
Abstract
Different mating systems can strongly affect the extent of genetic diversity and population structure among species. Given the increased effects of genetic drift on reduced population size, theory predicts that species undergoing self-fertilisation should have greater population structure than outcrossed species; however, demographic dynamics may affect this scenario. The mangrove killifish clade is composed of the two only known examples of self-fertilising species among vertebrates (Kryptolebias marmoratus and Kryptolebias hermaphroditus). A third species in this clade, Kryptolebias ocellatus, inhabits mangrove forests in southeast Brazil; however, its mating system and patterns of genetic structure have been rarely explored. Here, we examined the genetic structure and phylogeographic patterns of K. ocellatus along its distribution, using mitochondrial DNA and microsatellites to compare its patterns of genetic structure with the predominantly selfing and often-syntopic, K. hermaphroditus. Our results indicate that K. ocellatus reproduces mainly by outcrossing, with no current evidence of selfing, despite being an androdioecious species. Our results also reveal a stronger population subdivision in K. ocellatus compared to K. hermaphroditus, contrary to the theoretical predictions based on reproductive biology of the two species. Our findings indicate that, although morphologically similar, K. ocellatus and K. hermaphroditus had remarkably different evolutionary histories when colonising the same mangrove areas in southeastern Brazil, with other factors (e.g., time of colonisation, dispersal/establishment capacity) having more profound effects on the current population structuring of those species than differences in mating systems.
Similar content being viewed by others
Introduction
Differences in mating systems can have profound effects on the extent of genetic variation and of population structure (Charlesworth and Wright 2001). Theory predicts that selfing species should have deeper population structure than outcrossed species, given the stronger effects of genetic drift on reduced population size (Charlesworth 2003; Meunier et al. 2004). At a broader geographic scale, multiple geographically isolated selfing lineages should result in high levels of genetic diversity in selfing species (Avise and Tatarenkov 2015). However, this pattern can be influenced by temporal population dynamics, such as dispersal and colonisation capacity (Siol et al. 2007).
Although the impact of different mating systems on population structure has already been explored in plants (Willi and Määttänen 2011), this research has lagged behind in animal systems, particularly on vertebrates, where most species are dioicous and are obligate outcrossing (Jarne and Auld 2006). However, a unique diversity of mating systems exists in the mangrove killifish species of the genus Kryptolebias (Costa et al. 2010; Avise and Tatarenkov 2015). The mangrove killifish clade is composed of the only representatives among all rivulids (350+ species) living in brackish waters (Costa et al. 2010), and the only two known examples of self-fertilising hermaphroditism among vertebrates (Kryptolebias marmoratus and Kryptolebias hermaphroditus, species that form the ‘K. marmoratus species complex’, see Tatarenkov et al. 2017). A third species in the mangrove killifish clade is Kryptolebias ocellatus (previously known as Kryptolebias caudomarginatus, taxonomic nomenclature still under discussion (Costa 2011; Huber 2017, see Supplementary material). Kryptolebias ocellatus is endemic to intermittent mangrove microhabitats in southern and southeastern Brazil (Costa 2016).
Kryptolebias ocellatus has been historically bred in aquaria, and while behavioural observations indicate that it reproduces via outcrossing (Seegers 1984), its populations are composed of males and simultaneous hermaphrodites (Costa et al. 2010), leaving open the possibility that this species may also undergo self-fertilisation. However, while the genetic analysis of two populations in this species found no evidence for selfing (Tatarenkov et al. 2009), the possibility of self-fertilisation at a broader geographical scale cannot be ruled out, as rates of selfing and outcrossing are known to vary geographically in the mixed-mating Kryptolebias species (Berbel-Filho et al. 2019; Tatarenkov et al. 2011). In the northernmost part of its distribution (Guanabara and Sepetiba Bays, 22°S), K. ocellatus is often syntopic (i.e., coexisting at the same habitat at the same time) with K. hermaphroditus (Costa 2011, 2016), a species composed mostly of self-fertilising hermaphrodites and very rare males (Berbel-Filho et al. 2016; Costa 2016), resulting in occasional outcrossing but at very low frequencies (Berbel-Filho et al. 2019; Tatarenkov et al. 2017). Extremely low levels of genetic diversity in K. hermaphroditus, especially at the southernmost edge of its distribution (where it is syntopic with K. ocellatus), suggest relatively recent dispersal and colonisation of this species in southeastern Brazil (Tatarenkov et al. 2009, 2011, 2017).
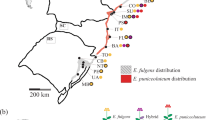
Kryptolebias ocellatus and K. hermaphroditus coexist in shallow mangrove microhabitats, such as temporary pools and crab burrows in discontinuous patches of mangrove forests in southeastern Brazil, and display very similar body shape and colour patterns (Costa 2016; Tatarenkov et al. 2017) (Fig. 1). For these reasons, morphologically based taxonomic classification of the two species has been historically difficult (Costa 2006, 2011, 2016; Huber 2017). However, phylogenetic studies indicate that K. ocellatus is the sister species of the clade containing the two selfing species from ‘K. marmoratus species complex’ (K. marmoratus and K. hermaphroditus) (Kanamori et al. 2016; Tatarenkov et al. 2009; Vermeulen and Hrbek 2005), suggesting that the current syntopy between congeners (K. hermaphroditus and K. ocellatus) in southeastern Brazil is more likely due to dispersal and colonisation rather than to local speciation.

Squares represent locations where K. ocellatus and K. hermaphroditus are syntopic, circles are for locations where only K. ocellatus is found, while triangle designates the site where only K. hermaphroditus is found. Labels for locations are described in Table 1.
Here, we investigate the population structure of K. ocellatus across its range using mitochondrial DNA and microsatellite markers. To test the potential role of different mating systems in determining the population structure, we compared the patterns of genetic structure and diversity of K. ocellatus with previously published data for the self-fertilising species K. hermaphroditus.
