Abstract
High-energy batteries for automotive applications require cells to endure well over a decade of constant use, making their long-term stability paramount. This is particularly challenging for emerging cell chemistries containing silicon, for which extended testing information is scarce. While much of the research on silicon anodes has focused on mitigating the consequences of volume changes during cycling, comparatively little is known about the time-dependent degradation of silicon-containing batteries. Here we discuss a series of studies on the reactivity of silicon that, collectively, paint a picture of how the chemistry of silicon exacerbates the calendar aging of lithium-ion cells. Assessing and mitigating this shortcoming should be the focus of future research to fully realize the benefits of this battery technology.
Similar content being viewed by others

Main
Lithium (Li)-ion batteries (LIBs) revolutionized the portable electronics market and are now key drivers in sectors such as stationary energy storage and electric mobility. In these applications, the long-term health of the battery is critical. The battery pack accounts for a sizeable portion of electric vehicle costs and is thus expected to remain functional during the entire lifespan of the car1. For stationary storage projects, economic feasibility depends decisively on the total service time2. In these scenarios, it is essential to consider that LIBs can age due to the action of time, even in the complete absence of cycling. The outcome of this time-dependent performance fade is known as calendar aging3. Present high-energy batteries containing graphite anodes can reportedly achieve over 15 years of calendar life under mild storage conditions at 20 °C to 40 °C (ref. 4), meaning that they would still retain 80% of their nominal capacity after remaining inactive for that period. As most batteries will experience a mix of active use and inactive storage, their overall aging is a superposition of the effects of cycling and time.
Calendar aging of LIBs causes capacity fade and impedance rise. Traditionally, most capacity fade due to calendar aging is caused by losses of Li+ to the solid-electrolyte interphase (SEI) of graphite anodes, and is largely a function of the electrochemical potential of the anode at the state of charge (SOC) of storage5. The lower the negative electrode potential (that is, the higher the cell SOC), the larger the driving force for electrolyte reduction and concomitant capacity loss6. In addition to these irreversible self-discharge processes, reversible self-discharge can cause apparent capacity losses, decreasing charge during storage that can be recovered after additional cycling7. Impedance rise can occur through more diverse pathways: from surface transformation at cathode particles8, pore inaccessibility due to substantial SEI growth at the anode3 and electrolyte depletion following sustained decomposition9. These aging mechanisms depend on storage temperature and chemical composition of the electrolyte, anode and cathode3.
Evaluating calendar aging is resource and time intensive. Given the multiyear lifetimes typical of today’s LIBs, calendar life is generally extrapolated from shorter tests rather than directly measured. Even the shorter experiments used for extrapolation extend through several months or years, often at elevated temperatures to accelerate aging5,10. The universal use of graphite anodes have helped make aging trends and life forecasting more predictable, notwithstanding that many uncertainties arise when moving to different electrode chemistries, such as the addition of silicon (Si).
During the past two decades, Si has been studied as a promising anode material that can increase cell energy while decreasing manufacturing costs. The Li–Si reactions lead to severe dimensional changes that contribute to rapid performance fade in Si-containing electrodes. Mitigating this and other Si-specific phenomena has been the focus of many battery scientists in recent years. Consequently, Si-containing batteries are now increasingly a reality in the market, with cells used in electric vehicles reportedly containing small amounts of Si as an additive to graphite anodes11, and many companies exploring Si-rich electrode compositions. Given that many effects of calendar aging arise at the anode, and the chemistry of Si is markedly different from that of graphite, this shift in anode composition has an impact on how the cells age as a function of time. As Si anodes approach technological maturity, a critical question must then be answered: how do chemical and electrochemical interactions between Si and the electrolyte affect calendar aging, especially compared with stable electrodes such as graphite?
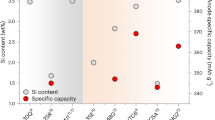
Information from the US Department of Energy (DOE) indicates that this remains an open question12. Figure 1a contains data from several leading manufacturers of Si-containing cells, demonstrating how cycle lives and energy densities have improved over the past decade and are quickly approaching performance targets set by the DOE. Conversely, Fig. 1b shows that the calendar lives from the latest iteration of these high-performing cells remains unsatisfactory, suggesting that strategies to improve cycling are insufficient to promote long-term stability.

a, Evolution of the cycle life achieved by leading manufacturers of Si- and SiOx-containing cells. Energy densities and cycle lifetimes have shown substantial progress over the past decade, quickly approaching the performance targets set by the US DOE. b, Calendar life of the state-of-the-art Si-containing cells shown in a, which fall short of the DOE’s performance targets, demonstrating a large technical gap that needs to be addressed. Calendar life data are shown only for cell formulations tested in 2020. Cycle life and calendar life are defined as the number of successive cycles or months of storage, respectively, at which the cells lose 20% of their initial capacity. Data from ref. 12.
