
Abstract
Evidence from human genetic pain disorders shows that voltage-gated sodium channel α-subtypes Nav1.7, Nav1.8 and Nav1.9 are important in the peripheral signalling of pain. Nav1.7 is of particular interest because individuals with Nav1.7 loss-of-function mutations are congenitally insensitive to acute and chronic pain, and there is considerable hope that phenocopying these effects with a pharmacological antagonist will produce a new class of analgesic drug. However, studies in these rare individuals do not reveal how and where voltage-gated sodium channels contribute to pain signalling, which is of critical importance for drug development. More than a decade of research utilizing rodent genetic models and pharmacological tools to study voltage-gated sodium channels in pain has begun to unravel the role of different subtypes. Here, we review the contribution of individual channel subtypes in three key physiological processes necessary for transmission of sensory information to the CNS: transduction of stimuli at peripheral nerve terminals, axonal transmission of action potentials and neurotransmitter release from central terminals. These data suggest that drugs seeking to recapitulate the analgesic effects of loss of function of Nav1.7 will need to be brain-penetrant — which most of those developed to date are not.
Similar content being viewed by others

Introduction
The history of local anaesthetic drugs is an interesting one. The coca leaf, Erythroxylum coca, was cultivated by several South American societies for hundreds of years Before Common Era, and was widely used in those societies for the treatment of many ailments1. The western world first became aware of the anaesthetic properties of the coca leaf extract from reports brought to Spain from their colonies. In 1653, the Spanish Jesuit Bernabé Cobo first described the use of coco extract to treat toothaches:
“And this happen’d to me once, that I repaired to a barber to have a tooth pull’d, that had work’d loose and ach’d, and the barber told me he would be sorry to pull it because it was sound and healthy; and a monk friend of mine who happen’d to be there and overhearing, advised me to chew for a few days on Coca. As I did, indeed, soon to find my toothache gone.”
The coco leaf itself, and later the active ingredient, cocaine, became quite fashionable in Europe, but much of the interest related to the euphoria produced. The first experimental studies on the anaesthetic properties of cocaine were undertaken by ophthalmologist Carl Koller, who worked alongside Sigmund Freud in Vienna. Freud, fascinated by the stimulatory effects of the drug, used it extensively on patients and himself and became addicted to it. Koller realized that the numbness that occurred when coco leaf was chewed might be exploited medically. He applied cocaine topically to the cornea of several animals and then, in 1884, he performed the first operation using it as a local anaesthetic on a patient with glaucoma2.
The modern era of local anaesthetics began early in the twentieth century with the introduction of novocaine. Lidocaine and other compounds followed and these, of course, have now been widely used in experimental and clinical settings. The effects are dramatic: local anaesthetics produce analgesia. Injection around a nerve trunk, for instance, in dentistry, usually produces complete analgesia, sufficient to allow painless surgery. Infiltration or topical application to peripheral tissues is similarly effective. Even in extreme pathology, local anaesthetics can give complete pain relief. In a series of patients with amputation pain, Vaso et al. reported that local anaesthetic treatment of sensory nerve fibres as they coursed towards the spinal cord was highly effective at temporarily relieving phantom limb pain3. Similarly, Haroutounian et al. showed that the neuropathic pain of trauma or diabetes (often refractory to treatment) can be completely reversed for a limited time by local anaesthetic injected around the appropriate peripheral nerves4.
Voltage-gated sodium channels (Navs) are critical for electrical signalling in neurons5,6, and local anaesthetics are blockers of Navs, but with little or no selectivity between the nine different α-Nav subtypes (Nav1.1–Nav1.9). One might argue that at the high concentrations used in some clinical studies, the agents have other actions, for instance, blocking voltage-gated calcium channels7,8, but in mammals the block of sensory conduction at low concentrations appears to be mediated exclusively or very largely through block of sodium channels. Complete block of action potential propagation, and thus transfer of sensory information, can be achieved when these agents are applied to the peripheral or central processes of first-order sensory neurons9,10. Thus, blocking Navs anywhere along the primary sensory axon is an effective way of blocking transmission of all sensory signals. Navs are widely expressed throughout the body and have other vital functional roles, for instance, Nav1.5 is expressed in the heart and supports electrical signalling in cardiomyocytes11. The non-selective action of local anaesthetics on Navs, therefore, limits their use outside acute nerve block and these agents are rarely administered systemically to provide pain relief because of the risk of disrupting cardiac and respiratory function as well.
There are a limited number of Nav subtypes expressed in sensory neurons (see Table 1 and below). In the past two decades there has been a significant effort to develop novel analgesic compounds that specifically target channels expressed in nociceptive neurons (see ref.12 for an update on drug development progress). This task has proved surprisingly difficult, but the effort has revealed numerous unexpected and counterintuitive mechanisms of Nav action that we review here. The mechanisms uncovered have considerable implications for the future development of Nav blockers for the treatment of pain.
Navs important in pain: human evidence
Nav channels comprise an ion-conducting pore-forming α-subunit that is associated with β-subunits, which can modulate the trafficking and gating properties of the α-subunits (see ref.13 for a review). Recent reports also suggest that some Navs can form dimers14,15 and can interact with other proteins, for example, collapsin response mediator protein 2, which can also modulate ion channel trafficking16,17. We have very limited clinical evidence for the importance of these ancillary proteins18, and therefore we focus our attention on the α-subunits. There are five different Nav α-subtypes normally expressed in adult mammalian sensory neurons (see Box 1): the tetrodotoxin-sensitive (TTX-S) subtypes Nav1.1, Nav1.6 and Nav1.7; and the TTX-resistant (TTX-R) subtypes Nav1.8 and Nav1.9 (ref.19). In general, the TTX-S Nav subtypes activate more rapidly and exhibit faster kinetics than the TTX-R subtypes20. The three major subtypes expressed in nociceptive neurons (Nav1.7, Nav1.8 and Nav1.9) have been the focus of study because mutations in the genes encoding these subtypes have been identified in humans who have abnormal sensitivity to pain21,22,23,24,25.

Nav1.7
Nav1.7 has been of particular interest because individuals with biallelic loss-of-function mutations are congenitally insensitive or indifferent to a wide range of stimuli that normally elicit pain. This includes not only acute stimuli (burns, abrasions and so on) but also trauma and disease-associated persistent pain states, such as childbirth and bone fractures24,26. These individuals, however, do produce a flare response in skin treated with topical capsaicin or mustard oil, but do not perceive it as painful or itchy27. Most other forms of sensation, including detection of innocuous temperature and tactile stimuli, remain intact in such individuals, with the only other reported sensory deficit being anosmia28. In contrast, Nav1.7 gain-of-function mutations cause extremely debilitating chronic pain conditions, such as inherited erythromelalgia and paroxysmal extreme pain disorder21,29,30. Individuals suffering from these conditions experience episodes of burning pain, which are often triggered by warmth in erythromelalgia. The association between these conditions of heightened pain and gain-of-function Nav1.7 mutations is well understood: the channel is expressed in nociceptive neurons, and hyperfunctional channels within these neurons cause increased excitability and spontaneous activity, which drive evoked and spontaneous pain symptoms, respectively21,29,31,32. Although we understand why these individuals suffer from heightened pain sensitivity, it is not clear why certain parts of the body are affected more than others, particularly in paroxysmal extreme pain disorder30.
The mechanism (or mechanisms) by which loss of Nav1.7 function leads to insensitivity to pain is still not completely understood. We discuss the different suggestions in some detail in this Review because of the obvious practical value in being able to pharmacologically phenocopy congenital analgesia.
One critical question is whether congenital analgesia is caused by abnormal development of nociceptors. One other well-described single gene mutation leading to congenital analgesia is loss of function of trkA (or its cognate ligand, NGF), which it transpires is essential for development and survival of small-diameter sensory nerve fibres33,34. However, this does not seem to be the case for Nav1.7 mutations, where nerve biopsies from Nav1.7-null humans show a normal size and distribution of fibres24,26. Although initial reports suggested that intra-epidermal nerve fibre (IENF) density was mostly normal in these individuals35,36, recent reports have found a substantial reduction in IENF density27,37, indicating that there may be a dying back of small fibres that contribute to the phenotype. This loss of IENFs may be age-dependent because an individual who showed normal IENF density was young (3 years old) and three individuals with substantial reductions in IENF were older (31–44 years old). As insensitivity to pain arises before such anatomical abnormalities in some of these individuals, it might contribute to but cannot explain the analgesic phenotype.
The majority of inactivating Nav1.7 mutations cause a complete loss of channel function; that is, when these mutant human channels are expressed in cell lines, they are unable to produce current24,27. However, some mutations that render channels hypofunctional (that is, the channels produce some current when expressed in cell lines) are reported to cause insensitivity to pain38,39. As channel function has not been characterized for some Nav1.7 mutations, we do not know the exact range of current loss necessary to produce analgesia. Electrophysiological recordings from nociceptive neurons derived from the stem cells of individuals with non-functional Nav1.7 show that these cells can still generate action potentials, albeit less readily than similar cells from unaffected individuals27. Recordings from the peripheral nerves of affected individuals via microneurography also reveal conduction in some C fibres, although they do not fit with the typical profile of those that are nociceptive. The simple idea that Nav1.7 loss-of-function mutations cause failure of all nociceptive signalling cannot be the case. This is further reinforced by the presence of flare responses in these individuals that are known to depend on action potential propagation in widely branched nociceptive neurons.