Materials and methods
Sampling collection
We sampled K. ocellatus in southern and southeastern Brazil, covering most of its known range (Costa 2016), between August and September 2017 (Fig. 1). Mangrove forests along this ~900-km- long coastal area are discontinuous and heavily fragmented by urbanisation (Barletta and Lima 2019; Branoff 2017). We collected the fish using hand nets in mangrove temporary pools and crab burrows (Fig. 1; Table 1). Sex (male or hermaphrodite) was inferred by body and fin coloration patterns, which are reliably used for sex differentiation in mangrove killifish species (Scarsella et al. 2018). In K. ocellatus, males were identified by a black spot on the dorsal part of the caudal fin (Costa 2016). In K. hermaphroditus, males were identified by the presence of a broad black margin along the whole caudal fin, bordered by a broad submarginal white zone as described in Costa (2016).
Genetic markers
A subset of 16 microsatellites from Mackiewicz et al. (2006) was amplified and genotyped following Tatarenkov et al. (2010). The mitochondrial gene cytochrome oxidase subunit I (cox1) was also used to investigate the genetic structure and major mtDNA lineage distributions.
A 618-bp region of the cox1 was amplified with FishCOI-F (5′-TCAACYAATCAYAAAGACATYGGCAC-3′) and FishCOI-R (5′-ACTTCYGGGTGTCCRAARAAYCA-3′) primers as in Tatarenkov et al. (2017). Both forward and reverse DNA strands were Sanger sequenced and assembled using Geneious v. 9.1.8 (www.geneious.comwww.geneious.com). Sequences were deposited in GenBank (accession numbers: K. ocellatus: MN400774–MN400902; K. hermaphroditus: MN400903–MN400963).
mtDNA and microsatellite datasets
We combined newly generated sequences and genotypes with data from previous studies (Tatarenkov et al. 2011, 2009) for the present genetic analyses (an update of the current taxonomic nomenclature for the study species, which changed in the last few years, is provided in Supplementary material).
The K. ocellatus dataset consisted of individuals from seven sampling locations, three of them (IRI, FUN and GUA in Fig. 1) where the species was found in syntopy with K. hermaphroditus in southeast Brazil, and four (PRT, PAR, SFR and FLO in Fig. 1), where only K. ocellatus is found. Overall, 200 K. ocellatus individuals were analysed, 119 (59.5%) of them were both sequenced for cox1 and genotyped for 16 microsatellites (Table 1). In addition, 10 individuals were sequenced only for cox1, and 71 individuals were only genotyped for microsatellites (Table 1), resulting in 129 individuals sequenced for cox1 and 190 individuals genotyped for microsatellites (Table 1).
In the case of Iriri population (IRI in Fig. 1), new cox1 K. ocellatus sequences were obtained for 22 of the 51 individuals previously genotyped for microsatellites in Tatarenkov et al. (2009) (Table 1). The K. ocellatus microsatellite dataset for Guaratiba (GUA in Fig. 1) consisted of 19 individuals sampled in 2017 (17 of them with cox1 data) and 24 genotypes from individuals sampled in 2007 (no cox1 data) reported in Tatarenkov et al. (2009) (Table 1).
To compare the patterns of genetic structure and diversity between K. ocellatus and K. hermaphroditus in southeastern Brazil, we generated a cox1 dataset for K. hermaphroditus consisting of 61 sequences from three locations in southeast Brazil (FUN, GUA and PIC in Fig. 1), two of them (FUN and GUA in Fig. 1) representing areas of syntopy for both species (Fig. 1; Table 1). We also used 35 K. hermaphroditus microsatellite genotypes from Tatarenkov et al. (2011), comparing both species for 14 of the 16 microsatellites amplified here for K. ocellatus (R34 and R112 were not genotyped in K. hermaphroditus). Kryptolebias hermaphroditus individuals had been sampled from two populations in southeastern Brazil (PIC and GUA in Fig. 1) in 2007.
mtDNA phylogenetic and phylogeographic analyses
A Bayesian coalescent reconstruction was carried out using BEAST v. 2.5.1 (Bouckaert et al. 2014). The sequences included the cox1 haplotypes found across 129 K. ocellatus individuals, as well as the following outgroups: the single haplotype found across 61 K. hermaphroditus individuals (see ‘Results’) in southeastern Brazil, two sequences of K. marmoratus (accession numbers: MF555022.1 and MF554974.1) and two sequences of the ‘Central clade’ lineage (accession numbers: MF555047.1 and MF555072.1), a selfing lineage present in Central America and Caribbean. The ‘Central clade’ is closely related to K. hermaphroditus (Tatarenkov et al. 2017); however, its formal taxonomic status is still under debate. The best-fit model of nucleotide substitution was selected according to the Akaike and Bayesian Criteria on jModelTest2 (Darriba et al. 2012). The substitution model indicated by jModelTest2 was the 3-paratemer model with unequal base frequencies and invariant sites (TPM1uf + I). To time-calibrate the phylogenetic reconstruction and allow for rate variation among lineages, a lognormal-relaxed molecular clock of 0.009 substitutions per site per million years was used, based on the cox1 Goodeidae fossil-calibrated molecular rate described in Webb et al. (2004). We performed three independent runs of 106 Markov Chain Monte Carlo (MCMC) steps, sampling every 103 steps. Tracer v. 1.7.1 (Rambaut et al. 2018) was used to assess convergence and effective sample sizes (≥200) among MCMC runs. The software TREEANNOTATOR v. 2.5.1 (Bouckaert et al. 2014) was used to discard the first 200 trees (20%) as burn-in, and to generate a consensus tree with posterior probability value for each clade.
For K. ocellatus, the number of haplotypes (H) and polymorphic sites (S), haplotype (h) and nucleotide diversities (π) for each sampling location and major mtDNA clades were calculated using DNAsp v. 6.10.04 (Rozas et al. 2017). For generating pairwise fixation indices (FST) among major clades and sampling locations, we used Arlequin v. 3.5.2.2 (Excoffier and Lischer 2010). We used Mega v. 7.0.26 (Kumar et al. 2016) to calculate Kimura-2-parameter (K2P) genetic distances among major clades (see Results) and sampled populations. To visualise haplotype distribution and divergence, we reconstructed a cox1 haplotype network using POPART (Leigh and Bryant 2015).
