
Abstract
Most genetic variants that contribute to disease1 are challenging to correct efficiently and without excess byproducts2,3,4,5. Here we describe prime editing, a versatile and precise genome editing method that directly writes new genetic information into a specified DNA site using a catalytically impaired Cas9 endonuclease fused to an engineered reverse transcriptase, programmed with a prime editing guide RNA (pegRNA) that both specifies the target site and encodes the desired edit. We performed more than 175 edits in human cells, including targeted insertions, deletions, and all 12 types of point mutation, without requiring double-strand breaks or donor DNA templates. We used prime editing in human cells to correct, efficiently and with few byproducts, the primary genetic causes of sickle cell disease (requiring a transversion in HBB) and Tay–Sachs disease (requiring a deletion in HEXA); to install a protective transversion in PRNP; and to insert various tags and epitopes precisely into target loci. Four human cell lines and primary post-mitotic mouse cortical neurons support prime editing with varying efficiencies. Prime editing shows higher or similar efficiency and fewer byproducts than homology-directed repair, has complementary strengths and weaknesses compared to base editing, and induces much lower off-target editing than Cas9 nuclease at known Cas9 off-target sites. Prime editing substantially expands the scope and capabilities of genome editing, and in principle could correct up to 89% of known genetic variants associated with human diseases.
Similar content being viewed by others

Main
The ability to make virtually any targeted change in the genome of any living cell or organism is a longstanding aspiration of the life sciences. Despite rapid advances in genome editing technologies, the majority of the more than 75,000 known disease-associated genetic variants in humans1 remain difficult to correct or install in most therapeutically relevant cell types (Fig. 1a). Programmable nucleases such as CRISPR–Cas9 make double-strand DNA breaks (DSBs) that can disrupt genes by inducing mixtures of insertions and deletions (indels) at target sites2,3,4. DSBs, however, are associated with undesired outcomes, including complex mixtures of products, translocations5, and activation of p536,7. Moreover, the vast majority of pathogenic alleles arise from specific insertions, deletions, or base substitutions that require more precise editing technologies to correct (Fig. 1a, Supplementary Discussion). Homology-directed repair (HDR) stimulated by DSBs8 has been widely used to install precise DNA changes. HDR, however, relies on exogenous donor DNA repair templates, typically generates an excess of indels from end-joining repair of DSBs, and is inefficient in most therapeutically relevant cell types (T cells and some types of stem cell being important exceptions)9,10. Whereas enhancing the efficiency and precision of DSB-mediated editing remains the focus of promising efforts11,12,13,14,15, these challenges motivate the exploration of alternative precision genome editing strategies.

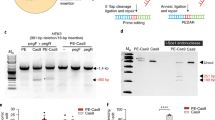
a, The 75,122 known pathogenic human genetic variants in ClinVar (accessed July, 2019), classified by type. b, A prime editing complex consists of a PE protein containing an RNA-guided DNA-nicking domain, such as Cas9 nickase, fused to an RT domain and complexed with a pegRNA. The PE–pegRNA complex enables a variety of precise DNA edits at a wide range of positions. spCas9, Streptococcus pyogenes Cas9. c, The PE–pegRNA complex binds the target DNA and nicks the PAM-containing strand. The resulting 3′ end hybridizes to the PBS, then primes reverse transcription of new DNA containing the desired edit using the RT template of the pegRNA. Equilibration between the edited 3′ flap and the unedited 5′ flap, cellular 5′ flap cleavage and ligation, and DNA repair results in stably edited DNA. d, In vitro primer extension assays with 5′-extended pegRNAs, pre-nicked dsDNA substrates containing 5′-Cy5-labelled PAM strands, dCas9, and a commercial M-MLV RT variant (RT, Superscript III). dCas9 was complexed with pegRNAs, then added to DNA substrates along with the indicated components. After 1 h, reactions were analysed by denaturing PAGE to visualize Cy5 fluorescence. e, Primer extension assays performed as in d using 3′-extended pegRNAs pre-complexed with dCas9 or Cas9(H840A) nickase, and pre-nicked or non-nicked dsDNA substrates. f, Yeast colonies transformed with GFP–mCherry fusion reporter plasmids edited in vitro with pegRNAs, Cas9 nickase, and RT. Plasmids containing nonsense or frameshift mutations between GFP and mCherry were edited with pegRNAs that restored mCherry translation via transversion, 1-bp insertion, or 1-bp deletion. GFP and mCherry double-positive cells (yellow) reflect successful editing. Images in d–f are representative of n = 2 independent replicates. For gel source data, see Supplementary Fig. 1.