We return below to other hypotheses regarding the critical role of Nav1.7 for pain perception that have been extensively explored recently in animal experiments.
Nav1.9
Gain-of-function mutations in Nav1.9 have been identified in individuals with clinical phenotypes associated with pain, such as painful peripheral neuropathy23 and familial episodic pain40. Individuals who are affected complain of tingling, numbness and pain, typically localized to the distal extremities. They also present autonomical abnormalities, such as dry eyes23. Electrophysiological studies of heterologously expressed mutant channels reveal pro-excitatory changes to their gating properties, manifest as a depolarization of the resting membrane potential by 3.5–5.5 mV and an increase in spontaneous and evoked firing. The resulting hyperexcitability and presence of the channel within human C-fibre neurons likely contributes towards the painful symptoms41.
Nav1.9 mutations are also reported to cause congenital insensitivity to pain, so far in a very small number of individuals25,42. The sensory phenotype in these individuals is markedly different from those with Nav1.7-inactivating mutations. Although these people do not appreciate somatic pain following, for instance, trauma, bone fracture and so on, they do experience spontaneous episodes of visceral pain and sometimes experience pruritus42. That is, their pain phenotype is partial, not complete. The patients exhibit self-inflicted skin lesions and are cognitively normal. Counterintuitively, the mutations to Nav1.9 identified in these individuals cause a large hyperpolarizing shift in the voltage dependence of activation, which initiates a gain in channel function25,43. There has been much discussion of how this might produce an insensitivity to pain. One likely possibility is that extreme depolarization of nociceptors leads to depolarization block — an inability to propagate action potentials43. Although academically fascinating, it would not be therapeutically useful to mimic this phenotype, even if that could be achieved pharmacologically. As Nav1.9 appears to be expressed in only some somatic nociceptors19,44,45,46, it is also puzzling that somatic insensitivity to pain should arise from a change in just this subset.
Nav1.8
Nav1.8 mutations have been identified in individuals who have been diagnosed with painful peripheral neuropathies22,47,48. Some of these mutations were reported to cause a gain of channel function and their expression within heterologous systems leads to hyperexcitability. It is likely that the painful symptoms in these individuals, which are typically localized to the distal extremities22, are in part due to the increased excitability of C-fibre neurons in which Nav1.8 is widely expressed41.
To date, loss-of-function mutations in Nav1.8 have not been identified in humans with a congenital insensitivity to pain phenotypes. However, a mutation that causes a moderate loss of function in Nav1.8 has been associated with reduced pain sensitivity49. The mutant channels displayed both pro-excitatory and anti-excitatory properties when characterized electrophysiologically, but when expressed in dorsal root ganglion (DRG) neurons the overall effect was hypoexcitability. Loss-of-function mutations to Nav1.8 are also associated with potentially lethal cardiac conditions such as Brugada syndrome50,51. How and where Nav1.8 channels function to control cardiac physiology is controversial. Reports on the channel’s contribution to excitability generation in intracardiac neurons and cardiomyocytes are conflicting52,53. Another theory posits that elements within the Nav1.8 gene, not the channel per se, function to regulate the expression of SCN5A (ref.54), the gene encoding Nav1.5 (critical for normal cardiac function).
Nav1.1 and Nav1.6
Nav1.1 and Nav1.6 mutations are not typically associated with human pain conditions, which is consistent with the known modest expression of these channels in the main nociceptive neuron subtypes (see Box 1). However, gain-of-function mutations in Nav1.1 have been linked to familial hemiplegic migraine55,56 and there is a single report of a gain of function in Nav1.6 associated with the painful condition trigeminal neuralgia57. The mechanisms of these disorders are not clear.
Loss-of-function mutations in Nav1.1 and Nav1.6 are reported but these are associated with CNS disorders58,59, particularly epilepsy60,61,62. The pain phenotype of these patients has not to our knowledge been reported, but it seems unlikely that a dramatic pain phenotype would have been overlooked.
To summarize the human genetic evidence: gain-of-function mutations strongly suggest that Nav1.7, Nav1.8 and Nav1.9 contribute to pain processing, whereas the data are less convincing for Nav1.1 and Nav1.6. Which of these channels are likely to hold the most promise for analgesic drug development is, however, likely to be indicated by observations on loss-of-function mutations. Here, there is a very strong case for the importance of Nav1.7, limited evidence for Nav1.8 and Nav1.9, and signs that Nav1.1 and Nav1.6 do not play a critical role in pain processing. Furthermore, the widespread expression of Nav1.1 and Nav1.6 throughout the CNS and peripheral nervous system makes them poor analgesic drug targets. Some caution is, of course, required because compensatory mechanisms might be in play in individuals who are affected. To further define the role and importance of individual channels, it is necessary to evaluate evidence from experimental methods that aim to manipulate channel expression or function, to which we now turn.
Navs in peripheral pain processing
Mechanistic studies in animals are essential for our understanding of how and where different Nav subtypes contribute towards the transmission of nociceptive information. To that end, one can study channel function using genetics or pharmacological tools (see Box 2 for further details).
In the following, we review the literature relating to the mechanisms outlined in Fig. 1. That is, the contribution of individual Nav subtypes to transduction, transmission and transmitter release in nociceptive neurons.

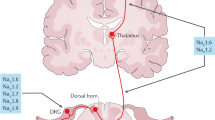
a | Schematic of a dorsal root ganglion neuron with examples of different innervation sites. b–d | Contribution of Nav1.1, Nav1.6, Nav1.7, Nav1.8 and Nav1.9 to the different processes of transduction (part b), transmission (part c) and transmitter release (part d) in nociceptors. The contribution of a particular channel to a particular process is indicated: ++, strong contribution; +, some contribution; –, negligible contribution66,67,68,69,70,71,72,73,74,75,76,77,84,86,87,91,92,93,100,101,102. Nav, voltage-gated sodium channel; TTX-R, tetrodotoxin-resistant; TTX-S, tetrodotoxin-sensitive.
Transduction
A key functional process in nociceptors is sensory transduction, the conversion of peripheral stimuli into electrical signals. Given the weight of genetic evidence, it is not surprising that the functional role of Nav1.7 in electrogenesis within nociceptive neurons has been examined extensively (see ref.63 for a recent review). The biophysical properties of the channel, specifically the activation at relatively hyperpolarized membrane potentials and slow closed-state inactivation64,65, allow it to conduct in response to sub-threshold, slowly depolarizing stimuli. As such, the channel might theoretically play an important role in bringing the membrane potential to the threshold for action potential generation. However, when tested, selective pharmacological inhibition of Nav1.7 at the terminal reduces, but does not abolish, responses induced by noxious heat and mechanical stimuli, and may be even less effective against cold stimuli66,67. Concordant findings have been found in Nav1.7 knockout mice, which demonstrate that Nav1.7 does not contribute to transduction in all nociceptive neurons68. Moreover, a significant proportion of cutaneous mechanical and heat-responsive nociceptors are insensitive to TTX, and therefore do not depend on Nav1.7 or another TTX-S channel for generating excitability67,69,70,71. For nociceptors inverting the viscera, although Nav1.7 is highly expressed in many colonic sensory neurons, channel blockade at the nerve terminals is not reported to affect responses to noxious stimulation66,72. Similar findings have been reported in vagal jugular nociceptors innervating the trachea, whereas there is evidence that Nav1.7 does appear to contribute to nodose ganglion nociceptors innervating the oesophagus and trachea73,74.
Together, these findings in rodents show that Nav1.7 has a significant but limited role in excitability generation at the peripheral terminals. Pharmacological studies imply that Nav1.7 partially contributes to excitability generation at the peripheral terminal in the population of cutaneous nociceptors that are TTX-S (and a substantial number of nociceptors are not, perhaps around 33–50%67). This is consistent with the observation, described above, of retention of flare responses in human skin with Nav1.7 mutations.
The ability of nociceptive terminals to transduce stimuli in the absence of Nav1.7 suggests that other Nav subtypes must be involved. As TTX abolishes nearly all of the responses to heat and mechanical stimuli in many C-fibre neurons, but only partial inhibition can be achieved with Nav1.7 blockers66,67, it seems likely that the other TTX-S channels expressed in sensory neurons, Nav1.1 and Nav1.6, could contribute. However, evidence from pharmacological studies examining the role of these channels in transduction at nociceptive terminals indicates that neither channel plays an important role in somatic C-fibre neurons75,76 (but see ref.77). For Nav1.6, these findings are, perhaps, somewhat surprising given a report that the channel is detected at free nerve endings (using immunohistochemistry)78 and is reported to contribute towards a third of the current in nociceptive neurons79. Future studies utilizing Nav1.6 selective blockers are warranted to elucidate any potential role of the channel in cutaneous nociceptive transduction. In the case of visceral afferents, however, there is stronger evidence for a role of Nav1.1 and Nav1.6 in transduction. Many visceral afferents express Nav1.1 and Nav1.6, and pharmacological studies have revealed a role for both channels in mechano-transduction66,72,77. The colonic afferents reported to express Nav1.1 are a subpopulation of high-threshold nociceptors, which implies that the channel contributes to pain signalling77.
