Abstract
Nanolattices exhibit attractive mechanical, energy conversion and optical properties, but it is challenging to fabricate large nanolattices while maintaining the dense regular nanometre features that enable their properties. Here we report a crack-free self-assembly approach for fabricating centimetre-scale nickel nanolattices with much larger crack-free areas than prior self-assembled nanolattices and many more unit cells than three-dimensionally printed nanolattices. These nickel nanolattices have a feature size of 100 nm, a grain size of 30 nm and a tensile strength of 260 MPa, which approaches the theoretical strength limit for porous nickel. The self-assembly method and porous metal mechanics reported in this work may advance the fabrication and applications of high-strength multifunctional porous materials.
Similar content being viewed by others


Main
Strong and lightweight porous materials are commonly used in industry, but the difficulties in controlling their physical and chemical structures during fabrication have limited their mechanical properties1. Nanolattices are porous materials with nanoscale features that promise to overcome these limitations using size-based effects2,3,4,5. Three-dimensional (3D) printing using two-photon polymerization is the most common nanolattice fabrication method2,3, but even with a record-high printing speed, it would take 64 days to fabricate a 20 × 20 × 0.1 mm3 woodpile nanolattice6. Alternatively, self-assembly methods have realized relatively fast nanolattice fabrication (typically several days for centimetre-scale face-centred cubic (fcc) nanolattices)7,8,9,10. Metallic nanolattices made by filling the voids of self-assembled colloidal templates10,11,12, in particular, have demonstrated 10 nm periodic features3,8, titanium-like compressive strength10, selective photonic absorption/emission12 and high temperature and chemical stability11,12,13. These properties show the advantages of nanolattices over traditional porous metals that have stochastic pore structures and thick struts or walls14. Self-assembled templates, however, are subject to dense cracks that, when filled with a material, form inverted-crack structures that divide the sample into small nanolattice domains9, cause stress concentrations, block fluid/gas transport15 and increase optical scattering16. Although many studies have focused on fabricating self-assembled nanolattices10,11,12,17, and several have attempted to eliminate inverted cracks16,18, no self-assembly fabrication approach can produce large-area metallic nanolattices without inverted cracks. A method to eliminate template cracks and precisely control metallic nanostructure across millions of units would realize nanolattices with unprecedented properties and enable applications in sensing, energy conversion and mechanics.
Additionally, there is a critical need to measure, predict and optimize nanolattice tensile properties to understand how these materials fracture and respond to complex loading2,19. Nearly all prior nanolattice mechanical characterization, however, has been done in compression with micro-/nanoindentation2,3,12,20,21,22,23,24 because of the limited size of 3D-printed nanolattices (typically <5 mm2)25,26, the small assembled nanolattice domains between inverted cracks (typically <0.01 mm2)9,10 and difficulties in reliably testing small nanolattices under tension3. The few studies that investigated nanolattice tensile behaviour used samples smaller than a few millimetres or with low absolute strength3,4,19,27,28, while no studies have explored the tensile properties of high-strength nanolattices at centimetre scales with millions of unit cells. To truly take advantage of the remarkable properties of nanolattices and further understand their large-scale fracture, it is essential to realize methods for fabricating macroscopic nanolattices and understand how their chemical and physical features affect their tensile properties.
We demonstrate a crack-free self-assembly approach to fabricate centimetre-scale multifunctional metallic nanolattices with 100 nm periodic features and 30 nm grain sizes, which corresponds to a 20,000 times increase in crack-free area and 1,000 times increase in the number of unit cells in the loaded direction, relative to prior nanolattices. These nanolattices have 257 MPa tensile strengths at 1.12% strain and a density of 2.67 g cm–3, which is 2.6 times the strength of the strongest porous metals with the same relative density at any scale. We eliminated cracks during self-assembly by maintaining a wet template and utilizing electrostatic forces to assist metal electrodeposition through the template. The resulting nickel nanolattices have excellent photonic colouration and approach their macroscopic theoretical tensile strength. The high absolute strength and the low density would allow nickel nanolattices to replace sandwich panel cores with 50% smaller volume than porous titanium, 50% lower mass than porous iron and, importantly, ten times less volume than other nanolattices.
Results
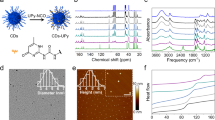
Conventional self-assembly approaches for fabricating metallic nanolattices lead to fully dense metal walls (inverted cracks) that surround nanolattice domains. Figure 1a shows the conventional method for growing fcc lattice templates from spherical nanoparticles (Methods). As a result of free energy minimization29,30, water evaporation drives nanoparticles to assemble into fcc lattices (also called opals)9 on a conductive substrate. During self-assembly, the top of the opal dries and cracks (Fig. 1a, inset), while the region closest to the receding waterline remains wet31 and has no cracks. The wet region typically maintains a constant height, while the dry, cracked region grows until all the water evaporates31. The cracked template is then converted into a nanolattice by sintering the particles, electrodepositing nickel into the voids and removing the template (Fig. 1b). Figure 1c shows a resulting nickel nanolattice with inverted cracks that separate nanolattice domains of typically less than 0.01 mm2 (Fig. 1c and Supplementary Figs. 8 and 9). Due to the energetic advantage of cracking in dried opals, our previous model confirmed the inevitable crack formation using the conventional fabrication method9.

