Abstract
Fatty acid metabolism is known to support tumorigenesis and disease progression as well as treatment resistance through enhanced lipid synthesis, storage and catabolism. More recently, the role of membrane fatty acid composition, for example, ratios of saturated, monounsaturated and polyunsaturated fatty acids, in promoting cell survival while limiting lipotoxicity and ferroptosis has been increasingly appreciated. Alongside these insights, it has become clear that tumour cells exhibit plasticity with respect to fatty acid metabolism, responding to extratumoural and systemic metabolic signals, such as obesity and cancer therapeutics, to promote the development of aggressive, treatment-resistant disease. Here, we describe cellular fatty acid metabolic changes that are connected to therapy resistance and contextualize obesity-associated changes in host fatty acid metabolism that likely influence the local tumour microenvironment to further modify cancer cell behaviour while simultaneously creating potential new vulnerabilities.
Similar content being viewed by others
Introduction
Cancer cells have distinctive metabolic features that allow the rapid manufacture of biomass to support cellular replication and other hallmarks of cancer, while managing redox homeostasis (see review1). In recent years, the field has developed an increasingly sophisticated understanding of cancer metabolism and, in particular, its heterogeneity with respect to cancer types2,3,4,5,6,7, grades8,9 and metastatic status10,11. In fact, it is clear that cancer cells exhibit considerable plasticity and flexibility in their metabolism to support rapid growth and survival in response to treatment and changes in environmental cues (see review12).
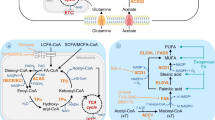
Fatty acid metabolism (Box 1) influences cancer cell biology in numerous ways, notably including the synthesis of lipid building blocks for membranes, that is, glycerophospholipids, and signalling intermediates such as phosphatidylinositol (4,5)-bisphosphate, diacylglycerol (DAG) and phosphatidate to facilitate mitogenic and/or oncogenic signalling13. Fatty acids are also substrates for mitochondrial ATP and NADH synthesis, eicosanoid production and post-translational protein–lipid modifications of signalling proteins (see review14). Cancer cells can acquire fatty acids from a range of intracellular and extracellular sources, and the altered metabolism of these fatty acids is a feature of both tumorigenesis and metastasis (Fig. 1; see review15). More recently, membrane lipid composition, as specified by fatty acyl saturation (for example, saturated, monounsaturated or polyunsaturated) and length, has received significant attention with emerging common tumour-associated features being identified16,17,18,19,20. The tumour lipidome characteristically includes increased proportions of saturated fatty acyl chains, and particularly monounsaturated fatty acyl chains, in glycerophospholipids from cancer cell lines and clinical tumour specimens, compared with non-malignant cells and benign tissues (see review14). Further, the clinical tumour lipidomes can distinguish malignant from normal tissue and reflect response and/or resistance to anticancer treatments. While data on clinical tumour metastases are lacking, comparisons between cell lines with different metastatic potential have identified increased DAGs and phosphatidylinositol lipids with greater levels of saturated and monounsaturated fatty acyl chains in metastatic as compared to non-metastatic and normal cell lines21,22. The increased proportions of phosphoinositide-based glycerophospholipids likely play key roles as membrane scaffolds and second messengers for oncogenic signalling pathways23,24. Beyond lipid and fatty acid abundance and desaturation, the elongation of fatty acid chains has been identified as a prominent feature of lung tumours25; however, its functional role remains to be defined. The importance of desaturation is of profound interest because it could be particularly advantageous to tumour cell survival by preventing both lipotoxicity from excess saturated fatty acyl chains (see review15) and ferroptosis triggered by the peroxidation of polyunsaturated fatty acyl chains (Box 2), as well as by reducing membrane permeability to promote chemoresistance (see Fatty acid metabolism in therapy resistance).

Cancer cell membranes are characterized by increased monounsaturated fatty acyl (MUFA) side chains to saturated fatty acyl (SFA) side chains and MUFA to polyunsaturated fatty acyl (PUFA) ratios that results in reduced lipotoxicity and susceptibility to ferroptosis. These traits are a result of increased uptake of extracellular fatty acids (FA) from the bloodstream and microenvironment via a range of mechanisms, including LDL receptor (LDLR), fatty acid transport protein (FATP) and CD36, and other mechanisms that contribute to the intracellular FA pool. Cancer cells also have increased de novo FA synthesis using a range of non-lipid substrates to produce palmitate, catalysed by acetyl-CoA carboxylase (ACC) and fatty acid synthase (FAS). Intracellular FAs are also mobilized via lipid droplet lipolysis, catalysed by adipose triacylglycerol lipase (ATGL), hormone sensitive lipase (HSL) and monoacylglycerol lipase (MAGL), and lipophagy. FAs are also released via the hydrolysis of glycerophospholipid by phospholipase As (PLAs) and phospholipase Bs to produce lysophospholipids. FAs are ‘activated’ by conversion to fatty acyl-CoAs (FA-CoAs) by long-chain acyl-CoA synthase (ACSL). FA-CoAs can be desaturated through the actions of stearoyl-CoA desaturase (SCD) or FA desaturases (FADS) and/or elongated by elongation of very long-chain fatty acid enzymes (ELOVLs) to increase MUFA-CoAs compared to PUFA-CoAs. These FA-CoAs are substrates for glycerolipid storage in lipid droplets and glycerophospholipid synthesis or remodelling via the acylation of lysophospholipids by lysophospholipid acyltransferase (LPLAT) to produce glycerophospholipids to maintain cellular membrane homeostasis. FA-CoAs can be oxidized in mitochondria and peroxisomes. Very long-chain FA (VLCFA)-CoAs are processed by peroxisomal oxidation to produce substrates for mitochondrial oxidation. Long-chain FA (LCFA)-CoAs are transported into the mitochondria by CPT1, whereas short-chain and medium-chain FA-CoAs passively diffuse across the membrane. Saturated FA-CoAs directly enter β-oxidation whereas the double bonds of unsaturated FA-CoAs (unFAs) are removed through the auxiliary pathway that includes Δ3, Δ2-enoyl-CoA isomerase (ECI) and 2,4-dienoyl CoA reductase 1 (DECR1) before returning to β-oxidation. These reactions fuel the electron transport chain (ETC). Overall, peroxisomal β-oxidation is reduced in cancer cells likely due to less very long-chain PUFA-CoAs, whereas long-chain PUFA-CoA β-oxidation is increased to lead to lower levels of PUFAs compared to MUFAs. ACLY, ATP-citrate lyase; DAG, diacylglycerol; MAG, monoacylglycerol; TAG, triacylglycerol; TCA, tricarboxylic acid.
Altered fatty acid metabolism is among a number of important potential mechanisms (see review26) that underpin the altered behaviour of many cancer types in patients with obesity, type 2 diabetes and/or metabolic syndrome27. In patients with obesity, it is likely that the combination of enhanced mitogenic and growth factor signalling in response to the altered hormonal milieu and the increased availability of carbon-rich nutrients, such as lipids and glucose, supports biomass production and proliferation, thereby accelerating disease progression and treatment resistance. Worldwide, obesity has nearly tripled since 1975 according to data by the World Health Organization, with a more substantial proportion of adults not only having obesity but also likely to have had obesity for a more extended portion of their lives compared to previous generations. Worryingly, the rate of mortality from obesity-associated cancers (for example, colorectal and breast cancer) has improved more slowly over the past 20 years than cancers not associated with obesity (for example, lung cancer and skin cancer)28. As such, the obesity-related impacts on cancer incidence, progression and treatment efficacy will increasingly challenge cancer management.
While precision oncology is largely considered in terms of genomic-driven treatment selection, the genomic alterations that define disease subtypes are invariably linked to altered metabolism14,24. Recent insights into the biological importance of lipidomic homeostasis have been reported and suggest a critical need for tumours to maintain optimal ratios of fatty acyl chain species (that is, monounsaturate to saturate and monounsaturate to polyunsaturate ratios) to avoid lipotoxicity and ferroptosis. In this Review, we focus on the role that fatty acid metabolism plays in responding to altered extratumoural or systemic signals from cancer therapies and the obese environment. We discuss the lipid characteristics and pathways that are common features of resistance to a range of treatment modalities. Additionally, we highlight obesity-associated changes in host fatty acid metabolism that likely influence the tumour microenvironment to affect cancer cell behaviour and response to therapy.

Fatty acid metabolism in therapy resistance
The concept of the tumour lipidome being reflective of changes in cancer cell behaviour extends to settings of extratumoural challenge, including in treatment-tolerant cancer cells as they rapidly adapt to enhance their survival and metastatic capacity (recently reviewed in detail12). Importantly, resistance to a range of cancer treatments is associated with changes in tumour cell fatty acid metabolism (Fig. 2).

Metabolism-based common features of therapy-resistant cells in different cancer or therapy settings can include increased lipid droplet (LD) number and size, increased LD and mitochondrial (mito) contacts that facilitate increased fatty acid (FA) oxidation, increased de novo FA synthesis catalysed by acetyl-CoA carboxylase (ACC) and FASN, all of which are associated with the altered expression of genes involved in FA metabolism. These changes also include increased levels of saturated fatty acyl side chains of membrane glycerophospholipids, leading to reduced membrane fluidity, endocytosis and passive diffusion of anticancer drugs as well as to reduced reactive oxygen species (ROS) production, ferroptosis and apoptosis. Finally, it also includes increased signalling domains that promote cell survival and multidrug resistance (MDR) drug pump-mediated drug efflux. ER, endoplasmic reticulum.