In rodents, it is reported that up to a third of cutaneous C fibres responsive to heat and mechanical stimuli are resistant to TTX67,69,70. In these afferents, Nav1.8 and/or Nav1.9 must play an important role in transduction. The relative importance of these two channels, however, remains somewhat contested. Nav1.9 channels activate at relatively hyperpolarized membrane potentials80,81, which suggests that channels are involved in bringing the membrane potential to the threshold. However, when the role of Nav1.9 in setting nociceptive thresholds was examined in Nav1.9 knockout mice, the results were somewhat conflicting80,82 (see ref.83 for a discussion). Application of a Nav1.8 blocker to the peripheral terminals of TTX-R neurons was reported to abolish responses to heat67. However, TTX-R heat responses have been recorded from C fibres in both Nav1.8 and Nav1.9 knockout mice, which suggests that some functional redundancy between Nav1.8 and Nav1.9 exists69. Nav1.8 channels are also reported to have a critical role in the transduction of stimuli at cold temperatures (<10 °C)70. At the visceral terminals, there is clearer evidence for the importance of Nav1.9 over Nav1.8 in afferents innervating the colon72,84, whereas Nav1.8 is reported to be important for transduction in vagal airway nociceptors originating from the jugular ganglion73.
It is important to note that the TTX block of transduction at the peripheral terminals of many nociceptors does not necessarily mean that Nav1.8 (which is expressed in most nociceptors) does not contribute to transduction in this population. In nociceptors, TTX-S channels are important for bringing the membrane potential to the threshold20, and blocking this current likely prevents the membrane potential reaching the more depolarized threshold required for activation of Nav1.8, which is the major driver of the upstroke of the action potential85. Of course, this is not the case for all nociceptive afferents because some are capable of generating action potentials without TTX-S channels69.
In summary, nociceptor transduction in general does not depend on a single Nav type. Rather, several different channels appear to contribute under different circumstances. For somatic afferents, around two-thirds of the terminals of C-fibre nociceptors are TTX-S, and in this population Nav1.7 makes a modest contribution to the transduction of stimuli. More experiments are required to determine the contribution of other TTX-S channels in this population. In the remaining third, which are almost entirely dependent on TTX-R channels, the evidence suggests that Nav1.8, and possibly Nav1.9, contributes to transduction. In contrast, at the terminals of visceral C-fibre afferents, Nav1.1, Nav1.6 and Nav1.9 are all reported to contribute to transduction of stimuli, whereas Nav1.7 and Nav1.8 do not appear to have a large role. One important conclusion is that congenital analgesia seen with loss-of-function mutations of Nav1.7 cannot be explained by its role in sensory transduction at the peripheral nerve terminal.
Transmission of action potentials
A second key function of nociceptors is to faithfully transmit action potentials over the considerable distance from the peripheral to the central terminal — more than a metre in some human axons, and considerably longer in some mammals. It is experimentally straightforward to test the role of different Nav subtypes using specific pharmacological agents. For instance, the application of TTX to peripheral sensory nerves has revealed that a proportion (30–50%) of nociceptive neurons can conduct action potentials in the absence of TTX-S Nav channels86,87,88,89. It is more difficult to assess the contribution of a particular channel to propagation in transgenic animals where a single axon cannot be studied ‘before’ and ‘after’ intervention (see Box 2). We know that Nav1.7 is not essential for all axonal propagation because nociceptive responses have been recorded in visceral and cutaneous peripheral nerves of Nav1.7 knockout mice using electrophysiology or calcium imaging66,68,90,91. The proportion failing to conduct has been estimated with C-compound action potential recordings, which suggest that propagation fails in about two-thirds of C fibres in Nav1.7 knockout mice91. A similar proportion (70–80%) was reported to fail following application of a selective concentration of Nav1.7 blocker Pro-TX-II92.
Does propagation fail in this remaining third of axons along the dorsal root? Although it has been reported that selective inhibition of Nav1.7 along the dorsal root blocked conduction in the majority of C and Aδ fibres93, numerous other studies have shown that there is still a significant proportion of C-fibre neurons that can conduct when Nav1.7 is blocked by TTX86,88,94. This population includes mechanosensitive and heat-sensitive nociceptors89,95.
Do these findings in animals apply to human nerves? A much greater portion (>50%) of C fibres in human sural nerves were reported to be resistant to TTX and therefore are independent of Nav1.7 for propagation96,97. However, nerve biopsies were taken from patients with neuropathy and it is not known what impact this might have had on the expression and distribution of Navs. Some short-range nociceptor conduction must occur in patients who are congenitally insensitive because of their retained flare responses. Microneurography data from three of these individuals reveals conduction in some C fibres, but these fibres have activity-dependent slowing profiles that fit with sympathetic or low-threshold C fibres27. However, it is plausible that nociceptive fibres were not identified in such individuals because activity-dependent slowing processes can change in the absence of Nav1.7 (ref.91).
The evidence shows that in the absence of Nav1.7, action potentials can be initiated and propagated in a substantial population of nociceptive neurons. Given that only a small number of nociceptors need to be activated for pain sensation98, the total loss of pain sensibility seen with Nav1.7 mutations must have some other explanation.
Do other TTX-S channels contribute to axonal propagation in nociceptors? Nav1.6 protein is reported to be expressed at nodes of Ranvier in myelinated neurons throughout the peripheral nervous system and CNS99, including some Nav1.8-positive neurons that are presumed nociceptive79, but its precise contribution to propagation in this population is not known. Nav1.6 was reported to contribute to propagation in peripheral C fibres of juvenile mice100. In contrast, pharmacological inhibition of Nav1.1 and Nav1.6 along peripheral nerve of rats 3–6 months old was reported to have minimal effect on propagation in C fibres, implying that neither channel makes a major contribution to propagation in adults87.
The TTX experiments described above have a further functional interpretation. That is, many (30–50%) nociceptive afferents must have axonal propagation critically dependent on Nav1.8 and Nav1.9 (as these are the only TTX-R channel types). In rodents, application of TTX to Nav1.8, but not Nav1.9, knockout mice blocked conduction in all C-fibre neurons, suggesting that Nav1.8 is the TTX-R subtype that is essential for supporting conduction in C-fibre neurons in the absence of TTX-S Nav channels86.
The contribution of different Nav subtypes to propagation in nerves specifically innervating deep structures, such as the muscle and viscera, is not well studied. Reports suggest that many nociceptive neurons innervating muscle do not require TTX-S Nav channels for propagation89,95. In vagal nociceptive fibres innervating the trachea, however, propagation is reported to be entirely dependent on Nav1.7 (refs73,101), whereas a proportion of jugular nociceptors are capable of propagating action potentials without TTX-S channels73.
To summarize, around two-thirds of DRG nociceptive neurons innervating the skin are critically dependent on a TTX-S channel for axonal propagation, which the evidence suggests is most likely to be Nav1.7. The remaining third, which can conduct action potentials in the absence of TTX-S Nav subtypes, are likely dependent mostly on Nav1.8.
Transmitter release
The third and final key function of a nociceptive neuron is the transmission of information to the CNS. This process occurs within synapses located within the superficial lamina of the spinal cord. The most parsimonious explanation consistent with the data presented above is that Nav1.7 is essential for normal pain transduction because of its importance for synaptic transmission in nociceptors. There is some direct evidence for this proposal: electrically evoked substance P and glutamate release is reduced by genetic knockdown and pharmacological inhibition of the channel68,93,102. But how does loss of Nav1.7 at this location lead to a reduction in transmitter release?
Do action potentials fail to propagate to the presynaptic terminal without Nav1.7? We know that, upon entering the spinal cord, C-fibre nociceptors can form extensive terminal arborizations that contain numerous en-passant boutons103,104,105,106. Such branch points and en-passant boutons can generate a considerable electrical load for the invading action potential and, thus, these regions are associated with a lower safety factor, meaning that the action potential is vulnerable to failure107,108,109,110. Nav1.7 could play a critical role in supporting action potential propagation in these areas where it is prone to failure. Computer models predict that reduced sodium ion conductance, a likely consequence of Nav1.7 removal, makes differential propagation more likely at branch points111. If the action potential fails to invade the presynaptic terminal, it is unlikely that the electrogenic spread of current would be sufficient to trigger the voltage-gated calcium channels required for transmitter release112.
Does Nav1.7 loss directly impact synaptic transmission? At the central synapses of nociceptive neurons, there is evidence that Nav1.7 channels support transmission of nociceptive information to the CNS. Inhibition of Nav1.7 in synaptosomes from peptidergic terminals of the spinal cord reduces evoked CGRP release, suggesting a direct role in supporting the release of neurotransmitters93. A recent report has claimed that channels are also located on the membrane of second-order dorsal horn neurons, where they function to support postsynaptic excitability113. This finding contrasts with that of a previous study reporting that the contribution of Nav1.7 to excitability was restricted to the presynaptic terminal93.