Despite this striking calendar life technical gap, very few studies have probed the long-term stability of Si-containing cells in the absence of cycling13,14,15. A rare example is the work by Zilberman et al., which examined cylindrical cells with ~3.5 wt% of Si in the anode that were stored for 11 months at 25 °C (ref. 7). The study found that most of the capacity fade observed at all SOCs was due to permanent losses of Si active material capacity. This active material loss is typically attributed to the electrical isolation of Si either from cracking or the formation of a resistive layer due to reactivity with the electrolyte, blocking Li+ transport. Conversely, traditional graphite anodes should remain stable under these same conditions, with Li+ trapping in the SEI as the main capacity fade mechanism. While the stability of the active material is of concern, there is also evidence suggesting that the Si SEI is less passivating compared with graphite. Kalaga et al. estimated the currents attributed to these parasitic processes and suggested that the rate of SEI ‘growth’ was higher on silicon–graphite anodes (15 wt% Si) compared with Si-free electrodes16. On the basis of these studies, active material loss, chemical instability and Li+ inventory consumption appear to be exacerbated with the addition of Si, contributing to increased self-discharge and the inadequate calendar lives observed in Fig. 1b.
In this Perspective, we examine studies on the chemistry and electrochemistry of Si that underlie this higher susceptibility to calendar aging. This problem stems from the fact that Si is substantially more reactive in the cell environment than graphite. We also examine strategies to improve the compatibility of Si with the electrolyte that have the potential to extend the calendar life of Si-containing cells. The historical research focus on the dimensional problems of Si has left other issues unattended, which must be addressed as this technology transitions into the market.
Si and its SEI are extremely reactive
The addition of Si to the anode promotes a variety of new side reactions, resulting in gassing, dissolution of the SEI and electrolyte decomposition, some of which can be traced back to the early periods of cell life. Figure 2 shows a summary of the different failure mechanisms, stemming from the reactivity of Si and its SEI, that may contribute to poor calendar life.

Schematic representation of the various issues that contribute to the poor calendar life observed in Si-containing cells. The SEI formed on Si undergoes continuous changes due to SEI reactivity with HF. This persistent electrolyte reactivity experienced by Si and its SEI can accelerate capacity fade, while also generating solid deposits that can block pores in the anode. After extended storage, both electrolyte consumption and pore obstruction can contribute to power fade. At a more fundamental level (inset), the SEI experiences constant morphological and compositional changes, affecting its ability to protect the Si core. Mechanical disruption of the surface layers is also possible during storage, as the particles will slowly contract due to self-discharge. The resulting exposure of Si to the electrolyte can continue to feed the hydrolytic cycle of PF6, generating additional HF that can be harmful to the cell. In addition, soluble products of electrolyte decomposition can diffuse and react at the surface of the cathode, causing unknown consequences to cell health. These processes may manifest themselves differently depending on temperature and SOC during storage as well as electrolyte/electrode composition, and are certainly exacerbated by the high surface areas of Si nanostructures.
When Si comes into contact with conventional carbonate electrolytes, surface changes occur as a result of chemical processes, even when a native oxide layer is present17,18. Complementary studies by Pekarek et al. and Seitzinger et al. demonstrated the reactivity of various materials and chemical groups by examining the solid-phase and gaseous components following exposure to electrolyte19,20, and found that different molecular coatings have varied levels of stability. Hydrogen-terminated Si, acidic SiO2 and silyl ester surface groups were found to be reactive, whereas basic SiO2 and silyl ethers were more chemically robust, demonstrating the importance of material choice and control of interfacial chemistry for Si anodes.
Furthermore, there is growing evidence suggesting that Si increases the anode reactivity even after the formation of the SEI. While the SEI on graphite can be made notably stable by electrolyte additives21, the Si SEI appears to be highly dynamic22 and inherently non-passivating (Fig. 2). It has been reported that the passivation layer of model Si wafer electrodes ‘breathes’ during cycling, becoming thinner and more inorganic during lithiation but thicker and more organic during delithiation with certain electrolytes23. These compositional changes are believed to originate from the precipitation and dissolution of SEI components such as lithium ethylene dicarbonate (LiEDC), LiPF6 decomposition products and polyethylene glycol (PEG) oligomeric species24. The addition of fluoroethylene carbonate (FEC) to the electrolyte, known for its beneficial properties to Si25, did not preclude this breathing but only reversed the order of observed thickness changes despite improving Si passivation26. Further evidence of the dynamic SEI formed in the presence of FEC and the reactivity of Si comes from the observed consumption of FEC with cycling27. Dynamic SEI thickness and composition are also observed on graphite, especially during formation28, but the graphite SEI eventually passivates, indicating that the relationship between a dynamic SEI and passivation is not fully understood.