a, Conventional self-assembly of lattice templates from the evaporation of negatively charged particle colloids on negatively charged surfaces. The inset is a top-view optical image of a cracked template in the dried area above the waterline. Scale bar, 20 µm. b, Fabrication steps to turn a lattice template into a metallic nanolattice. c, SEM images of nickel nanolattices with inverted cracks (white arrows) fabricated by the conventional method (also Supplementary Figs. 8 and 9). d, The WE method for fabricating metallic nanolattices without inverted cracks using positively charged particles and a glycerol additive. The inset is a top-view optical image of a wet opal template without cracks. Scale bar, 20 µm. e, SEM images of nickel nanolattices without inverted cracks, fabricated by the WE method (also Supplementary Figs. 10 and 11). f, Ratio of wet opal height to total opal height versus volume concentration of low-vapour-pressure alcohols added to the evaporating colloidal solution. The insets are images of templates made with pure water: top view of the vial after assembly (left) and top view of the sample (right). Scale bars, 1 cm. g,h, Schematics of electrostatic forces on negatively charged (g) and positively charged (h) templates during electrodeposition. V, voltage. Positive and negative signs denote charges on electrodes (green), particles (dark blue and orange) and ions (light blue and red).
We found that adding low-vapour-pressure alcohols to the colloidal solution can prevent assembled opals from drying, and therefore cracking, by concentrating the alcohols in the opal voids as water evaporates (Fig. 1d). We chose glycerol and ethylene glycol to prevent the opals from drying because they are soluble in water, are not volatile compared to water and only have hydroxyl groups that do not screen the charges on nanoparticles. As the assembled particles aggregated31, van der Waals forces between particles were strong enough to hold the template together to survive subsequent processing (Fig. 1b,d,e and Supplementary Section 12). Figure 1f shows the wet opal height, hwet, normalized by the full opal height, htotal, for glycerol and ethylene glycol at 0 to 1% initial volume concentrations. Without the alcohols (inset in Fig. 1f), the wet opal height was typically less than 10 mm (hwet/htotal = 0.45 in Fig. 1f). When 0.06% v/v glycerol was added, hwet/htotal increased to 1. For ethylene glycol, hwet/htotal was smaller than samples with glycerol but larger than pure water, as glycol has an evaporation rate higher than glycerol but lower than water. At low concentrations, glycerol and ethylene glycol did not impact the self-assembly order, whereas the assembled opal quality deteriorated when their concentrations increased beyond 1% v/v and 2% v/v (Supplementary Fig. 1). At this point, the alcohols’ local concentration at the assembly front was too high to densify particles into fcc packing (Supplementary Section 1). This experiment showed that 0.06% glycerol in the colloidal solution resulted in fully wet opals with high-quality packing and no cracks (Fig. 1d, inset).
Although keeping the opal wet prevented cracks, nickel could no longer be subsequently electrodeposited through the template due to particles’ negative surface charges. Negatively charged particles made from sulfated polystyrene (PS), sulfated polymethyl methacrylate (PMMA) and silica are commonly used because they are relatively easy to synthesize with high surface charge densities compared to positively charged particles17. During electrodeposition, however, the negatively charged electrode repels the particles’ negative charges and delaminates the opal from the substrate (Fig. 1g and Supplementary Sections 2 and 6). The conventional fabrication method uses sintering to prevent the delamination, but sintering requires a dry opal and exacerbates crack formation9.
We solved the delamination problem using positively charged particles, which reverses the electrostatic forces (Fig. 1h) so that nickel nanolattices without inverted cracks can be deposited (Fig. 1e and Supplementary Figs. 10 and 11). The positively charged particles, synthesized by dispersion polymerization (Methods), had amidine functional groups that fully protonated/ionized in water and created strong surface charges (67 to 82 mV zeta potentials; Supplementary Section 5). The strong charges were necessary to self-assemble particles into fcc packings29,30, whereas other weakly ionized, positively charged functional groups, such as amine groups, had low zeta potentials and resulted in random particle packings (Supplementary Sections 5 and 7). During electrodeposition, the strong electric field in the electrode double layer attracts positively charged particles after uncovering the ion cloud surrounding the particles (Fig. 1h; also Supplementary Section 6 for details)32, which allows nickel to deposit through the wet template without cracks (Fig. 1e and Supplementary Section 6). Figure 1b,d,h summarizes the developed method, which we call the wet–electrostatic (WE) method.