Which other Nav subtypes contribute towards synaptic transmission within nociceptive neurons? There is evidence that functional TTX-R channels are present in presynaptic neurons and, in the absence of TTX-S channel subtypes, can support propagation of signals in order to depolarize postsynaptic dorsal horn neurons in culture114,115. A report studying DRG/dorsal horn neuronal co-cultures concluded that Nav1.8 and Nav1.7 were necessary for central neurotransmission116. Whether the repertoire of channels expressed in these cultured DRG neurons obtained from embryonic or neonatal animals represents intact neurons in vivo is unknown (but see below).
The evidence suggests that central transmitter release may be a critical process that requires the participation of Nav1.7 in most or all nociceptors, including those that also utilize other channels in different nociceptor functions (that is, transduction and transmission). This conclusion would suggest that selective Nav1.7 antagonists would be effective analgesics in many pain states. However, for this to be the case, the drugs would need to cross the blood–brain barrier (BBB) to access the central terminals of nociceptors. The Nav1.7-selective drugs that have been tested so far in the clinic do not cross the BBB and this may explain the limited efficacy seen in clinical trials117,118, presumably representing the non-redundant role of Nav1.7 in peripheral compartments of the nociceptor.
Nav effects on gene expression
The enhancement of the endogenous opioid system has been proposed to contribute towards the analgesic phenotype associated with Nav1.7 loss-of-function mutations68,119,120,121. In Nav1.7 knockout mice, it has been reported that mRNA for PENK, the gene encoding the precursor of endogenous opioid peptide enkephalins, is upregulated119, and signalling through µ-opioid and δ-opioid receptors is potentiated120,121. Additionally, systemic administration of the opioid receptor antagonist naloxone is reported to restore pain sensation in Nav1.7 knockout mice, and partially in some individuals with loss-of-function Nav1.7 mutations68,119. Mechanistically, it is claimed that Nav1.7 loss of function causes the enhanced release of endogenous opioids via changes to intracellular sodium signalling120, and this is proposed to attenuate the central transmission of nociceptive information within the spinal cord68. However, this theory has recently been called into question. In a study examining human nociceptors derived from patients with Nav1.7 loss of function (with induced pluripotent stem cell technology), no upregulation of PENK mRNA was reported and treatment with the opioid antagonist naloxone did not reverse neuronal hypoexcitability27. Additionally, systemic administration of naloxone in a rat Nav1.7 loss-of-function model did not normalize pain hyposensitivity, indicating that endogenous opioids are not responsible for loss of acute pain sensation in this model122.
Changes to Nav function in pathology
The above discussion relates mainly to the functioning of the peripheral nociceptive system under normal physiological conditions. In many pathophysiological states associated with disease or injury, the peripheral nociceptive system becomes activated or sensitized and we know that this contributes to the clinical manifestations of pain and hyperalgesia. One question, therefore, is whether the role of different Navs, discussed above, is altered in these pathophysiological states. There is considerable evidence that the activity of different Navs can indeed be modified by inflammatory and neuropathic conditions.
Inflammatory states, inflammatory mediators and some of the intracellular signalling pathways they activate can sensitize sensory neurons via modulation of ion channels and receptors. Inflammatory conditions have been shown to increase TTX-S and TTX-R currents in nociceptive DRG neurons123,124. The mechanisms include altered phosphorylation of channels, altered trafficking and altered expression124,125,126,127,128, and undoubtedly play a very significant role in the increased excitability of nociceptors in clinically relevant conditions. In some cases, this might be exploited therapeutically. For instance, Nav1.8 currents are rapidly and markedly increased in various inflammatory conditions, suggesting that selective Nav1.8 channel blockers could be of use here. Nav1.8 knockout mice do show reduced inflammatory hyperalgesia129,130. However, do these changes represent a fundamental shift in the importance of different channels in pain signalling? Individuals with loss-of-function Nav1.7 variants experience various inflammatory conditions from trauma or disease. They do not appear to become pain-sensitive, suggesting that the altered function of other channel subtypes cannot substitute for Nav1.7, a similar conclusion reached from the study of transgenic mice90.
In neuropathic conditions involving nerve trauma, however, there are quite large changes in channel subtype expression. Most notably, Nav1.3 mRNA increases (from very low levels)131 whereas Nav1.8 and Nav1.9 mRNA decrease substantially132. Nav1.7 expression does not appear to shift markedly in damaged sensory neurons133,134. Does Nav1.3 take on an important functional role? The evidence we have from a knockout mouse suggests not135. There are alternative suggestions that the distribution of channels within the nociceptor might be functionally important. Following nerve injury, Navs accumulate at the site of injury (neuroma) and contribute towards the generation of ectopic activity in C- and A-fibre neurons136,137,138,139,140, which is one of the major drivers of neuropathic pain. Despite some claims that this ectopic firing depends on Nav1.8 (ref.141), ectopic firing in DRG neurons is apparently completely blocked by TTX142,143, suggesting that Nav1.6 or Nav1.7, but not Nav1.3 (as the knockout animal appears to show normal ectopic firing)135, is essential for ectopic firing. There are supporting data for a role of Nav1.6 in A-fibre neurons79. Redistribution of Nav1.8 to the axons in uninjured neurons was also reported to occur following nerve injury and this led to an increase in the proportion of C fibres capable of propagation in the presence of TTX144. However, whether this redistribution is important in driving neuropathic pain is unclear. Findings from Nav1.8 knockout mice indicate that the channel is not essential for the development of lowered sensory thresholds following neuropathic injury130,145. Certain types of neuropathic pain are reported to develop independently of Nav1.7 in mice146.
There has been long-standing interest in the role of the sympathetic nervous system in chronic pain states. In animal models of neuropathic pain, it is reported that sympathetic postganglionic neurons can become coupled to sensory neurons and exacerbate pain (see ref.147 for a review). Activity in sympathetic postganglionic fibres depends on Navs and this raises the question of whether specific subtypes are important in driving this aspect of neuropathic pain. The distribution of Nav1.7 is highly restricted in the body but is found in sympathetic as well as nociceptive neurons19. Studies in transgenic mice imply that sympathetic Nav1.7 is necessary for the full expression of neuropathic pain behaviours in some, but not all, models of neuropathic pain146.
The population of individuals with Nav1.7 loss of function is small, so we do not have a clear answer to whether they may or may not express neuropathic pain — although there is one case report suggesting that some signs of neuropathic pain might persist in the absence of Nav1.7 (ref.148).
In short, there may be significant quantitative changes in Nav channel function that contribute to pathological pain states. However, we do not have strong evidence for the emergence of novel channels that only contribute to these pathological states.
Concluding remarks and perspectives
The physiological mechanisms by which Navs contribute to peripheral pain signalling are far from straightforward. At the peripheral terminals of nociceptors, multiple Nav subtypes are involved in transduction, with Nav1.7, Nav1.8 and Nav1.9 contributing at somatic terminals and Nav1.1, Nav1.6 and Nav1.9 at the visceral terminals. Furthermore, some degree of functional redundancy exists between subtypes. Along the axons of cutaneous nerves, propagation is critically dependent on Nav1.7 in approximately two-thirds of C-fibre neurons, whereas Nav1.8 is essential in the remaining proportion. Less is known about the Nav subtypes that contribute towards the central transmission of nociceptive information, but here Nav1.7 has a crucial function in determining whether pain is perceived or not. Some general conclusions can be drawn about the likely effectiveness of targeting specific Nav subunits in different conditions.
Nav1.7 remains an excellent candidate for analgesic drug development but, as we have reviewed here, the precise location where the channel is targeted within the nociceptor could be of critical importance for providing effective analgesia. The peripherally restricted Nav1.7 blockers developed to date provide inadequate pain relief in patients with chronic pain117,149 and this likely reflects the less critical role of this Nav in the peripheral processes of nociceptors. That is, Nav1.7 is essential for propagation of action potentials in only about two-thirds of somatic nociceptors. Additionally, the process of nociceptor sensitization, known to be a major contributor to many clinical pain states, can arise independently of Nav1.7 (ref.68). However, action potential propagation in nociceptors that is not dependent on Nav1.7 appears to be largely dependent on Nav1.8, and this suggests that a dual-targeting strategy of peripheral Nav1.7 and Nav1.8 inhibition would be more effective in providing analgesia. Whether this strategy could also be effective in visceral pain conditions is not clear, and here the evidence suggests that targeting Nav1.9 could be more effective84,150.
To be fully effective analgesics, Nav1.7 inhibitors must cross the BBB to reach the central terminals of the spinal cord where the critical function of Nav1.7 in pain processing appears to reside. The unwanted effects of targeting Nav1.7 with BBB-penetrant inhibitors are likely to be anosmia151 and, possibly, weight gain owing to inhibition of Nav1.7 in hypothalamic neurons152 (although human individuals with Nav1.7 congenital indifference to pain do not have this problem). An outstanding question is what proportion of channels need to be inhibited before you start achieving analgesic efficacy. It seems unlikely that 100% channel block is necessary for analgesia because Nav1.7 mutations that render channels hypofunctional have been identified in individuals who are pain free38,39. Evidence from rodent Nav1.7 knockout models show that olfactory function is retained even when 50–70% of Nav1.7 protein is reduced, which indicates that at least an equivalent proportion of channels might need to be inhibited to achieve a functional effect in nociceptors151,153. Looking forward, gene therapy targeting Nav1.7 protein production in the DRG is a promising approach, which bypasses the issues surrounding BBB penetration associated with small-molecule inhibitors. However, gene therapy technology is still in its early days and it is not yet clear how practical this will be for pain treatment.