Genetic structuring and clustering analysis based on microsatellite data
For the microsatellite data, Micro-checker v. 2.2 (van Oosterhout et al. 2004) was used to check for errors in the data and presence of null alleles. To assess overall differentiation at the population level, we used FSTAT v. 2.9.3.2 (Goudet 1995) to calculate FST and conduct exact G tests based on 10,000 randomisations of alleles. FSTAT was also used to measure departures from Hardy–Weinberg equilibrium. P values for FIS for each locus were based on 2240 randomisations, and P values over all loci were calculated from a weighted average of the statistic obtained for each locus. Unbiased expected (HE) and observed heterozygosity (HO) were calculated using MSA v. 4.05 (Dieringer and Schlötterer 2003).
We generated a neighbour-joining tree with 1000 bootstrap replications using Poptree2 (Takezaki et al. 2010) based on a matrix of pairwise Nei’s genetic distances between sampling points. The overall genotypic associations of individuals were visualised with a factorial correspondence analysis (FCA) using the procedure implemented in GENETIX v. 4.04 (Belkhir 2004).
Based only on microssatelite data, we applied two different methods to estimate the most likely number of genetic clusters (K) across K. ocellatus distribution. First, using only microsatellite data, we ran STRUCTURE 2.3.4 (Pritchard et al. 2000) with the following parameters: K values ranging 1–10, 10 iterations per K, a total of 1,000,000 MCMC with 100,000 burn-in, admixture model and independent allele frequencies. To identify the uppermost hierarchical level of genetic structure, we chose the most likely K value using second-order rate of change of likelihood ΔK method (Evanno et al. 2005), implemented in Structure Harvester (Earl 2012). Independent STRUCTURE runs were aligned and plotted using CLUMPAK (Kopelman et al. 2015).
Given the uneven number of individuals in our sample, we also used STRUCTURESELECTOR (Li and Liu 2018), which provides four metrics of cluster estimates to identify the most likely number of genetic clusters (median of means (MedMeaK), maximum of means (MaxMeaK), median of medians (MedMedK) and maximum of medians (MaxMedK)) (Puechmaille 2016).
Genetic structuring based on mtDNA and microsatellite data
To integrate mtDNA, microsatellite data and spatial information, we used Geneland v. 4.0.8 (Guillot et al. 2008), which takes into account spatial information from each individual, also allowing for uncertainty in the positioning of sampled individuals. To identify spatial population distribution and assess individual assignment to the most likely K, we followed Guillot et al. (2005). Geneland allows for the inclusion of a particular individual even if nuclear or mtDNA data is missing. We combined mtDNA and microsatellite data for a total 200 K. ocellatus individuals, including individuals without mtDNA sequences (71 individuals) or microsatellite genotypes (10 individuals). To avoid any bias potentially introduced by introgression on genetic structure patterns of K. ocellatus, we removed microsatellite data for 11 potential hybrid individuals (see Results); however, we maintained their mtDNA for Geneland, as they showed no evidence of introgression and would represent the mtDNA of the parental individuals (see ‘Results'). Therefore, Geneland was run with information from 200 individuals, 129 with mtDNA information, 179 with microsatellite genotypes and 108 (after removal of microsatellite data from 11 hybrids) with information for both markers. We repeated the analysis excluding the hybrid individuals from the mtDNA to assess their contribution to the results. Geographical coordinates (geo-referenced according to the sampling points and with uncertainty of ±0.05 in both latitude and longitude) were included for all individuals. K ranges from 1 to 10. Ten multiple runs were performed with 10,000,000 MCMC iterations, sampled every 1000 iterations. Once the most likely K value was inferred from the modal value across ten multiple runs, we ran the MCMC again with other ten multiple runs and K fixed to the assigned value. These final ten runs were postprocessed (with a burn-in of 20%) in order to obtain posterior probabilities of population membership for each individual. All Geneland analyses were performed using ‘geneland' R package (Guillot et al. 2008).
Isolation-by-distance (IBD) in K. ocellatus
Given the discontinuous distribution of mangrove forest in southeastern Brazil, we tested the association between geographical and genetic distance in K. ocellatus. We estimated the pairwise geographical distance (straight line in kilometres) among sampling points in R v. 3.5.3. We used IBD v. 1.52 (Bohonak 2002), running a Mantel test between the matrices of pairwise FST between sampling points (both for mtDNA and microsatellites) and estimated geographical distance in kilometres.
Results
mtDNA phylogenetic and phylogeographic analysis
Twenty-two cox1 haplotypes (618-bp long) were recovered from 129 K. ocellatus individuals sequenced. In contrast, only one cox1 haplotype was found for K. hermaphroditus across 61 individuals (Table S1). Overall, our phylogenetic reconstruction grouped all K. ocellatus haplotypes in a monophyletic clade, with a sister clade composed of the selfing mangrove killifish species, namely K. hermaphroditus and K. marmoratus (Fig. 2). In K. ocellatus, a clear geographical pattern was found by the Bayesian reconstruction tree using cox1 haplotypes (Fig. 2). Three major lineages were found: a clade composed of haplotypes from sampling locations within Guanabara and Sepetiba’s Bays (IRI, FUN and GUA, hereafter called Northern clade), clustered with a clade containing haplotypes from the opposite side of Sepetiba Bay (PRT, hereafter called Parati clade), although the support for the grouping of Northern and Parati clades was low (PP: 0.75). The third clade was composed of haplotypes from sampling points in southern Brazil (PAR, SFR and FLO, hereafter called the Southern clade). These three major clades were also supported by NJ tree using microsatellite distances and the haplotype network (Figs. S1 and 2).

a Bayesian time-calibrated phylogenetic gene tree for 22 mitochondrial cytochrome oxidase 1 gene (cox1) haplotypes found across 129 specimens of Kryptolebias ocellatus. The single cox1 haplotype found for K. hermaphroditus in southeast Brazil, two cox1 haplotypes from ‘Central clade’, which is a lineage related to K. hermaphroditus (Tatarenkov et al. 2017) and two cox1 haplotypes from K. marmoratus were used as outgroups. The taxonomic status of the Central clade is in revision (Tatarenkov et al. 2020). Circles at nodes represent values of Bayesian posterior probability (PP). Only PP>0.75 is shown. Scale at the bottom in millions of years (Mya). b cox1 haplotype network for 129 specimens of K. ocellatus. Each circle represents a haplotype and its size is proportional to the frequency of the haplotype. Ticks on branches connecting the haplotypes indicate nucleotide mutations. Different colours are used for each sampling locality.