Chemotherapy
The response and resistance of tumour cells to chemotherapeutic agents have long been linked to altered lipid composition of cellular membranes. However, the field has been largely restricted to studies comparing resistant immortalized cell lines with parental lines or have looked at the acute effects of treatment on selected metabolic enzymes and/or pathways. Clinical data linking lipid metabolism in tumours to drug resistance remain elusive. Based on the available preclinical data, among the characteristics of chemoresistant cancer cell lines is a reduced fluidity of lipid bilayers in the membranes (Fig. 2). This reduced fluidity is based on the predominance of saturated fatty acyl chains in membrane lipids, particularly for lipogenic tumour cells17, and increased sphingomyelin and/or cholesterol content, for example, in chemotherapy-resistant ovarian and leukaemia cancer cell lines, compared to sensitive lines29,30. As a result of reduced fluidity, drug uptake via passive diffusion and/or endocytosis can be disrupted17,18,19,20. Furthermore, it results in the enhanced formation of detergent-resistant membrane domains, which can activate membrane-bound ATP-binding cassette (ABC) multidrug efflux transporters such as ATP-dependent translocase (also known as p-glycoprotein; ABCB1), thereby contributing to the multidrug resistance phenotype (see review31) that affects other anticancer drugs beyond chemotherapeutics. Intriguingly, pharmacological modulation of membrane fluidity (for example, via supplementation with polyunsaturated fatty acids) can alter ABCB1-mediated drug efflux32, suggesting that clinical lipid-modifying agents or dietary interventions could be promising chemosensitizing strategies.
With their relatively lower total cellular proportions of polyunsaturated to saturated fatty acyls, chemoresistant cancer cells are less susceptible than sensitive cancer cells to toxic lipid peroxidation (which can trigger apoptosis and ferroptosis), which occurs in response to the oxidative stress induced by many chemotherapeutic agents17,33. Indeed, chemoresistance has been linked to a dependency on glutathione peroxidase 4 (GPX4), a selenocysteine-containing enzyme that dissipates lipid peroxides and prevents ferroptotic cell death34,35 (see Box 2 for more detail). The decreased susceptibility to lipid peroxidation seems to be bolstered by enhanced antioxidant defences that are characteristic of chemoresistant cancer cells (reviewed in ref.36).
With the increasing body of evidence linking the above membrane changes to drug resistance, pharmacological intervention has focused on key pathways and enzymes driving the altered lipid features of cancer cells. As such, the pharmacological targeting of fatty acid synthase (FAS, encoded by FASN) sensitizes a range of cancer cell types to chemotherapy in vitro37,38, ex vivo37 and in vivo39,40, while the ectopic overexpression of FASN in breast cancer cells can confer broad chemoresistance in vitro38. Surprisingly, there has been limited focus on the mechanistic basis of FAS inhibition-mediated sensitization and the extent to which this reflects changes to fatty acid metabolism and lipid composition remains unclear, with only a single study demonstrating a rescue of in vitro chemosensitization of ovarian cancer cells by exogenous palmitate37. Interestingly, chemosensitization by the FAS inhibitor orlistat has been linked to the reduced expression of multidrug resistance proteins39, suggesting that altered membrane properties are likely to be important.
Targeting fatty acid oxidation has also received attention as a chemosensitization strategy given its key role in promoting tumour cell survival via energy generation and maintaining redox balance. Tumour tissue derived from patients with breast cancer that subsequently recurred exhibited enhanced expression of CPT1B mRNA compared to tumours that did not recur, and CPT1B mRNA was increased in chemoresistant versus primary breast tumours41, while tumoural CPT1A expression was associated with poorer overall survival in patients with gastric cancer42. Pharmacological inhibition of fatty acid oxidation using CPT1 inhibitors consistently chemosensitized tumour cells41,42,43.
The accumulation of lipid droplets is another characteristic though less well-studied phenotype of chemoresistant cancer cell lines44,45,46 (Fig. 2). Interestingly, triacsin C, a long-chain fatty acyl-CoA synthetase inhibitor that blocks fatty acid activation and thereby lipid droplet biogenesis, can chemosensitize colorectal cancer cells in vitro and in mouse xenografts46. Lipid droplets may directly contribute to chemoresistance by serving as an extra source of lipids for fatty acid oxidation under nutrient stress conditions, or as a ‘sink’ to sequester hydrophobic drugs47 (Fig. 2). Indeed, the total number of lipid droplets and the number colocalized with mitochondria were increased in a cell line model of chemoresistant breast cancer compared to the parental cells44. Subsequent profiling of these and clinically chemoresistant breast cancer cells revealed enhanced expression of the lipid droplet-localized protein PLIN4, which is involved in fatty acid mobilization from lipid droplets. Transcriptional silencing of PLIN4 reduced viability of the chemoresistant but not of the sensitive parental cells, indicating that lipid droplet-derived fatty acids are an important substrate for energy generation in the mitochondria of chemoresistant cancer cells. In chemoresistant colorectal cancer cells, marked lipid droplet accumulation was accompanied by induction of the lipid droplet-associated enzyme lysophosphatidylcholine acyltransferase 2 (LPCAT2), which catalyses the acylation of lysophosphatidylcholine to form phosphatidylcholine (PC), a component for lipid droplet biogenesis46. Enhanced synthesis of lipid droplets via LPCAT2 suppressed caspase activation and T cell infiltration in a syngeneic mouse tumour model due to the failure of dendritic cell maturation, both actions having the potential to promote resistance to chemotherapy and, potentially, immunotherapy46. Importantly, the level of expression of the lipid droplet-related genes PLIN4 or LPCAT2 were able to discriminate the degree of T cell infiltration in clinical colorectal cancer metastases and, while further detailed clinical validation is needed, provides encouraging evidence that the further study of lipid droplet biogenesis pathways will yield fruitful new targets.
Radiation therapy
Cancer cell lines that are resistant to radiation therapy commonly feature enhanced rates of fatty acid oxidation coupled with increased expression of CPT1A48,49,50,51, similar to chemoresistance41,42,43. Metabolic and expression analyses of radioresistant nasopharyngeal cancer (NPG) and breast cancer cells revealed enhanced fatty acid oxidation and CPT1A protein levels compared to radiosensitive cells, while inhibition of fatty acid oxidation (using genetic or pharmacological approaches) sensitized resistant cells in vitro to radiation48,49. The increase in fatty acid oxidation rate reported in radioresistant NPG cells was fuelled by an enhanced supply of fatty acids, facilitated by a greater number of contact sites between lipid droplets and mitochondria48. Similar findings have been reported in lung carcinoma cells, where combining etomoxir and radiation further reduced spheroid number and size compared to monotherapies52. The clinical significance of increased fatty acid oxidation and CPT1A expression in radioresistance is supported by the lower overall survival after radiation therapy of patients with NPG with higher levels of tumoural CPT1A expression48.
The potential for other metabolic processes beyond fatty acid oxidation to contribute to radioresistance has been reported in isogenic cell lines of head and neck squamous cell cancer53, where radioresistant cells exhibited reduced fatty acid uptake and enhanced glucose uptake compared to the sensitive cells. These resistant cells also upregulated FAS compared to the sensitive line, leading to enhanced fatty acid biosynthesis from glucose and enhanced oxidation of endogenous fatty acids54,55.
Targeted therapies
Biology-targeted therapies
With the findings that HER2 signalling activates the expression and/or activity of FAS to drive cancer cell proliferation56,57,58 and that a two-way crosstalk exists between these pathways59,60, FAS inhibition has been considered a rational strategy to overcome acquired resistance to the HER2-targeting therapeutics trastuzumab or lapatinib in preclinical cancer models59,60,61,62. For example, increased FASN expression in gastrointestinal stromal tumours from patients compared with normal tissues has been associated with shorter disease-free survival, while depletion of FASN or the inhibition of FAS (using C75) re-sensitized treatment-resistant gastrointestinal stromal tumour cell lines to the tyrosine kinase inhibitor imatinib63. However, mechanistically, C75 acted, at least in part, by reducing the transcription of the drug target (KIT) rather than by the predicted targeting of lipid synthesis and PI3K signalling63. Critically, no aspects of fatty acid metabolism were reported in this study and so it is challenging to determine whether the suppression of de novo fatty acid synthesis is central to overcoming trastuzumab resistance in this setting.
Similar to chemoresistance, cancer cells that survive pharmacological inhibition of the PI3K pathway have enhanced lipid droplet size and number as well as increased fatty acid oxidation, which sustains cell survival and tumour growth64. In this setting, ATG5-mediated autophagy and phospholipase A2 hydrolysis mobilized fatty acids from organelle glycerophospholipids to produce lysophospholipids, leading to enhanced fatty acid oxidation as well as spill over of fatty acids into lipid droplets for temporary storage. Together with the known hyperinsulinaemic and hypoglycaemic actions of PI3K inhibitors65, these mechanisms may contribute to the resistance to this class of agents observed clinically64. Interestingly, resistance to lapatinib (HER2 and EGFR inhibitor) in breast cancer cells in vitro most notably features transcriptional upregulation of the fatty acid transporter CD36 and, in turn, the uptake of fatty acids with concomitant lipid droplet accumulation66. The induction of CD36 was also evident in clinical breast cancer tissues after HER2 targeting therapy and tumours with higher levels of CD36 had poorer clinical outcomes, supporting the notion that fatty acid uptake and metabolism participate in drug resistance66.
Fatty acid oxidation is also an adaptive survival pathway in response to the targeted inhibition of heat shock protein 90 (HSP90)67. Using prostate cancer cells and patient-derived prostate tumours, we recently reported that culture with the HSP90 inhibitor luminespib significantly increased the abundance of proteins involved in oxidative phosphorylation and fatty acid metabolism. Further, combination treatment of luminespib with a clinical inhibitor of fatty acid oxidation, perhexiline, synergistically decreased the viability of prostate cancer cell lines and had significant efficacy in patient-derived tumour explants. Interestingly, this combination also attenuated the heat shock response (a known mediator of resistance), potentially through the regulation of intratumoural reactive oxygen species levels.