References
Calatayud, J. & Gonzalez, A. History of the development and evolution of local anesthesia since the coca leaf. Anesthesiology 98, 1503–1508 (2003).
Koller, C. On the use of cocaine for producing anæsthesia on the eye. Lancet 124, 990–992 (1884).
Vaso, A. et al. Peripheral nervous system origin of phantom limb pain. Pain 155, 1384–1391 (2014).
Haroutounian, S. et al. Primary afferent input critical for maintaining spontaneous pain in peripheral neuropathy. Pain 155, 1272–1279 (2014).
Hodgkin, A. L. & Huxley, A. F. Currents carried by sodium and potassium ions through the membrane of the giant axon of Loligo. J. Physiol. 116, 449–472 (1952).
Hodgkin, A. L. & Huxley, A. F. The dual effect of membrane potential on sodium conductance in the giant axon of Loligo. J. Physiol. 116, 497–506 (1952).
Xiong, Z. & Strichartz, G. R. Inhibition by local anesthetics of Ca2+ channels in rat anterior pituitary cells. Eur. J. Pharmacol. 363, 81–90 (1998).
Sugiyama, K. & Muteki, T. Local anesthetics depress the calcium current of rat sensory neurons in culture. Anesthesiology 80, 1369–1378 (1994).
Jaffe, R. A. & Rowe, M. A. Differential nerve block. Direct measurements on individual myelinated and unmyelinated dorsal root axons. Anesthesiology 84, 1455–1464 (1996).
Fink, B. R. & Cairns, A. M. Differential peripheral axon block with lidocaine: unit studies in the cervical vagus nerve. Anesthesiology 59, 182–186 (1983).
Papadatos, G. A. et al. Slowed conduction and ventricular tachycardia after targeted disruption of the cardiac sodium channel gene Scn5a. Proc. Natl Acad. Sci. USA 99, 6210–6215 (2002).
Alsaloum, M., Higerd, G. P., Effraim, P. R. & Waxman, S. G. Status of peripheral sodium channel blockers for non-addictive pain treatment. Nat. Rev. Neurol. 16, 689–705 (2020).
Hull, J. M. & Isom, L. L. Voltage-gated sodium channel β subunits:tThe power outside the pore in brain development and disease. Neuropharmacology 132, 43–57 (2018).
Ruhlmann, A. H. et al. Uncoupling sodium channel dimers restores the phenotype of a pain-linked Nav 1.7 channel mutation. Br. J. Pharmacol. 177, 4481–4496 (2020).
Clatot, J. et al. Voltage-gated sodium channels assemble and gate as dimers. Nat. Commun. 8, 2077 (2017).
Kanellopoulos, A. H. et al. Mapping protein interactions of sodium channel NaV1.7 using epitope-tagged gene-targeted mice. EMBO J. 37, 427–445 (2018).
Dustrude, E. T., Wilson, S. M., Ju, W., Xiao, Y. & Khanna, R. CRMP2 protein SUMOylation modulates NaV1.7 channel trafficking. J. Biol. Chem. 288, 24316–24331 (2013).
Alsaloum, M. et al. A gain-of-function sodium channel β2-subunit mutation in painful diabetic neuropathy. Mol. Pain 15, 1744806919849802 (2019).
Zeisel, A. et al. Molecular architecture of the mouse nervous system. Cell 174, 999–1014.e22 (2018).
Blair, N. T. & Bean, B. P. Roles of tetrodotoxin (TTX)-sensitive Na+ current, TTX-resistant Na+ current, and Ca2+ current in the action potentials of nociceptive sensory neurons. J. Neurosci. 22, 10277–10290 (2002).
Dib-Hajj, S. D. et al. Gain-of-function mutation in Nav1.7 in familial erythromelalgia induces bursting of sensory neurons. Brain 128, 1847–1854 (2005).
Faber, C. G. et al. Gain-of-function Nav1.8 mutations in painful neuropathy. Proc. Natl Acad. Sci. USA 109, 19444–19449 (2012).
Huang, J. et al. Gain-of-function mutations in sodium channel Na(v)1.9 in painful neuropathy. Brain 137, 1627–1642 (2014).
Cox, J. J. et al. An SCN9A channelopathy causes congenital inability to experience pain. Nature 444, 894–898 (2006).
Leipold, E. et al. A de novo gain-of-function mutation in SCN11A causes loss of pain perception. Nat. Genet. 45, 1399–1404 (2013). This paper reports on individuals with Nav1.9 mutations who are congenitally insensitive to pain and how these mutations are proposed to cause pain insensitivity.
Goldberg, Y. P. et al. Loss-of-function mutations in the Nav1.7 gene underlie congenital indifference to pain in multiple human populations. Clin. Genet. 71, 311–319 (2007).
McDermott, L. A. et al. Defining the functional role of NaV1.7 in human nociception. Neuron 101, 905–919.e8 (2019). This paper provides a clinical evaluation of three individuals who are insensitive to pain owing to Nav1.7 mutations, and provides a functional examination of their stem-cell derived nociceptors.
Weiss, J. et al. Loss-of-function mutations in sodium channel Nav1.7 cause anosmia. Nature 472, 186–190 (2011).
Dib-Hajj, S. D. et al. Paroxysmal extreme pain disorder M1627K mutation in human Nav1.7 renders DRG neurons hyperexcitable. Mol. Pain 4, 37 (2008).
Fertleman, C. R. et al. SCN9A mutations in paroxysmal extreme pain disorder: allelic variants underlie distinct channel defects and phenotypes. Neuron 52, 767–774 (2006).
Orstavik, K. & Jorum, E. Microneurographic findings of relevance to pain in patients with erythromelalgia and patients with diabetic neuropathy. Neurosci. Lett. 470, 180–184 (2010).
Namer, B. et al. Specific changes in conduction velocity recovery cycles of single nociceptors in a patient with erythromelalgia with the I848T gain-of-function mutation of Nav1.7. Pain 156, 1637–1646 (2015).
Indo, Y. Molecular basis of congenital insensitivity to pain with anhidrosis (CIPA): mutations and polymorphisms in TRKA (NTRK1) gene encoding the receptor tyrosine kinase for nerve growth factor. Hum. Mutat. 18, 462–471 (2001).
Einarsdottir, E. et al. A mutation in the nerve growth factor β gene (NGFB) causes loss of pain perception. Hum. Mol. Genet. 13, 799–805 (2004).
Nilsen, K. B. et al. Two novel SCN9A mutations causing insensitivity to pain. Pain 143, 155–158 (2009).
Klein, C. J. et al. Infrequent SCN9A mutations in congenital insensitivity to pain and erythromelalgia. J. Neurol. Neurosurg. Psychiatry 84, 386–391 (2013).
Marchi, M. et al. A novel SCN9A splicing mutation in a compound heterozygous girl with congenital insensitivity to pain, hyposmia and hypogeusia. J. Peripher. Nerv. Syst. 23, 202–206 (2018).
Emery, E. C. et al. Novel SCN9A mutations underlying extreme pain phenotypes: unexpected electrophysiological and clinical phenotype correlations. J. Neurosci. 35, 7674–7681 (2015).
Sun, J. et al. Novel SCN9A missense mutations contribute to congenital insensitivity to pain: unexpected correlation between electrophysiological characterization and clinical phenotype. Mol. Pain 16, 1744806920923881 (2020).
Zhang, X. Y. et al. Gain-of-function mutations in SCN11A cause familial episodic pain. Am. J. Hum. Genet. 93, 957–966 (2013).
Dib-Hajj, S. D. et al. Two tetrodotoxin-resistant sodium channels in human dorsal root ganglion neurons. FEBS Lett. 462, 117–120 (1999).
Woods, C. G., Babiker, M. O. E., Horrocks, I., Tolmie, J. & Kurth, I. The phenotype of congenital insensitivity to pain due to the NaV1.9 variant p.L811P. Eur. J. Hum. Genet. 23, 561–563 (2015).
Huang, J. et al. Sodium channel NaV1.9 mutations associated with insensitivity to pain dampen neuronal excitability. J. Clin. Invest. 127, 2805–2814 (2017).
Salvatierra, J. et al. A disease mutation reveals a role for NaV1.9 in acute itch. J. Clin. Invest. 128, 5434–5447 (2018).
Dib-Hajj, S., Black, J. A., Cummins, T. R. & Waxman, S. G. NaN/Nav1.9: a sodium channel with unique properties. Trends Neurosci. 25, 253–259 (2002).