In K. ocellatus, the overall haplotype diversity was 0.89, being the highest in the SFR (Southern clade) and the lowest in GUA (Northern clade) populations. Nucleotide diversity was generally low (π = 0.007), and followed a similar pattern to the haplotype diversity, being the highest at PAR (Southern clade) and the lowest at GUA (0.0004). The same pattern of haplotype and nucleotide diversity was seen when sampling locations were grouped according to the major mtDNA clades, with the Southern clade being the most diverse, followed by the Northern and Parati clades, respectively (Table S2).
The average FST value of all pairwise comparisons was 0.72 (sd = ±0.22) in K. ocellatus. All FST pairwise comparisons among sampling locations were significant, with the exception of the comparison between SFR and PAR (within the Southern clade). The highest FST value (0.92) was found in the comparison between FLO and PRT (Parati clade), while the lowest (0.14) was found between SFR and PAR (Table S3). All FST pairwise comparisons were significant when grouping sampling locations into the major mtDNA clades (mean = 0.80). K2P genetic distances followed a similar pattern to the FST values, with the highest value (1.4%) being observed between samples from the Southern and Parati clades, while the lowest value (0.2%) between samples of the same mtDNA clade (Table S3). K2P genetic distance between K. ocellatus and K. hermaphroditus was 11.2%.
Additional analysis revealed similar patterns of genetic diversity (Table S3) and genetic differentiation among populations (FST and genetic distances; Table S5) in a dataset excluding the cox1 sequences from hybrid individuals (see below), suggesting that the hybridisation has not affected the general patterns of genetic structure observed at the mtDNA.
Microsatellite variation within populations
We found evidence for possible hybridisation between K. ocellatus and K. hermaphroditus in two syntopic populations (FUN and GUA, Figs. 3 and S2). A Structure analysis using both K. ocellatus and K. hermaphroditus microsatellite data, indicated that the uppermost level of genetic structure was K = 3, with the two genetic clusters within K. ocellatus (corresponding to Northern and Southern populations, see below), and a third cluster with K. hermaphroditus individuals from GUA and PIC. Eleven individuals had admixed genetic background between K. ocellatus and K. hermaphroditus, five in FUN and six in GUA (only 2017 sampling) (Figs. 3 and S2). These potential hybrids exhibited K. ocellatus mtDNA haplotypes (haplotypes 4, 6, 7 and 8, Fig. 2; Table S1), all of them (except haplotype 7) commonly found in non-admixed individuals from other northern populations. To avoid any bias caused by these potential hybrids, we excluded them from all population genetics analyses based on microsatellite data.

a Most likely genetic cluster (K) value for Kryptolebias ocellatus using 16 microsatellites analysed in Structure and Geneland. K values determined by ΔK method of Evanno et al. (2005) and metrics of Puechmaille (2016) implemented in STRUCTURESELECTOR. Geneland analysis includes spatial and mtDNA information, in addition to the microsatellite’s genotypes. Each individual is represented by a bar, and each colour represents a genetic cluster. b–c Factorial correspondence analysis for all K. ocellatus individuals coloured and shaped according to their sampling sites.
On average, 22.4 K. ocellatus individuals were genotyped at 16 microsatellite loci per sampling location, but sample size varied considerably (from 5 to 51) (Table 2). Overall, there was a high level of variation at microsatellite loci in K. ocellatus. The number of alleles varied from 2 at locus R28 to 35 at locus R38, with an average of 17.6 alleles per locus considering all sampling locations combined. The mean expected heterozygosity (HE) was 0.56 (ranging from 0.47 to 0.60) for K. ocellatus. The K. ocellatus Northern clade populations (IRI, FUN and GUA) showed a higher average HE (0.59) than Parati (0.53) and Southern clades (0.53). Only one sampling point (GUA dataset for 2007) had significant heterozygote deficiency (Table 2). Examination of single-locus FIS values indicated that significant values of mean FIS were due to contribution of few loci and likely due to null alleles (Table S4). Since mean FIS was non-significant after the atypical loci were excluded, we consider that all studied populations of K. ocellatus are in Hardy–Weinberg equilibrium, and we kept all loci for further analyses (Table S4).
As expected for a selfing species, no loci were found to be under Hardy–Weinberg equilibrium for K. hermaphroditus. The number of alleles varied from 1 (at 50% of locus) to 10, with an average of 2.21 alleles per locus. No heterozygotes were found in both K. hermaphroditus populations. Significant heterozygote deficiency was detected for all loci that had more than one allele in K. hermaphroditus (Tables 2 and S4).
Genetic differentiation and clustering analysis
Classification of individuals using STRUCTURE provided consistent results for each K across the ten replicated runs. As expected in highly structured populations, the most divergent groups separate into distinct clusters first (Pritchard et al. 2000). Evanno’s ΔK method indicated that the uppermost level of genetic structure was K = 2. This analysis indicated one genetic cluster encompassing fish from the Northern populations (IRI, FUN, GUA and PRT) and another composed of fish from the southernmost sampling sites (PAR, SFR and FLO) (Figs. 1, 3). Outcomes of K = 5 (indicated as the most likely number of genetic clusters by all metrics in STRUCTURESELECTOR; Fig. S3) assigned all fish from GUA and PRT to their own genetic clusters. Two genetic lineages were found to be admixed in the sampling points of Guanabara Bay (IRI and FUN). Southernmost sampling points were assigned to the same genetic cluster (Fig. 3). Geneland results incorporating mtDNA, microsatellites and spatial data generally agreed with those from STRUCTURE. Posterior distributions of the number of genetic clusters (K) showed a mode at K = 5 across all ten replicated runs (Figs. 3, S3, and S4). Spatially, cluster 1 was composed of individuals from IRI and FUN, while individuals from GUA, PRT and PAR each represented a unique genetic cluster (clusters 2, 3 and 4, respectively). Cluster 5 was composed of the southernmost individuals from SFR and FLO (Fig. S4). No differences between GUA samples from 2007 to 2017 were found across any clustering analysis. An additional Geneland analysis using 108 individuals with data for both mtDNA and microsatellites (excluding the hybrids), suggested K = 4 as the most likely number of genetic clusters. Overall, the genetic clustering found in this analysis was similar to the one found using the whole dataset (Fig. 3), with the exception that individuals from GUA have clustered with individuals from other populations of the Northern clade (IRI and FUN) (Fig. S5).