Although the reactivity of Si with the electrolyte during cycling has been well documented, fewer studies have directly probed the stability of Si surfaces at open-circuit conditions experienced during calendar aging. Yin et al. investigated how the SEI of Si wafers evolved during rests at intermediate SOCs, and found that a decrease in passivation and increase in reactivity could already be observed after brief (1–10 h) periods18. Such behaviour persisted after hundreds of lithiation-rest cycles and was associated with the intrinsic reactivity of organic components of the SEI. Stetson et al. also observed the shedding of an organic-rich layer of the SEI after 45 h at open circuit, leaving a less resistive deposit at the Si surface29. These studies suggest that organic SEI components are unstable either because they continue to react or because they are soluble in the electrolyte, or both. A full understanding of the reactivity and reaction pathways of various Si SEI components has yet to be achieved30.
The examples above show that Si is chemically unstable with salts and organic solvents commonly used in LIB electrolytes and that this instability does not subside upon the formation of the SEI, as the ‘protective’ layer itself is not static (Fig. 2). Hence, sustained losses of Li+ inventory are expected in Si-containing anodes, which is highly problematic for the calendar life of batteries. This perennial state of reactivity of Si is demonstrated in Fig. 3, which shows the capacity passed through LiNi0.5Mn0.3Co0.2O2 (NMC532) cells containing the indicated anodes and electrolytes as they are held at 4.1 V after SEI formation cycles16. These capacities qualitatively reflect the parasitic processes at each anode: the greater the slope of capacity increase measured during the voltage hold, the higher the instantaneous rate of side reactions at the surface of the negative electrode16,31,32. These parasitic processes in Si-based anodes mainly originate from SEI reactivity, with possible contributions from other fade mechanisms depicted in Fig. 2. Clearly, addition of 15 wt% Si to graphite electrodes (shown in red in Fig. 3) increases the extent of side reactions from that of pure graphite (blue).

Integrated parasitic currents measured in NMC532/Si–graphite (15% Si) and NMC532/graphite cells. The data were collected at 30 °C during 600-h-long voltage holds at 4.1 V in coin cells after three formation cycles at C/20 rate. Data from ref. 16.
While anticipating the effects of Si surface instability on calendar aging is relatively straightforward, possible consequences of SEI ‘breathing’ are less clear. These cyclic compositional changes could lead to unexpected dependencies of calendar aging on SOC, in which case the knowledge acquired from graphite-based cells would not be directly transferable to Si anodes. Thus, there is a need to perform comprehensive calendar aging studies for Si-rich cells at multiple SOCs and temperatures to gain insight into the mechanism of SEI growth. Furthermore, although consequences of volumetric changes have largely been overcome to improve cycle life using morphology and particle-size controls (Fig. 1a), the dimensional change of Si may still interfere with typical calendar life experiments. Self-discharge and concomitant contraction during storage would slowly disrupt the SEI, exposing Si to the electrolyte (Fig. 2). Because of the combined reactivity and volume change of Si, such mechanical failures are more detrimental than on graphite. Similarly, perturbation to the Si SEI during periodic cycling to measure calendar life (reference performance tests) is more substantial than on graphite, leading to experimental outcomes that could depend on cycling frequency. Further studies are required to understand the contributions of mechanical degradation, as a function of SOC, to poor Si calendar life. Measuring stresses in Si and the SEI in situ over time could help quantify how mechanical failure relates to self-discharge.
Sustained Si reactivity accelerates power fade
Electrolyte decomposition can increase cell impedance in various ways. Persistent electrolyte reduction at the anode leads to a buildup of insoluble products that, after extensive aging, may slow down Li+ transport through the pore network of the electrode (Fig. 2)33. Localized pore obstruction can also be caused by formation of gas pockets in the cell, as reduction reactions often release gaseous by-products that can displace the electrolyte (Fig. 2)34. These latter processes cause de-wetting of electrodes due to electrolyte depletion9, with SEI instability effectively ‘drying up’ portions of the cell.
The constant SEI changes discussed in the previous section indicate that the consequences of long-term electrolyte reactivity for cell impedance could be more severe with the addition of Si. In fact, poor electrode utilization due to electrolyte consumption is believed to be one of the leading causes of the modest calendar lives reported in Fig. 1b12. This important effect can go undetected by the use of excess electrolyte volume in lab-scale test cells, which masks issues with electrolyte depletion. Awareness of this factor is essential to correctly assess the performance of new materials. Furthermore, on a particle level, inactivation of Si due to electrical isolation becomes likely at accelerated rates of surface reactivity. Although such effects are uncommon in graphite anodes maintained at mild temperatures, it has been observed in Si-containing cells after extended storage7. Transport limitations at all length scales caused by the chemical and electrochemical activity of Si need to be mitigated to approach the calendar life of incumbent technologies.
