
Abstract
Synthetic biology seeks to redesign biological systems to perform novel functions in a predictable manner. Recent advances in bacterial and mammalian cell engineering include the development of cells that function in biological samples or within the body as minimally invasive diagnostics or theranostics for the real-time regulation of complex diseased states. Ex vivo and in vivo cell-based biosensors and therapeutics have been developed to target a wide range of diseases including cancer, microbiome dysbiosis and autoimmune and metabolic diseases. While probiotic therapies have advanced to clinical trials, chimeric antigen receptor (CAR) T cell therapies have received regulatory approval, exemplifying the clinical potential of cellular therapies. This Review discusses preclinical and clinical applications of bacterial and mammalian sensing and drug delivery platforms as well as the underlying biological designs that could enable new classes of cell diagnostics and therapeutics. Additionally, we describe challenges that must be overcome for more rapid and safer clinical use of engineered systems.
Similar content being viewed by others
Introduction
Current methods for diagnosing and treating disease are hampered by their inability to respond locally and dynamically to disease states. Many diagnostic approaches necessitate invasive biopsies and subsequent pathological analysis1,2,3. Therapeutics face the challenge of administration without real-time knowledge of the internal diseased state. Despite recent advances in targeting different diseases, tissues or cell types of interest4, many biological-based therapeutics act systemically, thereby increasing the risk of off-target effects and potentially reducing patient compliance5.
Synthetic biology, a field that strives to engineer biology to perform user-defined functions, is well poised to meet the need for new classes of diagnostics and therapeutics. Early advances in synthetic biology led to the creation of prokaryotic cells capable of performing complex computations, whereby they produce differential output based on external signals6. Combining synthetic biology with concurrent advances in protein engineering led to the creation of cells that could use synthetic receptors to activate native pathways7. These systems laid the groundwork for building ‘theranostic’ cells, which can serve as both diagnostic tools and therapeutic delivery systems. Theranostic cells are engineered to express sensors that detect the presence of a disease marker (for example, a cell surface receptor that targets a ligand) and signalling machinery that precisely controls a cellular response (for example, therapeutic protein expression or cell killing)8. Relative to small molecules and biologics, which generally act systemically and in an untimed manner, these therapies enable more precise control as they should only activate upon sensing the target biomarker.
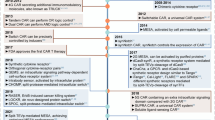
A major milestone in the field of theranostic cell engineering was the 2017 FDA approval of tisagenlecleucel (Kymriah), the first gene therapy to be approved in the USA9. Tisagenlecleucel is a chimeric antigen receptor (CAR) T cell therapy. It consists of immune cells taken from the patient, which are then engineered to express receptors that target B cell precursor acute lymphoblastic leukaemia. Since then, three other CAR T cell therapies — axicabtagene ciloleucel (Yescarta)10, brexucabtagene autoleucel (Tecartus)11 and lisocabtagene maraleucel (Breyanzi)12 — have been approved to treat different types of blood cancer. These therapies all demonstrate the potential of cell-based therapies as a new treatment modality. Building on this success, many academic laboratories and companies are developing cell therapies that are more effective, safe and applicable to a wide variety of diseases.
Our increased understanding of how cells function, combined with technological advances over the past decade, has expedited cell diagnostic and therapeutic development. For instance, research into the gut microbiome has illuminated the integral and complex role that microorganisms play in regulating physiology13, and advances in microbial engineering have enabled the creation of cells that can dynamically regulate this internal microbial ecosystem14. Similarly, genome editing techniques, such as CRISPR–Cas technologies, have led to more precise and potentially safer methods to introduce targeted edits into the human genome, a critical step for mitigating oncogenic adverse effects associated with random genomic integration of other gene editing methods such as viral vectors. More advanced cloning techniques such as Gibson assembly15 and dramatically reduced costs of DNA synthesis have enabled the development of new biological ‘parts’ in both prokaryotic and mammalian systems, significantly reducing the ‘design–build–test’ cycle. High-throughput sequencing has driven rapid and inexpensive organism characterization, and thus faster subsequent engineering16. Advances in robotics and high-throughput screening17 have helped to automate and streamline the construction and evaluation of engineered systems. Finally, advances in in vitro co-culture methods have enabled more robust and rapid characterization of the ways that different cell types interact with each other in a simulated complex environment18,19,20.
In this Review, we address recent advances in the applications of bacterial and mammalian cell diagnostics and therapeutics (Fig. 1). Whereas previous reviews have focused on these areas separately21,22,23,24, here we provide a broad overview across bacterial and mammalian systems and discuss systems that have been engineered for safer and more effective clinical use. We focus mainly on cellular applications but briefly touch on cell-free systems and viral therapies. First, we discuss bacterial diagnostics and therapeutics, focusing on engineering approaches that have enabled cells to function in the body over extended time periods, and give examples of engineered probiotics that have recently advanced to clinical trials. Then, we explore recent advances in mammalian cell engineering, focusing on ways that chimeric receptors can be engineered to create theranostic cells that modulate the immune system. We conclude by offering our outlook on the challenges that engineered cell diagnostics and therapeutics still face and the advances required for engineered cells to become a new pillar of modern diagnostics and therapeutics.

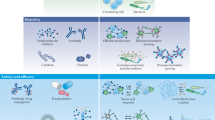
A | Clinical applications of synthetic biology. Microbial sensors can be used as diagnostics ex vivo, reporting on biomarker levels through easily detectable colour changes. Microorganisms can also report on in vivo biomarker levels: ingested bacterial sensors can report on biomarker levels in real time (through incorporation with biocompatible electronic systems) or through subsequent analysis of stool samples. Therapeutic cells can be used to target diseased states. Immune cells can be engineered to specifically target and kill cancer cells or to differentially modulate the immune system, and bacterial cells can be used to control the microbial and metabolic composition of the gut. B | Selection of circuit elements used to engineer cells. Ba | Transcription factors control cell output. A repressor binds to its cognate promoter to block expression of the gene of interest. Conversely, an activator binds to its cognate promoter to turn on expression of the gene of interest. Bb | A toggle switch uses two repressors to stably turn on and off gene expression. Each repressor binds to its cognate promoter, and inducers control the effective state of the cell. Upon addition of Inducer 2, Repressor 2 no longer inhibits expression from its cognate promoter (PRep,2), and Repressor 1 and the gene of interest are transcribed. Even if Inducer 2 is removed, the cell will stay in this state, until Inducer 1 is added to ‘switch’ the cells to an off state by turning on expression of Repressor 2 and, thus, repressing expression of the gene of interest. Bc | Example of an AND-gated genetic circuit. The circuit requires two activators (A and B) to be turned on. The gene of interest is only transcribed when both activators are present. Bd | Example of an OR-gated genetic circuit. The gene of interest will be transcribed when either activator A or activator B is present. To create different logic gates, various transcription factors can be used, and the promoter architecture can be altered by changing the layout of the transcription factor binding sites.
Bacterial diagnostics
The earliest work in synthetic biology used well-studied systems to engineer microorganisms to respond predictably to environmental changes25,26. Since then, a plethora of engineered sensors and more advanced genetic circuits have expanded the scope of compounds that microorganisms can sense and the computations that they can perform, resulting in microorganism-based systems with industrial, health and environmental applications27. More recently, bacterial sensors have been engineered to function in biological samples (for example, serum and urine) and even within the body, enabling them to serve as low-cost, minimally invasive diagnostics and theranostics that produce a therapeutic output upon sensing a diseased state. We differentiate between ex vivo diagnostics, which are used outside the body, and in vivo diagnostics, which are used inside the body.
Ex vivo diagnostics
Current ‘gold-standard’ methods to analyse compounds in the body such as ions, metabolites and peptides require the use of advanced machinery and extensive sample processing28,29. By harnessing microorganisms’ natural sense and respond machinery, biosensors offer a low-cost and potentially more accessible testing alternative. Microorganisms can be engineered to sense target compounds and produce visibly coloured changes in response, serving as a ‘litmus test’ for disease. Such sensors could enable fast and low-cost diagnoses, potentially at the point of care.
Whole-cell diagnostics
Nearly all ex vivo microbial diagnostics produce an easily detectable output — either a fluorescent protein or a visible pigment — upon recognition of a target signal. Multiple groups have engineered Escherichia coli cells to sense and respond to analytes such as micronutrients and sugars30,31,32 by harnessing transcription factors that naturally sense these molecules to control expression of colour-based reporters. For example, the zinc-responsive transcription factors Zur and ZntR can be used to control production of visible pigments, such that cells change to a different colour based on the zinc concentration in serum30,33 (Fig. 2Aa). Similarly, the sugar-responsive promoter PcpxP controls the production of fluorescent proteins and serves as the basis of a test for glycosuria (indicative of diabetes onset)32. To enable clinical use, the sensing systems can be tuned to respond to physiologically relevant concentrations of the target biomarker. For example, a biosensor for zinc deficiency initially responded to serum zinc levels that were far lower than those that are clinically useful. To shift the response to a higher zinc concentration, a transcriptional repressor was placed under control of a zinc-responsive promoter, such that the repressor is made (and thus the colour turned off) only at sufficiently high levels of zinc. The response threshold can be further tuned by modifying the half-life of the repressor: lower levels of the repressor correspond with higher response thresholds33. To enable tests to function in biological samples such as serum and urine, the form factor — that is, the way in which engineered cells are used for sample testing — of the test can be modified. For example, implantation of cells within a hydrogel prevents dilution and loss of signal in urine32. Similarly, using highly concentrated sensor cells prevents bacterial death in serum30.

A | Ex vivo diagnostics. Aa | Bacterial diagnostics for ex vivo diagnosis of zinc deficiency. Production of different pigments (lycopene, violacein and β-carotene) is controlled by zinc-responsive transcription factors. Cells can be lyophilized, rehydrated with serum and, after incubation at body temperature, turn different visible colours. Ab | Cell-free systems for ex vivo diagnosis of target nucleic acid sequences. Reactions consist of bacterial proteins, added reagents and the sensor plasmid. Upon addition of a sample, the reactions turn purple to indicate the presence of the target sequence. B | In vivo diagnostics. Ba | Toggle switch for analysis of gut inflammation. In the absence of an inflammatory stimulus (pink), the circuit is ‘off’ and no β-galactosidase (lacZ) is expressed from PR, a promoter that is repressed by cI. Upon exposure to an inflammatory stimulus (green), the protein Cro is expressed from an inflammatory-responsive promoter (Pinflam.). Cro represses cI expression from the promoter PRM and ‘flips’ the genetic switch into the ‘on’ state, leading to expression of β-galactosidase. A positive feedback loop ensures that the circuit stays in the ‘on’ state, even if the inflammatory stimulus is removed. The engineered bacteria can be orally administered to mice, and stool analysis reveals whether the mice have internal inflammation. Bb | Real-time monitoring of gut activity through bacterial-electronic systems. Bacteria are engineered to sense some internal signal (that is, blood) and then produce luciferase from a haem-responsive promoter (Phaem). These bacteria are embedded in a small electronic device that can be orally delivered to large mammals. The electronic device detects luciferase produced by the sensor bacteria and transmits the signal in real time via radio waves to electronic devices outside the body.