Endocrine-targeted therapies
Consistent with their anabolic actions, sex hormones such as oestrogens and androgens profoundly influence lipid metabolism in their target tissues and in hormone-dependent breast and prostate cancers68,69 (Box 3). The central role of endocrine therapies (targeting the production or action of sex hormones) in treating locally recurrent or metastatic disease reflects the dependence of breast and prostate cancer cells on these hormonal signalling pathways for survival; however, the development of resistance is common. In transcriptional or proteomic comparisons of hormone-naive versus endocrine-resistant breast and prostate cancers, lipid metabolism features prominently in analyses of upregulated pathways and processes in clinical samples70,71,72 and preclinical models45,73,74,75. Combination studies of lipid-altering agents (for example, CPT1 or FAS inhibitors) with endocrine therapies show promising preclinical efficacy in vitro and in mouse models of breast and prostate cancers (see below), but clinical support for these observations, particularly for breast cancer, is lacking.
In prostate cancer, endocrine therapy resistance (resulting in an incurable clinical state termed castrate-resistant prostate cancer; CRPC) is characterized by the reactivation of androgen receptor signalling76. Androgen receptor signalling coordinately controls the transcription of a suite of lipid metabolic genes in normal and malignant prostate epithelial cells72,77,78. Further, companion metabolic assays have demonstrated androgenic stimulation of de novo fatty acid synthesis, fatty acid uptake and oxidation, and aerobic glycolysis79,80,81,82. Intriguingly, there is evidence that, unlike the wild-type androgen receptor, which when activated primarily promotes lipid synthesis and glycolysis, signalling via the androgen receptor slice variant, AR-V7 — the predominant androgen receptor variant expressed in CRPC — promotes the utilization of citrate to favour amino acid biogenesis rather than lipid synthesis82. Thus, androgen receptor variants may not only activate canonical androgen receptor-directed pathways but could provide further metabolic plasticity as a survival advantage. Clinical CRPC tissues or experimental models typically feature the enhanced expression of androgen receptor-regulated metabolic genes compared to androgen-sensitive tumours or cell lines74,79, which has prompted strong interest in the therapeutic targeting of lipid metabolic processes, most notably de novo fatty acid synthesis but also, increasingly, fatty acid uptake and catabolism for the treatment of advanced prostate cancer83. Several recent studies have demonstrated the efficacy of targeting lipid metabolic enzymes as monotherapy in CRPC cell line and mouse models or, in combination, restoring sensitivity to androgen receptor-targeting agents74,84,85,86,87. For example, targeting fatty acid oxidation via CPT1 inhibition or targeting de novo fatty acid synthesis via FAS inhibition enhanced sensitivity to clinical androgen receptor antagonists in a range of preclinical models84,85,86. Mechanistically, targeting lipid synthesis has been shown to reduce the expression and/or activity of the androgen receptor86,88, with FAS inhibition also decreasing the expression of the constitutively active AR-V7 variant86. While a logical premise, it remains unclear whether crosstalk with androgen receptor expression and/or signalling is critical to the success of these re-sensitizing combinations or if other, as yet undefined, factors are at play.
A mechanistic link between the enhanced uptake of extracellular lipids and the development of CRPC has recently emerged, with androgen-sensitive prostate cancer cells revealing treatment-related increases in intracellular lipid content, notably glycerophospholipids and neutral lipids, as an adaptive response to androgen receptor targeting in vitro81,89. In particular, the development of therapy resistance in cell lines was accompanied by increases in glycerophospholipid species containing longer and more unsaturated fatty acyl chains89. The potential significance of this increased polyunsaturated fatty acid uptake in CRPC is underscored by recent work by us and others67,90 reporting that the gene encoding DECR1, which catalyses the rate-limiting step in polyunsaturated fatty acyl-CoA oxidation, is robustly overexpressed in clinical CRPC tissues compared to primary tumours and is associated with shorter relapse-free and overall survival. DECR1 knockdown in prostate cancer cells in vitro selectively inhibited β-oxidation of polyunsaturated fatty acyl-CoAs and inhibited the proliferation and migration of prostate cancer cells, including treatment-resistant lines, compared to DECR1-replete control cells. Collectively, these observations place mitochondrial polyunsaturated fatty acyl-CoA oxidation as a key mechanism in the generation of energy and in protecting against lipid peroxidation and ferroptosis (Box 2) to underpin the development of androgen receptor-targeted treatment resistance.
In breast cancer, the interplay between oestrogenic signalling and lipid metabolism is complicated by the presence of two cognate receptors (ERα and ERβ), each of which features distinct transcriptional programmes, and the multiple molecular disease subtypes that exist but have not yet been adequately modelled for metabolism. There is evidence that sterols can promote cancer growth and metastasis in preclinical models91 as they can act as ERα ligands92 and stimulate ER signalling73. A commonly reported feature of endocrine therapy-resistant breast cancer cell line models compared to isogenic sensitive lines is sterol regulatory element-binding protein 1 (SREBP)-driven upregulation of genes involved in lipid (notably cholesterol) biosynthesis45,73, and targeting of SREBP was effective in reducing the growth of these resistant cell lines45,73. However, the direct role of fatty acid metabolism in treatment resistance was not reported in these studies. One notable study reported that sublines of two invasive lobular breast cell lines that grew out after prolonged oestrogen deprivation all featured partial or complete loss of ERα activity and, interestingly, altered the expression of lipid metabolism genes, including increased SREBP1 and FASN, but also increased sensitivity to CPT1 inhibition of cell growth75. Again, little beyond gene expression was reported and these lipid phenotypes varied considerably between individual sublines, further emphasizing the caution that must be applied in interpreting the results from single treatment-resistant sublines, which dominate the literature. Nevertheless, analysis of RNA sequencing data from a neoadjuvant clinical trial of the aromatase inhibitor letrozole in breast cancer patients showed significant association between increased tumoural expression of SREBP1 post-treatment and a lack of clinical response, supporting the notion that this may be an important clinical mechanism of acquired resistance75.
Obesity and cancer progression
There remains a need for a detailed mechanistic understanding of the key metabolic switches that occur in response to therapy, the plasticity of such switching events and the metabolic impact of tumour heterogeneity in a more complex microenvironment. A key example of this is the influence of obesity, where it is commonly reported that cancer progression is altered in patients with obesity (see review93), including the development of treatment resistance. In this setting, tumour fatty acid metabolism adapts to ‘macro-level’ host attributes and, at the local microenvironment level, to influence disease behaviour.
Host physiology
The risk and cancer-related mortality of many cancer types are altered in populations with obesity93. This is supported by data arising from a range of preclinical cancer models fed an obesogenic high-fat diet94,95,96,97. The mechanisms associated with reduced cancer survival in patients with obesity remains to be defined but have been proposed to include hyperinsulinaemia, low-grade inflammation, altered adipokine levels, hyperglycaemia and dyslipidaemia (Fig. 3a; see review93). However, evidence that any of these mechanisms are viable therapeutic targets in patients with obesity is lacking.

a | Commonly proposed systemic changes that likely influence tumour biology in an obese host, including low-grade inflammation and altered circulating adipokine profiles, hyperinsulinaemia and growth factor levels, hyperglycaemia and dyslipidaemia. Altered mitogenic signalling and metabolism combine to promote tumour growth. b | Adipocyte-tumour fatty acid (FA) metabolic interactions that underpin obesity-influenced cancer progression. Local adipocytes respond to tumour-derived signals by increasing lipolysis, linked to increased adipose triacylglycerol (TAG) lipase (ATGL) levels, releasing FAs for tumour cells to take up. These adipocyte-derived FAs lead to increased TAG levels, lipid droplet expansion, and ATGL and hormone-sensitive lipase (HSL) levels as well as increased FA oxidation and CPT1 levels. These adipocyte-induced changes in tumour FA metabolism lead to increased cancer cell proliferation and migration. In the setting of obesity, in vitro experiments have shown that obese adipocytes co-cultured with cancer cells have increased ATGL expression and lipolytic rate, whereas cancer cells co-cultured with obese adipocytes have increased ATGL, HSL and CPT1 expression and FA oxidation rates compared to cells co-cultured with lean adipocytes, supporting enhanced cancer cell proliferation and migration. FAO, fatty acid oxidation; MAGL, monoacylglycerol lipase; PL, phospholipids.
Of direct relevance to this Review, the Paris Prospective Study of ~7,700 men reported that the highest quintile of circulating free fatty acids was associated with greatest all-cancer mortality98, and other studies have assessed the associations between fatty acid intake (that is, diet) and/or circulating fatty acid levels and cancer risk and/or mortality94,99,100,101. While this would suggest that greater fatty acid availability is linked to cancer, in general, these studies have failed to identify consistent associations or fatty acid species and/or total intake relationships (that is, food intake) or to unravel patterns that differed in populations with obesity compared to those without. To date, there is little, if any, direct functional evidence in preclinical or clinical settings that the increased in vivo availability of fatty acids alone or specific fatty acid species influence cancer cell behaviour in hosts with obesity. While this remains a major limitation in the field, one recent study reported a novel mechanism where tumour microenvironment levels of fatty acids are influenced by cancer cell fatty acid metabolism and thereby alter CD8+ T cell activity. Specifically, high-fat diet feeding of mice resulted in large tumours of syngeneic MC38 colorectal adenocarcinoma cells, E0771 breast adenocarcinoma, B16 melanoma and Lewis lung carcinoma compared to control diet and this was associated with increased fatty acid uptake and metabolism and reduced glycolysis in tumours96. Further, partitioning of fatty acids into tumours occurred at the expense of CD8+ T cells, with reduced T cell fatty acid content associated with impaired antitumour immunity. Critically, this partitioning was blocked by the overexpression of prolyl hydroxylase 3 (PHD3), which led to reduced tumour fatty acid oxidation and improved antitumour immune function in tumour-bearing mice fed a high-fat diet compared with mice bearing tumours expressing basal PHD3 levels. This observation suggests that the availability and competition for fatty acids between tumour and immune cells in the microenvironment supports tumour growth; however, the association with circulating fatty acids is lacking.