Fang, X. et al. Intense isolectin-B4 binding in rat dorsal root ganglion neurons distinguishes C-fiber nociceptors with broad action potentials and high Nav1.9 expression. J. Neurosci. 26, 7281–7292 (2006).
Han, C. et al. The G1662S NaV1.8 mutation in small fibre neuropathy: impaired inactivation underlying DRG neuron hyperexcitability. J. Neurol. Neurosurg. Psychiatry 85, 499–505 (2014).
Huang, J. et al. Small-fiber neuropathy Nav1.8 mutation shifts activation to hyperpolarized potentials and increases excitability of dorsal root ganglion neurons. J. Neurosci. 33, 14087–14097 (2013).
Duan, G. et al. A SCN10A SNP biases human pain sensitivity. Mol. Pain https://doi.org/10.1177/1744806916666083 (2016).
Behr, E. R. et al. Role of common and rare variants in SCN10A: results from the Brugada syndrome QRS locus gene discovery collaborative study. Cardiovasc. Res. 106, 520–529 (2015).
Hu, D. et al. Mutations in SCN10A are responsible for a large fraction of cases of Brugada syndrome. J. Am. Coll. Cardiol. 64, 66–79 (2014).
Verkerk, A. O. et al. Functional Nav1.8 channels in intracardiac neurons: the link between SCN10A and cardiac electrophysiology. Circ. Res. 111, 333–343 (2012).
Yang, T. et al. Blocking Scn10a channels in heart reduces late sodium current and is antiarrhythmic. Circ. Res. 111, 322–332 (2012).
van den Boogaard, M. et al. A common genetic variant within SCN10A modulates cardiac SCN5A expression. J. Clin. Invest. 124, 1844–1852 (2014).
Dichgans, M. et al. Mutation in the neuronal voltage-gated sodium channel SCN1A in familial hemiplegic migraine. Lancet 366, 371–377 (2005).
Gargus, J. J. & Tournay, A. Novel mutation confirms seizure locus SCN1A is also familial hemiplegic migraine locus FHM3. Pediatr. Neurol. 37, 407–410 (2007).
Tanaka, B. S. et al. A gain-of-function mutation in Nav1.6 in a case of trigeminal neuralgia. Mol. Med. 22, 338–348 (2016).
Trudeau, M. M., Dalton, J. C., Day, J. W., Ranum, L. P. & Meisler, M. H. Heterozygosity for a protein truncation mutation of sodium channel SCN8A in a patient with cerebellar atrophy, ataxia, and mental retardation. J. Med. Genet. 43, 527–530 (2006).
Wagnon, J. L. et al. Loss-of-function variants of SCN8A in intellectual disability without seizures. Neurol. Genet. 3, e170 (2017).
Wengert, E. R. et al. Biallelic inherited SCN8A variants, a rare cause of SCN8A-related developmental and epileptic encephalopathy. Epilepsia 60, 2277–2285 (2019).
Mantegazza, M. et al. Identification of an Nav1.1 sodium channel (SCN1A) loss-of-function mutation associated with familial simple febrile seizures. Proc. Natl Acad. Sci. USA 102, 18177–18182 (2005).
Lossin, C. et al. Epilepsy-associated dysfunction in the voltage-gated neuronal sodium channel SCN1A. J. Neurosci. 23, 11289–11295 (2003).
Bennett, D. L., Clark, A. J., Huang, J., Waxman, S. G. & Dib-Hajj, S. D. The role of voltage-gated sodium channels in pain signaling. Physiol. Rev. 99, 1079–1151 (2019).
Cummins, T. R., Howe, J. R. & Waxman, S. G. Slow closed-state inactivation: a novel mechanism underlying ramp currents in cells expressing the hNE/PN1 sodium channel. J. Neurosci. 18, 9607–9619 (1998).
Herzog, R. I., Cummins, T. R., Ghassemi, F., Dib-Hajj, S. D. & Waxman, S. G. Distinct repriming and closed-state inactivation kinetics of Nav1.6 and Nav1.7 sodium channels in mouse spinal sensory neurons. J. Physiol. 551, 3–50 (2003).
Hockley, J. R. et al. Visceral and somatic pain modalities reveal NaV 1.7-independent visceral nociceptive pathways. J. Physiol. 595, 2661–2679 (2017). This paper examines the contribution of Nav1.7 to transduction at visceral afferent terminals.
Kornecook, T. J. et al. Pharmacologic characterization of AMG8379, a potent and selective small molecule sulfonamide antagonist of the voltage-gated sodium channel NaV1.7. J. Pharmacol. Exp. Ther. 362, 146–160 (2017).
MacDonald, D. I. et al. The mechanism of analgesia in Nav1.7 null mutants. bioRxiv https://doi.org/10.1101/2020.06.01.127183 (2020).
Touska, F. et al. Heat-resistant action potentials require TTX-resistant sodium channels NaV1.8 and NaV1.9. J. Gen. Physiol. 150, 1125–1144 (2018).
Zimmermann, K. et al. Sensory neuron sodium channel Nav1.8 is essential for pain at low temperatures. Nature 447, 855–858 (2007).
Jonas, R. et al. TTX-resistant sodium channels functionally separate silent from polymodal C-nociceptors. Front. Cell Neurosci. 14, 13 (2020).
Feng, B., Zhu, Y., La, J. H., Wills, Z. P. & Gebhart, G. F. Experimental and computational evidence for an essential role of NaV1.6 in spike initiation at stretch-sensitive colorectal afferent endings. J. Neurophysiol. 113, 2618–2634 (2015).
Kollarik, M. et al. Different role of TTX-sensitive voltage-gated sodium channel (NaV 1) subtypes in action potential initiation and conduction in vagal airway nociceptors. J. Physiol. 596, 1419–1432 (2018).
Ru, F. et al. Stimulus intensity-dependent recruitment of NaV1 subunits in action potential initiation in nerve terminals of vagal C-fibers innervating the esophagus. Am. J. Physiol. Gastrointest. Liver Physiol. 319, G443–G453 (2020).
Jurcakova, D. et al. Voltage-gated sodium channels regulating action potential generation in itch-, nociceptive-, and low-threshold mechanosensitive cutaneous C-fibers. Mol. Pharmacol. 94, 1047–1056 (2018).
Israel, M. R. et al. NaV1.6 regulates excitability of mechanosensitive sensory neurons. J. Physiol. 597, 3751–3768 (2019).
Osteen, J. D. et al. Selective spider toxins reveal a role for the Nav1.1 channel in mechanical pain. Nature 534, 494–499 (2016).
Persson, A. K. et al. Sodium-calcium exchanger and multiple sodium channel isoforms in intra-epidermal nerve terminals. Mol. Pain 6, 84 (2010).
Chen, L. et al. Conditional knockout of NaV1.6 in adult mice ameliorates neuropathic pain. Sci. Rep. 8, 3845 (2018). This study demonstrates that Nav subtypes that are not typically associated with pain are involved in the development of neuropathic pain following nerve injury.
Priest, B. T. et al. Contribution of the tetrodotoxin-resistant voltage-gated sodium channel NaV1.9 to sensory transmission and nociceptive behavior. Proc. Natl Acad. Sci. USA 102, 9382–9387 (2005).
Cummins, T. R. et al. A novel persistent tetrodotoxin-resistant sodium current in SNS-null and wild-type small primary sensory neurons. J. Neurosci. 19, RC43 (1999).
Hoffmann, T. et al. Reduced excitability and impaired nociception in peripheral unmyelinated fibers from Nav1.9-null mice. Pain 158, 58–67 (2017).
Minett, M. S., Eijkelkamp, N. & Wood, J. N. Significant determinants of mouse pain behaviour. PLoS ONE 9, e104458 (2014).
Hockley, J. R. et al. Multiple roles for NaV1.9 in the activation of visceral afferents by noxious inflammatory, mechanical, and human disease-derived stimuli. Pain 155, 1962–1975 (2014).
Renganathan, M., Cummins, T. R. & Waxman, S. G. Contribution of Na(v)1.8 sodium channels to action potential electrogenesis in DRG neurons. J. Neurophysiol. 86, 629–640 (2001).
Klein, A. H. et al. Sodium channel Nav1.8 underlies TTX-resistant axonal action potential conduction in somatosensory C-fibers of distal cutaneous nerves. J. Neurosci. 37, 5204–5214 (2017).
Wilson, M. J. et al. μ-Conotoxins that differentially block sodium channels NaV1.1 through 1.8 identify those responsible for action potentials in sciatic nerve. Proc. Natl Acad. Sci. USA 108, 10302–10307 (2011).
Jeftinija, S. The role of tetrodotoxin-resistant sodium channels of small primary afferent fibers. Brain Res. 639, 125–134 (1994).
Steffens, H., Eek, B., Trudrung, P. & Mense, S. Tetrodotoxin block of A-fibre afferents from skin and muscle — a tool to study pure C-fibre effects in the spinal cord. Pflug. Arch. Eur. J. Physiol. 445, 607–613 (2003).
Gingras, J. et al. Global Nav1.7 knockout mice recapitulate the phenotype of human congenital indifference to pain. PLoS ONE 9, e105895 (2014). This paper describes the first global Nav1.7 knockout mouse and the behavioural, anatomical and electrophysiological changes that occur in these animals.