FCA confirmed the uppermost subdivisions detected by Evanno’s method (ΔK) from STRUCTURE (Fig. 3). The plot along the two main axes showed that the major division was between the southern and the northern populations along axis 1. Along axis 2, further genetic subdivision was found, matching Geneland results. PRT and PAR individuals formed separate clusters from Northern and Southern populations, respectively. Along axis 3, PAR individuals differentiated even further (Fig. 3).
Genetic differentiation between populations of K. ocellatus was high and significant in global tests and pairwise comparisons. For example, the average FST among all pairwise comparisons was 0.25 (P < 0.001). In pairwise comparisons, no difference was found between Guaratiba samples collected 10 years apart (FST = 0.00). FST in the remaining pairwise comparisons varied from 0.07 (between IRI and FUN, and SFR and FLO) to 0.38 (between IRI and PAR). The majority of pairwise FST was statistically significant after Bonferroni correction for multiple testing, with the exception of comparisons of FUN vs. GUA 2017 (FST = 0.072), and FUN vs. PAR (FST = 0.382), the latter most likely caused by small sample sizes in FUN and PAR (Table S7). Significant FST between PIC and GUA populations of K. hermaphroditus was also found (0.30, P = 0.01).
Strong evidence for isolation-by-distance was found in K. ocellatus using both mtDNA (R2 = 0.58, P = 0.01) and microsatellite (R2 = 0.84, P = 0.003) pairwise genetic distances. In particular, two loci (R9 and R18) showed evident pattern of regional geographic differentiation between Northern and Southern populations, with little-to-no overlap in allele distribution between Northern and Southern populations (Fig. S6).
Discussion
Theory predicts that, all else being equal, selfing should have magnified effects on genetic structure when compared to outcrossing as a consequence of reduced effective population size and increased inbreeding (Charlesworth 2003; Meunier et al. 2004). Here we found that, overall, K. ocellatus populations across much of known species distribution are under Hardy–Weinberg equilibrium, strongly suggesting that despite androdioecious, K. ocellatus is mostly an outcrossing species. This finding supports early behavioural (Costa et al. 2010; Seegers 1984) and genetic (Tatarenkov et al. 2009; for two populations only) indications of outcrossing as the main mating system in K. ocellatus, although the possibility that the species undergoes, even if only rarely, selfing cannot be fully discarded. It also remains to be established if hermaphrodites mate exclusively with males, or whether they can mate with each other. Our results also revealed deep population structuring in K. ocellatus, mostly following a pattern of isolation-by-distance (IBD), which generally contrasts with the high genetic homogeneity found in the morphologically similar, predominantly selfing and often-syntopic K. hermaphroditus across discontinuous mangrove forests along the Brazilian coast (Tatarenkov et al. 2009, 2011, 2017).
In the selfing species composing the K. marmoratus species complex, extensive genetic structure has been identified across (Tatarenkov et al. 2015, 2007) and within the same mangrove systems (Berbel-Filho et al. 2019; Ellison et al. 2012; Turko et al. 2018). An exception to this pattern of deep genetic structure is the high genetic homogeneity found among the selfing K. hermaphroditus populations across the Brazilian coast (Tatarenkov et al. 2011). As shown here, K. hermaphroditus from southeastern Brazil carries a single cox1 haplotype and is completely homozygous at polymorphic microsatellite loci. Geographically, this low genetic diversity scenario in K. hermaphroditus extends even further, with very little genetic differentiation in K. hermaphroditus populations from Southeast and Northeast Brazil, separated by ~2500 km along the coast (Tatarenkov et al. 2017). In contrast, in a much narrower geographic distribution (~900 km along the coast from Magé in the State of Rio de Janeiro to Florianópolis in Santa Catarina state), K. ocellatus showed a deeper genetic structure with division in two genetic clusters (Northern and Southern), and moderate internal genetic structure within these clusters. In its relatively narrow geographic distribution, K. ocellatus also showed more cox1 mtDNA haplotypes (22 vs. 18) and higher interclade mtDNA genetic distances (cox1 K2P distance: 1.1%) than the average genetic distance among clades (cox1 K2P distance: 0.98% between ‘Central’ and ‘Southern’ clades) in the widely distributed (Florida (29°N) to São Paulo (23°S)) K. marmoratus species complex (Tatarenkov et al. 2017). Although the K. ocellatus phylogenetic reconstruction was based on a single mtDNA gene, which may not accurately represent the species tree, it was highly concordant with the microsatellite tree (Fig. S1), supporting the existence of at least two major clades across K. ocellatus distribution, with further genetic subdivisions within them. Thus, our results indicate that the two Kryptolebias species in southeastern Brazil did not evolve by a sympatric speciation event in the region (Kanamori et al. 2016; Tatarenkov et al. 2017), and have remarkably different evolutionary history along the Brazilian coast, with K. hermaphroditus most likely being a recent coloniser of a mangrove area where K. ocellatus might have settled/originated much earlier.