The WE method extended the available materials for crack-free self-assembled nanolattices from only oxides to metals (Supplementary Fig. 34 for a Cu nanolattice), while simultaneously increasing the maximum nanolattice thickness by 10 times, allowing centimetre-scale samples, and maintaining excellent optical properties. Previousstudies that used coassembly to fabricate oxide nanolattices were limited to a thickness of 16 layers of particles, beyond which large internal stresses generated by oxide sintering cracked the sample18. By contrast, electrodeposition does not require a sintering process to transform or densify the solid phase as the as-deposited nickel is fully dense (8.908 ± 0.060 g cm–3; Supplementary Section 20), and the resulting nanolattice thickness is limited only by the opal thickness (up to 500 µm). Figure 2a shows a 90-μm-thick crack-free nickel nanolattice, which was ten times thicker than prior crack-free studies on oxide nanolattices18. The inset shows a centimetre-scale crack-free nickel nanolattice (>2 cm2), which is a 20,000 times increase in crack-free area compared to the conventional assembly method (0.01 mm2). Eliminating inverted cracks also decreased the surface roughness substantially (Supplementary Fig. 15), which improved the nanolattice optical functionality. Figure 2b shows a highly ordered (111) crystal plane on the top of a nickel nanolattice, along with a clear colour transition from blue to red as the angle, θ, between a camera and incident light increased (Methods). The regions having slight colour variation in the insets were due to variations in nanolattice grain orientations (Supplementary Fig. 14). The overall excellent colour uniformity throughout the sample further confirmed that the resulting nickel nanolattices had well-aligned polycrystalline structures separated by small-angle grain boundaries.

a, A cross-section SEM image of an inverted-crack-free and free-standing nickel nanolattice. The blue mark represents the prior thickness limit of inverted-crack-free silica nanolattices, equivalent to a thickness of 16 layers of particles18. The inset is an image of a large-area nickel nanolattice without inverted cracks. The blue and red columns denote sample thicknesses and do not relate to the colours of the inset. b, An SEM image of a high-quality inverted-crack-free nickel nanolattice with a (111) surface facet that selectively reflects colour based on the relative viewing angle. The left inset is the experimental set-up for the spectrum images on the right.
The WE method also enabled the macroscopic study of nanolattice tensile mechanical properties, which has been very challenging because of the difficulty in fabricating large (>1 cm2) samples2. Most prior studies measured compressive properties using micrometre-sized cubic or cylindrical samples2,3,5,10,20,21,22,23,24. Even for self-assembled nanolattices, the nanolattice properties could only be isolated with nanoindentation or nanocompression because of the small separation between inverted cracks9,10,12,20,21,22,23,24. Using the WE method on an indium-tin-oxide-coated (ITO-coated) substrate with a dog-bone pattern (Supplementary Fig. 18), we overcame these prior limits and fabricated free-standing crack-free nickel nanolattice dog bones with 2.4 cm sample length and four different internal pore sizes (determined by particle sizes; Supplementary Section 4). Figure 3a shows the dog-bone samples (Supplementary Fig. 21 for scanning electron microscopy (SEM) graphics). The coloured regions show the selective light reflection from the pure nickel nanolattices. ITO’s low adhesion to metals allowed easy peeling of the nanolattices from the substrates to form free-standing samples for tensile testing9.

a, Dog-bone samples of inverted-crack-free nickel nanolattices with different internal pore sizes. The coloured areas are nickel nanolattices. No false colouring was used. b, The fracture surface of a nickel nanolattice with a 649 ± 13 nm average pore size. The blue-coloured area shows a layer of solid nickel at the bottom of the sample. The majority of the fracture surfaces are {111} crystallographic planes. The insets are a dog-bone sample in tensile grips before (left), during (middle; the colour was excited by external light) and after (right) fracture. tnanolattice, nanolattice thickness; tNi, solid nickel layer thickness. c, Tensile stress (σ) versus strain (ϵ) of four typical samples with different pore sizes. d, Ultimate tensile strength of nickel nanolattice dog bones versus the fraction of solid nickel at the sample bottom, tNi/ttotal. The black dashed line is the linear fit for all data (Supplementary Fig. 30 for individual size data and fits). Error bars show s.d. R2, coefficient of determination.
Tensile testing of the dog bones showed an ultra-high ultimate tensile strength (UTS). Figure 3b shows a typical fracture surface of a nickel nanolattice after tensile failure. The insets show the full sample before, during and after testing. As indicated by the white dashed-line polygons in Fig. 3b, tensile fracture mainly occurred at {111} crystal planes. A thin solid nickel layer can be seen at the bottom of the fracture surface (blue region, typically 0–3 μm thick), which formed during the nucleation stage of electrodeposition. Although mechanical milling can remove the solid nickel layer (Supplementary Fig. 22), we tested the dog bones without milling to avoid the potential damage from milling. We treated the samples as laminated composites, and the dog-bone UTS is σUTS = σnanolattice(1 – x) + σNix, where σnanolattice and σNi are stresses in the nanolattice and in the solid nickel layer at failure; x = tNi/ttotal is the ratio of the solid nickel layer thickness to the total thickness.
Figure 3c shows four typical strain–stress measurements for samples with different pore sizes. The average fracture strain of the composite samples was 1.12% (Supplementary Table 6), and no obvious yielding stage was present. A 0.2% offset yield stress analysis indicated that there was very little plastic deformation before 0.9% strain (Supplementary Fig. 20). The strain in the figure was estimated using a simulation, which underestimated the strain at failure by 0.15% on average, and was accurate below 0.9% (Supplementary Section 14). The fracture strain of solid nickel was about 2.5% (Supplementary Fig. 24), suggesting that the nickel nanolattice fractured prior to the failure of the bottom solid nickel layer. Therefore, σnanolattice is the UTS of the nickel nanolattice, which can be obtained from a linear fit of composite dog-bone UTS measurements with respect to x. Figure 3d shows the UTS of composite dog bones versus tNi/ttotal. A larger tNi/ttotal led to a higher UTS, as expected. The data presented here were statistically analysed (over 50 samples) to prevent inaccuracy caused by sample misalignment during testing (Supplementary Section 23).