Base editing can efficiently install the four transition mutations (C→T, G→A, A→G, and T→C) without requiring DSBs in many cell types and organisms, including mammals16,17,18,19, but cannot currently perform the eight transversion mutations (C→A, C→G, G→C, G→T, A→C, A→T, T→A, and T→G), such as the T•A-to-A•T mutation needed to directly correct the most common cause of sickle cell disease (HBB(E6V)). In addition, no DSB-free method has been reported to perform targeted deletions, such as the removal of the four-base duplication that causes Tay-Sachs disease (HEXA1278+TATC), or targeted insertions, such as the three-base insertion required to directly correct the most common cause of cystic fibrosis (CFTR(ΔF508)). Targeted transversions, insertions, and deletions are therefore difficult to install or correct efficiently and without excess byproducts in most cell types, even though they collectively account for most known pathogenic alleles (Fig. 1a).
Here we describe the development of prime editing, a ‘search-and-replace’ genome editing technology that mediates targeted insertions, deletions, all 12 possible base-to-base conversions, and combinations thereof in human cells without requiring DSBs or donor DNA templates. Prime editors (PEs), initially exemplified by PE1, use a reverse transcriptase (RT) fused to an RNA-programmable nickase and a prime editing guide RNA (pegRNA) to copy genetic information directly from an extension on the pegRNA into the target genomic locus. PE2 uses an engineered RT to increase editing efficiencies, while PE3 nicks the non-edited strand to induce its replacement and further increase editing efficiency, typically to 20–50% with 1–10% indel formation in human HEK293T cells. Prime editing offers much lower off-target activity than Cas9 at known Cas9 off-target loci, far fewer byproducts and higher or similar efficiency compared to Cas9-initiated HDR, and complementary strengths and weaknesses compared to base editors. By enabling precise targeted insertions, deletions, and all 12 possible classes of point mutations without requiring DSBs or donor DNA templates, prime editing has the potential to advance the study and correction of the vast majority of pathogenic alleles.
Prime editing strategy
Cas9 targets DNA using a guide RNA containing a spacer sequence that hybridizes to the target DNA site2,3,4,20,21. We envisioned the generation of guide RNAs that both specify the DNA target and contain new genetic information that replaces target DNA nucleotides. To transfer information from these engineered guide RNAs to target DNA, we proposed that genomic DNA, nicked at the target site to expose a 3′-hydroxyl group, could be used to prime the reverse transcription of an edit-encoding extension on the engineered guide RNA (the pegRNA) directly into the target site (Fig. 1b, c, Supplementary Discussion).
These initial steps result in a branched intermediate with two redundant single-stranded DNA flaps: a 5′ flap that contains the unedited DNA sequence and a 3′ flap that contains the edited sequence copied from the pegRNA (Fig. 1c). Although hybridization of the perfectly complementary 5′ flap to the unedited strand is likely to be thermodynamically favoured, 5′ flaps are the preferred substrate for structure-specific endonucleases such as FEN122, which excises 5′ flaps generated during lagging-strand DNA synthesis and long-patch base excision repair. The redundant unedited DNA may also be removed by 5′ exonucleases such as EXO123.
We reasoned that preferential 5′ flap excision and 3′ flap ligation could drive the incorporation of the edited DNA strand, creating heteroduplex DNA containing one edited strand and one unedited strand (Fig. 1c). DNA repair to resolve the heteroduplex by copying the information in the edited strand to the complementary strand would permanently install the edit (Fig. 1c). On the basis of a similar strategy we developed to favourably resolve heteroduplex DNA during base editing16,17,18, we hypothesized that nicking the non-edited DNA strand might bias DNA repair to preferentially replace the non-edited strand.
Validation in vitro and in yeast
First, we tested whether the 3′ end of the protospacer-adjacent motif (PAM)-containing DNA strand cleaved by the RuvC nuclease domain of Cas9 was sufficiently accessible to prime reverse transcription. We designed pegRNAs by adding to single guide RNAs (sgRNAs) a primer binding site (PBS) that allows the 3′ end of the nicked DNA strand to hybridize to the pegRNA, and an RT template containing the desired edit (Fig. 1c). We constructed candidate pegRNAs by extending sgRNAs on either end with a PBS sequence (5–6 nucleotides (nt)) and an RT template (7–22 nt), and confirmed that 5′-extended pegRNAs support Cas9 binding to target DNA in vitro and that both 5′-extended and 3′-extended pegRNAs support Cas9-mediated DNA nicking in vitro and DNA cleavage in mammalian cells (Extended Data Fig. 1a–c). Next, we tested the compatibility of these candidate pegRNAs with reverse transcription using pre-nicked 5′-Cy5-labelled double-stranded DNA (dsDNA) substrates, catalytically dead Cas9 (dCas9), and a commercial Moloney murine leukaemia virus (M-MLV) RT variant (Extended Data Fig. 1d). When all components were present, the labelled DNA strand was efficiently converted into longer DNA products with gel mobilities consistent with reverse transcription along the RT template (Fig. 1d, Extended Data Fig. 1d, e). Omission of dCas9 led to nick translation products that resulted from RT-mediated DNA polymerization on the DNA template, with no pegRNA information transfer. No DNA polymerization products were observed when the pegRNA was replaced by a conventional sgRNA (Fig. 1d). These results demonstrate that nicked DNA exposed by dCas9 is competent to prime reverse transcription from a pegRNA.