Free fatty acid availability in obesity is complex, with studies consistently reporting that total plasma free fatty acid levels are not increased in patients with obesity102,103 and are not associated with BMI98. There is certainly nuance surrounding adipose lipolysis and fatty acid turnover in patients with obesity informed by studies using stable isotope tracing techniques and accounting for differences in adipose mass102,104. Nonetheless, it is commonly reported that patients with obesity have increased plasma triacylglycerol (TAG) levels102,103. Additionally, it is important to acknowledge that the size of the circulating TAG pool is much greater than the free fatty acid pool and is further increased in patients with obesity compared with individuals without obesity103. These insights therefore suggest that increased systemic fatty acid availability to cancer cells in patients with obesity arises from lipoprotein-contained TAGs and not from adipose-derived free fatty acids. Since lipoprotein-contained TAGs are taken up by cells via multiple mechanisms (see reviews14,15), the increased availability of fatty acids to cancer cells in the circulation of patients with obesity is likely to involve a diverse array of uptake mechanisms that can introduce redundancy and flexibility to the system.
Underpinning the proposed mechanisms that link reduced cancer survival in patients with obesity, including fatty acid availability, is the assumption that obesity is a homogenous environment, defined by hyperinsulinaemia, low-grade inflammation, altered adipokine levels, hyperglycaemia and dyslipidaemia. However, it has been estimated that one-third of patients with obesity are metabolically healthy, with the remaining being metabolically unhealthy105, highlighting the metabolic diversity within a population defined as having obesity by BMI. While currently there is no universally accepted criteria for identifying metabolically (un)healthy individuals, generally it includes a combination of the presence of adiposity, insulin sensitivity and inflammation as well as the levels of circulating glucose and lipids106,107. As such, by determining whether ‘metabolically healthy obesity’ influences cancer behaviour the same way as ‘metabolically unhealthy obesity’ or whether effects are similar to ‘lean, metabolically unhealthy’ individuals, we believe that important insights can be made into whether disease behaviour is influenced by expanded adipose mass alone or other metabolic factors. Interestingly, overall cancer risk in older adults is lower among those with overweight/obesity who are metabolically healthy than among those with overweight/obesity who are metabolically unhealthy108 but it is not clear whether similar patterns are evident in terms of cancer progression. Of relevance for this review, a range of mechanisms that result in impaired lipid storage and circulating levels of lipids have been proposed to distinguish between these subtypes106,107. It remains to be determined whether circulating lipid levels (for example, lipoprotein TAG) or other yet to be identified mechanisms alter cancer progression in obese populations or in metabolically unhealthy populations. Nonetheless, tumour behaviour is heavily influenced by host physiology and, therefore, the presence of obesity implies effects on systemic metabolic drivers as well as substrate availability.
Adipocyte–tumour interaction
A commonly proposed mechanism linking obesity and altered tumour biology is an interaction between local (stromal) adipocytes and cancer cells. Many tumours co-localize with adipose tissue at various stages of the disease. For example, breast cancer arises in adipose-rich mammary tissue109, prostate cancer invades into periprostatic adipose tissue110, ovarian cancer metastasises into mesenteric adipose tissue111, pancreatic cancer invades local adipose tissue112 and many cancers metastasize to the bone, which is rich in bone marrow adipocytes113. Adipocytes are the predominant cell type of adipose tissue, and those adipocytes that closely localize to tumours are smaller compared to those distal to the tumour–adipocyte interface112,114,115. This suggests that tumours delipidate nearby adipocytes; in fact, it has been demonstrated that adipocyte-derived fatty acids do accumulate in cancer cells in vitro (recent examples include refs4,115,116,117,118). The ability of tumours to influence peritumoral adipocytes results in a modified phenotype (cancer-associated adipocytes)119. However, we recently reported that we did not observe any meaningful difference in ex vivo periprostatic adipose tissue biology, including basal and stimulated lipolysis rates, the profile of fatty acid species secreted, and adipocyte size, that associated with the aggressiveness of localized prostate cancer or obesity120. These findings do not negate the possibility that the in vivo milieu, influenced by local and systemic signals, including tumour-derived signals, will result in altered lipolytic flux and other lipid attributes of periprostatic adipose tissue.
Adipocytes can influence cancer cell behaviour in vitro4,112,114,115,116,117,118,121,122,123. Numerous adipocyte-derived factors have been proposed to mediate these effects, including adipokines, adipocytokines, hormones, proteases and lipids109, and we showed that adipocyte lipolysis was required for adipocyte-mediated effects on breast cancer cell proliferation4. The accumulation of adipocyte-derived fatty acids in cancer cells is facilitated by increased levels of a range of fatty acid uptake-related proteins that are required for the pro-growth effects of adipocytes116,117,118 (Fig. 3b). Adipocyte-derived fatty acids can act as substrates for lipid synthesis and storage in cancer cells4,117,118,121. Interestingly, gastric cancer cells co-cultured with adipocytes accumulated monounsaturated oleoyl-acyl chains in cellular lipids but not saturated palmitoyl or stearoyl-acyl chains114. This was likely due to either the selective uptake of adipocyte-derived oleate and/or uptake of palmitate and stearate, alongside oleate, which were then elongated and desaturated into oleoyl-CoA. This enrichment in oleoyl-acyl chains in lipid droplets is likely to play a major role in maintaining membrane monounsaturated to saturated and monounsaturated to polyunsaturated fatty acyl side chain ratios. Further, the accumulation of lipid droplets in breast cancer cells co-cultured with adipocytes was associated with changes in cancer cell protein levels of adipose TAG lipase (ATGL) and hormone-sensitive lipase (HSL)117, which can hydrolyse TAG-contained fatty acids and contribute to the intracellular fatty acid pool117,121. Silencing of ATGL in breast cancer cells impaired the migration ability of cells co-cultured with adipocytes117,121, suggesting that adipocyte-derived fatty acids influence cancer cell biology via actions at the lipid droplet.
The pro-growth and migration effects of adipocytes on cancer cells involves mitochondrial fatty acid oxidation (see review124). Adipocytes stimulate long-chain fatty acid oxidation in a range of cancer cells4,116,118,121,123, which is associated with increased protein levels of CPT1A4,121 or CPT1B116 (Fig. 3b). Importantly, CPT1A expression in breast cancer cells was required to metabolize adipocyte-derived fatty acids and thereby supported the increased invasion and epithelial-to-mesenchymal transition induced by adipocytes121. The increase in fatty acid oxidation following adipocyte co-culture may also arise from the increased phosphorylation of AMPK and acetyl-CoA carboxylase, leading to reduced allosteric inhibition of CPT1 (ref.121). Downstream of CPT1, we reported increased protein levels of mitochondrial electron transport chain complex subunits in breast cancer cells4, which was likely due to increased mitochondrial number as has been observed in melanoma cancer cells co-cultured with adipocytes123. However, others did not see these changes in similar conditions121.
While there is a growing body of evidence that adipocytes in the tumour microenvironment are active participants, many studies that have explored this relationship have done so using an in vitro experimental design of minus/plus adipocytes. This in vitro experimental design may model the commonly observed juxtapositioning of cancer cells and adipocytes observed in invasive melanoma118, prostate125 and ovarian cancers126, as examples, but it is questionable whether co-culture models are physiologically representative of an obese setting and, by inference, whether cells cultured without adipocytes are representative of a lean setting. It is important to also highlight that adipose tissue is a heterogeneous mix of cell types comprising mature adipocytes, resident immune cells (such as macrophages), fibroblasts, and the stem cell population termed ‘preadipocytes’109 and that the common changes in the adipose tissue microenvironment during body-weight gain and its potential influence on tumour initiation and progression have recently been discussed (see review127). The question here is whether fatty acid metabolism of obese adipocytes alters cancer cell biology beyond that observed with lean adipocytes. We and others have shown that culture with obese adipocytes (either in vitro models or adipose tissue from obese, high-fat diet-fed mice) enhances cancer cell fatty acid oxidation, lipid storage, and cell proliferation and migration compared to cancer cells cultured with lean adipocytes4,122,128,129,130. Interestingly, the pro-growth effects of obese adipose tissue from obese ZDF rats on MCF-7 breast cancer cells were reversed by the supplementation of rats with resveratrol prior to adipose tissue harvesting and conditioned media generation130. The resveratrol-stimulated reduction in MCF-7 cell proliferation was associated with changes in the adiponectin to leptin ratio, which was similar to that of lean animals. These observations suggest that targeting patient physiology, including adipose tissue alongside altering growth factor and hormone signalling65, has potential for cancer control, including obesity-stimulated cancer progression.
Obesity and treatment resistance
Obesity is associated with poorer clinical survival benefit from therapy for a range of cancers131,132,133,134. Mechanistically, this link is likely multifactorial, with some differences related to the systemic effects of obesity on drug pharmacokinetics and metabolism, reduced dosage due to poorer health, or lack of dosage adjustment for increased body weight135. Moreover, numerous agents are sequestered and metabolized in adipocytes136,137, while increased adipocyte size and hypoxia reduces blood flow and enhances inflammation138, potentially limiting the effective levels of drug exposure in patients with higher adiposity. However, drug availability and dosage factors cannot fully account for treatment resistance in obesity.