Linear regression analysis of each dataset in Fig. 3d (Supplementary Fig. 30 for details) determined the UTS values to be 247.5 ± 9.3, 261.3 ± 7.8, 256.9 ± 6.7 and 264.9 ± 3.7 MPa for samples with pore sizes of 494 ± 10, 649 ± 13, 754 ± 12 and 844 ± 9 nm, respectively, at a 0.298 ± 0.006 relative density (Supplementary Section 22), which corresponded to 0.0935 ± 0.006, 0.0988 ± 0.006, 0.0971 ± 0.005 and 0.100 ± 0.004 MPa/(kg m−3) specific strengths, respectively. The measured relative density was higher than the ideal value, 0.26 (ref. 20), because of particle size variation and defects in the nanolattices (Supplementary Fig. 25). These effects can also broaden the nano strut size variation and potentially cause weak spots in nanolattices, which could be a reason for the limited pore-size dependence in Fig. 3d. Our observations agree with previous compression measurements, which showed no strong size dependence with struts larger than 60 nm (the smallest strut size here is 76 nm)10. The linear fit for all the data (black dashed line) gave an overall UTS of 257.3 ± 3.9 MPa, and indicated the stress in the solid nickel layer, σNi, at fracture was 781 ± 53 MPa, agreeing with the solid nickel stress at 1.12% strain (732 ± 17 MPa; Supplementary Fig. 24). Supplementary Tables 6 and 7 summarize all the tensile testing data.
The fabricated nickel nanolattices approached their theoretical UTS limit and realized a combination of material properties that outperformed other porous metals. A well-known formula to predict the porous material UTS, σUTS, is
where σb and ρ*/ρ are the bulk counterpart UTS and the relative density of the porous metal33. ρ* and ρ are the densities of porous metal and bulk metal, respectively. The constant, C (typically, ≤1), characterizes the porous geometric structure33. A larger C corresponds to a porous structure allowing a more even stress distribution and smaller internal stress concentrations.
Figure 4a summarizes the UTS of several porous metals (Ni (refs. 34,35,36), Fe (refs. 37,38,39,40), Ti (refs. 39,41), Al (ref. 42), Cu (refs. 43,44), Au (refs. 45,46,47) and Ag (refs. 48,49)) versus relative density with the dashed lines following equation (1) at different Cσb values. Prior fabrication techniques, including particle sintering37,40,41,48,49, dealloying36,45,46,47, Gasar38,43,44, slurry foaming34,39 and syntactic foaming42, encountered a Cσb limit of 0.6 GPa (black dashed line). This empirical limit has been set due to compromises in porous metal manufacturing. For example, a small Cσb can result from poor geometric control of internal pores (small C) or from the inability to tune the material microstructure (small σb). The solid dots in the figure show the nickel nanolattice dog bones in this study. Our nanolattices broke the Cσb = 0.6 GPa limit by a factor of 2.6, with a Cσb of 1.57 GPa (overall σUTS = 257.3 MPa, and ρ*/ρ = 0.298). The high strength was due to the fcc-arranged nanostructures with 100 nm periodic features (large C) and 30 nm average grain size tuned by electrodeposition (large σb; Supplementary Sections 9 and 24).
We compared this performance to the theoretical limit by substituting the strongest bulk nickel UTS (2 GPa in electrodeposited nickel with 26 nm grain size50) into the bulk strength in equation (1). Using σb = 2 GPa and C = 1, equation (1) predicts that the theoretical UTS limit of porous nickel is 325 MPa at a relative density of 0.298, only 26% higher than our nickel nanolattices (257 MPa). When compared to other nanolattices4,19,27,28, nanoporous graphene51,52,53 and aerogels54, the nickel nanolattices had an order of magnitude higher UTS (Fig. 4b). This agrees with prior simulations showing that the inverse opal structure can outperform octet trusses and isotropic trusses in terms of Young’s modulus, bulk modulus and shear modulus20. Compared to other porous metals’ tensile failure strains (0.1–7.3% with 0.16–0.84 relative densities)36,39,46,47,49, our nickel nanolattices’ average failure strain (1.12%) did not show a substantial drawback. Overall, the presented nanolattice has excellent tensile strength compared to prior porous metals because of the unique combination of pore shape and material microstructure.

a, UTS versus relative density of porous Ag, Al, Au, Cu, Fe, Ni and Ti. Dashed lines follow equation (1) with Cσb from 0 to 1.8 GPa. The black dashed line denotes the empirical limit of prior fabrication methods, including particle sintering, dealloying, Gasar, slurry foaming and syntactic foaming. b, Ultimate tensile strength versus relative density of nanolattices, nanoporous graphene and aerogels. c, Characteristic volume (σUTS−1/2) versus characteristic mass (ρ*σUTS−1/2) of a rectangular beam to bear a bending moment for different porous materials. The dashed lines are defined by specific tensile strengths between 0.005 and 0.08 MPa (kg m−3)−1. d, Specific tensile strength versus the ratio of sample loaded length to unit-cell length of nanolattices. Refs. 19,47, upper and lower bounds used; ALD, atomic layer deposition; CVD, chemical vapour deposition; NP, nanoporous.