Next, we tested non-nicked dsDNA substrates with a Cas9(H840A) nickase that nicks the PAM-containing strand2. In these reactions, 5′-extended pegRNAs generated reverse transcription products inefficiently (Extended Data Fig. 1f), but 3′-extended pegRNAs enabled efficient Cas9 nicking and reverse transcription (Fig. 1e). The use of 3′-extended pegRNAs generated only a single apparent product, despite the theoretical possibility that reverse transcription could terminate anywhere within the pegRNA. DNA sequencing of reactions with Cas9 nickase, RT, and 3′-extended pegRNAs revealed that the complete RT template sequence was reverse transcribed into the DNA substrate (Extended Data Fig. 1g). These experiments establish that 3′-extended pegRNAs can direct Cas9 nickase and template reverse transcription in vitro.
To evaluate the eukaryotic cell DNA repair outcomes of 3′ flaps produced by pegRNA-programmed reverse transcription in vitro, we performed in vitro prime editing on reporter plasmids, then transformed the reaction products into yeast cells (Extended Data Fig. 2). We constructed reporter plasmids encoding EGFP and mCherry separated by a linker containing an in-frame stop codon, +1 frameshift, or −1 frameshift. When plasmids were edited in vitro with Cas9 nickase, RT, and 3′-extended pegRNAs encoding a transversion that corrects the premature stop codon, 37% of yeast transformants expressed both GFP and mCherry (Fig. 1f, Extended Data Fig. 2). Reactions edited with 5′-extended pegRNAs yielded fewer GFP and mCherry double-positive colonies (9%). Productive editing was also observed using 3′-extended pegRNAs that insert a single nucleotide (15%) or delete a single nucleotide (29%) to correct frameshift mutations (Fig. 1f, Extended Data Fig. 2). These results demonstrate that DNA repair in eukaryotic cells can resolve 3′ DNA flaps from prime editing to incorporate precise transversions, insertions, and deletions.
Prime editor 1
Encouraged by these observations, we sought to develop a prime editing system with a minimum number of components that could edit genomic DNA in mammalian cells. We transfected HEK293T cells with one plasmid encoding a fusion of the wild-type M-MLV RT through a flexible linker to either terminus of the Cas9(H840A) nickase, and a second plasmid encoding a pegRNA (Extended Data Fig. 3a). Initial attempts led to no detectable editing.
Extension of the PBS in the pegRNA to 8–15 bases, however, led to detectable installation of a transversion at the HEK293 site 3 (hereafter referred to as HEK3) target site, with higher efficiencies when the RT was fused to the C terminus of Cas9 nickase than when it was fused to the N terminus (Extended Data Fig. 3b). These results suggest that wild-type M-MLV RT fused to Cas9 requires longer PBS sequences for genome editing in human cells compared to what is required in vitro using the commercial variant of M-MLV RT supplied in trans. We designated this M-MLV RT fused to the C terminus of Cas9(H840A) nickase as PE1.
We tested the ability of PE1 to introduce transversion point mutations at four additional genomic sites specified by the pegRNA (Fig. 2a). Editing efficiency at these sites was dependent on PBS length, with maximal editing efficiencies reaching 0.7–5.5% (Fig. 2a). Indels from PE1 were minimal, averaging 0.2 ± 0.1% (mean ± s.d.) for the five sites under conditions that maximized each site’s editing efficiency (Extended Data Fig. 3a–f). PE1 also mediated targeted insertions and deletions with 4–17% efficiency at the HEK3 locus (Fig. 2a). These findings show that PE1 can directly install targeted transversions, insertions, and deletions without requiring DSBs or DNA templates.

a, Use of an engineered M-MLV reverse transcriptase (D200N, L603W, T306K, W313F, T330P) in PE2 substantially improves prime editing efficiencies at five genomic sites in HEK293T cells, and small insertion and small deletion edits at HEK3. b, PE2 editing efficiencies with varying RT template lengths at five genomic sites in HEK293T cells. Editing efficiencies reflect sequencing reads that contain the intended edit and do not contain indels among all treated cells, with no sorting. Mean ± s.d. of n = 3 independent biological replicates.