Hoffmann, T. et al. NaV1.7 and pain: contribution of peripheral nerves. Pain 159, 496–506 (2018). This paper provides a detailed examination of the electrophysiological changes that occur in peripheral nerve nociceptors of Nav1.7 knockout mice.
Schmalhofer, W. A. et al. ProTx-II, a selective inhibitor of NaV1.7 sodium channels, blocks action potential propagation in nociceptors. Mol. Pharmacol. 74, 1476–1484 (2008).
Alexandrou, A. J. et al. Subtype-selective small molecule inhibitors reveal a fundamental role for Nav1.7 in nociceptor electrogenesis, axonal conduction and presynaptic release. PLoS ONE 11, e0152405–e0152405 (2016). This paper provides an in vitro and ex vivo examination of Nav1.7 function in nociceptors using selective pharmacological inhibitors of the channel.
Pinto, V., Derkach, V. A. & Safronov, B. V. Role of TTX-sensitive and TTX-resistant sodium channels in Aδ- and C-fiber conduction and synaptic transmission. J. Neurophysiol. 99, 617–628 (2008).
Lambertz, D., Hoheisel, U. & Mense, S. Distribution of synaptic field potentials induced by TTX-resistant skin and muscle afferents in rat spinal segments L4 and L5. Neurosci. Lett. 409, 14–18 (2006).
Quasthoff, S., Grosskreutz, J., Schroder, J. M., Schneider, U. & Grafe, P. Calcium potentials and tetrodotoxin-resistant sodium potentials in unmyelinated C fibres of biopsied human sural nerve. Neuroscience 69, 955–965 (1995).
Grosskreutz, J., Quasthoff, S., Kuhn, M. & Grafe, P. Capsaicin blocks tetrodotoxin-resistant sodium potentials and calcium potentials in unmyelinated C fibres of biopsied human sural nerve in vitro. Neurosci. Lett. 208, 49–52 (1996).
Torebjork, H. E., Schady, W. & Ochoa, J. L. A new method for demonstration of central effects of analgesic agents in man. J. Neurol. Neurosurg. Psychiatry 47, 862–869 (1984).
Caldwell, J. H., Schaller, K. L., Lasher, R. S., Peles, E. & Levinson, S. R. Sodium channel Na(v)1.6 is localized at nodes of ranvier, dendrites, and synapses. Proc. Natl Acad. Sci. USA 97, 5616–5620 (2000).
Black, J. A., Renganathan, M. & Waxman, S. G. Sodium channel Na(v)1.6 is expressed along nonmyelinated axons and it contributes to conduction. Brain Res. Mol.Brain Res. 105, 19–28 (2002).
Muroi, Y. et al. Selective silencing of Na(V)1.7 decreases excitability and conduction in vagal sensory neurons. J. Physiol. 589, 5663–5676 (2011).
Minett, M. S. et al. Distinct Nav1.7-dependent pain sensations require different sets of sensory and sympathetic neurons. Nat. Commun. 3, 791 (2012).
Sugiura, Y., Lee, C. L. & Perl, E. R. Central projections of identified, unmyelinated (C) afferent fibers innervating mammalian skin. Science 234, 358–361 (1986).
Sugiura, Y., Terui, N. & Hosoya, Y. Difference in distribution of central terminals between visceral and somatic unmyelinated (C) primary afferent fibers. J. Neurophysiol. 62, 834–840 (1989).
Rethelyi, M. Preterminal and terminal axon arborizations in the substantia gelatinosa of cat’s spinal cord. J. Comp. Neurol. 172, 511–521 (1977).
Sugiura, Y. et al. Quantitative analysis of central terminal projections of visceral and somatic unmyelinated (C) primary afferent fibers in the guinea pig. J. Comp. Neurol. 332, 315–325 (1993).
Luscher, C., Streit, J., Quadroni, R. & Luscher, H. R. Action potential propagation through embryonic dorsal root ganglion cells in culture. I. Influence of the cell morphology on propagation properties. J. Neurophysiol. 72, 622–633 (1994).
Streit, J., Luscher, C. & Luscher, H. R. Depression of postsynaptic potentials by high-frequency stimulation in embryonic motoneurons grown in spinal cord slice cultures. J. Neurophysiol. 68, 1793–1803 (1992).
Luscher, H. R. & Shiner, J. S. Simulation of action potential propagation in complex terminal arborizations. Biophys. J. 58, 1389–1399 (1990).
Luscher, H. R. & Shiner, J. S. Computation of action potential propagation and presynaptic bouton activation in terminal arborizations of different geometries. Biophys. J. 58, 1377–1388 (1990).
Stockbridge, N. & Stockbridge, L. L. Differential conduction at axonal bifurcations. I. Effect of electrotonic length. J. Neurophysiol. 59, 1277–1285 (1988).
Bao, J., Li, J. J. & Perl, E. R. Differences in Ca2+ channels governing generation of miniature and evoked excitatory synaptic currents in spinal laminae I and II. J. Neurosci. 18, 8740–8750 (1998).
Alles, S. R. A. et al. Sensory neuron-derived NaV1.7 contributes to dorsal horn neuron excitability. Sci. Adv. 6, eaax4568 (2020).
Medvedeva, Y. V., Kim, M. S., Schnizler, K. & Usachev, Y. M. Functional tetrodotoxin-resistant Na+ channels are expressed presynaptically in rat dorsal root ganglia neurons. Neuroscience 159, 559–569 (2009).
Gu, J. G. & MacDermott, A. B. Activation of ATP P2X receptors elicits glutamate release from sensory neuron synapses. Nature 389, 749–753 (1997).
Vysokov, N., McMahon, S. B. & Raouf, R. The role of NaV channels in synaptic transmission after axotomy in a microfluidic culture platform. Sci. Rep. 9, 12915 (2019).
McDonnell, A. et al. Efficacy of the Nav1.7 blocker PF-05089771 in a randomised, placebo-controlled, double-blind clinical study in subjects with painful diabetic peripheral neuropathy. Pain 159, 1465–1476 (2018).
Siebenga, P. et al. Lack of detection of the analgesic properties of PF-05089771, a selective Nav 1.7 inhibitor, using a battery of pain models in healthy subjects. Clin. Transl. Sci. 13, 318–324 (2020).
Minett, M. S. et al. Endogenous opioids contribute to insensitivity to pain in humans and mice lacking sodium channel Nav1.7. Nat. Commun. 6, 8967 (2015). This study details the proposed mechanism that endogenous opioids are responsible for Nav1.7 loss-of-function analgesia.
Pereira, V. et al. Analgesia linked to Nav1.7 loss of function requires μ- and δ-opioid receptors. Wellcome Open Res. 3, 101 (2018).
Isensee, J. et al. Synergistic regulation of serotonin and opioid signaling contributes to pain insensitivity in Nav1.7 knockout mice. Sci Signal 10, eaah4874 (2017).
Chen, L. et al. Pharmacological characterization of a rat Nav1.7 loss-of-function model with insensitivity to pain. Pain 161, 1350–1360 (2020). This study reports that endogenous opioids are not responsible for analgesia in a rat Nav1.7 loss-of-function insensitivity to pain model.
Gold, M. S., Reichling, D. B., Shuster, M. J. & Levine, J. D. Hyperalgesic agents increase a tetrodotoxin-resistant Na+ current in nociceptors. Proc. Natl Acad. Sci. USA 93, 1108–1112 (1996).
Black, J. A., Liu, S., Tanaka, M., Cummins, T. R. & Waxman, S. G. Changes in the expression of tetrodotoxin-sensitive sodium channels within dorsal root ganglia neurons in inflammatory pain. Pain. 108, 237–247 (2004).
Docherty, R. J. & Farrag, K. J. The effect of dibutyryl cAMP on tetrodotoxin-sensitive and -resistant voltage-gated sodium currents in rat dorsal root ganglion neurons and the consequences for their sensitivity to lidocaine. Neuropharmacology 51, 1047–1057 (2006).
Gold, M. S., Levine, J. D. & Correa, A. M. Modulation of TTX-R/Na by PKC and PKA and their role in PGE2-induced sensitization of rat sensory neurons in vitro. J. Neurosci. 18, 10345–10355 (1998).
Akopian, A. N. et al. Trans-splicing of a voltage-gated sodium channel is regulated by nerve growth factor. FEBS Lett. 445, 177–182 (1999).
Akin, E. J. et al. Building sensory axons: delivery and distribution of NaV1.7 channels and effects of inflammatory mediators. Sci. Adv. 5, eaax4755 (2019).
Akopian, A. N. et al. The tetrodotoxin-resistant sodium channel SNS has a specialized function in pain pathways. Nat. Neurosci. 2, 541–548 (1999).
Kerr, B. J., Souslova, V., McMahon, S. B. & Wood, J. N. A role for the TTX-resistant sodium channel Nav 1.8 in NGF-induced hyperalgesia, but not neuropathic pain. Neuro Report 12, 3077–3080 (2001).