There is increasing evidence of adipocyte-driven mechanisms being involved in acquired treatment resistance. Specifically, haematological or solid tumour cells co-cultured with adipocytes developed resistance to a range of chemotherapies and targeted therapies137,139,140,141,142, while induced obesity promotes chemoresistance in animal models of cancer (summarized in ref.143). Notably, this behaviour was most common in cancer types that are intimately co-located with adipocytes in primary or secondary tumour growth sites (see above; reviewed in ref.144). Much attention has focused on the importance of secreted adipokines, such as leptin or IL-6, in promoting chemoresistance in cancer cells41, at least in part by altering cancer cell fatty acid uptake and oxidation41. Together, the observations that cancer cells stimulate adipocyte lipolysis and transfer of fatty acids to cancer cells4,111 and adipocytes stimulate cancer cell fatty acid uptake and oxidation4,41, in addition to the fatty acid metabolism features of treatment-resistant cells, which include increased fatty acid oxidation, lipid droplet expansion and changes in membrane composition (Fig. 2), imply that cancer cell fatty acid metabolism drives treatment resistance in the setting of obesity. Additionally, chemotherapeutic agents have direct effects on adipocyte lipid metabolism, resulting in enhanced free fatty acid availability that promotes cancer cell survival in animal models of cancer (see review143). On the other hand, co-culturing cancer cells with adipocytes has been linked to altered subcellular distribution of the chemotherapeutic doxorubicin into vesicles in breast cancer cells, culminating in enhanced drug efflux mediated by the major vault protein142. Importantly, in light of the preceding section, the effect of adipocytes on promoting treatment resistance is exacerbated in adipocytes derived from donors with obesity versus lean donors142,145. This is further supported by observations in a diet-induced obesity model of breast cancer, which features enhanced lipogenesis and lipolysis in tumour cells and increased resistance to doxorubicin146. As such, it is conceivable that the enhanced tumour fatty acid metabolic activity that occurs in the lipid-rich obese setting2,3,4 likely plays a central role in obesity-induced treatment resistance.
Conclusion and perspectives
In recent years, there has been a growing appreciation that fatty acid metabolism profoundly influences tumour progression beyond ATP production via β-oxidation and bulk availability for glycerophospholipid synthesis. Specifically, this includes the maintenance of fatty acid homeostasis with respect to redox stress, thereby preventing ferroptosis as well as influencing membrane fluidity and permeability to promote motility and metastasis. Many of these changes in fatty acid metabolism are also implicated in acquired treatment resistance, including in obesity-associated resistance, and may underpin the changes in cancer cell behaviour reported in patients with obesity. Importantly, the many recent reports of targeting fatty acid metabolism to overcome treatment resistance point to the likelihood that co-targeting strategies are a viable future approach and may be particularly crucial in a setting of obesity and metabolic dysfunction. All of these outcomes are reliant on future investigations involving emerging pharmacological agents that overcome some of the known deficiencies and off-target effects of current experimental and clinical inhibitors. Moreover, using more complex three-dimensional and patient-derived model systems and clinical specimens in these investigations is critical if co-targeting strategies are to be effectively employed in clinical practice. Finally, we believe that valuable opportunities remain to integrate the genomic classification of tumours with environmental factors, including diet and systemic metabolism, to improve patient prognostication and to design more wholistic precision medicine strategies.
References
Faubert, B., Solmonson, A. & DeBerardinis, R. J. Metabolic reprogramming and cancer progression. Science 368, eaaw5473 (2020).
Balaban, S. et al. Heterogeneity of fatty acid metabolism in breast cancer cells underlies differential sensitivity to palmitate-induced apoptosis. Mol. Oncol. 12, 1623–1638 (2018).
Balaban, S. et al. Extracellular fatty acids are the major contributor to lipid synthesis in prostate cancer. Mol. Cancer Res. 17, 949–962 (2019).
Balaban, S. et al. Adipocyte lipolysis links obesity to breast cancer growth: adipocyte-derived fatty acids drive breast cancer cell proliferation and migration. Cancer Metab. 5, 1 (2017).
Li, H. et al. The landscape of cancer cell line metabolism. Nat. Med. 25, 850–860 (2019). This study reported the metabolome of 928 cell lines from more than 20 cancer types in the Cancer Cell Line Encyclopedia and the heterogeneity of this panel of cancer cells.
Chen, P. H. et al. Metabolic diversity in human non-small cell lung cancer cells. Mol. Cell 76, 838–851.e5 (2019).
Hakimi, A. A. et al. An integrated metabolic atlas of clear cell renal cell carcinoma. Cancer Cell 29, 104–116 (2016).
Garg, G. et al. Targeted metabolomic profiling of low and high grade serous epithelial ovarian cancer tissues: a pilot study. Metabolomics 14, 154 (2018).
Lin, H. M. et al. A distinct plasma lipid signature associated with poor prognosis in castration-resistant prostate cancer. Int. J. Cancer 141, 2112–2120 (2017).
Christen, S. et al. Breast cancer-derived lung metastases show increased pyruvate carboxylase-dependent anaplerosis. Cell Rep. 17, 837–848 (2016).
Elia, I., Doglioni, G. & Fendt, S. M. Metabolic hallmarks of metastasis formation. Trends Cell Biol. 28, 673–684 (2018).
Fendt, S. M., Frezza, C. & Erez, A. Targeting metabolic plasticity and flexibility dynamics for cancer therapy. Cancer Discov. 10, 1797–1807 (2020).
Zhu, J. & Thompson, C. B. Metabolic regulation of cell growth and proliferation. Nat. Rev. Mol. Cell Biol. 20, 436–450 (2019).
Butler, L. et al. Lipids and cancer: emerging roles in pathogenesis, diagnosis and therapeutic intervention. Adv. Drug Deliv. Rev. 159, 245–293 (2020).
Nagarajan, S. R., Butler, L. M. & Hoy, A. J. The diversity and breadth of cancer cell fatty acid metabolism. Cancer Metab. 9, 2 (2021).
Harayama, T. & Riezman, H. Understanding the diversity of membrane lipid composition. Nat. Rev. Mol. Cell Biol. 19, 281–296 (2018).
Rysman, E. et al. De novo lipogenesis protects cancer cells from free radicals and chemotherapeutics by promoting membrane lipid saturation. Cancer Res. 70, 8117–8126 (2010).
Peetla, C. et al. Drug resistance in breast cancer cells: biophysical characterization of and doxorubicin interactions with membrane lipids. Mol. Pharm. 7, 2334–2348 (2010).
Callaghan, R., van Gorkom, L. C. & Epand, R. M. A comparison of membrane properties and composition between cell lines selected and transfected for multi-drug resistance. Br. J. Cancer 66, 781–786 (1992).
Zong, L. et al. Liquid extraction surface analysis nanospray electrospray ionization based lipidomics for in situ analysis of tumor cells with multidrug resistance. Rapid Commun. Mass. Spectrom. 32, 1683–1692 (2018).
Roy, J., Dibaeinia, P., Fan, T. M., Sinha, S. & Das, A. Global analysis of osteosarcoma lipidomes reveal altered lipid profiles in metastatic versus nonmetastatic cells. J. Lipid Res. 60, 375–387 (2019).
Kim, H. Y. et al. Discovery of potential biomarkers in human melanoma cells with different metastatic potential by metabolic and lipidomic profiling. Sci. Rep. 7, 8864 (2017).
Hoxhaj, G. & Manning, B. D. The PI3K-AKT network at the interface of oncogenic signalling and cancer metabolism. Nat. Rev. Cancer 20, 74–88 (2020).
Hopkins, B. D., Goncalves, M. D. & Cantley, L. C. Insulin-PI3K signalling: an evolutionarily insulated metabolic driver of cancer. Nat. Rev. Endocrinol. 16, 276–283 (2020).
Marien, E. et al. Phospholipid profiling identifies acyl chain elongation as a ubiquitous trait and potential target for the treatment of lung squamous cell carcinoma. Oncotarget 7, 12582–12597 (2016). By profiling phospholipids in a large clinical cohort of lung cancers, this study revealed fatty acid elongation as a cancer-related lipidomic feature that could be targeted via the elongase ELOVL6.
Park, J., Morley, T. S., Kim, M., Clegg, D. J. & Scherer, P. E. Obesity and cancer–mechanisms underlying tumour progression and recurrence. Nat. Rev. Endocrinol. 10, 455–465 (2014).
Balaban, S., Lee, L. S., Schreuder, M. & Hoy, A. J. Obesity and cancer progression: is there a role of fatty acid metabolism? Biomed. Res. Int. 2015, 274585 (2015).
Avery, C. L., Howard, A. G. & Nichols, H. B. Comparison of 20-year obesity-associated cancer mortality trends with heart disease mortality trends in the US. JAMA Netw. Open 4, e218356 (2021).
Veldman, R. J. et al. Altered sphingolipid metabolism in multidrug-resistant ovarian cancer cells is due to uncoupling of glycolipid biosynthesis in the Golgi apparatus. FASEB J. 16, 1111–1113 (2002).
Mannechez, A., Reungpatthanaphong, P., de Certaines, J. D., Leray, G. & Le Moyec, L. Proton NMR visible mobile lipid signals in sensitive and multidrug-resistant K562 cells are modulated by rafts. Cancer Cell Int. 5, 2 (2005).
Kopecka, J. et al. Phospholipids and cholesterol: inducers of cancer multidrug resistance and therapeutic targets. Drug Resist. Updat. 49, 100670 (2019).