Prime editor 2
We hypothesized that engineering the RT in PE1 might improve the efficiency of DNA synthesis during prime editing. M-MLV RT mutations that increase thermostability24,25, processivity24, and DNA–RNA substrate affinity26, and that inactivate RNaseH activity27, have been reported. We constructed 19 variants of PE1 containing a variety of RT mutations to evaluate their editing efficiency in human cells.
First, we investigated M-MLV RT variants that support reverse transcription at elevated temperatures24. Introduction of D200N, L603W and T330P into M-MLV RT, hereafter referred to as M3, led to a 6.8-fold average increase in transversion and insertion editing efficiency across five genomic loci in HEK293T cells compared to PE1 (Extended Data Fig. 4).
We tested additional RT mutations that have been shown to enhance binding to the template–PBS complex, enzyme processivity, and thermostability26. Among the 14 additional mutants analysed, addition of T306K and W313F to M3 improved editing efficiency an additional 1.3-fold to 3.0-fold for six transversion or insertion edits across five genomic sites (Extended Data Fig. 4). This pentamutant RT incorporated into PE1 (Cas9(H840A)–M-MLV RT(D200N/L603W/T330P/T306K/W313F)) is hereafter referred to as prime editor 2 (PE2).
PE2 installs single-nucleotide transversion, insertion, and deletion mutations with substantially higher efficiency than PE1, and is compatible with shorter PBS sequences, consistent with enhanced engagement of transient genomic DNA–PBS complexes (Fig. 2a). On average, PE2 led to a 1.6- to 5.1-fold improvement in the efficiency of prime editing point mutations over PE1. PE2 also performed targeted insertions and deletions more efficiently than PE1 (Fig. 2a, Extended Data Fig. 4d).
Optimization of pegRNAs
We systematically probed the relationship between pegRNA structure and PE2 editing efficiency. Priming regions with lower G/C content generally required longer PBS sequences, consistent with the energetic requirements of hybridization of the nicked DNA strand to the pegRNA PBS (Fig. 2a). No PBS length or G/C content level was strictly predictive of editing efficiency, suggesting that other factors such as DNA primer or RT template secondary structure also influence editing activity. We recommend starting with a PBS length of about 13 nt, and testing different PBS lengths during optimization, especially if the priming region deviates from about 40–60% G/C content.
Next, we systematically evaluated pegRNAs with RT templates 10–20 nt long at five genomic target sites using PE2 (Fig. 2b), and with RT templates up to 31 nt at three genomic sites (Extended Data Fig. 5a–c). As with PBS length, RT template length could also be varied to maximize prime editing efficiency, although many RT template lengths of ten or more nucleotides performed comparably. As some target sites preferred longer RT templates (more than 15 nt; FANCF, EMX1), whereas other loci preferred shorter RT templates (HEK3 and HEK293 site 4, hereafter referred to as HEK4) (Fig. 2b), we recommend starting with about 10–16 nt and testing shorter and longer RT templates during pegRNA optimization.
Notably, the use of RT templates that place a C adjacent to the 3′ hairpin of the sgRNA scaffold generally resulted in lower editing efficiency (Extended Data Fig. 5a–c). We speculate that a C as the first nucleotide of the 3′ extension can disrupt guide RNA structure by pairing with G81, which normally forms a pi stack with Y1356 in Cas9 and a non-canonical base pair with A68 of the sgRNA28. Because many RT template lengths support prime editing, we recommend designing pegRNAs so that the first base of the 3′ extension is not C.
Prime editor 3 systems
The resolution of heteroduplex DNA from PE2 containing one edited and one non-edited strand determines long-term editing outcomes. To optimize base editing we previously used Cas9 nickase to nick the non-edited strand, directing DNA repair to that strand using the edited strand as a template16,17,18. To apply this strategy to enhance prime editing, we tested nicking the non-edited strand using the Cas9(H840A) nickase already present in PE2 and a simple sgRNA (Fig. 3a). As the edited DNA strand is also nicked to initiate prime editing, we tested a variety of nick locations on the non-edited strand to minimize DSBs that lead to indels.

a, Overview of prime editing by PE3. After initial synthesis of the edited strand, 5′ flap excision leaves behind a DNA heteroduplex containing one edited strand and one non-edited strand. Mismatch repair resolves the heteroduplex to give either edited or non-edited products. Nicking the non-edited strand favours repair of that strand, resulting in preferential generation of duplex DNA containing the desired edit. b, The effect of complementary strand nicking on prime editing efficiency and indel formation. ‘None’ refers to PE2 controls, which do not nick the complementary strand. c, Comparison of editing efficiencies with PE2, PE3, and PE3b (edit-specific complementary strand nick). Editing efficiencies reflect sequencing reads that contain the intended edit and do not contain indels among all treated cells, with no sorting. Mean ± s.d. of n = 3 independent biological replicates.