Porous materials are often used to reduce the weight of structural materials subjected to bending and buckling, such as sandwich panel cores, and our nickel nanolattice shows excellent promise for bearing bending loads with a low volume and low mass. Previous nanolattice studies emphasized specific strength2,3, but it is also important to consider the total mass and volume required to resist a load. For example, the strongest 3D-printed nanolattice (UTS = 27.4 MPa)28 requires ten times more cross-sectional area (or volume) than a steel wire (UTS > 400 MPa) to hold the same weight. Here, we consider how much volume and mass a material needs to resist failure when being bent. For a rectangular beam with variable height, ρ*σUTS−1/2 and σUTS−1/2 characterize the minimum beam mass and volume needed to resist failure (Supplementary Section 25 for derivation)55. Figure 4c shows σUTS−1/2 versus ρ*σUTS−1/2 for different porous metals, nanolattices and nanoporous materials. The dashed lines denote different specific strengths and show that our nickel nanolattices have the highest specific strength among porous metals. The high UTS and low density allow the nickel nanolattices to resist a bending load with 50% smaller volume than porous titanium, 50% lower mass than porous iron and, importantly, ten times less volume than nanoporous graphene and other nanolattices.
Moreover, the presented fabrication approach can create much larger nanolattices than prior 3D-printing-based approaches. The challenge with nanolattice fabrication is to maintain precise nanoscale dimensions at the unit-cell level, which enable the enhanced properties, while simultaneously connecting millions of unit cells in a reasonable time with minimal defects or imperfections, such as big voids and missing struts, that could substantially decrease the strength. Thus, a technique resulting in fewer such imperfections should produce higher-strength nanolattices at larger scales. Here, we define the fabrication method’s scalability as the ratio of a sample’s loaded length to unit-cell length. Figure 4d shows the specific strength versus the scalability of nanolattices. For our samples, the loaded length was the equivalent length (Supplementary Fig. 19), while for prior work, we used the total distance between grips. The specific strengths of the nickel nanolattices outperform most other nanolattices, and the self-assembly technique allows a scalability that is three orders of magnitude larger than 3D printing. The large sample size and the high specific strength of the nickel nanolattices show that the self-assembly fabrication approach is capable of minimizing imperfections while maintaining nanoscale features across millions of unit cells.
Conclusions
In summary, this work presents a crack-free self-assembly approach to fabricate large-area multifunctional metallic nanolattices with an ultra-high 257 MPa tensile strength, which is 2.6 times the strength of prior porous metals at 0.298 relative density. We found that the key to eliminating cracks during self-assembly was to keep the template wet with 0.06% glycerol. Additionally, synthesized, positively charged PS particles allowed subsequent electrodeposition through the thick, wet opals due to electrostatic forces. Benefiting from the scalability of this fabrication approach, we grew large-area nickel nanolattices without inverted cracks and measured the tensile properties with macroscopic testing equipment. The resulting nickel nanolattices had excellent photonic colouration, approached the theoretical limit of the upper tensile strength, achieved a combination of strength and relative density that outperformed other porous metals and nanolattices and could resist bending fractures with a lower volume and mass than most porous materials. The developed methods and findings in this work will further the design and fabrication of lightweight porous metals with the promising combination of high strength, electrical and thermal conductivity, structural colouration and high specific surface area, which may enhance the performance of many applications such as high-power-density batteries, efficient heat and mass exchangers and selective infiltration membranes.
Methods
We prepared amidine-functionalized PS particles with positive charges by dispersion polymerization with 2,2′-azobis(2-methylpropionamidine) dihydrochloride and poly(diallyl dimethyl ammonium chloride) as the initiator and the stabilizer, respectively. Supplementary Fig. 3 shows the experimental set-up and synthesis process. Synthesis recipes, particle size distributions and particle zeta potentials at a self-assembly pH of 7 and electrodeposition pH of 4 are in Supplementary Sections 3–6. The detailed synthesis steps can be found in Supplementary Section 3. In general, a flask was filled with ethanol, aqueous poly(diallyl dimethyl ammonium chloride) and styrene, sequentially. The mixture was deoxygenated for 10 min by bubbling nitrogen gas and heated to 70 °C. Deoxygenated aqueous 2,2′-azobis(2-methylpropionamidine) dihydrochloride was then injected into the flask. The final mixture was kept at 70 °C and magnetically stirred for 20 h. The synthesized particles were centrifuged, washed by Milli-Q water and redispersed in water by sonication five times before use. The average particle diameters were characterized by SEM and image processing in MATLAB. Delsa Nano C particle analyser characterized the particle zeta potentials. MicroParticles and Thermo Fisher supplied negatively charged PS colloid suspensions functionalized with sulfate groups and PS particles functionalized with amine groups. Supplementary Table 8 lists the source, grade and concentration of all the chemicals.