Mangrove forests are typically associated with intertidal zones along rivers, estuaries and bay areas with brackish water (Ball 1988; Hamilton and Casey 2016). This association forms an overall discontinuous distribution of mangrove patches (Hamilton and Casey 2016). Further contributing for mangrove forest fragmentation is human activity, in which its effects are particularly pronounced in heavily urbanised areas, such as southeast Brazil (Branoff 2017; Ferreira and Lacerda 2016). The fragmented distribution of mangrove forests may have contributed for the pattern of IBD found here for K. ocellatus, with geographically more distant populations also being the most genetically dissimilar, both at mtDNA and microsatellite markers. The IBD pattern of genetic structure has also been found for highly selfing populations of K. marmoratus in Florida (Tatarenkov et al. 2015), indicating that, in some occasions (but see below), long-distance dispersal in mangrove killifishes is limited. Mangrove killifishes are the only rivulid species living in brackish waters (Costa et al. 2010) and rarely share mangrove microhabitats with other fish species permanently (Taylor 2012). Therefore, making inferences between the genetic structure of mangrove killifishes and other mangrove-dwelling fish species is challenging. Studies of various mangrove tree species showed weak genetic structure among estuaries in southeastern Brazil, with a general north–south pattern of dispersal, guided by the Brazilian ocean current (Francisco et al. 2018; Mori et al. 2015; Pil et al. 2011). This high gene-flow scenario among different estuaries has also been observed in other mangrove-dwelling species in the same region, which disperse through pelagic larvae, such as crabs (Britto et al. 2018; de Oliveira-Neto et al. 2008; Oliveira-Neto et al. 2007). The strong genetic subdivision found in K. ocellatus between Northern and Southern estuaries in southwestern Atlantic (with particularly high FST values at the mtDNA), contrasts with the general pattern of panmixia pattern mentioned above for mangrove-associated species in the same region. Although our data indicate that K. ocellatus reproduces mostly via outcrossing, we cannot discard that the high genetic differentiation between Northern and Southern populations could have been amplified by geographical variation on ancestral events of selfing. In addition, given the strong indication of IBD, the high differentiation between Northern and Southern populations may have been magnified by the lack of sampling in more intermediate locations (e.g., along São Paulo state coast, Fig. 1). Finally, further research is needed to indicate whether hybridisation (and potential ancestral introgression) between K. ocellatus and K. hermaphroditus may have influenced the allele distribution and population differentiation of K. ocellatus in the Northern populations.
The deep genetic structure and limited dispersal between estuaries of K. ocellatus also contrasts with the long-distance dispersal capacity observed in K. hermaphroditus along the Brazilian coast (Tatarenkov et al. 2017), which could be due to differences in colonisation success as a result of their different mating systems. Previous research in plants indicated that selfing is associated with increased dispersal capacity and colonisation success (de Waal et al. 2014). Mangrove killifishes are poor swimmers, but their long-term dispersal can be facilitated by adhesive eggs transported via floating material (Tatarenkov et al. 2012; Turko and Wright 2015). In K. hermaphroditus, self-fertilisation provides the possibility for a single individual to find a new population after a long-distance dispersal event, while K. ocellatus would require at least two individuals to breed. This hypothesis is supported by the large combined geographic range of the selfing mangrove killifishes (K. marmoratus and K. hermaphroditus), extending from Florida (23°N) to south Brazil (29°S), although further research is needed to investigate how the differences in mating systems between K. ocellatus and K. hermaphroditus can influence their colonisation capacities.
Conclusions
Contrary to the theoretical predictions that selfing species should result in high population structuring given its reduced effective population size due to inbreeding, we found that the outcrossing species K. ocellatus had stronger population structure in a narrower geographical range than its morphologically similar and often-syntopic selfing species K. hermaphroditus. These findings highlight that other factors, such as colonisation time, extent of gene flow, dispersal and colonisation success, may have more profound effects on the current patterns of population structure than differences in mating systems between selfing and outcrossing species.
Data availability
cox1 mtDNA sequences are deposited in GenBank (accession numbers: K. ocellatus: MN400774–MN400902; K. hermaphroditus: MN400903–MN400963). Microsatellite genotypes are available at Dryad Digital Repository: https://doi.org/10.5061/dryad.m0cfxpp1n.
References
Avise JC, Tatarenkov A (2015) Population genetics and evolution of the mangrove rivulus Kryptolebias marmoratus, the world’s only self-fertilizing hermaphroditic vertebrate. J Fish Biol 87:519–538
Ball MC (1988) Ecophysiology of mangroves. Trees 2:129–142
Barletta M, Lima ARA (2019) Systematic review of fish ecology and anthropogenic impacts in South American estuaries: setting priorities for ecosystem conservation. Front Mar Sci 6:237
Belkhir K (2004) GENETIX 4.05, logiciel sous Windows TM pour la génétique des populations. http://www.genetixuniv-montp2fr/genetix/genetixhtm.