We first tested this strategy, designated PE3, at five genomic sites in HEK293T cells using sgRNAs that induce nicks 14–116 nt away from the site of the pegRNA-induced nick. In four of the five sites tested, nicking the non-edited strand increased editing efficiency by 1.5- to 4.2-fold compared to PE2, to as high as 55% (Fig. 3b). Although the optimal nicking position varied depending on the genomic site (Supplementary Discussion), nicks positioned 3′ of the edit about 40–90 bp from the pegRNA-induced nick generally increased editing efficiency (averaging 41%) without excess indel formation (6.8% average indels for the sgRNA with the highest editing efficiency) (Fig. 3b). We recommend starting with non-edited strand nicks about 50 bp from the pegRNA-mediated nick, and testing alternative nick locations if indel frequencies exceed acceptable levels.
Nicking the non-edited strand only after resolution of the edited strand flap should minimize the presence of concurrent nicks, thereby minimizing formation of DSBs and indels. To achieve this goal, we designed sgRNAs with spacers that matched the edited strand, but not the original allele. Using this strategy, denoted PE3b, mismatches between the spacer and the unedited allele should disfavour sgRNA nicking until after editing of the PAM strand has taken place. PE3b resulted in a 13-fold decrease in the average number of indels (0.74%) compared to PE3, without any evident decrease in editing efficiency (Fig. 3c). When the edit lies within a second protospacer, we recommend the PE3b approach.
Together, these findings establish that PE3 systems improve editing efficiencies about threefold compared with PE2, albeit with a higher range of indels than PE2. When it is possible to nick the non-edited strand with an sgRNA that requires editing before nicking, the PE3b system offers PE3-like editing levels while greatly reducing indel formation.
To demonstrate the targeting scope and versatility of prime editing with PE3, we performed all 24 possible single-nucleotide substitutions across the +1 to +8 positions (counting the first base 3′ of the pegRNA-induced nick as position +1) of the HEK3 target site using PE3 and pegRNAs with 10-nt RT templates (Fig. 4a). These 24 edits collectively cover all 12 possible transition and transversion mutations, and proceeded with average editing efficiencies (containing no indels) of 33 ± 7.9%, with 7.5 ± 1.8% average indels.

a, All 12 types of single-nucleotide edit from position +1 to +8 of the HEK3 site using a 10-nt RT template, counting the first nucleotide following the pegRNA-induced nick as position +1. b, Long-range PE3 edits at HEK3 using a 34-nt RT template. c–e, PE3-mediated transition and transversion edits at the specified positions for RNF2 (c), RUNX1 (d), and VEGFA (e). f, Targeted 1- and 3-bp insertions, and 1- and 3-bp deletions with PE3 at seven endogenous genomic loci. g, Targeted precise deletions of 5–80 bp at HEK3. h, Combination edits at three endogenous genomic loci. Editing efficiencies reflect sequencing reads that contain the intended edit and do not contain indels among all treated cells, with no sorting. Mean ± s.d. of n = 3 independent biological replicates.
Notably, long-distance RT templates can also give rise to efficient prime editing. Using PE3 with a 34-nt RT template, we installed point mutations at positions +12, +14, +17, +20, +23, +24, +26, +30, and +33 in the HEK3 locus with 36 ± 8.7% average efficiency and 8.6 ± 2.0% indels (Fig. 4b). Other RT templates of 30 or more nucleotides at three other genomic sites also supported prime editing (Extended Data Fig. 5a–c). As an NGG PAM on either DNA strand occurs on average every 8 bp, far less than edit-to-PAM distances that support efficient prime editing, prime editing is not substantially constrained by the availability of a nearby PAM sequence, in contrast to other precision editing methods11,15,16. Given the presumed relationship between RNA secondary structure and prime editing efficiency, when designing pegRNAs for long-range edits we recommend testing RT templates of various lengths and, if necessary, sequence compositions (for example, using synonymous codons).
To further test the scope and limitations of PE3 for introducing point mutations, we tested 72 additional edits covering all possible types of point mutation across six additional genomic target sites (Fig. 4c–e, Extended Data Fig. 5d–f). Editing efficiency averaged 25 ± 14%, while indel formation averaged 8.3 ± 7.5%. Because the pegRNA RT template includes the PAM sequence, prime editing can induce changes in the PAM sequence. In these cases, we observed higher editing efficiency (averaging 39 ± 9.7%) and lower indel generation (averaging 5.0 ± 2.9%; Fig. 4, mutations at +5 or +6), potentially due to the inability of Cas9 nickase to re-bind and nick the edited strand before the repair of the complementary strand. We recommend editing the PAM, in addition to other desired changes, whenever possible.