Si promotes runaway hydrolytic HF cycle
Generation of hydrofluoric acid (HF) is a known consequence of using LiPF6-based electrolytes, as this salt is hydrolysed by residual moisture introduced during cell assembly35. Many components in traditional LIBs are comparatively inert to HF, including commercial binders, graphite anodes and polyolefin separators36. Thus, batteries with these traditional components typically tolerate the HF generated by water contamination up to 1,000 ppm without substantial performance degradation37. However, a completely different scenario occurs when Si-based materials are present in the cell. In this case, HF reacts with any oxide-containing Si species to generate more water as well as gaseous and soluble products38:
Generation of water allows further hydrolytic production of HF, which can continue indefinitely if the oxide-containing Si species are accessible to HF (Fig. 2). The consequences of this chemical attack to surface Si–O groups were highlighted in a recent paper by Ha et al., which showed that as little as 50 ppm of moisture in the electrolyte is sufficient to pit the surface of Si wafers38. Such reactions were observed to expose fresh surfaces to the electrolyte, leading to additional parasitic reactions and resulting in thick, non-uniform SEI layers.
This runaway cycle of etching and HF regeneration could have severe implications for the calendar life of cells with Si electrodes. PF6– hydrolysis is a relatively slow process35, and thus the effective concentration of HF and its by-products in the electrolyte is expected to increase over time until achieving equilibrium, while the LiPF6 necessary for cycling the cell is simultaneously depleted. The susceptibility of the oxygenated Si surfaces to acidolysis can eventually generate problems of interfacial incompatibility in the anode. Much of the research on binders for Si electrodes have explored these functional groups as anchoring points for polymer chains39, which would slowly vanish due to fluorination. Given the large dimensional changes of Si during cycling, these weakened interfaces could eventually cause electrode delamination and active material loss. In addition, while HF etching of Si is typically limited to the oxide-containing surface layers in aqueous solutions, the non-aqueous electrolyte and extreme electrochemical potentials that are present in batteries result in a different chemical environment. As such, it may be possible for HF and its by-products to etch and dissolve away the Si active material itself, as evidenced by deep pitting of Si wafers extending far below the native surface oxide layers38. Finally, it is possible that enriching the electrolyte with gaseous and soluble Si species (such as SiF4 and H2SiF6) can impair cell health due to the ‘cross-talk’. Silica-like deposits have been detected at the cathode surface in Si-based full cells40, indicating that these compounds can travel through the separator to be oxidized in the positive electrode, becoming a possible source of impedance rise. Most of the reactions above are chemical in nature and can occur independent of electrochemical cycling, making them likely causes of both active material loss and impedance rise during calendar aging of Si-containing cells.
Mitigation strategies and challenges
Several strategies originally conceived to improve the cyclic stability of Si could potentially be useful to mitigate rapid calendar aging (Fig. 4), though effective gains in long-term stability have yet to be demonstrated. Surface modification of the Si active material is a promising direction, as the resulting outer layer provides chemical compatibility with the electrolyte (Fig. 4a). Molecular coatings on the Si surface can decrease its intrinsic reactivity19, effectively lowering the rate of Li+ trapping at the SEI. In addition, electrolyte additives can be engineered to form protective layers in situ41, creating a buffer between the active material and the electrolyte. An example of this approach is the use of small amounts of Mg2+ salts to form a Li–Mg–Si Zintl-phase surface layer on lithiated Si that is more redox stable than lithiated Si at low potentials due to a subtle shift in surface structure (Fig. 4a)42.

a, Molecular surface coatings to improve chemical compatibility with the electrolyte. Certain functional groups are less susceptible to chemical attack by the electrolyte, while in situ formation of surface layers (such as Li–Mg–Si compounds) have been shown to render Si particles more inert in the cell environment. b, Physically shielding Si from the electrolyte using coatings. Fabrication of core– and yolk–shell structures with carbon or embedding Si nanodomains within a more inert matrix can prevent parasitic reactions by negating electrolyte access to the Si surfaces. c, Decreasing the reactive surface area that engages in parasitic reactions. Smaller surface areas require larger Si feature sizes that exacerbate issues with dimension stability that must be addressed by other strategies, such as those shown in a or b. d, Addition of water scavengers that can prevent HF generation. Successful ideas must strike a balance between improvements in cycle and calendar lives and must employ protection mechanisms that persist as the cell ages.