We functionalized substrates to adjust surface charges before using them for self-assembly. ITO-coated glass slides were sonicated in methanol, acetone and isopropyl alcohol for 15 min each. After that, soaking the ITO slides in a ‘base piranha’ solution (H2O2/NH4OH/H2O with the volume ratio of 1:1:5) at 80 °C for 2 h functionalized the ITO surface with hydroxyl groups. Then, soaking the substrates in 1.0 wt% 3-(trihydroxysilyl) propane-1-sulfonic acid methanol solution, or 1.0 wt% N-(3-(trimethoxysilyl)propyl)-N,N,N-trimethylammonium chloride methanol solution, for 24 h functionalized the substrates with negative/positive charges. The substrates were rinsed with Milli-Q water thoroughly before being used for growing opals. For fabricating dog-bone samples, ITO slides were patterned by an ultraviolet laser (IPG Photonics IX-255) before functionalization (Supplementary Section 14 for the pattern dimensions).
Figure 1a,b shows the conventional nickel nanolattice fabrication process using self-assembly. ITO slides functionalized with negative charges were placed in Nalgene vials filled with 0.5 wt% negatively charged PS colloid suspensions. The vials were then heated to 55 °C to assemble opals for 24–36 h. After self-assembly, opals were sintered at 95 °C for 3 h to increase the adhesion to the substrate, followed by electrodepositing nickel at −1.5 V with nickel as a counter electrode in an Elevate Nickel 5910 bath (Supplementary Table 3 for the chemical composition). After electrodeposition, samples were soaked in toluene for over 12 h to dissolve the PS templates.
Figure 1b,d shows the WE (crack-free) fabrication approach for nickel nanolattices without inverted cracks. The self-assembly process followed the conventional method with some important changes. Instead of negatively charged particles and ITO slides, we used amidine-functionalized PS particles and positively charged ITO substrates. The positively charged PS colloid solution was 1 wt% PS with 0.06% v/v of added glycerol. The crack-free self-assembled template could be made as thick as 500 µm. After self-assembly, the wet opals were quickly transferred to the electrodeposition bath following the steps in Supplementary Section 12 to electrodeposit nickel at –1.5 V versus a nickel counter electrode. After electrodeposition, samples were mechanically peeled from their substrates (Supplementary Fig. 18) and then soaked in toluene for over 12 h to dissolve the PS template. For the dog-bone sample fabrication, only the dog-bone pattern was connected to the power source so that the deposited nickel nanolattice automatically formed the dog-bone shape (Supplementary Fig. 17).
A camera (Nikon D7100) mounted on top of a sample measured the structural colour while the incident light angle was adjusted from 61° to 71° with respect to the camera (Supplementary Fig. 13). An Instron mechanical tester (68SC-2) performed mechanical testing with side-action tensile grips and flat surface specimen holders (Fig. 3b, middle inset and Supplementary Fig. 20). During gripping, the specimen was adjusted several times until there was no visible sample distortion, to minimize the effect of misalignment. The initial distance between grips was 16.36 mm. Only dog-bone samples where the crack-free nickel nanolattices filled the majority of the area between grips were used for mechanical characterization. Preparation of dog-bone samples was carried out with caution to ensure that no inverted cracks appeared in the gauge sections (Supplementary Sections 12 and 15). The crosshead speed was 1 μm s–1, equivalent to a strain rate of 8 × 10−5 s−1. Only the samples that fractured in the gauge section were analysed and reported. The sample thicknesses were characterized in an SEM instrument (Supplementary Section 17) to calculate the stress (the sample width was 1.95 mm). The strain was estimated by dividing the grip displacement by 12.62 mm, which was determined by simulation and verified using image processing with time-series sample images (Supplementary Section 14). Electrodeposited nickel density and nickel nanolattice relative density were calculated from mass and volume measurements (Supplementary Sections 20 and 22). Rigaku MiniFlex X-ray diffraction characterized the nickel grain size (Supplementary Section 24).
Data availability
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
References
Zhao, B. et al. A review on metallic porous materials: pore formation, mechanical properties, and their applications. Int. J. Adv. Manuf. Technol. 95, 2641–2659 (2018).
Bauer, J. et al. Nanolattices: an emerging class of mechanical metamaterials. Adv. Mater. 29, 1701850 (2017).
Zhang, X., Wang, Y., Ding, B. & Li, X. Design, fabrication, and mechanics of 3D micro-/nanolattices. Small 16, e1902842 (2020).
Zheng, X. et al. Multiscale metallic metamaterials. Nat. Mater. 15, 1100–1106 (2016).
Vyatskikh, A. et al. Additive manufacturing of 3D nano-architected metals. Nat. Commun. 9, 593 (2018).
Geng, Q., Wang, D., Chen, P. & Chen, S.-C. Ultrafast multi-focus 3-D nano-fabrication based on two-photon polymerization. Nat. Commun. 10, 2179 (2019).
Hsueh, H.-Y. et al. Nanoporous gyroid nickel from block copolymer templates via electroless plating. Adv. Mater. 23, 3041–3046 (2011).
Bagal, A. et al. Large-area nanolattice film with enhanced modulus, hardness, and energy dissipation. Sci. Rep. 7, 9145 (2017).