Waxman, S. G., Kocsis, J. D. & Black, J. A. Type III sodium channel mRNA is expressed in embryonic but not adult spinal sensory neurons, and is reexpressed following axotomy. J. Neurophysiol. 72, 466–470 (1994).
Boucher, T. J. et al. Potent analgesic effects of GDNF in neuropathic pain states. Science 290, 124–127 (2000).
Renthal, W. et al. Transcriptional reprogramming of distinct peripheral sensory neuron subtypes after axonal injury. Neuro 108, 128–144 (2020).
Nguyen, M. Q., Le Pichon, C. E. & Ryba, N. Stereotyped transcriptomic transformation of somatosensory neurons in response to injury. eLife 8, e49679 (2019).
Nassar, M. A. et al. Nerve injury induces robust allodynia and ectopic discharges in Nav1.3 null mutant mice. Mol. Pain 2, 33 (2006).
Devor, M., Govrin-Lippmann, R. & Angelides, K. Na+ channel immunolocalization in peripheral mammalian axons and changes following nerve injury and neuroma formation. J.Neurosci. 13, 1976–1992 (1993).
Devor, M., Wall, P. D. & Catalan, N. Systemic lidocaine silences ectopic neuroma and DRG discharge without blocking nerve conduction. Pain 48, 261–268 (1992).
Welk, E., Leah, J. D. & Zimmermann, M. Characteristics of A- and C-fibers ending in a sensory nerve neuroma in the rat. J. Neurophysiol. 63, 759–766 (1990).
Rivera, L., Gallar, J., Pozo, M. A. & Belmonte, C. Responses of nerve fibres of the rat saphenous nerve neuroma to mechanical and chemical stimulation: an in vitro study. J. Physiol. 527, 305–313 (2000).
Black, J. A., Nikolajsen, L., Kroner, K., Jensen, T. S. & Waxman, S. G. Multiple sodium channel isoforms and mitogen-activated protein kinases are present in painful human neuromas. Ann. Neurol. 64, 644–653 (2008).
Roza, C., Laird, J. M., Souslova, V., Wood, J. N. & Cervero, F. The tetrodotoxin-resistant Na+ channel Nav1.8 is essential for the expression of spontaneous activity in damaged sensory axons of mice. J.Physiol 550, 921–926 (2003).
Matzner, O. & Devor, M. Hyperexcitability at sites of nerve injury depends on voltage-sensitive Na+ channels. J. Neurophysiol. 72, 349–359 (1994).
Omana-Zapata, I., Khabbaz, M. A., Hunter, J. C., Clarke, D. E. & Bley, K. R. Tetrodotoxin inhibits neuropathic ectopic activity in neuromas, dorsal root ganglia and dorsal horn neurons. Pain 72, 41–49 (1997).
Gold, M. S. et al. Redistribution of NaV1.8 in uninjured axons enables neuropathic pain. J. Neurosci. 23, 158–166 (2003).
Nassar, M. A., Levato, A., Stirling, L. C. & Wood, J. N. Neuropathic pain develops normally in mice lacking both Nav1.7 and Nav1.8. Mol. Pain 1, 24 (2005).
Minett, M. S. et al. Pain without nociceptors? Nav1.7-independent pain mechanisms. Cell Rep. 6, 301–312 (2014).
Knudsen, L. F., Terkelsen, A. J., Drummond, P. D. & Birklein, F. Complex regional pain syndrome: a focus on the autonomic nervous system. Clin. Auton. Res. 29, 457–467 (2019).
Wheeler, D. W., Lee, M. C., Harrison, E. K., Menon, D. K. & Woods, C. G. Case report: neuropathic pain in a patient with congenital insensitivity to pain. F1000Res 3, 135 (2014).
Price, N. et al. Safety and efficacy of a topical sodium channel inhibitor (TV-45070) in patients with postherpetic neuralgia (PHN): a randomized, controlled, proof-of-concept, crossover study, with a subgroup analysis of the Nav1.7 R1150W genotype. Clin. J. Pain 33, 310–318 (2017).
Hockley, J. R. et al. P2Y receptors sensitize mouse and human colonic nociceptors. J. Neurosci. 36, 2364–2376 (2016).
Shields, S. D. et al. Insensitivity to pain upon adult-onset deletion of Nav1.7 or its blockade with selective inhibitors. J. Neurosci. 38, 10180–10201 (2018). This paper shows that adult-onset deletion of Nav1.7 produces analgesia in mice, suggesting that Nav1.7 loss-of-function analgesia is not developmental.
Branco, T. et al. Near-perfect synaptic integration by Nav1.7 in hypothalamic neurons regulates body weight. Cell 165, 1749–1761 (2016).
Grubinska, B. et al. Rat NaV1.7 loss-of-function genetic model: deficient nociceptive and neuropathic pain behavior with retained olfactory function and intra-epidermal nerve fibers. Mol. Pain 15, 1744806919881846 (2019).
Nagi, S. S. et al. An ultrafast system for signaling mechanical pain in human skin. Sci. Adv. 5, eaaw1297 (2019).
Abrahamsen, B. et al. The cell and molecular basis of mechanical, cold, and inflammatory pain. Science 321, 702–705 (2008).
Tsukamoto, T. et al. Differential binding of tetrodotoxin and its derivatives to voltage-sensitive sodium channel subtypes (Nav 1.1 to Nav 1.7). Br. J. Pharmacol. 174, 3881–3892 (2017).
Bricelj, V. M. et al. Sodium channel mutation leading to saxitoxin resistance in clams increases risk of PSP. Nature 434, 763–767 (2005).
Sangameswaran, L. et al. A novel tetrodotoxin-sensitive, voltage-gated sodium channel expressed in rat and human dorsal root ganglia. J. Biol. Chem. 272, 14805–14809 (1997).
Akopian, A. N., Sivilotti, L. & Wood, J. N. A tetrodotoxin-resistant voltage-gated sodium channel expressed by sensory neurons. Nature 379, 257–262 (1996).
Jarvis, M. F. et al. A-803467, a potent and selective Nav1.8 sodium channel blocker, attenuates neuropathic and inflammatory pain in the rat. Proc. Natl Acad. Sci. USA 104, 8520–8525 (2007).
Payne, C. E. et al. A novel selective and orally bioavailable Nav 1.8 channel blocker, PF-01247324, attenuates nociception and sensory neuron excitability. Br. J. Pharmacol. 172, 2654–2670 (2015).
Lin, Z., Santos, S., Padilla, K., Printzenhoff, D. & Castle, N. A. Biophysical and pharmacological characterization of Nav1.9 voltage dependent sodium channels stably expressed in HEK-293 cells. PLoS ONE 11, e0161450 (2016).
Chisholm, K. I., Khovanov, N., Lopes, D. M., La Russa, F. & McMahon, S. B. Large scale in vivo recording of sensory neuron activity with GCaMP6. eNeuro https://doi.org/10.1523/eneuro.0417-17.2018 (2018). This study demonstrates how in vivo optical imaging techniques can be used to further our understanding of peripheral pain signalling mechanisms.
Wang, F. et al. Sensory afferents use different coding strategies for heat and cold. Cell Rep. 23, 2001–2013 (2018).
Luiz, A. P. et al. Cold sensing by NaV1.8-positive and NaV1.8-negative sensory neurons. Proc. Natl Acad. Sci. USA 116, 3811–3816 (2019).
Acknowledgements
The authors thank E. Stevens for useful comments on the manuscript.
Author information
Authors and Affiliations
Contributions
G.G. and S.B.M. contributed equally to this work.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information
Nature Reviews Neuroscience thanks Ewan St. John Smith and the other anonymous reviewer(s) for their contribution to the peer review of this manuscript.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Glossary
- Paroxysmal extreme pain disorder
-
A type of peripheral neuropathy characterized by skin redness, warmth and attacks of severe pain in various parts of the body.
- Pruritus
-
Severe itching of the skin.
- Brugada syndrome
-
A genetic disorder that can cause a dangerous irregular heartbeat.
- Microneurography
-
A neurophysiological technique for recording electrical activity from peripheral nerve fibres.
- Hyperalgesia
-
Increased pain from a stimulus that normally provokes pain.
- Neuroma
-
A disorganized growth of nerve fibres at the site of nerve injury.
Rights and permissions
About this article

Cite this article
Goodwin, G., McMahon, S.B. The physiological function of different voltage-gated sodium channels in pain. Nat Rev Neurosci 22, 263–274 (2021). https://doi.org/10.1038/s41583-021-00444-w
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41583-021-00444-w
This article is cited by
-
Anxiety and dysautonomia symptoms in patients with a NaV1.7 mutation and the potential benefits of low-dose short-acting guanfacine
Clinical Autonomic Research (2024)
-
Fabrication of lidocaine-loaded polymer dissolving microneedles for rapid and prolonged local anesthesia
Biomedical Microdevices (2024)
-
Naphthylisoquinoline alkaloids, a new structural template inhibitor of Nav1.7 sodium channel
Acta Pharmacologica Sinica (2023)
-
Cannabidiol inhibits Nav channels through two distinct binding sites
Nature Communications (2023)
-
Peripheral Mechanism of Cancer-Induced Bone Pain
Neuroscience Bulletin (2023)