Another strategy that can have beneficial side effects on calendar aging is creating physical barriers that shield the active Si core from the electrolyte (Fig. 4b). This can be achieved by coating particles with carbon during synthesis43, or by post-treatment by techniques such as atomic layer deposition44, for example. Naturally, these coatings must be stable, ionically conductive and able to withstand the dimensional changes of Si during cycling; otherwise, their benefits to calendar aging can be quickly negated. This latter point is essential to understand why calendar aging remains a problem when many of the synthesis strategies for the electrodes shown in Fig. 4b are now commonplace in the literature. The data shown in Fig. 1 probably originated from cells that used some or all these methods, indicating that the solution to calendar aging will need to be multifaceted and that current surface protection schemes are still insufficient to provide enduring protection against calendar aging. Coatings and techniques that worked well for cycling may not perform on longer timescales and thus may need to be re-optimized to simultaneously improve cycle and calendar life.
Alternatively, decreasing the surface area of Si-based materials could help improve long-term chemical and electrochemical stability. The use of Si architectures with small surface areas (Fig. 4c) is a strategy that contradicts much recent research aiming to use nanoscale structures to improve capacity retention during cycling. The use of nanoscale Si to improve cycling stability probably comes at the expense of calendar aging, as the nanoscale features expose a substantial surface area to the electrolyte, and the two competing effects must be balanced in future research. As such, possible benefits from using smaller surface areas by increasing particle size are only maintained if the larger Si feature sizes exhibit sufficient transport properties and do not excessively fragment to expose new surfaces to electrolyte during cycling. Such a strategy may prove challenging to implement, as cycling with traditional LiPF6–carbonate electrolytes has been shown to reduce primary feature sizes in Si particles to less than 10 nm, suggesting that any Si larger than that will increase in surface area during cycling45.
Just as discussed for Fig. 4b, maintaining the area that is exposed to the electrolyte after cycling is just as important as improving particle design. Minimizing Si surface area will have to work in conjunction with other strategies such as the formation of a compliant SEI that continues to protect Si throughout the cell life. As an example, recent reports have shown that atypical electrolyte compositions can promote stable cycling of multi-micrometre-sized Si particles46. Despite the obvious challenges, this approach holds promise to address the intrinsic reactivity of Si surfaces.
Finally, the deleterious effects of HF on silicon can be mitigated by interrupting the hydrolytic cycle using additives that scavenge H2O or HF from the electrolyte (Fig. 4d)47. In theory, the hydrolytic cycle could be prevented by removing the LiPF6 or SiO2 reactants involved. Indeed, fluorine-free electrolytes that do not react with water to form HF are another promising research direction48, although LiPF6 is a challenging electrolyte component to replace due to its ability to passivate the aluminium current collector of the cathode40.
Outlook
Much of the research on Si anodes has focused on the consequences of volume change, so there is a need to increase emphasis on other aspects of cell aging. Although advances in materials (for example, SiOx particles) and in prelithiation techniques have helped cells achieve and maintain high energy, they did not directly address issues with time-dependent stability. Comparing the promises of Fig. 4 with the reality of Fig. 1 shows that achieving holistic solutions that balance cycling and calendar aging is still an open problem for Si anodes. Clearly, having cycle life as the sole metric to judge Si structures is an incomplete approach, and we hope that this view will be explicitly considered in future research.
There are several research opportunities that could help further the understanding of Si calendar aging. For example, there is a vast knowledge of how formation protocols influence the composition and properties of the graphite SEI, and extending this expertise to Si could help establish best practices for extending long-term stability. In addition, cell-stack pressure can also affect SEI properties, and understanding its influence on passivation would undoubtedly be useful. As we highlighted in many examples above, the Si SEI, or parts of it, dissolves away even in the absence of cycling. Hence, tracking how properties such as SEI composition, porosity and conductivity evolve as a function of time rather than cycle number will be invaluable for addressing silicon’s calendar life problem.
Additional opportunities exist to investigate shortcomings of the potential mitigation strategies discussed above. For example, coatings on Si particles must be evaluated for not only enabling cycling but also their ability to remain conformal after successive expansion/contraction. Identifying when, why and how much Si is exposed to the electrolyte could help tune fabrication processes to yield materials with improved long-term stability. Similarly, methods for quantifying the effective surface area of Si particles after cycling and how much of the active material remains electronically connected must be developed; this would require advanced characterization efforts at all levels, from in situ to post mortem.