Berbel-Filho WM, de Leaniz CG, Morán P, Cable J, Lima SM, Consuegra S (2019) Local parasite pressures and host genotype modulate epigenetic diversity in a mixed-mating fish. Ecol Evol 9:8736–8748
Berbel-Filho WM, Espirito-Santo HMV, Lima SMQ (2016) First record of a male of Kryptolebias hermaphroditus Costa, 2011 (Cyprinodontiformes: Cynolebiidae). Neotrop Ichthyol 14:e160024
Bohonak A (2002) IBD (isolation by distance): a program for analyses of isolation by distance. J Heredity 93:153–154
Bouckaert R, Heled J, Kuhnert D, Vaughan T, Wu CH, Xie D et al. (2014) BEAST 2: a software platform for Bayesian evolutionary analysis. PLoS Comput Biol 10:e1003537
Branoff BL (2017) Quantifying the influence of urban land use on mangrove biology and ecology: a meta‐analysis. Glob Ecol Biogeogr 26:1339–1356
Britto FB, Schmidt AJ, Carvalho AM, Vasconcelos CC, Farias AM, Bentzen P et al. (2018) Population connectivity and larval dispersal of the exploited mangrove crab Ucides cordatus along the Brazilian coast. PeerJ 6:e4702
Charlesworth D (2003) Effects of inbreeding on the genetic diversity of populations. Philos Trans R Soc B 358:1051–1070
Charlesworth D, Wright SI (2001) Breeding systems and genome evolution. Curr Opin Gen Dev 11:685–690
Costa WJEM (2006) Redescription of Kryptolebias ocellatus (Hensel) and K. caudomarginatus (Seegers) (Teleostei: Cyprinodontiformes: Rivulidae), two killifishes from mangroves of south-eastern Brazil. Aqua 11:5–13
Costa WJEM (2011) Identity of Rivulus ocellatus and a new name for a hermaphroditic species of Kryptolebias from south-eastern Brazil (Cyprinodontiformes: Rivulidae). Ichthyol Explor Freshw 22:185–192
Costa WJEM (2016) Colouration, taxonomy and geographical distribution of mangrove killifishes, the Kryptolebias marmoratus species group, in southern Atlantic coastal plains of Brazil (Cyprinodontiformes: Rivulidae). Ichthyol Explor Freshw 27:183–192
Costa WJEM, Lima SMQ, Bartolette R (2010) Androdioecy in Kryptolebias killifish and the evolution of self-fertilizing hermaphroditism. Biol J Linn Soc 99:344–349
Darriba D, Taboada GL, Doallo R, Posada D (2012) jModelTest 2: more models, new heuristics and parallel computing. Nat Methods 9:772
de Oliveira-Neto JF, Pie MR, Chammas MA, Ostrensky A, Boeger WA (2008) Phylogeography of the blue land crab, Cardisoma guanhumi (Decapoda: Gecarcinidae) along the Brazilian coast. J Mar Biol Assoc 88:1417–1423
de Waal C, Rodger JG, Anderson B, Ellis AG (2014) Selfing ability and dispersal are positively related, but not affected by range position: a multispecies study on southern African Asteraceae. J Evol Biol 27:950–959
Dieringer D, Schlötterer C (2003) Microsatellite analyser (MSA): a platform independent analysis tool for large microsatellite data sets. Mol Ecol Notes 3:167–169
Earl DA (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4:359–361
Ellison A, Wright P, Taylor DS, Cooper C, Regan K, Currie S et al. (2012) Environmental diel variation, parasite loads, and local population structuring of a mixed-mating mangrove fish. Ecol Evol 2:682–1695
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620
Excoffier L, Lischer HE (2010) Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Res 10:564–567
Ferreira AC, Lacerda LD (2016) Degradation and conservation of Brazilian mangroves, status and perspectives. Ocean Coast Manag 125:38–46
Francisco PM, Mori GM, Alves FM, Tambarussi EV, de Souza AP (2018) Population genetic structure, introgression, and hybridization in the genus Rhizophora along the Brazilian coast. Ecol Evol 8:3491–3504
Goudet J (1995) FSTAT (version 1.2): a computer program to calculate F-statistics. J Heredity 86:485–486
Guillot G, Estoup A, Mortier F, Cosson JF (2005) A spatial statistical model for landscape genetics. Genetics 170:1261–1280
Guillot G, Santos F, Estoup A (2008) Analysing georeferenced population genetics data with Geneland: a new algorithm to deal with null alleles and a friendly graphical user interface. Bioinformatics 24:1406–1407
Hamilton SE, Casey D (2016) Creation of a high spatio‐temporal resolution global database of continuous mangrove forest cover for the 21st century (CGMFC‐21). Glob Ecol Biogeogr 25:729–738
Huber J (2017) Reanalysis of single type of Rivulus ocellatus Hensel, 1880 in Berlin museum pending its putative molecular analysis, with the proposal of revalidation of Rivulus caudomarginatus. Kill-Data Ser 2016–2017:4–12
Jarne P, Auld JR (2006) Animals mix it up too: the distribution of self‐fertilization among hermaphroditic animals. Evolution 60:1816–1824
Kanamori A, Sugita Y, Yuasa Y, Suzuki T, Kawamura K, Uno Y et al.(2016) A genet map only self-fertilizing vertebrate. G3 6:1095–1106
Kopelman NM, Mayzel J, Jakobsson M, Rosenberg NA, Mayrose I (2015) CLUMPAK: a program for identifying clustering modes and packaging population structure inferences across K. Mol Ecol Res 15:1179–1191
Kumar S, Stecher G, Tamura K (2016) MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol 33:1870–1874
Leigh JW, Bryant D (2015) popart: full‐feature software for haplotype network construction. Methods Ecol Evol 6:1110–1116
Li YL, Liu JX (2018) StructureSelector: a web‐based software to select and visualize the optimal number of clusters using multiple methods. Mol Ecol Res 18:176–177
Mackiewicz M, Tatarenkov A, Perry A, Martin JR, Elder JF, Bechler DL et al. (2006) Microsatellite documentation of male-mediated outcrossing between inbred laboratory strains of the self-fertilizing mangrove killifish (Kryptolebias marmoratus). J Heredity 97:508–513
Meunier C, Hurtrez‐Bousses S, Durand P, Rondelaud D, Renaud F (2004) Small effective population sizes in a widespread selfing species, Lymnaea truncatula (Gastropoda:Pulmonata). Mol Ecol 13:2535–2543
Mori GM, Zucchi MI, Souza AP (2015) Multiple-geographic-scale genetic structure of two mangrove tree species: the roles of mating system, hybridization, limited dispersal and extrinsic factors. PLoS ONE 10:e0118710
Oliveira-Neto JF, Boeger WA, Pie MR, Ostrensky A, Hungria DB (2007) Genetic structure of populations of the mangrove crab Ucides cordatus (Decapoda: Ocypodidae) at local and regional scales. Hydrobiologia 583:69–76
Pil MW, Boeger MR, Muschner VC, Pie MR, Ostrensky A, Boeger WA (2011) Postglacial north–south expansion of populations of Rhizophora mangle (Rhizophoraceae) along the Brazilian coast revealed by microsatellite analysis. Am J Bot 98:1031–1039