Beyond detecting molecular biomarkers, bacterial sensors can report on the presence of pathogenic bacteria via quorum-sensing systems, which bacteria naturally use to coordinate population-level responses34. For example, E. coli cells engineered to express quorum-sensing proteins from Vibrio cholerae can be used to monitor the presence and proliferation of V. cholerae35. Similarly, yeast GPCR pheromone sensors have been used to report on the presence of pathogenic fungi36. However, despite reported laboratory successes, ex vivo microbial diagnostics have yet to be used clinically, in part because of regulatory challenges associated with using engineered organisms as diagnostics37.
Cell-free diagnostics
Cell-free systems, which consist of a mixture of nucleic acids, metabolites and proteins, have recently emerged as another biological-based sensing platform38. Cell-free systems have the same fundamental transcription and translation machinery as whole cells and can be engineered to detect diverse biomarkers and produce results within minutes of sample addition39 (Fig. 2Ab). These have been used to detect viral biomarkers, such as nucleic acids derived from Ebola39, Zika40 and SARS-CoV-2 (ref.41) as well as small molecules such as zinc (which reflects nutrition levels)42 or quorum-sensing molecules secreted from pathogenic bacteria (which indicate the degree of infection)43.
An advantage of both microbial and cell-free systems for ex vivo analysis is their ability to function in diverse environments and to produce easily detectable outputs. Sensors can be lyophilized and stored at ambient temperatures for long periods of time, and upon reconstitution with a biological sample they can produce visibly coloured reporters30,36,39. This supports the use of these diagnostics in low-resource settings, as they can be shipped to remote regions of the world or easily sold from a pharmacy, then used and interpreted with minimal or no equipment. The safety and logistical considerations to such use will be discussed in the later part of this Review.
In vivo diagnostics
As bacteria naturally live in symbiosis with the human body, they can be harnessed to serve as in vivo diagnostics, reporting on internal biomarkers in a minimally invasive fashion. Current in vivo diagnostics have been used to detect cancer44 and inflammation45 and to monitor gut function and regulation in real time46.
Microbiome diagnostics
The gut microbiome has become an engineering hotspot, as the growing pool of microbiome research has revealed its critical role in maintaining proper immune and digestive function and in drug metabolism47. As bacteria naturally colonize the gastrointestinal tract (termed the gut), they have the potential to serve as stable and long-term reporters of its state. Gut inflammation is a hallmark of diseases such as inflammatory bowel disease and Crohn’s disease, but real-time monitoring of inflammation has been difficult, in part, because of a lack of reliable biomarkers in easily accessible samples48: traditional markers of inflammation such as CRP49 (analysed from blood samples) and calprotectin50 (analysed from stool) are not specific to inflammation of the gut and have high variability. Biomarkers indicative of the reactive oxygen species (ROS) produced in the gut during inflammation51 would be more valuable, but indicators of ROS, such as tetrathionate52, are transient and cannot be detected without invasive procedures. To monitor inflammation in the mouse gut in a non-invasive way, a commensal murine strain of E. coli (NGF1) was engineered to internally sense and record tetrathionate exposure53. This engineering approach connects a tetrathionate sensor to a transcriptional element that then continually produces the reporter β-galactosidase. When stool samples from mice that have ingested engineered bacteria are collected and plated, they show β-galactosidase activity based on mouse gut inflammation (Fig. 2Ba).
Information on the time course of disease progression could be valuable both for better understanding the pathogenesis of gut inflammation and for developing more efficient treatments. To this end, the repressilator, a fundamental synthetic biology tool, was harnessed to create a ‘bacterial clock’ that provides information on cellular activity in the gut. The repressilator functions by using three orthogonal promoter–repressor pairs to control three differentially fluorescent proteins; expression of each protein turns on in a controlled and predictable fashion54. When fed to mice and subsequently analysed, these engineered bacteria can report on cellular growth rate and abnormal conditions (such as gut inflammation) that can disrupt standard transcriptional oscillations55. Similar systems could be used to dynamically modulate the gut microbiome; the resulting theranostics are described in subsequent sections.
Real-time reporting
The interplay between nanotechnology and biotechnology has led to the development of devices that can transmit signals from inside the body, generating real-time health reports. For example, bacteria were engineered to produce luciferase upon detection of clinically relevant biomarkers, such as haemoglobin, thiosulfate and molecules indicative of specific bacterial strains46. These engineered bacteria were embedded in an ingestible electronic capsule that processed the light produced from the bacteria and transmitted the information via radio waves to a phone or computer outside the body (Fig. 2Bb). The capsule can safely migrate through the digestive tract, providing real-time information on the insults encountered through the capsule’s journey. This approach has been successfully used to assess blood in the gastrointestinal tract of a pig but has yet to be tested in humans.
Bacterial therapeutics
Naturally occurring bacteria have been used extensively as probiotics for years, and synthetic biology has enabled the creation of engineered probiotics that can treat specific diseases or conditions. Bacteria can be programmed to release therapeutics upon sensing a target compound. In this manner, bacteria have been used to modulate cancer progression, metabolic disorders and microbiome dysbiosis. Several bacteria-based therapeutic systems have advanced to clinical trials.
Cancer therapeutics
Bacteria for tumour targeting
Bacteria have long been explored as potential cancer treatments: in the 1800s, an injection of streptococcal bacteria shrank a malignant tumour56, and in the 1970s bacillus Calmette–Guérin, an attenuated strain of Mycobacterium bovis, was approved to treat bladder cancer. More recently, Salmonella typhimurium has gained attention because it preferentially colonizes necrotic and hypoxic tumour microenvironments. The oxygen-deprived, immune-privileged environment57 is conducive to anaerobic bacterial growth, which subsequently induces host immune responses58 to target the bacteria and tumours in a cancer antigen-independent fashion59. In the past, S. typhimurium has been involved in numerous phase I trials to treat cancers such as melanoma. However, the treatments were ineffective in humans, and failures were attributed to poor tumour targeting and dose-related toxicity60. More recently, treatments utilizing S. typhimurium in combination with chemotherapy drugs have been investigated, and one that targets pancreatic cancer has advanced to a phase II clinical study (NCT04589234). Additional genetically tractable obligate and facultative anaerobes, including Bifidobacterium, Escherichia and Clostridium, were genetically modified to increase tumour specificity61 by, for example, expressing tumour-targeting peptides or antibodies on the cell surface62.
Bacteria can deliver various anticancer effectors upon sensing a diseased state. In general, these therapies function by placing the gene encoding an effector molecule under the control of a promoter that responds to a tumour-specific signal (Fig. 3a). S. typhimurium was engineered to produce a cytolysin protein HlyE upon sensing hypoxia, which resulted in reduced tumour volume when tested in vivo63. Similarly, E. coli strains have been engineered to produce antitumour proteins upon sensing a specific cell density, low oxygen levels64 or decreasing glucose gradients65. These sensors have been coupled to additional effector molecules such as prodrug-cleaving enzymes66 or short interfering RNAs that suppress tumour growth67. A phase I clinical trial (NCT01562626) is currently testing whether Bifidobacterium longum that expresses the prodrug-converting enzyme cytosine deaminase enhances the efficacy of flucytosine-based treatment of solid tumours; the cytosine deaminase is expected to convert flucytosine into the standard chemotherapy drug 5-fluorouracil at the site of the tumour. Although these therapeutic systems are relatively straightforward, tuning activity is an ongoing challenge, as it is critical that they respond to the appropriate signal threshold and generate appropriate levels of effector molecules.

a | Facultative anaerobic bacteria have been engineered to colonize tumour environments, which may include tumour-specific microbiome communities (represented as blue and green rectangles), by sensing various tumour-specific signals, such as hypoxia or decreasing glucose concentrations. This triggers activation of host immune defences, which facilitate destruction of tumour cells (step 1). Additional circuits have been employed to specifically trigger autolysis when a certain bacterial density is reached (step 2). This allows constitutively expressed effector molecules to be delivered within the tumour microenvironment. b | As a proposed treatment for phenylketonuria disorders caused by defects in phenylalanine-metabolizing enzymes, Escherichia coli has been metabolically engineered to increase assimilation of l-phenylalanine (l-Phe) to form trans-cinnamate, lowering blood l-Phe levels in mouse models79. Expression of the periplasm-associated enzyme L-amino acid deaminase also lowers blood L-Phe levels by converting L-Phe into phenylpyruvate. This provided the basis of a clinical trial that investigated the application of using engineered bacteria to treat phenylketonuria disorders in patients with defects in L-Phe-metabolizing enzymes. c | E. coli Nissle was demonstrated to decrease viability of pathogenic Pseudomonas aeruginosa in a co-culture experiment and to impair pathogenic P. aeruginosa colonization in the mouse gut80. In the presence of the target-derived quorum-sensing molecules, this ‘sense-and-kill’ strain expresses several effector genes placed under the control of a PluxR promoter. Specifically, DspB disrupts the target biofilm. The antibacterial agent pyocin S5 is produced and released into the environment after the E7 lysis proteins lyse the host cell. For biocontainment, the engineered strain requires exogenous d-alanine for growth owing to deletions of the alanine racemase genes alr and dadX. d | E. coli was engineered to deliver a plasmid encoding RNA-guide nuclease to cleave an antibiotic-resistance gene in enterohaemorrhagic E. coli. In this particular study, a type II CRISPR–Cas system was used to cleave the target DNA sequence74. AHL, acyl-homoserine lactone.
Dynamic delivery of anticancer drugs
To effectively and safely treat cancer, bacteria must be able to deliver the anticancer payload in a controlled fashion and to autoregulate their replication rates. Inducible autolysis has been explored as a strategy to both release a drug and maintain a stable bacterial population68,69. This approach harnesses quorum-sensing systems. When the concentration of acyl-homoserine lactone (AHL), a quorum-sensing molecule, is low, the cells divide and produce an anticancer drug. As the bacterial density increases, the concentration of AHL reaches a threshold that activates autolysis, releasing the anticancer protein into the tumour microenvironment (Fig. 3a). Mice injected with these engineered cells showed significant reduction of tumour volume compared with effector alone and cell-only controls68.
In conclusion, the preliminary bacteria-based anticancer treatments discussed here hold promise to specifically target and kill cancerous cells. Although therapies that use more advanced genetic circuits are still in preclinical development, many bacteria-based cancer therapies have advanced through phase I clinical trials (Table 1).
Limitations to bacterial cancer therapies
Bacteria-based treatments that yield effective results in humans, especially strains with complex, engineered gene networks, remain limited. Balancing the fitness of the bacteria, maintaining stability of the introduced gene circuit, attenuating virulence and increasing target specificity in vivo remain grand challenges to developing bacteria-based cancer therapies. Furthermore, bacterial treatment of cancers (such as leukaemia) that do not form solid tumours conducive to bacterial colonization would likely be ineffective and dangerous, as such treatments would require high concentrations of bacteria in the bloodstream. Treatments in these cases would likely rely on employing engineered mammalian cells, such as those discussed in subsequent sections. Finally, in some cases it is known that tumours contain their own natural microbiome that influences cancer progression. These tumour-specific microbial communities are highly variable between patients70,71, and their potentially different effects on therapeutic performance must be taken into consideration during strain selection and engineering70.