More fundamentally, a central point is that Si and graphite reduce electrolytes at the same potential but have completely different passivation behaviour. The lack of a comprehensive modelling framework capable of describing this difference is a major roadblock in developing Si electrodes with sufficient calendar life. The complexity of SEI layers have hindered the development of accurate SEI models. Part of the challenge is the need to combine modelling at different length scales, from atomistic simulations to develop reaction kinetic mechanisms up to electrode-level modelling that captures transport effects. Usually, models at these different scales are developed in isolation and not collectively, limiting their usefulness. Recent improvements in time and length resolution of characterization techniques can support the development of more accurate SEI models for Si, building on pseudo-continuum scale models that have been around since 2004 for graphite49. A well-informed Si SEI model could provide insights into the root cause of poor SEI stability during calendar aging, providing suggestions for strategies to improve specific SEI properties.
A central challenge with calendar aging is that diagnostics and solution development are very time consuming, making the divide between ideas and validation unreasonably large. A common practice to expedite such studies is to use elevated temperatures, as aging processes are accelerated and can be identified after shorter experiments. There is always a risk that these higher temperatures can introduce aging mechanisms that are not relevant at milder conditions50, and this effect could be even more problematic for Si. Acceleration of electrolyte reactions releasing HF that can react with Si surfaces could disproportionately penalize cells with a higher Si content, affecting the ability of these shorter experiments to predict calendar lives. The development of fast chemical and electrochemical assays to reliably assess long-term cell stability could be critical to support the deployment of Si by the battery industry.
References
Nykvist, B. & Nilsson, M. Rapidly falling costs of battery packs for electric vehicles. Nat. Clim. Change 5, 329–332 (2015).
He, G. N., Ciez, R., Moutis, P., Kar, S. & Whitacre, J. F. The economic end of life of electrochemical energy storage. Appl. Energy 273, 115151 (2020).
Dubarry, M., Qin, N. & Brooker, P. Calendar aging of commercial Li-ion cells of different chemistries—a review. Curr. Opin. Electrochem. 9, 106–113 (2018).
Harlow, J. E. et al. A wide range of testing results on an excellent lithium-ion cell chemistry to be used as benchmarks for new battery technologies. J. Electrochem. Soc. 166, A3031–A3044 (2019).
Keil, P. et al. Calendar aging of lithium-ion batteries I. Impact of the graphite anode on capacity fade. J. Electrochem. Soc. 163, A1872–A1880 (2016).
Single, F., Latz, A. & Horstmann, B. Identifying the mechanism of continued growth of the solid–electrolyte interphase. ChemSusChem 11, 1950–1955 (2018).
Zilberman, I., Sturm, J. & Jossen, A. Reversible self-discharge and calendar aging of 18650 nickel-rich, silicon-graphite lithium-ion cells. J. Power Sources 425, 217–226 (2019).
Rodrigues, M. T. F., Kalaga, K., Trask, S. E., Shkrob, I. A. & Abraham, D. P. Anode-dependent impedance rise in layered-oxide cathodes of lithium-ion cells. J. Electrochem. Soc. 165, A1697–A1705 (2018).
Deng, Z. et al. Ultrasonic scanning to observe wetting and “unwetting” in Li-ion pouch cells. Joule 4, 2017–2029 (2020).
Naumann, M., Schimpe, M., Keil, P., Hesse, H. C. & Jossen, A. Analysis and modeling of calendar aging of a commercial LiFePO4/graphite cell. J. Energy Storage 17, 153–169 (2018).
Ding, Y. L., Cano, Z. P., Yu, A. P., Lu, J. & Chen, Z. W. Automotive Li-ion batteries: current status and future perspectives. Electrochem. Energy Rev. 2, 1–28 (2019).
Cunningham, B. Silicon and Intermetallic Anode Portfolio Strategy Overview Annual Merit Review (US Department of Energy, 2020).
Zilberman, I., Ludwig, S. & Jossen, A. Cell-to-cell variation of calendar aging and reversible self-discharge in 18650 nickel-rich, silicon–graphite lithium-ion cells. J. Energy Storage 26, 100900 (2019).
De Sutter, L. et al. Comprehensive aging analysis of volumetric constrained lithium-ion pouch cells with high concentration silicon-alloy anodes. Energies 11, 2948 (2018).
Lu, W., Zhang, L., Qin, Y. & Jansen, A. Calendar and cycle life of lithium-ion batteries containing silicon monoxide anode. J. Electrochem. Soc. 165, A2179–A2183 (2018).
Kalaga, K., Rodrigues, M. T. F., Trask, S. E., Shkrob, I. A. & Abraham, D. P. Calendar-life versus cycle-life aging of lithium-ion cells with silicon-graphite composite electrodes. Electrochim. Acta 280, 221–228 (2018).