Next, we performed 28 targeted small insertions and small deletions at seven genomic sites using PE3 (Fig. 4f). Targeted 1-bp and 3-bp insertions proceeded with an average efficiency of 32 ± 9.8% and 39 ± 16%, respectively. Targeted 1-bp and 3-bp deletions were also efficient, averaging 29 ± 14% and 32 ± 11% editing, respectively. Indel generation (beyond the target insertion or deletion) averaged 6.8 ± 5.4%. Because insertions and deletions between positions +1 and +6 alter the location or structure of the PAM, we speculate that insertions or deletions at these positions are more efficient because they prevent re-engagement of the edited strand.
We also tested PE3 for its ability to mediate larger precise deletions of 5–80 bp at the HEK3 site (Fig. 4g). We observed very high editing efficiencies (52–78%) for precise 5-, 10-, 15-, 25-, and 80-bp deletions, with indels averaging 11 ± 4.8%. Finally, we tested the ability of PE3 to mediate 12 combinations of insertions, deletions, and/or point mutations across three genomic sites. These combination edits were also very efficient, averaging 55% editing with 6.4% indels (Fig. 4h). Together, the 156 distinct edits in Fig. 4 and Extended Data Fig. 5d–f establish the versatility, precision, and targeting flexibility of PE3 systems.
Prime editing compared with base editing
Cytidine base editors (CBEs) and adenine base editors (ABEs) can install transition mutations efficiently and with few indels16,17,18. The application of base editing can be limited by unwanted bystander edits from the presence of multiple cytidine or adenine bases within the base editing activity window16,17,18,29, or by the absence of a PAM positioned about 15 ± 2 nt from the target nucleotide16,30. We anticipated that prime editing could complement base editing when bystander edits are unacceptable or when the target site lacks a suitably positioned PAM.
We compared PEs and CBEs at three genomic loci that contain multiple target cytosines in the canonical base editing window (protospacer positions 4–8, counting the PAM as positions 21–23) using current-generation CBEs31 without or with nickase activity (BE2max and BE4max, respectively), or using analogous PE2 and PE3 prime editing systems. Among the nine total cytosines within the base editing windows of the three sites, BE4max yielded 2.2-fold higher average total C•G-to-T•A conversion than PE3 for bases in the centre of the base editing window (protospacer positions 5–7, Extended Data Fig. 6a). However, PE3 outperformed BE4max by 2.7-fold at cytosines positioned outside the centre of the base editing window. Overall, indel frequencies for PE2 were very low (averaging 0.86 ± 0.47%), and for PE3 were similar to or modestly higher than that of BE4max (PE3: 2.5–21%; BE4max: 2.5–14%) (Extended Data Fig. 6b).
For the installation of precise edits (with no bystander editing), the efficiency of prime editing greatly exceeded that of base editing at the above sites, which, like most genomic DNA sites, contain multiple cytosines within the base editing window. BE4max generated few products containing only the single target base-pair conversion with no bystander edits. By contrast, prime editing at this site could be used to selectively install a C•G-to-T•A edit at any position or combination of positions (Extended Data Fig. 6c).
We also compared nicking and non-nicking adenine base editors (ABEs) with PE3 and PE2, with similar results (Extended Data Fig. 6d–f, Supplementary Discussion). Collectively, these results indicate that base editing and prime editing offer complementary strengths and weaknesses for making targeted transition mutations. When a single target nucleotide is present within the base editing window, or when bystander edits are acceptable, current base editors are typically more efficient and generate fewer indels than prime editors. When multiple cytosines or adenines are present and bystander edits are undesirable, or when PAMs that position target nucleotides for base editing are not available, prime editors offer substantial advantages.
Off-target prime editing
Prime editing requires target DNA–pegRNA spacer complementarity for the Cas9 domain to bind, target DNA–pegRNA PBS complementarity to initiate pegRNA-templated reverse transcription, and target DNA–RT product complementarity for flap resolution. To test whether these three distinct DNA hybridization steps reduce off-target prime editing compared to editing methods that require only target–guide RNA complementarity, we treated HEK293T cells with PE3 or PE2 and 16 pegRNAs that target four genomic loci, each of which has at least four well-characterized Cas9 off-target sites32,33. We also treated cells with Cas9 nuclease and the same 16 pegRNAs, or with Cas9 and four sgRNAs targeting the same four protospacers (Supplementary Table 1).
Consistent with previous studies32, Cas9 and sgRNAs targeting HEK3, HEK4, EMX1, and FANCF modified the top four known Cas9 off-target loci for each sgRNA with average frequencies of 16 ± 16%, 60 ± 26%, 48 ± 28%, and 4.3 ± 5.6%, respectively (Extended Data Fig. 6g). Cas9 with pegRNAs modified on-target sites with similar efficiency as Cas9 with sgRNAs, whereas Cas9 with pegRNAs modified off-target sites at 4.4-fold lower average efficiency than Cas9 with sgRNAs.