Hajjaji, N. & Bougnoux, P. Selective sensitization of tumors to chemotherapy by marine-derived lipids: a review. Cancer Treat. Rev. 39, 473–488 (2013).
Yang, W. H. et al. A TAZ-ANGPTL4-NOX2 axis regulates ferroptotic cell death and chemoresistance in epithelial ovarian cancer. Mol. Cancer Res. 18, 79–90 (2020).
Viswanathan, V. S. et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature 547, 453–457 (2017).
Hangauer, M. J. et al. Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature 551, 247–250 (2017). Viswanathan et al.34 and Hangauer et al.35 reported a mechanism of therapy resistance in cancer cells that arises from a dependency on the antioxidant enzyme glutathione peroxidase, which protects cells from a lipid peroxidation-induced form of cell death called ferroptosis.
Alexa-Stratulat, T., Pesic, M., Gasparovic, A. C., Trougakos, I. P. & Riganti, C. What sustains the multidrug resistance phenotype beyond ABC efflux transporters? Looking beyond the tip of the iceberg. Drug Resist. Updat. 46, 100643 (2019).
Bauerschlag, D. O. et al. Fatty acid synthase overexpression: target for therapy and reversal of chemoresistance in ovarian cancer. J. Transl. Med. 13, 146 (2015).
Wu, X. et al. FASN regulates cellular response to genotoxic treatments by increasing PARP-1 expression and DNA repair activity via NF-kappaB and SP1. Proc. Natl Acad. Sci. USA 113, E6965–E6973 (2016).
Kant, S., Kumar, A. & Singh, S. M. Tumor growth retardation and chemosensitizing action of fatty acid synthase inhibitor orlistat on T cell lymphoma: implication of reconstituted tumor microenvironment and multidrug resistance phenotype. Biochim. Biophys. Acta 1840, 294–302 (2014).
Papaevangelou, E., Almeida, G. S., Box, C., deSouza, N. M. & Chung, Y. L. The effect of FASN inhibition on the growth and metabolism of a cisplatin-resistant ovarian carcinoma model. Int. J. Cancer 143, 992–1002 (2018).
Wang, T. et al. JAK/STAT3-regulated fatty acid beta-oxidation is critical for breast cancer stem cell self-renewal and chemoresistance. Cell Metab. 27, 136–150 e135 (2018).
He, W. et al. MSC-regulated lncRNA MACC1-AS1 promotes stemness and chemoresistance through fatty acid oxidation in gastric cancer. Oncogene 38, 4637–4654 (2019).
Luo, J. et al. An indispensable role of CPT-1a to survive cancer cells during energy stress through rewiring cancer metabolism. Tumour Biol. 37, 15795–15804 (2016).
Sirois, I. et al. A unique morphological phenotype in chemoresistant triple-negative breast cancer reveals metabolic reprogramming and PLIN4 expression as a molecular vulnerability. Mol. Cancer Res. 17, 2492–2507 (2019).
Hultsch, S. et al. Association of tamoxifen resistance and lipid reprogramming in breast cancer. BMC Cancer 18, 850 (2018).
Cotte, A. K. et al. Lysophosphatidylcholine acyltransferase 2-mediated lipid droplet production supports colorectal cancer chemoresistance. Nat. Commun. 9, 322 (2018). This study provided new mechanistic insights into the role of lipid droplet accumulation, driven by LPCAT2, in cancer cell chemoresistance and reduced immunogenicity.
Dubey, R. et al. Lipid droplets can promote drug accumulation and activation. Nat. Chem. Biol. 16, 206–213 (2020).
Tan, Z. et al. Targeting CPT1A-mediated fatty acid oxidation sensitizes nasopharyngeal carcinoma to radiation therapy. Theranostics 8, 2329–2347 (2018).
Han, S. et al. CPT1A/2-mediated FAO enhancement-a metabolic target in radioresistant breast cancer. Front. Oncol. 9, 1201 (2019).
Du, Q. et al. PGC1alpha/CEBPB/CPT1A axis promotes radiation resistance of nasopharyngeal carcinoma through activating fatty acid oxidation. Cancer Sci. 110, 2050–2062 (2019).
Wan, H., Xu, B., Zhu, N. & Ren, B. PGC-1alpha activator-induced fatty acid oxidation in tumor-infiltrating CTLs enhances effects of PD-1 blockade therapy in lung cancer. Tumori 106, 55–63 (2019).
Dheeraj, A. et al. A novel approach to target hypoxic cancer cells via combining beta-oxidation inhibitor etomoxir with radiation. Hypoxia 6, 23–33 (2018).
Mims, J. et al. Energy metabolism in a matched model of radiation resistance for head and neck squamous cell cancer. Radiat. Res. 183, 291–304 (2015).
Chuang, H. Y., Lee, Y. P., Lin, W. C., Lin, Y. H. & Hwang, J. J. Fatty acid inhibition sensitizes androgen-dependent and -independent prostate cancer to radiotherapy via FASN/NF-kappaB pathway. Sci. Rep. 9, 13284 (2019).
Zhan, N., Li, B., Xu, X., Xu, J. & Hu, S. Inhibition of FASN expression enhances radiosensitivity in human non-small cell lung cancer. Oncol. Lett. 15, 4578–4584 (2018).
Li, N. et al. Fatty acid synthase regulates proliferation and migration of colorectal cancer cells via HER2-PI3K/Akt signaling pathway. Nutr. Cancer 64, 864–870 (2012).
Jin, Q. et al. Fatty acid synthase phosphorylation: a novel therapeutic target in HER2-overexpressing breast cancer cells. Breast Cancer Res. 12, R96 (2010).
Long, X. H. et al. Lapatinib alters the malignant phenotype of osteosarcoma cells via downregulation of the activity of the HER2-PI3K/AKT-FASN axis in vitro. Oncol. Rep. 31, 328–334 (2014).
Grunt, T. W. et al. Interaction between fatty acid synthase- and ErbB-systems in ovarian cancer cells. Biochem. Biophys. Res. Commun. 385, 454–459 (2009).
Vazquez-Martin, A., Colomer, R., Brunet, J. & Menendez, J. A. Pharmacological blockade of fatty acid synthase (FASN) reverses acquired autoresistance to trastuzumab (Herceptin by transcriptionally inhibiting ‘HER2 super-expression’ occurring in high-dose trastuzumab-conditioned SKBR3/Tzb100 breast cancer cells. Int. J. Oncol. 31, 769–776 (2007).
Blancafort, A. et al. Dual fatty acid synthase and HER2 signaling blockade shows marked antitumor activity against breast cancer models resistant to anti-HER2 drugs. PLoS ONE 10, e0131241 (2015).
Puig, T. et al. A novel inhibitor of fatty acid synthase shows activity against HER2+ breast cancer xenografts and is active in anti-HER2 drug-resistant cell lines. Breast Cancer Res. 13, R131 (2011).
Li, C. F. et al. Overexpressed fatty acid synthase in gastrointestinal stromal tumors: targeting a progression-associated metabolic driver enhances the antitumor effect of imatinib. Clin. Cancer Res. 23, 4908–4918 (2017).
Lue, H. W. et al. Metabolic reprogramming ensures cancer cell survival despite oncogenic signaling blockade. Genes Dev. 31, 2067–2084 (2017).
Hopkins, B. D. et al. Suppression of insulin feedback enhances the efficacy of PI3K inhibitors. Nature 560, 499–503 (2018).
Feng, W. W. et al. CD36-mediated metabolic rewiring of breast cancer cells promotes resistance to HER2-targeted therapies. Cell Rep. 29, 3405–3420.e5 (2019).
Nassar, Z. D. et al. Fatty acid oxidation is an adaptive survival pathway induced in prostate tumors by heat shock protein 90 inhibition. Mol. Cancer Res. 18, 1500–1511 (2020).
Butler, L. M., Centenera, M. M. & Swinnen, J. V. Androgen control of lipid metabolism in prostate cancer: novel insights and future applications. Endocr. Relat. Cancer 23, R219–R227 (2016).
Harrelson, J. P. & Lee, M. W. Expanding the view of breast cancer metabolism: promising molecular targets and therapeutic opportunities. Pharmacol. Ther. 167, 60–73 (2016).
Latonen, L. et al. Integrative proteomics in prostate cancer uncovers robustness against genomic and transcriptomic aberrations during disease progression. Nat. Commun. 9, 1176 (2018).
Iglesias-Gato, D. et al. The proteome of prostate cancer bone metastasis reveals heterogeneity with prognostic implications. Clin. Cancer Res. 24, 5433–5444 (2018).
Sharma, N. L. et al. The androgen receptor induces a distinct transcriptional program in castration-resistant prostate cancer in man. Cancer Cell 23, 35–47 (2013).
Nguyen, V. T. et al. Differential epigenetic reprogramming in response to specific endocrine therapies promotes cholesterol biosynthesis and cellular invasion. Nat. Commun. 6, 10044 (2015).
Han, W. et al. Reactivation of androgen receptor-regulated lipid biosynthesis drives the progression of castration-resistant prostate cancer. Oncogene 37, 710–721 (2018).
Du, T. et al. Key regulators of lipid metabolism drive endocrine resistance in invasive lobular breast cancer. Breast Cancer Res. 20, 106 (2018).
Karantanos, T. et al. Understanding the mechanisms of androgen deprivation resistance in prostate cancer at the molecular level. Eur. Urol. 67, 470–479 (2015).
Swinnen, J. V., Ulrix, W., Heyns, W. & Verhoeven, G. Coordinate regulation of lipogenic gene expression by androgens: evidence for a cascade mechanism involving sterol regulatory element binding proteins. Proc. Natl Acad. Sci. USA 94, 12975–12980 (1997).
