad (Bufo exsul). Mol Ecol 18(18):3847–3856
Werneck FP (2011) The diversification of eastern South American open vegetation biomes: historical biogeography and perspectives. Quat Sci Rev 30:1630–1648
Werneck FP, Costa GC, Colli GR, Prado DE, Sites Jr JW (2011) Revisiting the historical distribution of Seasonally Dry Tropical Forests: new insights based on palaeodistribution modelling and palynological evidence. Global Ecol Biogeogr 20(2):272–288
Werneck FP, Gamble T, Colli GR, Rodrigues MT, Sites JJW (2012) Deep diversification and long-term persistence in the South American ‘dry diagonal’: integrating continent-wide phylogeography and distribution modeling of geckos. Evolution 66(10):3014–3034
Werneck FP, Leite RN, Geurgas SR, Rodrigues MT (2015) Biogeographic history and cryptic diversity of saxicolous Tropiduridae lizards endemic to the semiarid Caatinga. BMC Evol Biol 15(1):94
Wilson MF, O’Connell B, Brown C, Guinan JC, Grehan AJ (2007) Multiscale terrain analysis of multibeam bathymetry data for habitat mapping on the continental slope. Mar Geod 30(1-2):3–35
Wright S (1943) Isolation by distance. Genetics 28(2):114
Zeller KA, McGarigal K, Whiteley AR (2012) Estimating landscape resistance to movement: a review. Landscape Ecol 27(6):777–797
Zellmer A, Knowles LL (2009) Disentangling the effects of historic vs. contemporary landscape structure on population genetic divergence. Mol Ecol 18(17):3593–3602
Acknowledgements
We thank researchers and curators of the following Brazilian herpetological collections for providing tissue samples and distribution records: Universidade Católica do Salvador (UCSAL), Universidade Federal do Ceará (UFC), and Universidade de São Paulo (USP). We also thank LO Drummond and PH Medeiros for their assistance in the field expeditions; and E Myers, X Chen, A McKelvy, and S Ruane from the Burbrink laboratory for assistance in laboratorial works. EFO and VASP thank Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) for providing graduate fellowships, and Instituto Chico Mendes de Conservação da Biodiversidade (ICMBio) for issuing collecting permit (no. 26255-1). This work was funded by two grants from Conselho Nacional de Desenvolvimento Científico e Tecnológico—CNPq (SISBIOTA Grant CNPq no. 563352/2010-8 to GCC, and ICMBio/CNPq Grant no. 552031/2011-9 to AAG), and two from CAPES (23038.005577/2012-28 and 23038.009565/2013-53 to AAG). AAG thanks CNPq for financial support (no. 309531/2015-2 and 431433/2016-0). DOM thanks CAPES for a post-doctorate fellowship and CNPq for a research fellowship (no. 303610/2014-0). This research was supported in part by a US National Science Foundation grant (DEB 1257926) to FTB. GRC thanks CAPES, CNPq, Fundação de Apoio à Pesquisa do Distrito Federal (FAPDF), and the United States Agency for International Development (USAID) Partnerships for Enhanced Engagement in Research (PEER) program, administered by the US National Academy of Sciences (NAS), for financial support. GCC thanks CNPq grants 474392/2013-9, 201413/2014-0, and 302297/2015-4.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Oliveira, E.F., Martinez, P.A., São-Pedro, V.A. et al. Climatic suitability, isolation by distance and river resistance explain genetic variation in a Brazilian whiptail lizard. Heredity 120, 251–265 (2018). https://doi.org/10.1038/s41437-017-0017-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41437-017-0017-2
This article is cited by
-
Effects of Environmental Variation in Structuring Population Genetic Variation in the False-Water Cobras (Xenodontinae: Hydrodynastes)
Evolutionary Biology (2023)
-
Geography vs. past climate: the drivers of population genetic structure of the Himalayan langur
BMC Ecology and Evolution (2022)
-
Idiosyncratic responses to drivers of genetic differentiation in the complex landscapes of Isthmian Central America
Heredity (2021)
-
The riverine thruway hypothesis: rivers as a key mediator of gene flow for the aquatic paradoxical frog Pseudis tocantins (Anura, Hylidae)
Landscape Ecology (2021)
-
Conservation genetics of an island-endemic lizard: low Ne and the critical role of intermediate temperatures for genetic connectivity
Conservation Genetics (2021)