Strikingly, PE3 or PE2 with the same 16 pegRNAs containing these four target spacers resulted in detectable off-target editing at only 3 out of 16 off-target sites, with only 1 of 16 showing an off-target editing efficiency of 1% or more (Extended Data Fig. 6h). Average off-target prime editing for pegRNAs targeting HEK3, HEK4, EMX1, and FANCF at the top four known Cas9 off-target sites for each protospacer was <0.1%, <2.2 ± 5.2%, <0.1%, and <0.13 ± 0.11%, respectively (Extended Data Fig. 6h). Notably, at the HEK4 off-target 3 site that was edited by Cas9 with pegRNA1 at 97% efficiency, PE2 with pegRNA1 resulted in only 0.2% off-target editing despite sharing the same pegRNA, demonstrating how the two additional hybridization events required for prime editing can greatly reduce off-target modification. Together, these results suggest that prime editing induces much lower off-target editing than Cas9 at known Cas9 off-target sites.
Reverse transcription of 3′-extended pegRNAs in principle can proceed into the guide RNA scaffold, resulting in scaffold sequence insertion that contributes to indels at the target locus. We analysed 66 PE3 editing experiments at four loci in HEK293T cells and observed 1.7 ± 1.5% average total insertion of any number of pegRNA scaffold nucleotides (Extended Data Fig. 7). We speculate that inaccessibility of the guide RNA scaffold to reverse transcription due to Cas9 domain binding, and cellular excision of the mismatched 3′ end of 3′ flaps that extend into the pegRNA scaffold, minimize products that incorporate pegRNA scaffold nucleotides.
The presence of endogenous human RTs from retroelements34 and telomerase suggests that RT activity is not inherently toxic to human cells. Indeed, we observed no differences in the viability of HEK293T cells expressing dCas9, Cas9(H840A) nickase, PE2, or PE2 with R110S and K103L mutations (PE2-dRT) that inactivate the RT and abolish prime editing35 (Extended Data Fig. 8a, b). To evaluate changes in the cellular transcriptome that result from prime editing, we performed RNA sequencing (RNA-seq) on HEK293T cells expressing PE2, PE2-dRT, or Cas9(H840A) nickase together with a PRNP-targeting or HEXA-targeting pegRNA (Extended Data Fig. 8c–k), and observed that active PE2 minimally perturbed the transcriptome relative to Cas9 nickase or a control lacking active RT (Supplementary Discussion).
Prime editing pathogenic mutations
We tested the ability of PE3 to directly install or correct in human cells transversion, insertion, and deletion mutations that cause genetic diseases. Sickle cell disease is caused by a A•T-to-T•A transversion mutation in HBB, resulting in an E6V mutation in β-globin (Supplementary Discussion). We used PE3 to install this HBB mutation into HEK293T cells with 44% efficiency and 4.8% indels (Fig. 5a) and isolated from a single prime editing experiment six HEK293T cell lines that were homozygous (triploid) for the mutated HBB allele (Supplementary Note 1). To correct the mutant HBB allele to wild-type HBB, we treated HEK293T cells homozygous for mutant HBB with PE3 and a pegRNA programmed to directly revert the HBB mutation to wild-type HBB. All 14 tested pegRNAs mediated efficient correction of mutant HBB to wild-type HBB (26–52% efficiency), and indel levels averaged 2.8 ± 0.70% (Extended Data Fig. 9a). Introduction of a PAM-modifying silent mutation improved editing efficiency and product purity to 58% correction with 1.4% indels (Fig. 5a).

a, Installation (via T•A-to-A•T transversion) and correction (via A•T-to-T•A transversion) of the pathogenic E6V-coding mutation in HBB in HEK293T cells. Correction either to wild-type HBB, or to HBB containing a PAM-disrupting silent mutation, is shown. b, Installation (via 4-bp insertion) and correction (via 4-bp deletion) of the pathogenic HEXA1278+TATC allele in HEK293T cells. Correction either to wild-type HEXA, or to HEXA containing a PAM-disrupting silent mutation, is shown. c, Installation of the protective G127V-coding variant in PRNP in HEK293T cells via G•C-to-T•A transversion. d, Installation of a G•C-to-T•A transversion in DNMT1 of mouse primary cortical neurons using a split-intein PE3 lentivirus system (see Methods). Sorted values reflect editing or indels from GFP-positive nuclei, while unsorted values are from all nuclei. e, f, PE3 editing and indels (e) or Cas9-initiated HDR editing and indels (f) at endogenous genomic loci in HEK293T, K562, U2OS, and HeLa cells. g, Targeted insertion of a His6 tag (18 bp), Flag epitope tag (24 bp), or extended loxP site (44 bp) in HEK293T cells by PE3. Editing efficiencies reflect sequencing reads that contain the intended edit and do not contain indels among all treated cells, with no sorting, except where specified in e. Mean ± s.d. of n = 3 independent biological replicates.