Schneier, D. et al. Elucidation of the spontaneous passivation of silicon anodes in lithium battery electrolytes. J. Electrochem. Soc. 166, A4020–A4024 (2019).
Yin, Y. L. et al. Nonpassivated silicon anode surface. ACS Appl. Mater. Interfaces 12, 26593–26600 (2020).
Pekarek, R. T. et al. Intrinsic chemical reactivity of solid–electrolyte interphase components in silicon-lithium alloy anode batteries probed by FTIR spectroscopy. J. Mater. Chem. A 8, 7897–7906 (2020).
Seitzinger, C. L. et al. Intrinsic chemical reactivity of silicon electrode materials: gas evolution. Chem. Mater. 32, 3199–3210 (2020).
Aurbach, D. et al. On the use of vinylene carbonate (VC) electrolyte solutions for Li-ion as an additive to batteries. Electrochim. Acta 47, 1423–1439 (2002).
Yoon, I., Abraham, D. P., Lucht, B. L., Bower, A. F. & Guduru, P. R. In situ measurement of solid electrolyte interphase evolution on silicon anodes using atomic force microscopy. Adv. Energy Mater. 6, 1600099 (2016).
Veith, G. M. et al. Direct determination of solid–electrolyte interphase thickness and composition as a function of state of charge on a silicon anode. J. Phys. Chem. C 119, 20339–20349 (2015).
Hasa, I. et al. Electrochemical reactivity and passivation of silicon thin-film electrodes in organic carbonate electrolytes. ACS Appl. Mater. Interfaces 12, 40879–40890 (2020).
Shkrob, I. A., Wishart, J. F. & Abraham, D. P. What makes fluoroethylene carbonate different? J. Phys. Chem. C 119, 14954–14964 (2015).
Veith, G. M. et al. Determination of the solid electrolyte interphase structure grown on a silicon electrode using a fluoroethylene carbonate additive. Sci. Rep. 7, 6326 (2017).
Jung, R. et al. Consumption of fluoroethylene carbonate (FEC) on Si-C composite electrodes for Li-ion batteries. J. Electrochem. Soc. 163, A1705–A1716 (2016).
Bryngelsson, H., Stjerndahl, M., Gustafsson, T. & Edström, K. How dynamic is the SEI? J. Power Sources 174, 970–975 (2007).
Stetson, C. et al. Temperature-dependent solubility of solid electrolyte interphase on silicon electrodes. ACS Energy Lett. 4, 2770–2775 (2019).
Hou, T. Z. et al. The influence of FEC on the solvation structure and reduction reaction of LiPF6/EC electrolytes and its implication for solid electrolyte interphase formation. Nano Energy 64, 103881 (2019).
Gao, H. et al. Parasitic reactions in nanosized silicon anodes for lithium-ion batteries. Nano Lett. 17, 1512–1519 (2017).
Tornheim, A., Trask, S. E. & Zhang, Z. C. Evaluation of electrolyte oxidation stability on charged LiNi0.5Co0.2Mn0.3O2 cathode surface through potentiostatic holds. J. Electrochem. Soc. 163, A1717–A1722 (2016).
Barre, A. et al. A review on lithium-ion battery ageing mechanisms and estimations for automotive applications. J. Power Sources 241, 680–689 (2013).
Xu, H. et al. Roll-to-roll prelithiation of Sn foil anode suppresses gassing and enables stable full-cell cycling of lithium ion batteries. Energ. Environ. Sci. 12, 2991–3000 (2019).
Stich, M., Gottlinger, M., Kurniawan, M., Schmidt, U. & Bund, A. Hydrolysis of LiPF6 in carbonate-based electrolytes for lithium-ion batteries and in aqueous media. J. Phys. Chem. C 122, 8836–8842 (2018).
Wiemers-Meyer, S., Jeremias, S., Winter, M. & Nowak, S. Influence of battery cell components and water on the thermal and chemical stability of LiPF6 based lithium ion battery electrolytes. Electrochim. Acta 222, 1267–1271 (2016).
Burns, J. C. et al. The impact of intentionally added water to the electrolyte of Li-ion cells. J. Electrochem. Soc. 160, A2281–A2287 (2013).
Ha, Y. Y. et al. Effect of water concentration in LiPF6-based electrolytes on the formation, evolution, and properties of the solid electrolyte interphase on Si anodes. ACS Appl. Mater. Interfaces 12, 49563–49573 (2020).
Jung, C. H., Kim, K. H. & Hong, S. H. Stable silicon anode for lithium-ion batteries through covalent bond formation with a binder via esterification. ACS Appl. Mater. Interfaces 11, 26753–26763 (2019).