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Puechmaille SJ (2016) The program structure does not reliably recover the correct population structure when sampling is uneven: subsampling and new estimators alleviate the problem. Mol Ecol Res 16:608–627
Rambaut A, Drummond AJ, Xie D, Baele G, Suchard MA (2018) Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst Biol 67:901–904
Rozas J, Ferrer-Mata A, Sánchez-DelBarrio JC, Guirao-Rico S, Librado P, Ramos-Onsins SE et al. (2017) DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol Biol Evol 34:3299–3302
Scarsella G, Gresham J, Earley R (2018) Relationships between external sexually dimorphic characteristics and internal gonadal morphology in a sex‐changing fish. J Zool 305:133–140
Seegers L (1984) Zur revision der Rivulus-arten sudost-Brasiliens, mit einer Neubeschreibung von Rivulus luelingi n. spp. und. Rivulus caudomarginatus n. spp.(Pisces: Cyprinodontidae: Rivulinae). Bonn Zool Beitr 28:271–320
Siol M, Bonnin I, Olivieri I, Prosperi J, Ronfort J (2007) Effective population size associated with self‐fertilization: lessons from temporal changes in allele frequencies in the selfing annual Medicago truncatula. J Evol Biol 20:2349–2360
Takezaki N, Nei M, Tamura K (2010) POPTREE2: software for constructing population trees from allele frequency data and computing other population statistics with Windows interface. Mol Biol Evol 27:747–752
Tatarenkov A, Earley RL, Perlman BM, Taylor DS, Turner BJ, Avise JC (2015) Genetic subdivision and variation in selfing rates among Central American populations of the mangrove rivulus, Kryptolebias marmoratus. J Heredity 106:276–284
Tatarenkov A, Earley RL, Taylor DS, Avise JC (2012) Microevolutionary distribution of isogenicity in a self-fertilizing fish (Kryptolebias marmoratus) in the Florida keys. Integr Comp Biol 52:743–752
Tatarenkov A, Earley RL, Taylor DS, Davis WP, Avise JC (2020) Extensive hybridization and past introgression between divergent lineages in a quasi‐clonal hermaphroditic fish: ramifications for species concepts and taxonomy. J Evol Biol. https://doi.org/10.1111/jeb.13624
Tatarenkov A, Gao H, Mackiewicz M, Taylor DS, Turner BJ, Avise JC (2007) Strong population structure despite evidence of recent migration in a selfing hermaphroditic vertebrate, the mangrove killifish (Kryptolebias marmoratus). Mol Ecol 16:2701–2711
Tatarenkov A, Lima SM, Earley RL, Berbel-Filho WM, Vermeulen FB, Taylor DS et al. (2017) Deep and concordant subdivisions in the self-fertilizing mangrove killifishes (Kryptolebias) revealed by nuclear and mtDNA markers. Biol J Linn Soc 122:558–578
Tatarenkov A, Lima SMQ, Avise JC (2011) Extreme homogeneity and low genetic diversity in Kryptolebias ocellatus from south-eastern Brazil suggest a recent foundation for this androdioecious fish population. J Fish Biol 79:2095–2105
Tatarenkov A, Lima SMQ, Taylor DS, Avise JC (2009) Long-term retention of self-fertilization in a fish clade. Proc Natl Acad Sci USA 106:14456–14459
Tatarenkov A, Ring BC, Elder JF, Bechler DL, Avise JC (2010) Genetic composition of laboratory stocks of the self-fertilizing fish Kryptolebias marmoratus: a valuable resource for experimental research. PLoS ONE 5:e12863
Taylor DS (2012) Twenty-four years in the mud: what have we learned about the natural history and ecology of the mangrove rivulus, Kryptolebias marmoratus? Integr Comp Biol 52:724–736
Turko AJ, Tatarenkov A, Currie S, Earley RL, Platek A, Taylor DS et al. (2018) Emersion behaviour underlies variation in gill morphology and aquatic respiratory function in the amphibious fish Kryptolebias marmoratus. J Exp Biol 221:8
Turko AJ, Wright PA (2015) Evolution, ecology and physiology of amphibious killifishes (Cyprinodontiformes). J Fish Biol 87:815–835
van Oosterhout C, Hutchinson WF, Wills DP, Shipley P (2004) MICRO‐CHECKER: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes 4:535–538
Vermeulen FB, Hrbek T (2005) Kryptolebias sepia n. sp.(Actinopterygii: Cyprinodontiformes: Rivulidae), a new killifish from the Tapanahony River drainage in southeast Surinam. Zootaxa 928:1–20
Webb SA, Graves JA, Macias-Garcia C, Magurran AE, Foighil DO, Ritchie MG (2004) Molecular phylogeny of the livebearing Goodeidae (Cyprinodontiformes). Mol Phylogenet Evol 30:527–544
Willi Y, Määttänen K (2011) The relative importance of factors determining genetic drift: mating system, spatial genetic structure, habitat and census size in Arabidopsis lyrata. New Phytol 189:1200–1209
Acknowledgements
We are grateful to ICMbio for providing help with accommodation and facilities, especially the teams working at Parque Estadual Serra do Mar: Núcleo Picinguaba and Parque Estadual Serra do Mar: Estação Ecológica Juréia-Itatins. We are thankful to Dr. Ingo Schlupp from Oklahoma University for his friendly review. We also thank Dr. Joana Robalo and two other anonymous reviewers whose comments and suggestions substantially improved the paper.
Funding
This work was supported by the National Geographic/Waitt program [W461-16] and by the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) [233161/2014-7]. SMQL receives research productivity grant issued by CNPq [313644/2018-7]. AT is grateful for support from the funds provided by the University of California at Irvine to Prof. John C. Avise. HMVE-S received a postdoctoral fellowship from Coordenação de Aperfeiçoamento de Pessoal de Nível Superior-Brasil (CAPES).
Author information
Authors and Affiliations
Contributions
SC, WMB-F, AT, SMQL and CGL conceived the study and obtained the funding. WMB-F, HMVE-S, MGL and SMQL collected the samples. WMB-F and AT carried out the genetic analyses. WMB-F wrote the paper with participation of all authors.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This work followed the Swansea ethics committee guidelines (SU-Ethics-Student-250717/245). Sampling work was carried out under license ICMBio/SISBIO 57145-1/2017.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Berbel-Filho, W.M., Tatarenkov, A., Espírito-Santo, H.M.V. et al. More than meets the eye: syntopic and morphologically similar mangrove killifish species show different mating systems and patterns of genetic structure along the Brazilian coast. Heredity 125, 340–352 (2020). https://doi.org/10.1038/s41437-020-00356-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41437-020-00356-y