Gut therapeutics
Engineered microorganisms can modulate the gut microbiome by sensing biomarker levels, providing potential treatments for gut dysbiosis, inflammation and metabolic diseases72. Most current therapies require ingestion of engineered bacteria, but efforts are being made to modify microorganisms in vivo73,74,75, which could expand the scope of therapeutic applications.
Gut modulation with engineered bacteria
The gut is a prime target for bacterial therapeutics because bacteria naturally colonize the gut and because the gut microbiome plays an important role in modulating diseases such as obesity, diabetes, inflammatory diseases and cancer76. E. coli Nissle 1917 is a popular chassis for therapeutic engineering because it is non-pathogenic and easy to engineer, and has a naturally positive effect on the gut microbiome. Other strains, such as Lactobacillus, Clostridium and Bacteroides, have also shown promise in therapeutic development77.
Metabolic diseases are a prime target for dynamic modulation, as bacteria can be readily engineered to process the accumulated metabolite. However, these efforts have had mixed results. Hyperammonaemia is a disease characterized by excess ammonia accumulation in the blood, resulting from defective enzymes in the urea cycle. An E. coli Nissle strain was engineered to assimilate ammonia and sequester the nitrogen into the amino acid l-arginine78. Administration of this engineered bacteria to mice with hyperammonaemia reduced blood ammonia levels and improved survival. It completed phase I clinical trials (NCT03179878), but was terminated owing to ineffectiveness in lowering blood ammonia in humans. A similar strategy was used to address phenylketonuria, a genetic disease caused by an inability to metabolize l-phenylalanine (l-Phe) (Fig. 3b). E. coli Nissle engineered to convert l-Phe into other metabolites resulted in increased l-Phe metabolism in monkeys79, a strategy that recently passed phase I clinical trials (NCT03516487) and is on track for testing in phase II trials.
Engineered bacteria could also be used to control the composition of the gut microbiome and eliminate pathogenic bacteria. Commensal E. coli Nissle were engineered to target Pseudomonas aeruginosa, a bacterium that can cause serious infection80. The E. coli cells contain a genetic circuit encoding antimicrobial peptides and a biofilm-degrading enzyme. Upon detecting the P. aeruginosa quorum-sensing compound, the engineered cells produce the peptide and enzyme (Fig. 3c). Co-culture of the two strains reduces P. aeruginosa viability and biofilm content. In a mouse infection model, administering the engineered E. coli led to ~70% reduction of P. aeruginosa colonization, providing a viable antimicrobial strategy to combat antibiotic-resistant pathogens.
Gene delivery and gene expression modulation
Gut therapeutics can function by delivering gene circuits to bacteria that are already present in the gut, which can enable precise editing and modification of the gut microbiome. For example, gut bacteria have been engineered to deliver CRISPR-based tools into recipient pathogenic cells to reduce host drug resistance or deactivate virulence genes74,75 (Fig. 3d). This strategy could be used to create novel antibiotics, as it can eliminate pathogenic bacteria or decrease their pathogenic effects. Alternatively, phages can be used to modulate bacterial gene expression in the gut. Non-lytic, temperate phages can deliver catalytically inactive (‘dead’) Cas9 (dCas9) and CRISPR RNAs in situ, which alters gene expression of infected bacteria. This strategy could enable the development of phage therapy to modulate pathogen gene expression by, for example, suppressing the expression of virulence factors81.
Engineered bacteria can also control gene expression in mammalian cells. For example, commensal bacteria were engineered to modify mammalian cells that overexpress cyclooxygenase 2 (COX2), which is characteristic of inflammatory diseases such as Crohn’s disease and ulcerative colitis73. These bacteria invade cells in the colon mucosa and transfer plasmids expressing short interfering RNAs that downregulate expression of COX2. This strategy has been demonstrated to attenuate the inflammatory responses in mouse models73, but the general approach still faces challenges to clinical translation; primarily, there is currently little control over specific entry into mammalian cells, which could cause detrimental off-target effects if bacteria were to centre healthy cells. Additionally, it is difficult to control the rate of circuit delivery into recipient cells, which could lead to non-uniform levels of gene knockdown.
Biocontainment and safety of engineered bacteria
Using engineered bacteria in a safe and contained way is a top priority in therapeutic development and is required to obtain regulatory approval. For example, in both the European Union and the USA, regulatory agencies require extensive demonstration of the bacteria’s safety, genome stability, colonization time and ability to be removed77,82,83. To effectively engineer bacteria to meet these criteria, it is critical to attenuate pathogenicity, control bacterial survival and replication, and minimize the risk of mutation.
Engineering safe and containable strains
Various engineering strategies can be used to make bacteria safe and to ensure that they do not survive outside their intended environments. To ensure safety, virulence genes can be readily removed via standard gene editing approaches84. Further, ‘suicide genes’ can be incorporated so that engineered bacteria can be selectively removed from the population. A quorum-sensing system that prompts self-destruction upon reaching a certain density threshold is one example of an effective suicide gene85, but other strategies can offer more external control. For instance, the use of auxotrophic bacteria allows growth only when an exogenous nutrient (for example, an unnatural amino acid) is supplied86, enabling easy removal of engineered bacteria through withholding of the amino acid.
Maintenance of genetic stability
Another safety concern for engineered bacteria is ensuring that they do not mutate over time. This can happen through mutations in the sensor-encoding or effector-encoding genetic circuit, which can reduce treatment effectiveness or cause unwanted adverse effects. Circuits with minimal burden on engineered cells have been shown to be genetically stable in the gut environment53, and engineering approaches can further stabilize systems. For instance, synthetic communities composed of multiple bacterial strains engineered to sense and replace a mutating subpopulation have increased circuit stability87. Bacterial cells are also subject to horizontal gene transfer, whereby genetic material from other cells or viruses centre and can alter cell function in an unpredictable fashion88. To prevent horizontal gene transfer, bacteria can be genetically re-coded to impair expression of viral proteins or replication of foreign plasmids, minimizing the risk of major mutations89.
Mammalian diagnostics
Compared with bacterial engineering, mammalian synthetic biology faces the added complexity of eukaryotic cell biology and associated gene regulation7, but recent technological advances have improved our ability to control eukaryotic cell output at the transcriptional, translational and post-translational level90,91,92. Although bacterial cells have been the primary chassis for whole-cell diagnostics, a handful of mammalian cell diagnostics have been developed93,94. These show potential for diagnosing conditions with biomarkers that mammalian cells can recognize more easily than bacterial cells, such as inflammatory molecules produced by the immune system.
Ex vivo mammalian diagnostics
One prominent example of an ex vivo mammalian diagnostic is a whole-cell sensor for personalized, precise profiling of allergies95. Allergen profiling is normally done with intrusive skin pricks that expose patients to allergens and induce immune reactions in the skin. As an alternative, HEK293 cells were engineered to robustly respond to histamine, a compound secreted by immune cells that indicates an allergic reaction95. When a blood sample taken from a patient is exposed to an allergen, immune effector cells secrete histamine as usual, and the engineered sensor cells can detect and score the amount of histamine produced. These cells could be the basis of a high-throughput assay for allergic responses, which could replace the traditional skin prick test.
In vivo mammalian diagnostics
In vivo mammalian cell diagnostics are less common, primarily because most mammalian cells engineered to respond to diseases in vivo also serve as therapeutics, which we discuss in detail in the next section. However, one example of a purely diagnostic mammalian cell system is a sensor for hypercalcaemia96. High levels of calcium are a result of hormone-mediated dysregulation of bone resorption and are associated with asymptomatic cancers97. HEK293 cells were engineered to serve as sentinel cells for cancer by continuously monitoring calcium levels. When calcium in the blood surpasses a target threshold, the engineered cells produce melanin, a pigment that is visible through the skin. When encapsulated in alginate beads and injected under the skin of mice, these aptly named HEKTattoo cells function well as calcium reporters. However, they have not been tested in humans, presumably because of the immunogenicity associated with cell implantation if the cells were to leak out of the alginate capsules.
Mammalian therapeutics
The primary focus in the field of mammalian synthetic biology has been the creation of theranostic cells that can simultaneously recognize a diseased state and respond to it in vivo8. These systems harness the innate ability of mammalian cells to respond to a wide variety of stimuli, and thus show promise for real-time regulation of complex diseased states. In this section, we explore recent progress in the field of mammalian theranostic cell therapies. We first discuss engineered T cells as prime examples of theranostic cell engineering, focusing mainly on CAR T cells. We describe new protein engineering approaches to build CAR cells with improved specificity and safety profiles. Finally, we briefly describe ways that other types of theranostic cell are being engineered for diverse applications.
Synthetic TCR T cell therapeutics
T cell receptor (TCR)-modified T cells were first developed as a strategy to harness the potent therapeutic effects of cytotoxic T cells for anticancer therapies. TCRs recognize antigen peptides displayed on major histocompatibility complex (MHC) proteins, and they can be engineered to target specific, researcher-defined antigen peptides98. These antigen peptides can originate from membrane-bound or intracellular proteins, giving researchers a wide range of potential targets. However, TCRs can only recognize certain peptide–MHC complexes, and MHC genes are highly polymorphic in the general population, which limits these therapies to patients expressing a given MHC haplotype. Nevertheless, clinical trials of TCR-engineered T cells have been successful in treating myeloma and melanoma99,100, and many others are currently underway101.
CAR T cells for cancer treatment
Similar to TCR cell therapies, CAR T cells were engineered to sense cancer biomarkers and elicit a downstream cytotoxic response (Fig. 4A). However, unlike TCRs, CARs (previously referred to as T bodies and immunoreceptors) are artificial receptors that combine the antigen-binding specificity of an antibody and the T cell-activating signalling domains of the TCR without MHC restriction102,103. The extracellular domain of a CAR is a single-chain variable fragment (scFv), which confers antigen-binding specificity, and the intracellular domain contains elements that activate T cell signalling104. Upon extracellular target recognition, the intracellular domain activates the T cell response, which produces co-stimulatory signals necessary for T cell function, proliferation and persistence104, leading to killing of cells that have the targeted receptor. Multiple generations of CARs have been created to stimulate the optimal combination of intracellular signalling, T cell activation and T cell persistence105 (Fig. 4A). Currently approved CAR T cell therapies are autologous cell therapies. T cells harvested from the patient are first expanded and engineered ex vivo with a viral vector encoding the CAR protein for long-term expression; the engineered T cells are then infused back into the patient, where they home to tumours expressing an antigen of interest106. Patients with previously non-responsive B cell cancers have experienced complete remission upon treatment with CAR T cells, and they are currently being developed for the treatment of many other cancer types, including solid tumours107.