Bareno, J., Shkrob, I. A., Gilbert, J. A., Klett, M. & Abraham, D. P. Capacity fade and its mitigation in Li-ion cells with silicon-graphite electrodes. J. Phys. Chem. C 121, 20640–20649 (2017).
Shin, J., Kim, T. H., Lee, Y. & Cho, E. Key functional groups defining the formation of Si anode solid–electrolyte interphase towards high energy density Li-ion batteries. Energy Storage Mater. 25, 764–781 (2020).
Han, B. H. et al. Using mixed salt electrolytes to stabilize silicon anodes for lithium-ion batteries via in situ formation of Li–M–Si ternaries (M = Mg, Zn, Al, Ca). ACS Appl. Mater. Interfaces 11, 29780–29790 (2019).
Liu, N. et al. A yolk–shell design for stabilized and scalable Li-ion battery alloy anodes. Nano Lett. 12, 3315–3321 (2012).
Hy, S. et al. Stabilizing nanosized Si anodes with the synergetic usage of atomic layer deposition and electrolyte additives for Li-ion batteries. ACS Appl. Mater. Interfaces 7, 13801–13807 (2015).
Wetjen, M. et al. Morphological changes of silicon nanoparticles and the influence of cutoff potentials in silicon-graphite electrodes. J. Electrochem. Soc. 165, A1503–A1514 (2018).
Chen, J. et al. Electrolyte design for LiF-rich solid–electrolyte interfaces to enable high-performance microsized alloy anodes for batteries. Nat. Energy 5, 386–397 (2020).
Han, J. G., Kim, K., Lee, Y. & Choi, N. S. Scavenging materials to stabilize LiPF6-containing carbonate-based electrolytes for Li-ion batteries. Adv. Mater. 31, 1804882 (2019).
Hernandez, G. et al. Elimination of fluorination: the influence of fluorine-free electrolytes on the performance of LiNi1/3Mn1/3Co1/3O2/silicon-graphite Li-ion battery cells. ACS Sustain. Chem. Eng. 8, 10041–10052 (2020).
Christensen, J. & Newman, J. A mathematical model for the lithium-ion negative electrode solid electrolyte interphase. J. Electrochem. Soc. 151, A1977–A1988 (2004).
Gewald, T., Lienkamp, M., Lehmkuhl, D. & Hahn, A. Accelerated aging characterization of lithium-ion cells: limitation of arrhenius dependency. In Fourteenth International Conference on Ecological Vehicles and Renewable Energies 1–10 (IEEE, 2019).
Acknowledgements
This research was supported by the US Department of Energy (DOE)’s Vehicle Technologies Office under the Silicon Consortium Project. This work was conducted in part by the Alliance for Sustainable Energy, LLC, the manager and operator of the National Renewable Energy Laboratory for the DOE under contract no. DE-AC36-08GO28308. The submitted manuscript has been created by UChicago Argonne, LLC, Operator of Argonne National Laboratory (‘Argonne’). Argonne, a US DOE Office of Science laboratory, is operated under contract no. DE-AC02-06CH11357. This manuscript has been authored by UT-Battelle, LLC, under contract no. DE-AC05-00OR22725 with the US DOE. Sandia National Laboratories is a multimission laboratory managed and operated by National Technology and Engineering Solutions of Sandia, LLC, a wholly owned subsidiary of Honeywell International Inc., for the US DOE’s National Nuclear Security Administration under contract DE-NA0003525. Lawrence Berkeley National Laboratory is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the US DOE under contract no. DE-AC02-05CH11231. The views expressed in the article do not necessarily represent the views of the DOE or the US Government. The US Government retains and the publisher, by accepting the article for publication, acknowledges that the US Government retains a non-exclusive, paid-up, irrevocable, worldwide license to publish or reproduce the published form of this work, or allow others to do so, for US Government purposes.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Energy thanks Jun-Tao Li and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
McBrayer, J.D., Rodrigues, MT.F., Schulze, M.C. et al. Calendar aging of silicon-containing batteries. Nat Energy 6, 866–872 (2021). https://doi.org/10.1038/s41560-021-00883-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41560-021-00883-w
This article is cited by
-
High voltage electrolytes for lithium-ion batteries with micro-sized silicon anodes
Nature Communications (2024)
-
Aging Mechanisms and Evolution Patterns of Commercial LiFePO4 Lithium-Ion Batteries
Journal of Electronic Materials (2024)
-
Interfacial structure changes between amorphous silicon anode/liquid electrolyte using a highly dense and flat model electrode
Journal of Solid State Electrochemistry (2024)
-
Designing electrolytes and interphases for high-energy lithium batteries
Nature Reviews Chemistry (2023)
-
Revealing the aging process of solid electrolyte interphase on SiOx anode
Nature Communications (2023)