Jiang, Z., Hsain, Z. & Pikul, J. H. Thick free-standing metallic inverse opals enabled by new insights into the fracture of drying particle films. Langmuir 36, 7315–7324 (2020).
Pikul, J. H. et al. High strength metallic wood from nanostructured nickel inverse opal materials. Sci. Rep. 9, 719 (2019).
Zhang, C. et al. Enhanced capillary-fed boiling in copper inverse opals via template sintering. Adv. Funct. Mater. 28, 1803689 (2018).
Zhang, R., Cohen, J., Fan, S. & Braun, P. V. Electrodeposited high strength, thermally stable spectrally selective rhenium nickel inverse opals. Nanoscale 9, 11187–11194 (2017).
Pikul, J. H., Zhang, H. G., Cho, J., Braun, P. V. & King, W. P. High-power lithium ion microbatteries from interdigitated three-dimensional bicontinuous nanoporous electrodes. Nat. Commun. 4, 1732 (2013).
Kränzlin, N. & Niederberger, M. Controlled fabrication of porous metals from the nanometer to the macroscopic scale. Mater. Horiz. 2, 359–377 (2015).
Pham, Q. N., Barako, M. T., Tice, J. & Won, Y. Microscale liquid transport in polycrystalline inverse opals across grain boundaries. Sci. Rep. 7, 10465 (2017).
Phillips, K. R. et al. Nanocrystalline precursors for the co-assembly of crack-free metal oxide inverse opals. Adv. Mater. 30, e1706329 (2018).
Vogel, N., Retsch, M., Fustin, C.-A., del Campo, A. & Jonas, U. Advances in colloidal assembly: the design of structure and hierarchy in two and three dimensions. Chem. Rev. 115, 6265–6311 (2015).
Hatton, B., Mishchenko, L., Davis, S., Sandhage, K. H. & Aizenberg, J. Assembly of large-area, highly ordered, crack-free inverse opal films. Proc. Natl Acad. Sci. USA 107, 10354–10359 (2010).
Montemayor, L. C., Wong, W. H., Zhang, Y. W. & Greer, J. R. Insensitivity to flaws leads to damage tolerance in brittle architected meta-materials. Sci. Rep. 6, 20570 (2016).
do Rosário, J. J., Berger, J. B., Lilleodden, E. T., McMeeking, R. M. & Schneider, G. A. The stiffness and strength of metamaterials based on the inverse opal architecture. Extrem. Mech. Lett. 12, 86–96 (2017).
Schmalbach, K. M. et al. Temperature-dependent mechanical behavior of three-dimensionally ordered macroporous tungsten. J. Mater. Res. 35, 2556–2566 (2020).
Ramachandramoorthy, R. et al. Dual-templated electrodeposition and characterization of regular metallic foam based microarchitectures. Appl. Mater. Today 20, 100667 (2020).
do Rosário, J. J., Häntsch, Y., Schneider, G. A. & Lilleodden, E. T. A combined compression and indentation study of mechanical metamaterials based on inverse opal coatings. Acta Mater. 195, 98–108 (2020).
do Rosário, J. J. et al. Self-assembled ultra high strength, ultra stiff mechanical metamaterials based on inverse opals. Adv. Eng. Mater. 17, 1420–1424 (2015).
Meza, L. R., Das, S. & Greer, J. R. Strong, lightweight, and recoverable three-dimensional ceramic nanolattices. Science 345, 1322–1326 (2014).
Jang, D., Meza, L. R., Greer, F. & Greer, J. R. Fabrication and deformation of three-dimensional hollow ceramic nanostructures. Nat. Mater. 12, 893–898 (2013).
Bauer, J., Schroer, A., Schwaiger, R. & Kraft, O. The impact of size and loading direction on the strength of architected lattice materials. Adv. Eng. Mater. 18, 1537–1543 (2016).
Mateos, A. J., Huang, W., Zhang, Y.-W. & Greer, J. R. Discrete-continuum duality of architected materials: failure, flaws, and fracture. Adv. Funct. Mater. 29, 1806772 (2019).
Manoharan, V. N. Colloidal matter: packing, geometry, and entropy. Science 349, 1253751 (2015).
Li, Q., Jonas, U., Zhao, X. & Kappl, M. The forces at work in colloidal self-assembly: a review on fundamental interactions between colloidal particles. Asia Pac. J. Chem. Eng. 3, 255–268 (2008).
Goehring, L., Clegg, W. J. & Routh, A. F. Solidification and ordering during directional drying of a colloidal dispersion. Langmuir 26, 9269–9275 (2010).
Guglielmi, N. Kinetics of the deposition of inert particles from electrolytic baths. J. Electrochem. Soc. 119, 1009–1012 (1972).
Gibson, L. J. & Ashby, M. F. Cellular Solids: Structure and Properties (Cambridge Univ. Press, 1999).
Aly, M. S. Tensile properties of open-cell nickel foams. Mater. Des. 31, 2237–2240 (2010).
Lee, K. & Lewandowski, J. J. Effects of microstructural characteristics on mechanical properties of open-cell nickel foams. Mater. Sci. Technol. 21, 1355–1358 (2005).
Kashani, H. & Chen, M. W. Flaw-free nanoporous Ni for tensile properties. Acta Mater. 166, 402–412 (2019).