A | Chimeric antigen receptors (CARs) consist of four domains: the antigen-recognition domain (single-chain variable fragment (scFv)), the spacer domain (hinge), the transmembrane domain and the intracellular signalling domain. There are four generations of CAR designs that differ by the number and type of intracellular signalling domains. First-generation CARs contain one signalling domain for T cell activation, usually the CD3ζ chain of the endogenous T cell receptor (TCR)102. Second-generation and third-generation CARs contain one or two additional co-stimulatory domains, for example, CD28, 4-1BB, OX40 or CD27, which improve cytokine production, proliferation and persistence of CAR T cells105,160. Currently approved CAR T cells are second-generation designs directed towards the B cell antigen CD19. Fourth-generation CAR T cells (also known as T cells redirected for universal cytokine-mediated killing (TRUCKs))161 are designed to deliver a transgenic protein (such as cytokines) upon CAR signalling, which improves persistence relative to earlier generations. B | Recent synthetic biology innovations to improve the safety and efficacy of CAR T cells seek to better regulate CAR T cell activation in vivo compared with older generations. Ba | Suicide genes cause CAR T cell apoptosis in the presence of a small-molecule drug. Bb | Antigen-specific inhibitory CARs (iCARs)114 prevent CAR T cell activity against healthy cells. Bc | A ligand-induced degradation (LID) domain allows the selective degradation of CAR surface molecules in a small-molecule dose-dependent manner. Bd | SynNotch-gated CARs119 require recognition of two antigens in a sequential fashion. After recognition of the first antigen, an intracellular transcription factor is cleaved, translocates to the nucleus and triggers transcription of the second CAR. Be | Split, universal and programmable (SUPRA) CARs125 are designed to be modular and can be controlled through injection of different protein fusions that target different antigens. Bf | AvidCARs122 are lower-affinity, single antigen-binding domain CARs that must bind antigen and dimerize to activate T cell effector function. HSV-TK, herpes simplex virus thymidine kinase; iCasp9, inducible caspase 9.
Despite the clinical success of CAR T cells in treating haematological cancers, there are still major obstacles to safe and efficacious CAR T cell treatment of diverse cancer types. In both haematological and solid tumours, there is a dearth of tumour-specific antigens, and on-target off-tumour killing of healthy cells can occur107. Tumour cells can also downregulate expression of the antigen targeted by CAR T cells, a process known as antigen escape, allowing the tumour to grow again unchecked by the immune response108. Additionally, many patients experience major adverse effects during treatment, such as neurotoxicity and cytokine release syndrome, which results from constitutive CAR activation109. Ongoing efforts to modify CARs aim to overcome the above challenges, and thus improve treatment safety and efficacy. Specifically, research has demonstrated ways that CARs can be designed to enhance tumour specificity and to control the spatio-temporal profile of inflammatory cytokines. Novel modifications to CARs should both improve treatment safety and maximize the on-target immune response.
Improving CAR T cell safety
Unlike traditional small-molecule drugs where the dose is controlled during administration, the activity and proliferation of CAR T cells is largely uncontrollable once the therapy is administered. Thus, much research has focused on engineering control systems that allow in vivo CAR T cell modulation to improve the safety of the therapy. Many of these systems use small molecules to modulate T cell function105,110,111. One approach is the development of cells engineered to have inducible suicide function so that they self-destruct upon addition of a small-molecule regulator112,113. Two suicide genes that have been effectively used are an inducible caspase 9 (iCasp9)112, which initiates downstream apoptotic pathways once activated by a dimerizing small molecule, and herpes simplex virus thymidine kinase (HSV-TK)113, which inhibits DNA synthesis when activated with the small molecule ganciclovir (Fig. 4Ba). A second approach to improving safety is expression of co-receptors that inhibit CAR T cell action against healthy cells114. Antigen-specific inhibitory CARs114 contain an scFv directed to antigens expressed on healthy cells fused to the signalling domains of T cell inhibitory receptors, CTLA4 and PD1 (Fig. 4Bb). When bound to antigens indicative of healthy cells, CAR T cell action is inhibited. However, this approach is limited, as it is challenging to find cell surface markers that are unique to healthy cells. A third approach to controlling function is to modulate levels of functional CAR proteins at the cell surface115,116. A prime example of CAR surface expression control is the development of CAR T cells that can be reversibly paused after administration of a small-molecule ligand117. These T cells express second-generation CARs fused to a ligand-induced degradation (LID) domain (Fig. 4Bc). Binding of the ligand to the LID domain induces the release of a cryptic degron, which results in selective CAR degradation; however, the T cells themselves still remain, unlike inducible suicide gene systems. Thus, the CAR T cells can resume activity when the ligand is removed, enabling precise and reversible control of CAR T cell function in a ligand concentration-dependent manner.
Improving CAR T cell efficacy
The ideal CAR T cell will only be active upon recognition of cancer-specific antigens, but the lack of tumour-specific antigens107 and antigen-independent activation of CARs (termed tonic signalling)118 can lead to unwanted CAR T cell activation. Additionally, tumours can lose expression of antigens targeted by CARs108, rendering the treatment useless. One approach for overcoming antigen specificity challenges is the use of cells that can recognize multiple antigens simultaneously, for example, using SynNotch-gated CARs119. These comprise AND-gated Boolean logic gates, which means they are only activated after binding two tumour antigens (Fig. 4Bd). Binding of one scFv to its targeted tumour antigen triggers the translocation of a synthetic transcription factor to the nucleus, which causes expression of a CAR directed to a second tumour antigen.
Affinity or avidity tuning of chimeric proteins is another form of AND-gating in both receptor and ligand design120 that can diminish the targeting of healthy antigen-presenting cells121. CARs dimerize to effect their signalling in the cell; however, tonic signalling, whereby CARs dimerize without the presence of antigen, is an issue that currently approved CARs face78. One example of avidity tuning is the AvidCAR T cell platform122, which prevents unwanted dimerization and cell activation. This system employs monomeric CARs with low-affinity, single-domain antigen-binding domains (instead of an scFv) that rely on bivalent antigen engagement for dimerization and activation (Fig. 4Be). Reduced affinity of the single-domain antigen-binding domain prevents constitutive CAR dimerization, and CAR signalling and effector function are only active when antigens are co-expressed on the same cell. This platform is thus an easily controllable and combinatorial system that better targets tumour cells co-expressing antigens rather than healthy surrounding tissue. Aside from AND gates, other logic gates, such as OR and NOT gates, have been engineered to recognize different combinations of surface antigens to increase tumour targeting over the targeting of healthy cells105,118,123.
Finally, an alternative approach to combat antigen escape is the development of universal CAR T cells, which can be altered to detect different antigens without having to entirely re-engineer the CAR124,125,126,127. These systems split the antigen-recognition domain and the co-stimulatory domains of conventional CARs into separate components. The first component is an engineered T cell expressing a universal CAR construct consisting of intracellular signalling domains and an extracellular adapter (instead of an scFv directed to the antigen of interest). The second component is a complementary adapter molecule that confers antigen-binding specificity and the modularity of the platform. One example of such an approach is the split, universal and programmable CAR (SUPRA CAR) platform125, which concurrently addresses specificity, safety and ease of design (Fig. 4Bf). The SUPRA CAR system consists of a T cell that only expresses the T cell signalling domains of a CAR linked to an extracellular leucine zipper domain (zipCAR) and an adapter molecule, which is an scFv linked to a complementary leucine zipper (zipFv). The zipFv confers the antigen specificity of the CAR T cell, and different scFv leucine zippers can be easily injected into a patient without reinfusing cells. This modularity allows combinatorial logic and inhibition of CAR function depending on the zipCARs and zipFvs used.
CAR T cells for solid tumours
Although this review focuses on CAR T cells engineered to treat haematological cancers, CAR T cells are also being explored to treat solid tumours107. These approaches have not been as successful in generating remission107, and there are no currently approved CAR T cells that target solid tumours. Beyond the challenges discussed above, these therapies must overcome the immunosuppressive tumour microenvironment and impaired trafficking of CAR T cells into the tumour mass107. Examples of CAR T cell modifications to circumvent these challenges are the expression of inflammatory cytokines, such as IL-12, that improve CAR persistence and activity and the co-expression of chemokine receptors, such as CCR4 (ref.107). Additionally, CARs can be used in combination with oncolytic viruses, which can cause direct tumour cell lysis or can generate an inflammatory immune response128,129, but these systems are beyond the scope of this Review.
Other immunomodulatory CAR cells
The majority of recent T cell engineering has focused on developing novel cancer therapeutics, yet CARs also have the potential to treat autoimmune disorders or to regulate ageing cells. By targeting cells that secrete inflammatory cytokines, CARs can dampen an overactive immune response. Recently, ‘senolytic’ CAR T cells were used to recognize and eliminate senescent cells130. Eliminating senescent cells reduces inflammation and tissue damage and increases the healthspan131,132,133, suggesting that senolytic T cells could be a potential anti-ageing therapeutic.
Additionally, introducing CARs into immune cells beyond T cells leverages the diversity of effector functions found across immune cell types (Box 1). The resulting therapies could overcome some of the current limitations of CAR T cells and also broaden the scope of CAR cell applications to other immune disorders.

Theranostic cells for other applications
Whereas recent mammalian theranostics have centred on therapeutic T cells, mammalian theranostic cells have also been developed for an array of other diseases91,93,134,135. Although these have not yet been developed for clinical use, they have shown great potential in preclinical studies, particularly for the treatment of autoimmune diseases.
Cells to modulate the immune system
To engineer cells with novel functions, synthetic biologists can piece together genetic elements from diverse cell types. A prime example of such engineering is the development of HEK293 cells for the treatment of psoriasis. These cells express anti-inflammatory cytokines upon recognition of psoriasis-specific inflammatory cytokines136 (Fig. 5a). The cytokines TNF and IL-22 are characteristic of psoriasis, but HEK293 cells only endogenously express one-half of the IL-22 receptor (IL-10RB). To endow HEK293 cells with the ability to recognize IL-22, the endogenous TNF-responsive pathway was engineered to control production of the other half of the IL-22 receptor (IL-22RA)136. Then, binding of IL-22 to the expressed receptor triggers the IL-22 signalling cascade. This pathway was rewired to control production of the two anti-inflammatory cytokines IL-4 and IL-10 (Fig. 5a). The resulting cells successfully reduce inflammation upon sensing the psoriatic phenotype, and in mouse models they prevent onset of psoriatic flares and attenuate acute psoriasis136. Thus, the researchers rewired an endogenous pathway (TNF signalling) to express a component (IL-22 receptor) from a different cell type to generate novel responses in the theranostic cell. Using similar engineering tactics, other theranostic cells have been developed to treat diverse diseased states such as diabetes137,138, methicillin-resistant Staphylococcus aureus allergy139 and inflammation140,141.