Heemers, H. V., Verhoeven, G. & Swinnen, J. V. Androgen activation of the sterol regulatory element-binding protein pathway: Current insights. Mol. Endocrinol. 20, 2265–2277 (2006).
Massie, C. E. et al. The androgen receptor fuels prostate cancer by regulating central metabolism and biosynthesis. EMBO J. 30, 2719–2733 (2011).
Tennakoon, J. B. et al. Androgens regulate prostate cancer cell growth via an AMPK-PGC-1alpha-mediated metabolic switch. Oncogene 33, 5251–5261 (2014).
Tousignant, K. D. et al. Lipid uptake is an androgen-enhanced lipid supply pathway associated with prostate cancer disease progression and bone metastasis. Mol. Cancer Res. 17, 1166–1179 (2019).
Shafi, A. A. et al. Differential regulation of metabolic pathways by androgen receptor (AR) and its constitutively active splice variant, AR-V7, in prostate cancer cells. Oncotarget 6, 31997–32012 (2015).
Zadra, G. & Loda, M. Metabolic vulnerabilities of prostate cancer: diagnostic and therapeutic opportunities. Cold Spring Harb. Perspect. Med. 8, a030569 (2018).
Schlaepfer, I. R. et al. Lipid catabolism via CPT1 as a therapeutic target for prostate cancer. Mol. Cancer Ther. 13, 2361–2371 (2014).
Zadra, G. et al. A novel direct activator of AMPK inhibits prostate cancer growth by blocking lipogenesis. EMBO Mol. Med. 6, 519–538 (2014).
Zadra, G. et al. Inhibition of de novo lipogenesis targets androgen receptor signaling in castration-resistant prostate cancer. Proc. Natl Acad. Sci. USA 116, 631–640 (2019).
Itkonen, H. M. et al. Lipid degradation promotes prostate cancer cell survival. Oncotarget 8, 38264–38275 (2017).
Kong, Y. et al. Inhibition of cholesterol biosynthesis overcomes enzalutamide resistance in castration-resistant prostate cancer (CRPC). J. Biol. Chem. 293, 14328–14341 (2018).
Tousignant, K. D. et al. Therapy-induced lipid uptake and remodeling underpin ferroptosis hypersensitivity in prostate cancer. Cancer Metab. 8, 11 (2020).
Blomme, A. et al. 2,4-dienoyl-CoA reductase regulates lipid homeostasis in treatment-resistant prostate cancer. Nat. Commun. 11, 2508 (2020).
Nelson, E. R. et al. 27-Hydroxycholesterol links hypercholesterolemia and breast cancer pathophysiology. Science 342, 1094–1098 (2013).
Perone, Y. & Magnani, L. Going off the grid: ERα breast cancer beyond estradiol. J. Mol. Endocrinol. 57, F1–F5 (2016).
Hopkins, B. D., Goncalves, M. D. & Cantley, L. C. Obesity and cancer mechanisms: cancer metabolism. J. Clin. Oncol. 34, 4277–4283 (2016).
Labbe, D. P. et al. High-fat diet fuels prostate cancer progression by rewiring the metabolome and amplifying the MYC program. Nat. Commun. 10, 4358 (2019).
Chang, H. H. et al. Incidence of pancreatic cancer is dramatically increased by a high fat, high calorie diet in KrasG12D mice. PLoS ONE 12, e0184455 (2017).
Ringel, A. E. et al. Obesity shapes metabolism in the tumor microenvironment to suppress anti-tumor immunity. Cell 183, 1848–1866.e26 (2020). This study reported that tumours and local immune cells compete for fatty acids in high-fat diet-fed animal models and that blocking tumour fatty acid oxidation increased fatty acid uptake by T cells, which led to their activation.
Broadfield, L. A. et al. Fat induces glucose metabolism in non-transformed liver cells and promotes liver tumorigenesis. Cancer Res. 81, 1988–2001 (2021).
Charles, M. A. et al. High plasma nonesterified fatty acids are predictive of cancer mortality but not of coronary heart disease mortality: results from the Paris Prospective Study. Am. J. Epidemiol. 153, 292–298 (2001).
Brennan, S. F., Woodside, J. V., Lunny, P. M., Cardwell, C. R. & Cantwell, M. M. Dietary fat and breast cancer mortality: a systematic review and meta-analysis. Crit. Rev. Food Sci. Nutr. 57, 1999–2008 (2017).
Van Blarigan, E. L. et al. Dietary fat intake after colon cancer diagnosis in relation to cancer recurrence and survival: CALGB 89803 (Alliance). Cancer Epidemiol. Biomarkers Prev. 27, 1227–1230 (2018).
Yammine, S. et al. Dietary and circulating fatty acids and ovarian cancer risk in the European Prospective Investigation into Cancer and Nutrition. Cancer Epidemiol. Biomarkers Prev. 29, 1739–1749 (2020).
McQuaid, S. E. et al. Downregulation of adipose tissue fatty acid trafficking in obesity: a driver for ectopic fat deposition? Diabetes 60, 47–55 (2011).
Jocken, J. W. et al. Effect of beta-adrenergic stimulation on whole-body and abdominal subcutaneous adipose tissue lipolysis in lean and obese men. Diabetologia 51, 320–327 (2008).
Horowitz, J. F. & Klein, S. Whole body and abdominal lipolytic sensitivity to epinephrine is suppressed in upper body obese women. Am. J. Physiol. Endocrinol. Metab. 278, E1144–E1152 (2000).
Ahima, R. S. & Lazar, M. A. Physiology. The health risk of obesity–better metrics imperative. Science 341, 856–858 (2013).
Stefan, N., Schick, F. & Haring, H. U. Causes, characteristics, and consequences of metabolically unhealthy normal weight in humans. Cell Metab. 26, 292–300 (2017).
Iacobini, C., Pugliese, G., Blasetti Fantauzzi, C., Federici, M. & Menini, S. Metabolically healthy versus metabolically unhealthy obesity. Metabolism 92, 51–60 (2019).
Moore, L. L., Chadid, S., Singer, M. R., Kreger, B. E. & Denis, G. V. Metabolic health reduces risk of obesity-related cancer in framingham study adults. Cancer Epidemiol. Biomarkers Prev. 23, 2057–2065 (2014).
Hoy, A. J., Balaban, S. & Saunders, D. N. Adipocyte-tumor cell metabolic crosstalk in breast cancer. Trends Mol. Med. 23, 381–392 (2017).
Kapoor, J. et al. Extraprostatic extension into periprostatic fat is a more important determinant of prostate cancer recurrence than an invasive phenotype. J. Urol. 190, 2061–2066 (2013).
Nieman, K. M. et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat. Med. 17, 1498–1503 (2011). This study demonstrated metabolic symbiosis between a range of cancer cells and adipocytes.
Cai, Z. et al. Cancer-associated adipocytes exhibit distinct phenotypes and facilitate tumor progression in pancreatic cancer. Oncol. Rep. 42, 2537–2549 (2019).
Cha, Y. J. & Koo, J. S. Roles of omental and bone marrow adipocytes in tumor biology. Adipocyte 8, 304–317 (2019).
Xiang, F. et al. Omental adipocytes enhance the invasiveness of gastric cancer cells by oleic acid-induced activation of the PI3K-Akt signaling pathway. Int. J. Biochem. Cell Biol. 84, 14–21 (2017).
Laurent, V. et al. Periprostatic adipose tissue favors prostate cancer cell invasion in an obesity-dependent manner: role of oxidative stress. Mol. Cancer Res. 17, 821–835 (2019). This study reported that adipocytes from obese donors further promotes cancer cell progression above that of adipocytes from lean donors.
Tan, Y. et al. Adipocytes fuel gastric cancer omental metastasis via PITPNC1-mediated fatty acid metabolic reprogramming. Theranostics 8, 5452–5468 (2018).
Yang, D. et al. Utilization of adipocyte-derived lipids and enhanced intracellular trafficking of fatty acids contribute to breast cancer progression. Cell Commun. Signal. 16, 32 (2018).
Zhang, M. et al. Adipocyte-derived lipids mediate melanoma progression via FATP proteins. Cancer Discov. 8, 1006–1025 (2018).
Dirat, B. et al. Cancer-associated adipocytes exhibit an activated phenotype and contribute to breast cancer invasion. Cancer Res. 71, 2455–2465 (2011).
Miladinovic, D. et al. Assessment of periprostatic and subcutaneous adipose tissue lipolysis and adipocyte size from men with localized prostate cancer. Cancers 12, 1385 (2020).
Wang, Y. Y. et al. Mammary adipocytes stimulate breast cancer invasion through metabolic remodeling of tumor cells. JCI Insight 2, e87489 (2017).
Laurent, V. et al. Periprostatic adipocytes act as a driving force for prostate cancer progression in obesity. Nat. Commun. 7, 10230 (2016).
Lazar, I. et al. Adipocyte exosomes promote melanoma aggressiveness through fatty acid oxidation: a novel mechanism linking obesity and cancer. Cancer Res. 76, 4051–4057 (2016).
Attane, C. & Muller, C. Drilling for oil: tumor-surrounding adipocytes fueling cancer. Trends Cancer 6, 593–604 (2020).
Nassar, Z. D. et al. Peri-prostatic adipose tissue: the metabolic microenvironment of prostate cancer. BJU Int. 121(Suppl. 3), 9–21 (2018).
Nieman, K. M., Romero, I. L., Van Houten, B. & Lengyel, E. Adipose tissue and adipocytes support tumorigenesis and metastasis. Biochim. Biophys. Acta 1831, 1533–1541 (2013).