Furumoto, T. et al. Permeability and strength of a porous metal structure fabricated by additive manufacturing. J. Mater. Process. Tech. 219, 10–16 (2015).
Kashihara, M. et al. Fabrication of lotus-type porous carbon steel via continuous zone melting and its mechanical properties. Mater. Sci. Eng. A 524, 112–118 (2009).
Kato, K. et al. Cytocompatibility and mechanical properties of novel porous 316 L stainless steel. Mater. Sci. Eng. C 33, 2736–2743 (2013).
Falkowska, A., Seweryn, A. & Szusta, J. Predicting the fatigue strength and life of 316L steel sinters of varying porosity for implants in a uniaxial loading state. Eng. Fract. Mech. 200, 146–165 (2018).
Kelly, C. N. et al. Fatigue behavior of as-built selective laser melted titanium scaffolds with sheet-based gyroid microarchitecture for bone tissue engineering. Acta Biomater. 94, 610–626 (2019).
Tao, X. F. & Zhao, Y. Y. Compressive failure of Al alloy matrix syntactic foams manufactured by melt infiltration. Mater. Sci. Eng. A 549, 228–232 (2012).
Nakajima, H. Fabrication, properties and application of porous metals with directional pores. Prog. Mater. Sci. 52, 1091–1173 (2007).
Simone, A. E. & Gibson, L. J. The tensile strength of porous copper made by the GASAR process. Acta Mater. 44, 1437–1447 (1996).
Gwak, E. J., Jeon, H., Song, E. J., Kang, N. R. & Kim, J. Y. Twinned nanoporous gold with enhanced tensile strength. Acta Mater. 155, 253–261 (2018).
Xia, R. et al. Microtensile tests of mechanical properties of nanoporous Au thin films. J. Mater. Sci. 44, 4728–4733 (2009).
Badwe, N., Chen, X. Y. & Sieradzki, K. Mechanical properties of nanoporous gold in tension. Acta Mater. 129, 251–258 (2017).
Ahmed, H. S. T. & Jankowski, A. F. Stiffening of sub-micro-porous silver membranes under tensile deformation. Mater. Sci. Eng. B 177, 43–47 (2012).
Gadaud, P., Caccuri, V., Bertheau, D., Carr, J. & Milhet, X. Ageing sintered silver: relationship between tensile behavior, mechanical properties and the nanoporous structure evolution. Mater. Sci. Eng. A 669, 379–386 (2016).
Budrovic, Z., Swygenhoven, H. V., Derlet, P. M., Petegem, S. V. & Schmitt, B. Plastic deformation with reversible peak broadening in nanocrystalline nickel. Science 304, 273–276 (2004).
Sun, H., Xu, Z. & Gao, C. Multifunctional, ultra-flyweight, synergistically assembled carbon aerogels. Adv. Mater. 25, 2554–2560 (2013).
Kashani, H., Ito, Y., Han, J., Liu, P. & Chen, M. Extraordinary tensile strength and ductility of scalable nanoporous graphene. Sci. Adv. 5, eaat6951 (2019).
Xu, Z., Zhang, Y., Li, P. & Gao, C. Strong, conductive, lightweight, neat graphene aerogel fibers with aligned pores. ACS Nano 6, 7103–7113 (2012).
Parmenter, K. E. & Milstein, F. Mechanical properties of silica aerogels. J. Non Cryst. Solids 223, 179–189 (1998).
Betts, C. Benefits of metal foams and developments in modelling techniques to assess their materials behaviour: a review. Mater. Sci. Technol. 28, 129–143 (2012).
Acknowledgements
We thank J. L. Bassani for insightful discussions on mechanics, D. Lee and J. Rosenfeld for helpful discussions on zeta potential characterization and L. Yang for discussions on particle synthesis. This work was partially funded by the pilot grant programme from the Center for Innovation and Precision Dentistry at the University of Pennsylvania, by the National Science Foundation under CAREER Grant no. 1943243 and by the American Society of Mechanical Engineers (ASME) Applied Mechanics Division Haythornthwaite Foundation Research Initiation Grant (J.H.P. and Z.J.). This work was carried out in part at the Singh Center for Nanotechnology, which is supported by the National Science Foundation National Nanotechnology Coordinated Infrastructure Program under grant NNCI-1542153.
Author information
Authors and Affiliations
Contributions
Z.J. and J.H.P conceived the idea. Z.J. designed and performed the experiments, and J.H.P directed the project. All authors analysed the data and wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Materials thanks the anonymous reviewers for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary Figs. 1–34, Sections 1–28 and Tables 1–8.
Rights and permissions
About this article

Cite this article
Jiang, Z., Pikul, J.H. Centimetre-scale crack-free self-assembly for ultra-high tensile strength metallic nanolattices. Nat. Mater. 20, 1512–1518 (2021). https://doi.org/10.1038/s41563-021-01039-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41563-021-01039-7
This article is cited by
-
Macroscopic photonic single crystals via seeded growth of DNA-coated colloids
Nature Communications (2023)
-
Dislocation-strained MoS2 nanosheets for high-efficiency hydrogen evolution reaction
Nano Research (2022)
-
Nano goes the distance
Nature Materials (2021)