a | Cytokine converter cells sense the presence of two inflammatory cytokines expressed in psoriasis, TNF and IL-22, and express anti-inflammatory cytokines, IL-4 and IL-10, in response. The endogenous TNF receptor (TNFR)/NF-κB signalling pathway of HEK293 cells is rewired to activate expression of IL-22 receptor-α (IL-22RA) in the presence of TNF. When IL-22RA is expressed and IL-22 is present, IL-22RA and endogenous IL-10RB dimerize and activate the endogenous JAK–STAT pathway. STAT3 signalling is rewired to activate expression of anti-inflammatory cytokines IL-4 and IL-10 that can improve the psoriatic phenotype by calming inflammation. b | Highlighted strategies for engineering native therapeutic pathways in mesenchymal stem cells include expression of receptor intracellular domains to induce a pro-erythropoietic phenotype (step 1), transgenic expression of vascular endothelial growth factor (VEGF) (step 2) and upregulation of the native isoforms of VEGF to promote angiogenesis (step 3).
Engineered stem cells for regenerative medicine
Genetically engineered mesenchymal stem cells are powerful tools for tissue regeneration and gene therapy, and using synthetic biology approaches to control their activity could expand their regenerative applications. For example, engineering cells to overexpress IL-1 receptor antagonist (IL-1RA) can dampen an overactive, inflammatory immune response, and simultaneously expressing vascular endothelial growth factor (VEGF) promotes angiogenesis, both critical components of tissue regeneration142. Whereas delivery of VEGF-encoding DNA produces angiogenic effects143,144,145, stimulation of the native VEGF promoter triggers more robust angiogenesis146. This is thought to be due to post-transcriptional processing leading to multiple splice variants of VEGF, which provide a more comprehensive set of angiogenic stimuli. In line with these results, transgenic expression of survivin, an enhancer of VEGF production, accelerates myocardial healing post infarction147. Because mammalian cells have complex post-transcriptional processing that is difficult to control (that is, alternative RNA splicing)148, stimulating native cytokine expression may not be sufficient to achieve the desired phenotype; alternatively, exogenous expression of cytokines can be more rationally engineered and more tightly controlled (Fig. 5b).
Ectopic transcription factor expression (expression of transcription factors not normally present in a cell type) can also modify cell behaviour, most potently demonstrated by the use of Yamanaka factors for generating induced pluripotent stem cells149. Since then, expression of other transcription factors has expanded the scope of mesenchymal stem cell therapies. For example, overexpression of HIF1α enhances haematopoietic growth factor production, which could increase the success of bone marrow mesenchymal stem cell transplants150. As engineered receptors, transcription factors and cytokines are connected and regulated in more complex pathways, mammalian synthetic biologists will be able to better control cell function and create new types of therapeutic cell with more potent regenerative capabilities.
Conclusions and perspectives
Although engineered cells show great promise for changing treatment paradigms, many challenges must be addressed to ensure their widespread clinical approval and success. A primary challenge for engineered bacterial cells is the current dearth of sensors available for many physiologically relevant compounds. Most bacterial diagnostic and theranostic approaches rely on finding existing proteins that interact with and respond to the molecule of interest. This bioprospecting approach works well when sensors are available for a given target, but it is not widely generalizable. A platform that uses modular sensors such as aptamers or antibody fragments, which can be evolved to specifically bind a target molecule with a user-defined affinity151,152, could greatly expand the scope of bacterial diagnostics and theranostics. Another challenge for bacterial theranostics is the precise control of cellular output. Bacteria can be engineered to produce and secrete small-molecule and protein drugs, but it is difficult to tightly control the amounts of drugs that are produced153; such control is critical, especially for drugs that have small therapeutic windows. Cells that integrate production over time or can sense external levels of the produced drug could be used to effectively titrate drug levels. As in silico design approaches make it easier to build robust and complex circuits154, it is becoming more feasible to build such complex systems.
In mammalian cell engineering, the high cost and logistical challenges associated with CAR T cell treatment, in addition to potentially lethal adverse effects, have limited their use to a ‘last-resort’ option. Manufacturing a single dose of tisagenlecleucel costs upwards of US$40,000, and other costs associated with treatment have driven the cost to $475,000 per person, making it the most expensive cancer therapy to date155. The need to individually engineer each patient’s cells ex vivo is largely responsible for the high cost. A promising alternative, currently in clinical trials, is the use of allogeneic T cells, which could serve as ‘off-the-shelf’ cell therapies, eliminating the need to engineer custom therapies for each patient156,157. However, the use of allogeneic cells increases the risk of graft-versus-host disease104, necessitating further genetic engineering to limit rejection. Another potential way to reduce therapy costs and manufacturing challenges would be to directly inject genetic circuits into patients, rather than into their isolated cells, which is feasible when the tissue can be accessed directly; for example, in the case of retinal tissue, voretigene neparvovec (Luxturna) is directed directly into the eye and was recently approved as a gene therapy against vision loss158. Advances in targeting adeno-associated virus vectors to specific tissues and cell types could help provide similarly specific gene delivery to tissues that are inaccessible by injections159.
As theranostic cells become more potent, it will be critical to monitor their impact on the surrounding tissue. For example, incorporation of CARs into diverse immune cell types will lead to changes in the local microenvironment around the target cell, which are good for the immediate treatment goal but could have potentially detrimental long-term consequences. As these therapies become more developed, it will be critical to assess the reversibility of these changes and then develop engineering approaches to enable better control.
Finally, both bacterial and mammalian cell therapies must overcome negative public opinion, which is fuelled both by the stigma of using genetically modified organisms and by previous failures and breaches of scientific ethics during the use of genetic therapies. Positive branding and public campaigns highlighting the safety and health benefits of diagnostic and therapeutic bacteria could help to alter public opinion on the use of engineered cells. Expanding mammalian cell-based treatments to non-terminal diseases will require extensive demonstration of their safety and benefits over more traditional treatments.
Taken together, recently developed cellular diagnostics and therapeutics have shown that synthetic biology has real potential to transform health-care paradigms. Cell-based therapies have rapidly progressed through clinical trials and regulatory approval, and are thus emerging as an alternative treatment modality to existing small-molecule drugs and protein biologics. The expanding cellular engineering toolbox will lead to more sensitive diagnostics and novel therapeutics for previously intractable diseases.
References
Hornick, J. L. Limited biopsies of soft tissue tumors: the contemporary role of immunohistochemistry and molecular diagnostics. Mod. Pathol. 32, 27–37 (2019).
Litwin, M. S. & Tan, H.-J. The diagnosis and treatment of prostate cancer: a review. JAMA 317, 2532–2542 (2017).
Smetherman, D. H. Screening, imaging, and image-guided biopsy techniques for breast cancer. Surg. Clin. North. Am. 93, 309–327 (2013).
Robbins, P. F. et al. A pilot trial using lymphocytes genetically engineered with an NY-ESO-1-reactive T-cell receptor: long-term follow-up and correlates with response. Clin. Cancer Res. 21, 1019–1027 (2015).
Seebacher, N. A., Stacy, A. E., Porter, G. M. & Merlot, A. M. Clinical development of targeted and immune based anti-cancer therapies. J. Exp. Clin. Cancer Res. 38, 156 (2019).
Cameron, D. E., Bashor, C. J. & Collins, J. J. A brief history of synthetic biology. Nat. Rev. Microbiol. 12, 381–390 (2014).
Way, J. C., Collins, J. J., Keasling, J. D. & Silver, P. A. Integrating biological redesign: where synthetic biology came from and where it needs to go. Cell 157, 151–161 (2014).
Kojima, R., Aubel, D. & Fussenegger, M. Toward a world of theranostic medication: programming biological sentinel systems for therapeutic intervention. Adv. Drug Deliv. Rev. 105, 66–76 (2016).
Braendstrup, P., Levine, B. L. & Ruella, M. The long road to the first FDA-approved gene therapy: chimeric antigen receptor T cells targeting CD19. Cytotherapy 22, 57–69 (2020).
FDA. FDA approves CAR-T cell therapy to treat adults with certain types of large B-cell lymphoma. US Food and Drug Administration https://www.fda.gov/news-events/press-announcements/fda-approves-car-t-cell-therapy-treat-adults-certain-types-large-b-cell-lymphoma (2017).
FDA. FDA approves brexucabtagene autoleucel for relapsed or refractory mantle cell lymphoma. US Food and Drug Administration https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-brexucabtagene-autoleucel-relapsed-or-refractory-mantle-cell-lymphoma (2020).
FDA. FDA approves lisocabtagene maraleucel for relapsed or refractory large B-cell lymphoma. US Food and Drug Administration https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-lisocabtagene-maraleucel-relapsed-or-refractory-large-b-cell-lymphoma (2021).
Fan, Y. & Pedersen, O. Gut microbiota in human metabolic health and disease. Nat. Rev. Microbiol. 19, 55–71 (2020).
Lu, T. K., Mimee, M., Citorik, R. J. & Pepper, K. Engineering the Microbiome for Human Health Applications. The Chemistry of Microbiomes: Proceedings of a Seminar Series (National Academies Press (US), 2017).
Gibson, D. G. et al. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6, 343–345 (2009).
Goodwin, S., McPherson, J. D. & McCombie, W. R. Coming of age: ten years of next-generation sequencing technologies. Nat. Rev. Genet. 17, 333–351 (2016).
Sarnaik, A., Liu, A., Nielsen, D. & Varman, A. M. High-throughput screening for efficient microbial biotechnology. Curr. Opin. Biotechnol. 64, 141–150 (2020).
Dijkstra, K. K. et al. Generation of tumor-reactive T cells by co-culture of peripheral blood lymphocytes and tumor organoids. Cell 174, 1586–1598.e12 (2018).
Harimoto, T. et al. Rapid screening of engineered microbial therapies in a 3D multicellular model. Proc. Natl Acad. Sci. USA 116, 9002–9007 (2019).
Jalili-Firoozinezhad, S. et al. A complex human gut microbiome cultured in an anaerobic intestine-on-a-chip. Nat. Biomed. Eng. 3, 520–531 (2019).
Fischbach, M. A., Bluestone, J. A. & Lim, W. A. Cell-based therapeutics: the next pillar of medicine. Sci. Transl Med. 5, 179ps7 (2013).
Lim, W. A. & June, C. H. The principles of engineering immune cell to treat cancer. Cell 168, 724–740 (2017).
Riglar, D. T. & Silver, P. A. Engineering bacteria for diagnostic and therapeutic applications. Nat. Rev. Microbiol. 16, 214–225 (2018).
Landry, B. P. & Tabor, J. J. Engineering diagnostic and therapeutic gut bacteria. Microbiol. Spectr. 5, 5 (2017).
Gardner, T. S., Cantor, C. R. & Collins, J. J. Construction of a genetic toggle switch in Escherichia coli. Nature 403, 339–342 (2000).
Elowitz, M. B. & Leibler, S. A synthetic oscillatory network of transcriptional regulators. Nature 403, 335–338 (2000).
Khalil, A. S. & Collins, J. J. Synthetic biology: applications come of age. Nat. Rev. Genet. 11, 367–379 (2010).