Quail, D. F. & Dannenberg, A. J. The obese adipose tissue microenvironment in cancer development and progression. Nat. Rev. Endocrinol. 15, 139–154 (2019).
Clement, E. et al. Adipocyte extracellular vesicles carry enzymes and fatty acids that stimulate mitochondrial metabolism and remodeling in tumor cells. EMBO J. 39, e102525 (2020).
Nimri, L., Peri, I., Yehuda-Shnaidman, E. & Schwartz, B. Adipocytes isolated from visceral and subcutaneous depots of donors differing in BMI crosstalk with colon cancer cells and modulate their invasive phenotype. Transl. Oncol. 12, 1404–1415 (2019).
Theriau, C. F., Sauve, O. S., Beaudoin, M. S., Wright, D. C. & Connor, M. K. Proliferative endocrine effects of adipose tissue from obese animals on MCF7 cells are ameliorated by resveratrol supplementation. PLoS ONE 12, e0183897 (2017).
Incio, J. et al. Obesity-induced inflammation and desmoplasia promote pancreatic cancer progression and resistance to chemotherapy. Cancer Discov. 6, 852–869 (2016). This study implicated aggravation of desmoplasia as a key mechanism of obesity-promoted pancreatic cancer progression, indicating that clinically available antifibrotic/inflammatory agents could improve the treatment response of pancreatic cancer in obese hosts.
Iwase, T. et al. Quality and quantity of visceral fat tissue are associated with insulin resistance and survival outcomes after chemotherapy in patients with breast cancer. Breast Cancer Res. Treat. 179,435–443 (2020).
Guiu, B. et al. Visceral fat area is an independent predictive biomarker of outcome after first-line bevacizumab-based treatment in metastatic colorectal cancer. Gut 59, 341–347 (2010).
Osman, M. A. & Hennessy, B. T. Obesity correlation with metastases development and response to first-line metastatic chemotherapy in breast cancer. Clin. Med. Insights Oncol. 9, 105–112 (2015).
Horowitz, N. S. & Wright, A. A. Impact of obesity on chemotherapy management and outcomes in women with gynecologic malignancies. Gynecol. Oncol. 138, 201–206 (2015).
Sheng, X. et al. Adipocytes sequester and metabolize the chemotherapeutic daunorubicin. Mol. Cancer Res. 15, 1704–1713 (2017). This study reported a mechanism of chemoresistance in which adipocytes metabolize and inactivate daunorubicin and thereby clear it from the tumour microenvironment.
Behan, J. W. et al. Adipocytes impair leukemia treatment in mice. Cancer Res. 69, 7867–7874 (2009).
Engin, A. Adipose tissue hypoxia in obesity and its impact on preadipocytes and macrophages: hypoxia hypothesis. Adv. Exp. Med. Biol. 960, 305–326 (2017).
Su, F., Ahn, S., Saha, A., DiGiovanni, J. & Kolonin, M. G. Adipose stromal cell targeting suppresses prostate cancer epithelial-mesenchymal transition and chemoresistance. Oncogene 38, 1979–1988 (2019).
Herroon, M. K. et al. Prostate tumor cell-derived IL1beta induces an inflammatory phenotype in bone marrow adipocytes and reduces sensitivity to docetaxel via lipolysis-dependent mechanisms. Mol. Cancer Res. 17, 2508–2521 (2019).
Liu, Z. et al. Mature adipocytes in bone marrow protect myeloma cells against chemotherapy through autophagy activation. Oncotarget 6, 34329–34341 (2015).
Lehuede, C. et al. Adipocytes promote breast cancer resistance to chemotherapy, a process amplified by obesity: role of the major vault protein (MVP). Breast Cancer Res. 21, 7 (2019).
Mentoor, I., Engelbrecht, A. M., van Jaarsveld, P. J. & Nell, T. Chemoresistance: intricate interplay between breast tumor cells and adipocytes in the tumor microenvironment. Front. Endocrinol. 9, 758 (2018).
Cao, Y. Adipocyte and lipid metabolism in cancer drug resistance. J. Clin. Invest. 129, 3006–3017 (2019).
Bougaret, L. et al. Adipocyte/breast cancer cell crosstalk in obesity interferes with the anti-proliferative efficacy of tamoxifen. PLoS ONE 13, e0191571 (2018).
Mentoor, I. et al. Decreased efficacy of doxorubicin corresponds with modifications in lipid metabolism markers and fatty acid profiles in breast tumors from obese vs. Lean mice. Front. Oncol. 10, 306 (2020).
Dixon, S. J. et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149, 1060–1072 (2012).
Stockwell, B. R. et al. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell 171, 273–285 (2017).
Zou, Y. et al. A GPX4-dependent cancer cell state underlies the clear-cell morphology and confers sensitivity to ferroptosis. Nat. Commun. 10, 1617 (2019).
Doll, S. et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 13, 91–98 (2017).
Kagan, V. E. et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat. Chem. Biol. 13, 81–90 (2017).
Bersuker, K. et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 575, 688–692 (2019).
Doll, S. et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 575, 693–698 (2019).
Yang, W. S. et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 156, 317–331 (2014).
Yang, W. S. et al. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc. Natl Acad. Sci. USA 113, E4966–E4975 (2016).
Magtanong, L. et al. Exogenous monounsaturated fatty acids promote a ferroptosis-resistant cell state. Cell Chem. Biol. 26, 420–432.e9 (2019). This study demonstrated the influence of MUFA abundance on the sensitivity of cancer cells to ferroptotic cell death.
Lee, J. Y. et al. Polyunsaturated fatty acid biosynthesis pathway determines ferroptosis sensitivity in gastric cancer. Proc. Natl Acad. Sci. USA 117, 32433–32442 (2020).
Dixon, S. J. et al. Human haploid cell genetics reveals roles for lipid metabolism genes in nonapoptotic cell death. ACS Chem. Biol. 10, 1604–1609 (2015).
Nassar, Z. D. et al. Human DECR1 is an androgen-repressed survival factor that regulates PUFA oxidation to protect prostate tumor cells from ferroptosis. eLife 9, e54166 (2020).
Luiken, J. J. et al. Insulin induces the translocation of the fatty acid transporter FAT/CD36 to the plasma membrane. Am. J. Physiol. Endocrinol. Metab. 282, E491–E495 (2002).
Coleman, R. A. & Mashek, D. G. Mammalian triacylglycerol metabolism: synthesis, lipolysis, and signaling. Chem. Rev. 111, 6359–6386 (2011).
Zhang, J. et al. EGFR modulates monounsaturated fatty acid synthesis through phosphorylation of SCD1 in lung cancer. Mol. Cancer 16, 127 (2017).
Gallagher, E. J. & LeRoith, D. Hyperinsulinaemia in cancer. Nat. Rev. Cancer 20, 629–644 (2020).
Acknowledgements
The authors apologise to colleagues whose work they were unable to discuss due to space constraints. Research in the Hoy lab related to the subject of this review is supported by a Robinson Fellowship and funding from the University of Sydney and Sydney Catalyst. L.M.B. is supported by a Principal Cancer Research Fellowship produced with the financial and other support of Cancer Council SA’s Beat Cancer Project on behalf of its donors and the State Government of South Australia through the Department of Health, and the US Department of Defense. The Hoy and Butler labs are supported by a Movember Revolutionary Team Award from the Movember Foundation and the Prostate Cancer Foundation of Australia (MRTA3).
Author information
Authors and Affiliations
Contributions
A.J.H., S.R.N. and L.M.B. researched and discussed the relevant research literature, wrote the manuscript, and drafted the figures.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interest.
Additional information
Peer review information
Nature Reviews Cancer thanks R. Perry, A. Schulze and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Related links
Data by the World Health Organization: http://www.who.int/news-room/fact-sheets/detail/obesity-and-overweight
Glossary
- Tumour lipidome
-
The full lipid complement of the tumour that captures the levels of lipid classes and species.
- Macropinocytosis
-
An endocytic process that involves the engulfment of extracellular content, including soluble molecules, nutrients and antigens, in vesicles known as macropinosomes.
- Type 2 diabetes
-
A progressive metabolic condition in which the body becomes resistant to the normal effects of insulin and/or gradually loses the capacity to produce enough insulin in the pancreas.
- Metabolic syndrome
-
A cluster of conditions, including abdominal obesity, high blood pressure, high blood glucose, high serum triglycerides and low serum high-density lipoprotein, that occur together and increase the risk of heart disease, stroke and type 2 diabetes.
- Lipophagy
-
The autophagic degradation of intracellular lipid droplets.
- Resveratrol
-
A phenolic compound of the stilbene family present in wines and various parts of the grape that exhibits antioxidant and antiproliferative activities.
Rights and permissions
About this article

Cite this article
Hoy, A.J., Nagarajan, S.R. & Butler, L.M. Tumour fatty acid metabolism in the context of therapy resistance and obesity. Nat Rev Cancer 21, 753–766 (2021). https://doi.org/10.1038/s41568-021-00388-4
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41568-021-00388-4
This article is cited by
-
Identification of fatty acids synthesis and metabolism-related gene signature and prediction of prognostic model in hepatocellular carcinoma
Cancer Cell International (2024)
-
Reprogramming of lipid metabolism in the tumor microenvironment: a strategy for tumor immunotherapy
Lipids in Health and Disease (2024)
-
Cancer cell metabolism and antitumour immunity
Nature Reviews Immunology (2024)
-
RBM45 reprograms lipid metabolism promoting hepatocellular carcinoma via Rictor and ACSL1/ACSL4
Oncogene (2024)
-
Hsa_circ_0021205 enhances lipolysis via regulating miR-195-5p/HSL axis and drives malignant progression of glioblastoma
Cell Death Discovery (2024)