Jannetto, P. J. & Fitzgerald, R. L. Effective use of mass spectrometry in the clinical laboratory. Clin. Chem. 62, 92–98 (2016).
Anderson, N. L. The clinical plasma proteome: a survey of clinical assays for proteins in plasma and serum. Clin. Chem. 56, 177–185 (2010).
McNerney, M. P., Michel, C. L., Kishore, K., Standeven, J. & Styczynski, M. P. Dynamic and tunable metabolite control for robust minimal-equipment assessment of serum zinc. Nat. Commun. 10, 5514 (2019). This article demonstrates ways that bacterial diagnostics can be tuned to respond to physiologically relevant concentrations and to function in biological samples.
McNerney, M. P., Piorino, F., Michel, C. L. & Styczynski, M. P. Active analyte import improves the dynamic range and sensitivity of a vitamin B12 biosensor. ACS Synth. Biol. 9, 402–411 (2020).
Courbet, A., Endy, D., Renard, E., Molina, F. & Bonnet, J. Detection of pathological biomarkers in human clinical samples via amplifying genetic switches and logic gates. Sci. Transl Med. 7, 289ra83 (2015).
Watstein, D. M. & Styczynski, M. P. Development of a pigment-based whole-cell zinc biosensor for human serum. ACS Synth. Biol. 7, 267–275 (2018).
Mukherjee, S. & Bassler, B. L. Bacterial quorum sensing in complex and dynamically changing environments. Nat. Rev. Microbiol. 17, 371–382 (2019).
Holowko, M. B., Wang, H., Jayaraman, P. & Poh, C. L. Biosensing Vibrio cholerae with genetically engineered Escherichia coli. ACS Synth. Biol. 5, 1275–1283 (2016).
Ostrov, N. et al. A modular yeast biosensor for low-cost point-of-care pathogen detection. Sci. Adv. 3, e1603221 (2017).
Carter, S. R., Rodemeyer, M., Garfinkel, M. S. & Friedman, R. M. Synthetic biology and the U.S. biotechnology regulatory system: challenges and options. US Department of Energy Office of Scientific and Technical Information https://www.osti.gov/biblio/1169537 (2014)
Silverman, A. D., Karim, A. S. & Jewett, M. C. Cell-free gene expression: an expanded repertoire of applications. Nat. Rev. Genet. 21, 151–170 (2020).
Pardee, K. et al. Paper-based synthetic gene networks. Cell 159, 940–954 (2014).
Pardee, K. et al. Rapid, low-cost detection of zika virus using programmable biomolecular components. Cell 165, 1255–1266 (2016).
Joung, J. et al. Point-of-care testing for COVID-19 using SHERLOCK diagnostics. Preprint at medRxiv https://doi.org/10.1101/2020.05.04.20091231 (2020).
McNerney, M. P. et al. Point-of-care biomarker quantification enabled by sample-specific calibration. Sci. Adv. 5, eaax4473 (2019).
Wen, K. Y. et al. A cell-free biosensor for detecting quorum sensing molecules in P. aeruginosa-infected respiratory samples. ACS Synth. Biol. 6, 2293–2301 (2017).
Danino, T. et al. Programmable probiotics for detection of cancer in urine. Sci. Transl Med. 7, 289ra84 (2015).
Daeffler, K. N.-M. et al. Engineering bacterial thiosulfate and tetrathionate sensors for detecting gut inflammation. Mol. Syst. Biol. 13, 923 (2017).
Mimee, M. et al. An ingestible bacterial-electronic system to monitor gastrointestinal health. Science 360, 915–918 (2018). This article demonstrates that bacterial sensors interface with an electronic capsule to provide real-time reporting of the gut composition.
Cho, I. & Blaser, M. J. The human microbiome: at the interface of health and disease. Nat. Rev. Genet. 13, 260–270 (2012).
Sands, B. E. Biomarkers of inflammation in inflammatory bowel disease. Gastroenterology 149, 1275–1285.e2 (2015).
Vermeire, S., Van Assche, G. & Rutgeerts, P. C-reactive protein as a marker for inflammatory bowel disease. Inflamm. Bowel Dis. 10, 661–665 (2004).
Lehmann, F. S., Burri, E. & Beglinger, C. The role and utility of faecal markers in inflammatory bowel disease. Ther. Adv. Gastroenterol. 8, 23–36 (2015).
Tian, T., Wang, Z. & Zhang, J. Pathomechanisms of oxidative stress in inflammatory bowel disease and potential antioxidant therapies. Oxid. Med. Cell. Longev. 2017, 4535194 (2017).
Winter, S. E. et al. Gut inflammation provides a respiratory electron acceptor for Salmonella. Nature 467, 426–429 (2010).
Riglar, D. T. et al. Engineered bacteria can function in the mammalian gut long-term as live diagnostics of inflammation. Nat. Biotechnol. 35, 653–658 (2017). This article demonstrates that bacterial cells can respond to inflammation and ‘remember’ exposure to inflammation for long periods of time, enabling diagnosis of gut inflammation from plating of stool samples from mice fed the engineered bacteria.
Potvin-Trottier, L., Lord, N. D., Vinnicombe, G. & Paulsson, J. Synchronous long-term oscillations in a synthetic gene circuit. Nature 538, 514–517 (2016).
Riglar, D. T. et al. Bacterial variability in the mammalian gut captured by a single-cell synthetic oscillator. Nat. Commun. 10, 4665 (2019).
McCarthy, E. F. The toxins of William B. Coley and the treatment of bone and soft-tissue sarcomas. Iowa Orthop. J. 26, 154–158 (2006).
Forbes, N. S. Engineering the perfect (bacterial) cancer therapy. Nat. Rev. Cancer 10, 785–794 (2010).
Gniadek, T. J. et al. A phase I, dose escalation, single dose trial of oral attenuated salmonella typhimurium containing human IL-2 in patients with metastatic gastrointestinal cancers. J. Immunother. 43, 217–221 (2020). This report provides a current example of a genetically modified Salmonella strain expressing human IL-2 that is being tested for treating metastic cancer.
Leventhal, D. S. et al. Immunotherapy with engineered bacteria by targeting the STING pathway for anti-tumor immunity. Nat. Commun. 11, 2739 (2020).
Toso, J. F. et al. Phase I study of the intravenous administration of attenuated Salmonella typhimurium to patients with metastatic melanoma. J. Clin. Oncol. 20, 142–152 (2002).
Chien, T., Doshi, A. & Danino, T. Advances in bacterial cancer therapies using synthetic biology. Curr. Opin. Syst. Biol. 5, 1–8 (2017).
Piñero-Lambea, C. et al. Programming controlled adhesion of E. coli to target surfaces, cells, and tumors with synthetic adhesins. ACS Synth. Biol. 4, 463–473 (2015).
Ryan, R. M. et al. Bacterial delivery of a novel cytolysin to hypoxic areas of solid tumors. Gene Ther. 16, 329–339 (2009).
Anderson, J. C., Clarke, E. J., Arkin, A. P. & Voigt, C. A. Environmentally controlled invasion of cancer cells by engineered bacteria. J. Mol. Biol. 355, 619–627 (2006).
Panteli, J. T. & Forbes, N. S. Engineered bacteria detect spatial profiles in glucose concentration within solid tumor cell masses. Biotechnol. Bioeng. 113, 2474–2484 (2016).
Zheng, J. H. et al. Two-step enhanced cancer immunotherapy with engineered Salmonella typhimurium secreting heterologous flagellin. Sci. Transl Med. 9, eaak9537 (2017).
Duong, M. T.-Q., Qin, Y., You, S.-H. & Min, J.-J. Bacteria-cancer interactions: bacteria-based cancer therapy. Exp. Mol. Med. 51, 1–15 (2019).
Din, M. O. et al. Synchronized cycles of bacterial lysis for in vivo delivery. Nature 536, 81–85 (2016). This study uses an elegant synthetic circuit to affect population-level behaviour such that an anticancer payload is delivered to tumours upon bacterial accumulation in the tumour microenvironment.
Chowdhury, S. et al. Programmable bacteria induce durable tumor regression and systemic antitumor immunity. Nat. Med. 25, 1057–1063 (2019).
Nejman, D. et al. The human tumor microbiome is composed of tumor type-specific intracellular bacteria. Science 368, 973–980 (2020).
Riquelme, E. et al. Tumor microbiome diversity and composition influence pancreatic cancer outcomes. Cell 178, 795–806.e12 (2019).
Ozdemir, T., Fedorec, A. J. H., Danino, T. & Barnes, C. P. Synthetic biology and engineered live biotherapeutics: toward increasing system complexity. Cell Syst. 7, 5–16 (2018).
Spisni, E. et al. Cyclooxygenase-2 silencing for the treatment of colitis: a combined in vivo strategy based on RNA interference and engineered Escherichia coli. Mol. Ther. 23, 278–289 (2015).
Citorik, R. J., Mimee, M. & Lu, T. K. Sequence-specific antimicrobials using efficiently delivered RNA-guided nucleases. Nat. Biotechnol. 32, 1141–1145 (2014).
Bikard, D. et al. Exploiting CRISPR–Cas nucleases to produce sequence-specific antimicrobials. Nat. Biotechnol. 32, 1146–1150 (2014).
Flint, H. J., Scott, K. P., Louis, P. & Duncan, S. H. The role of the gut microbiota in nutrition and health. Nat. Rev. Gastroenterol. Hepatol. 9, 577–589 (2012).
Charbonneau, M. R., Isabella, V. M., Li, N. & Kurtz, C. B. Developing a new class of engineered live bacterial therapeutics to treat human diseases. Nat. Commun. 11, 1738 (2020).
Kurtz, C. B. et al. An engineered E. coli Nissle improves hyperammonemia and survival in mice and shows dose-dependent exposure in healthy humans. Sci. Transl Med. 11, eaau7975 (2019).
Isabella, V. M. et al. Development of a synthetic live bacterial therapeutic for the human metabolic disease phenylketonuria. Nat. Biotechnol. 36, 857–864 (2018). This study develops a bacterial therapeutic to treat phenylketonuria, which is the basis of a treatment currently in phase II clinical trials.
Hwang, I. Y. et al. Engineered probiotic Escherichia coli can eliminate and prevent Pseudomonas aeruginosa gut infection in animal models. Nat. Commun. 8, 15028 (2017).
Hsu, B. B., Way, J. C. & Silver, P. A. Stable neutralization of a virulence factor in bacteria using temperate phage in the mammalian gut. mSystems 5, 1 (2020).
Center for Biologics Evaluation and Research. Early clinical trials with live biotherapeutic products: chemistry, manufacturing, and control information; guidance for industry. US Food and Drug Administration https://www.fda.gov/regulatory-information/search-fda-guidance-documents/early-clinical-trials-live-biotherapeutic-products-chemistry-manufacturing-and-control-information (2018).
The European Pharmacopoeia Commission. Live Biotherapeutic Products for Human Use Vol. 9 (Council of Europe, 2019).
Dang, L. H., Bettegowda, C., Huso, D. L., Kinzler, K. W. & Vogelstein, B. Combination bacteriolytic therapy for the treatment of experimental tumors. Proc. Natl Acad. Sci. USA 98, 15155–15160 (2001).
Miano, A., Liao, M. J. & Hasty, J. Inducible cell-to-cell signaling for tunable dynamics in microbial communities. Nat. Commun. 11, 1193 (2020).
Mandell, D. J. et al. Biocontainment of genetically modified organisms by synthetic protein design. Nature 518, 55–60 (2015).
Liao, M. J., Din, M. O., Tsimring, L. & Hasty, J. Rock-paper-scissors: engineered population dynamics increase genetic stability. Science 365, 1045–1049 (2019).
Soucy, S. M., Huang, J. & Gogarten, J. P. Horizontal gene transfer: building the web of life. Nat. Rev. Genet. 16, 472–482 (2015).
Ma, N. J. & Isaacs, F. J. Genomic recoding broadly obstructs the propagation of horizontally transferred genetic elements. Cell Syst. 3, 199–207 (2016).
Lienert, F., Lohmueller, J. J., Garg, A. & Silver, P. A. Synthetic biology in mammalian cells: next generation research tools and therapeutics. Nat. Rev. Mol. Cell Biol. 15, 95–107 (2014).
Kitada, T., DiAndreth, B., Teague, B. & Weiss, R. Programming gene and engineered-cell therapies with synthetic biology. Science 359, eaad1067 (2018).
Xie, M. & Fussenegger, M. Designing cell function: assembly of synthetic gene circuits for cell biology applications. Nat. Rev. Mol. Cell Biol. 19, 507–525 (2018).
Sedlmayer, F., Aubel, D. & Fussenegger, M. Synthetic gene circuits for the detection, elimination and prevention of disease. Nat. Biomed. Eng. 2, 399–415 (2018).
Hicks, M., Bachmann, T. T. & Wang, B. Synthetic biology enables programmable cell-based biosensors. ChemPhysChem 21, 132–144 (2020).
Ausländer, D. et al. A designer cell-based histamine-specific human allergy profiler. Nat. Commun. 5, 4408 (2014).
Tastanova, A. et al. Synthetic biology-based cellular biomedical tattoo for detection of hypercalcemia associated with cancer. Sci. Transl Med. 10, eaap8562 (2018).
Goldner, W. Cancer-related hypercalcemia. J. Oncol. Pract. 12, 426–432 (2016).
Clay, T. M. et al. Efficient transfer of a tumor antigen-reactive TCR to human peripheral blood lymphocytes confers anti-tumor reactivity. J. Immunol. 163, 507 (1999).
Rapoport, A. P. et al. NY-ESO-1–specific TCR-engineered T cells mediate sustained antigen-specific antitumor effects in myeloma. Nat. Med. 21, 914–921 (2015).
Morgan, R. A. et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science 314, 126 (2006).
Ping, Y., Liu, C. & Zhang, Y. T-cell receptor-engineered T cells for cancer treatment: current status and future directions. Protein Cell 9, 254–266 (2018).
Eshhar, Z., Waks, T., Gross, G. & Schindler, D. G. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the γ or ζ subunits of the immunoglobulin and T-cell receptors. Proc. Natl Acad. Sci. USA 90, 720–724 (1993).
Gross, G., Gorochov, G., Waks, T. & Eshhar, Z. Generation of effector T cells expressing chimeric T cell receptor with antibody type-specificity. Transplant. Proc. 21, 127–130 (1989).
Sadelain, M., Rivière, I. & Riddell, S. Therapeutic T cell engineering. Nature 545, 423–431 (2017).
Hong, M., Clubb, J. D. & Chen, Y. Y. Engineering CAR-T cells for next-generation cancer therapy. Cancer Cell 38, 473–488 (2020).
Levine, B. L., Miskin, J., Wonnacott, K. & Keir, C. Global manufacturing of CAR T cell therapy. Mol. Ther. Methods Clin. Dev. 4, 92–101 (2017).
Majzner, R. G. & Mackall, C. L. Clinical lessons learned from the first leg of the CAR T cell journey. Nat. Med. 25, 1341–1355 (2019).
Shah, N. N. & Fry, T. J. Mechanisms of resistance to CAR T cell therapy. Nat. Rev. Clin. Oncol. 16, 372–385 (2019).
Perales, M.-A., Kebriaei, P., Kean, L. S. & Sadelain, M. Building a safer and faster CAR: seatbelts, airbags, and CRISPR. Biol. Blood Marrow Transpl. 24, 27–31 (2018).
Caliendo, F., Dukhinova, M. & Siciliano, V. Engineered cell-based therapeutics: synthetic biology meets immunology. Front. Bioeng. Biotechnol. 7, 43 (2019).
Esensten, J. H., Bluestone, J. A. & Lim, W. A. Engineering therapeutic T cells: from synthetic biology to clinical trials. Annu. Rev. Pathol. Mech. Dis. 12, 305–330 (2017).
Diaconu, I. et al. Inducible caspase-9 selectively modulates the toxicities of CD19-specific chimeric antigen receptor-modified T cells. Mol. Ther. 25, 580–592 (2017).
Casucci, M. et al. Extracellular NGFR spacers allow efficient tracking and enrichment of fully functional CAR-T cells co-expressing a suicide gene. Front. Immunol. 9, 507 (2018).
Fedorov, V. D., Themeli, M. & Sadelain, M. PD-1- and CTLA-4-based inhibitory chimeric antigen receptors (iCARs) divert off-target immunotherapy responses. Sci. Transl Med. 5, 215ra172 (2013).
Chakravarti, D., Caraballo, L. D., Weinberg, B. H. & Wong, W. W. Inducible gene switches with memory in human T cells for cellular immunotherapy. ACS Synth. Biol. 8, 1744–1754 (2019).
Wu, C.-Y., Roybal, K. T., Puchner, E. M., Onuffer, J. & Lim, W. A. Remote control of therapeutic T cells through a small molecule–gated chimeric receptor. Science 350, aab4077 (2015).
Richman, S. A. et al. Ligand-induced degradation of a CAR permits reversible remote control of CAR T cell activity in vitro and in vivo. Mol. Ther. J. Am. Soc. Gene Ther. 28, 1600–1613 (2020). This article demonstrates an approach to improving safety of CAR T cells, through engineering a CAR that contains a ligand-induced degradation domain so that it can be reversibly downregulated.
Guedan, S., Calderon, H., Posey, A. D. & Maus, M. V. Engineering and design of chimeric antigen receptors. Mol. Ther. Methods Clin. Dev. 12, 145–156 (2019).
Roybal, K. T. et al. Precision tumor recognition by T cells with combinatorial antigen-sensing circuits. Cell 164, 770–779 (2016).
Lee, J. et al. Rational design of a bifunctional AND-gate ligand to modulate cell–cell interactions. ACS Synth. Biol. 9, 191–197 (2020).
Stoiber, S. et al. Limitations in the design of chimeric antigen receptors for cancer therapy. Cells 8, 472 (2019).
Salzer, B. et al. Engineering AvidCARs for combinatorial antigen recognition and reversible control of CAR function. Nat. Commun. 11, 4166 (2020). This study shows a way to enhance CAR T cell specificity, through development of a low-affinity CAR that requires bivalent antigen binding and thus prevents off-tumour CAR activation.
Cho, J. H. et al. Engineering advanced logic and distributed computing in human CAR immune cells. Nat. Commun. 12, 792 (2021).
Cartellieri, M. et al. Switching CAR T cells on and off: a novel modular platform for retargeting of T cells to AML blasts. Blood Cancer J. 6, e458 (2016).
Cho, J. H., Collins, J. J. & Wong, W. W. Universal chimeric antigen receptors for multiplexed and logical control of T cell responses. Cell 173, 1426–1438.e11 (2018). This article presents a modular CAR system, composed of a universal receptor and adapter scFvs, that enables targeting of different antigens without having to entirely re-engineer the CAR.
Urbanska, K. et al. A universal strategy for adoptive immunotherapy of cancer through use of a novel T-cell antigen receptor. Cancer Res. 72, 1844–1852 (2012).
Loff, S. et al. Rapidly switchable universal CAR-T cells for treatment of CD123-positive leukemia. Mol. Ther. Oncolytics 17, 408–420 (2020).
Rosewell Shaw, A. & Suzuki, M. Oncolytic viruses partner with T-cell therapy for solid tumor treatment. Front. Immunol. 9, 2103 (2018).
Ajina, A. & Maher, J. Prospects for combined use of oncolytic viruses and CAR T-cells. J. Immunother. Cancer 5, 90 (2017).
Amor, C. et al. Senolytic CAR T cells reverse senescence-associated pathologies. Nature 583, 127–132 (2020).
Kirkland, J. L. & Tchkonia, T. Cellular senescence: a translational perspective. EBioMedicine 21, 21–28 (2017).
Baker, D. J. et al. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 479, 232–236 (2011).
Franceschi, C., Garagnani, P., Vitale, G., Capri, M. & Salvioli, S. Inflammaging and ‘Garb-aging’. Trends Endocrinol. Metab. 28, 199–212 (2017).
Higashikuni, Y., Chen, W. C. & Lu, T. K. Advancing therapeutic applications of synthetic gene circuits. Curr. Opin. Biotechnol. 47, 133–141 (2017).
Ho, P. & Chen, Y. Y. Mammalian synthetic biology in the age of genome editing and personalized medicine. Curr. Opin. Chem. Biol. 40, 57–64 (2017).
Schukur, L., Geering, B., Hamri, G. C.-E. & Fussenegger, M. Implantable synthetic cytokine converter cells with AND-gate logic treat experimental psoriasis. Sci. Transl Med. 7, 318ra201 (2015). This article develops theranostic cells to sense pro-inflammatory cytokines associated with psoriasis and to respond by expressing therapeutic anti-inflammatory cytokines.
Ye, H. et al. Self-adjusting synthetic gene circuit for correcting insulin resistance. Nat. Biomed. Eng. 1, 005 (2017).
Liu, Y. et al. Immunomimetic designer cells protect mice from MRSA infection. Cell 174, 259–270.e11 (2018).
Chassin, H. et al. Sensing and responding to allergic response cytokines through a genetically encoded circuit. Nat. Commun. 8, 1101 (2017).
Qudrat, A., Mosabbir, A. A. & Truong, K. Engineered proteins program mammalian cells to target inflammatory disease sites. Cell Chem. Biol. 24, 703–711.e2 (2017).
Smole, A., Lainšcˇek, D., Bezeljak, U., Horvat, S. & Jerala, R. A synthetic mammalian therapeutic gene circuit for sensing and suppressing inflammation. Mol. Ther. 25, 102–119 (2017).
Nowakowski, A., Walczak, P., Janowski, M. & Lukomska, B. Genetic engineering of mesenchymal stem cells for regenerative medicine. Stem Cell Dev. 24, 2219–2242 (2015).
Moon, H.-H. et al. MSC-based VEGF gene therapy in rat myocardial infarction model using facial amphipathic bile acid-conjugated polyethyleneimine. Biomaterials 35, 1744–1754 (2014).
Lai, T. et al. Over-expression of VEGF in marrow stromal cells promotes angiogenesis in rats with cerebral infarction via the synergistic effects of VEGF and Ang-2. J. Huazhong Univ. Sci. Technol. Med. Sci. 32, 724–731 (2012).
Serra, J. et al. Engineering of human mesenchymal stem/stromal cells with vascular endothelial growth factor-encoding minicircles for angiogenic ex vivo gene therapy. Hum. Gene Ther. 30, 316–329 (2019).
Rebar, E. J. et al. Induction of angiogenesis in a mouse model using engineered transcription factors. Nat. Med. 8, 1427–1432 (2002).
Fan, L. et al. Transplantation with survivin-engineered mesenchymal stem cells results in better prognosis in a rat model of myocardial infarction. Eur. J. Heart Fail. 11, 1023–1030 (2009).
Sakharkar, M. K., Chow, V. T. K. & Kangueane, P. Distributions of exons and introns in the human genome. Silico Biol. 4, 387–393 (2004).
Takahashi, K. & Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663–676 (2006).
Kiani, A. A. et al. Over expression of HIF-1α in human mesenchymal stem cells increases their supportive functions for hematopoietic stem cells in an experimental co-culture model. Hematol. Amst. Neth. 19, 85–98 (2014).
Ku, T.-H. et al. Nucleic acid aptamers: an emerging tool for biotechnology and biomedical sensing. Sensors 15, 16281–16313 (2015).
McMahon, C. et al. Yeast surface display platform for rapid discovery of conformationally selective nanobodies. Nat. Struct. Mol. Biol. 25, 289–296 (2018).
McNerney, M. P., Watstein, D. M. & Styczynski, M. P. Precision metabolic engineering: the design of responsive, selective, and controllable metabolic systems. Metab. Eng. 31, 123–131 (2015).
Nielsen, A. A. K. et al. Genetic circuit design automation. Science 352, aac7341 (2016).
Lin, J. K. et al. Cost effectiveness of chimeric antigen receptor T-cell therapy in relapsed or refractory pediatric B-cell acute lymphoblastic leukemia. J. Clin. Oncol. 36, 3192–3202 (2018).
Qasim, W. et al. Molecular remission of infant B-ALL after infusion of universal TALEN gene-edited CAR T cells. Sci. Transl Med. 9, eaaj2013 (2017).
Benjamin, R. et al. Genome-edited, donor-derived allogeneic anti-CD19 chimeric antigen receptor T cells in paediatric and adult B-cell acute lymphoblastic leukaemia: results of two phase 1 studies. Lancet 396, 1885–1894 (2020).
FDA. FDA approves novel gene therapy to treat patients with a rare form of inherited vision loss. US Food and Drug Administration https://www.fda.gov/news-events/press-announcements/fda-approves-novel-gene-therapy-treat-patients-rare-form-inherited-vision-loss (2020).
Wang, D., Tai, P. W. L. & Gao, G. Adeno-associated virus vector as a platform for gene therapy delivery. Nat. Rev. Drug Discov. 18, 358–378 (2019).
Sadelain, M., Brentjens, R. & Rivière, I. The promise and potential pitfalls of chimeric antigen receptors. Curr. Opin. Immunol. 21, 215–223 (2009).
Chmielewski, M., Kopecky, C., Hombach, A. A. & Abken, H. IL-12 release by engineered T cells expressing chimeric antigen receptors can effectively muster an antigen-independent macrophage response on tumor cells that have shut down tumor antigen expression. Cancer Res. 71, 5697–5706 (2011).
Vivier, E., Tomasello, E., Baratin, M., Walzer, T. & Ugolini, S. Functions of natural killer cells. Nat. Immunol. 9, 503–510 (2008).
Liu, E. L. et al. Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors. N. Engl. J. Med. 382, 545–553 (2020).
Xie, G. et al. CAR-NK cells: a promising cellular immunotherapy for cancer. EBioMedicine 59, 102975 (2020).
Yang, Y. et al. Phase I study of random healthy donor-derived allogeneic natural killer cell therapy in patients with malignant lymphoma or advanced solid tumors. Cancer Immunol. Res. 4, 215–224 (2016).
Lupo, K. B. & Matosevic, S. Natural killer cells as allogeneic effectors in adoptive cancer immunotherapy. Cancers 11, 769 (2019).
Li, Y., Hermanson, D. L., Moriarity, B. S. & Kaufman, D. S. Human iPSC-derived natural killer cells engineered with chimeric antigen receptors enhance anti-tumor activity. Cell Stem Cell 23, 181–192.e5 (2018).
Lim, R. M., Rong, L., Zhen, A. J. & Xie, J. M. A universal CAR-NK cell targeting various epitopes of HIV-1 gp160. ACS Chem. Biol. 15, 2299–2310 (2020).
Klichinsky, M. et al. Human chimeric antigen receptor macrophages for cancer immunotherapy. Nat. Biotechnol. 38, 947–953 (2020). This work describes the therapeutic effect of CAR macrophages, and demonstrates the ability of CAR-engineered cells to target solid tumours.
Murphy, K. & Weaver, C. in Janeway’s Immunobiology 9th edn 355–365 (Garland Science, 2017).
Rana, J. & Biswas, M. Regulatory T cell therapy: current and future design perspectives. Cell. Immunol. 356, 104193 (2020).
Arce-Sillas, A. et al. Regulatory T cells: molecular actions on effector cells in immune regulation. J. Immunol. Res. 2016, 1720827 (2016).
Grossman, W. J. et al. Human T regulatory cells can use the perforin pathway to cause autologous target cell death. Immunity 21, 589–601 (2004).
Brusko, T. M. et al. Human antigen-specific regulatory T cells generated by T cell receptor gene transfer. PLoS ONE 5, e11726 (2010).
Mohseni, Y. R. et al. The future of regulatory T cell therapy: promises and challenges of implementing CAR technology. Front. Immunol. 11, 13 (2020).
MacDonald, K. G. et al. Alloantigen-specific regulatory T cells generated with a chimeric antigen receptor. J. Clin. Invest. 126, 1413–1424 (2016).
Acknowledgements
The authors acknowledge members of the Silver laboratory for helpful conversations and acknowledge support from Harvard Medical School’s Dean’s Initiative Award and DARPA (140D0420C0057).
Author information
Authors and Affiliations
Contributions
M.P.M., K.E.D., T.L.N. and T.Z.C. researched the literature and wrote the article. All authors contributed to discussions of the content and reviewed and/or edited the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information
Nature Reviews Genetics thanks the anonymous reviewers for their contribution to the peer review of this work.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Glossary
- Off-target effects
-
Unintended therapeutic consequences that occur when a drug binds to molecules in the body that are not the intended target of the drug.
- Synthetic biology
-
A multidisciplinary field of biological research that seeks to design and engineer biology akin to other engineering disciplines. Goals of the field are the development of molecules, cells and even organisms with novel functions.
- Chimeric antigen receptor
-
(CAR). A synthetic receptor that combines the specific, extracellular antigen-binding ability of an antibody and the T cell-activating abilities of the T cell receptor (intracellular stimulatory domains) to redirect immune cell action towards a disease antigen of interest.
- Cell-free systems
-
Mixtures of proteins, nucleic acids, salts and metabolites that resemble the cytoplasm of cells and can be used to enact genetic circuits.
- Probiotics
-
Beneficial microorganisms that are used to promote health. Probiotics can be naturally occurring microorganisms or microorganisms that have been engineered to sense and respond to a specific condition.
- Genetic circuits
-
DNA systems in which the presence of different combinations of signals leads to differential gene expression. Circuits usually consist of an input (that is, surface receptor binding to a ligand) and an output (that is, expression of therapeutics) that is enacted only after the input condition is satisfied.
- Transcription factors
-
Proteins that control the expression of a gene, generally either by activating or repressing its expression.
- Quorum-sensing systems
-
Cell to cell communication systems, prominent in prokaryotes, that use extracellular small molecules as signalling cues.
- Dysbiosis
-
An imbalance of the gut microbiome community. Disruption to gut bacterial homeostasis has been associated with human diseases such as inflammatory bowel diseases and irritable bowel syndrome.
- Chassis
-
The organism or cell line that houses a genetic circuit and is engineered to perform specific tasks.
- Biofilm
-
A collection of microorganisms associated with a primarily polysaccharide-based matrix. Biofilms are recalcitrant to antibiotic treatments.
- Alginate beads
-
Beads used to encapsulate cells to allow their implantation into the body while keeping them isolated from the host immune system. In addition to preventing an immune response, alginate encapsulation forms a semipermeable barrier to allow the diffusion of nutrients and gases.
- T cell receptor
-
(TCR). The antigen-binding complex natively expressed on the surface of T cells. Binding of antigen to the TCR is necessary for T cell activation.
- Antigen peptides
-
Short amino acid sequences that can be recognized by T cell receptors (TCRs) or antibodies expressed by cells of the adaptive immune system. Binding of an antigen to cell receptors or antibodies can trigger an immune response.
- Single-chain variable fragment
-
(scFV). A fusion protein consisting of the variable domains of the heavy and light chains of an antibody connected by a small linker. scFvs confer antigen-binding specificity to the chimeric antigen receptor (CAR).
- Autologous
-
A term to denote cells or tissue derived from a patient for the treatment of that same patient.
- Cytokine release syndrome
-
A potentially deadly adverse effect of chimeric antigen receptor (CAR) T cell therapy, resulting from the mass release of pro-inflammatory cytokine expression from activated T cells.
- Cytokines
-
Secreted proteins that bind to receptors on other cells that induce a change in that target cell, generally categorized as pro-inflammatory or anti-inflammatory.
- Boolean logic gates
-
Systems of binary, on/off signals used to generate complex behaviours in a system (for example, an AND-gated circuit necessitates the presence of two inputs before expression of the output).
- Allogeneic
-
A term to describe cells or tissue derived from one person used to treat a genetically different person (allotransplantation).
Rights and permissions
About this article

Cite this article
McNerney, M.P., Doiron, K.E., Ng, T.L. et al. Theranostic cells: emerging clinical applications of synthetic biology. Nat Rev Genet 22, 730–746 (2021). https://doi.org/10.1038/s41576-021-00383-3
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41576-021-00383-3
This article is cited by
-
Programmable synthetic receptors: the next-generation of cell and gene therapies
Signal Transduction and Targeted Therapy (2024)
-
Synthetic biology-inspired cell engineering in diagnosis, treatment, and drug development
Signal Transduction and Targeted Therapy (2023)
-
Bacterial therapies at the interface of synthetic biology and nanomedicine
Nature Reviews Bioengineering (2023)
-
Myoglobin-loaded gadolinium nanotexaphyrins for oxygen synergy and imaging-guided radiosensitization therapy
Nature Communications (2023)
-
Improved cryptic plasmids in probiotic Escherichia coli Nissle 1917 for antibiotic-free pathway engineering
Applied Microbiology and Biotechnology (2023)