The most common mutation that causes Tay-Sachs disease is a 4-bp insertion in HEXA (HEXA1278+TATC). We used PE3 to install this 4-bp insertion into HEXA with 31% efficiency and 0.8% indels (Fig. 5b), and isolated two HEK293T cell lines that were homozygous for HEXA1278+TATC (Supplementary Note 1). We used these cells to test 43 pegRNAs and three nicking sgRNAs with PE3 or PE3b systems for correction of the pathogenic insertion in HEXA (Extended Data Fig. 9b). Nineteen of the 43 pegRNAs tested resulted in editing with an efficiency of 20% or more. Correction to wild-type HEXA with the best pegRNA proceeded with 33% efficiency and 0.32% indels using PE3b (Fig. 5b, Extended Data Fig. 9b).
Finally, we used PE3 to install a protective G•C-to-T•A transversion into PRNP (resulting in PRNP(G127V)) into HEK293T cells, introducing a mutant allele that confers resistance to prion disease in humans36 and mice37 (Supplementary Discussion). We evaluated four pegRNAs and three nicking sgRNAs. The most effective pegRNA with PE3 resulted in 53% installation of G127V, with 1.7% indels (Fig. 5c). Together, these results establish the ability of prime editing in human cells to install or correct transversion, insertion, or deletion mutations that cause or confer resistance to disease efficiently, and with few byproducts.
Other cell lines and primary neurons
Next, we tested prime editing at endogenous sites in three additional human cell lines (Extended Data Fig. 10a, Supplementary Discussion). In K562 cells, PE3 achieved three transversion edits and a His6 tag insertion with 15–30% editing efficiency and 0.85–2.2% indels (Extended Data Fig. 10a). In U2OS cells, we installed transversion mutations, as well as a 3-bp insertion and His6 tag insertion, with 7.9–22% editing efficiency and 0.13–2.2% indels (Extended Data Fig. 10a). Finally, in HeLa cells we performed a 3-bp insertion with 12% average efficiency and 1.3% indels (Extended Data Fig. 10a). Collectively, these data indicate that cell lines other than HEK293T support prime editing, although editing efficiencies vary by cell type and are generally less efficient than in HEK293T cells. Editing:indel ratios remained favourable in all human cell lines tested.
To determine whether prime editing is possible in post-mitotic, terminally differentiated primary cells, we transduced primary cortical neurons from E18.5 mice with a PE3 lentiviral delivery system in which PE2 protein components were expressed from the neuron-specific synapsin promoter38 along with a GFP marker (see Methods). Nuclei were isolated two weeks after transduction and sequenced directly, or sorted for GFP expression before sequencing. We observed 7.1% average prime editing of DNMT1 with 0.58% average indels in sorted cortical neuron nuclei (Fig. 5d). Cas9 nuclease in the same lentivirus system resulted in 31% average indels among sorted nuclei (Fig. 5d). These data indicate that post-mitotic, terminally differentiated primary cells can support prime editing.
Prime editing compared with HDR
Finally, we compared the performance of PE3 with that of optimized Cas9-initiated HDR11,14 in mitotic cell lines that support HDR14. We treated HEK293T, HeLa, K562 and U2OS cells with Cas9 nuclease, an sgRNA, and a single-stranded DNA (ssDNA) donor template designed to install a variety of transversion and insertion edits (Fig. 5e, f, Extended Data Fig. 10). Cas9-initiated HDR in all cases successfully installed the desired edit, but with far higher levels of indel byproducts than with PE3, as expected given that Cas9 induces DSBs. In HEK293T cells, the ratio of editing to indels for installation or correction of the allele encoding HBB(E6V) or installation of the allele encoding PRNP(G127V) was on average 270-fold higher for PE3 than for Cas9-initiated HDR.
Comparisons between PE3 and HDR in human cell lines other than HEK293T showed similar results, although with lower PE3 editing efficiencies (Fig. 5e, f, Supplementary Discussion). Collectively, these data indicate that HDR typically results in similar or lower editing efficiencies than PE3 with far more indels in four tested human cell lines (Extended Data Fig. 10).
Discussion and future directions
The ability to insert arbitrary DNA sequences with single-nucleotide precision is an especially promising capability of prime editing. For example, we used PE3 in HEK293T cells to precisely insert into HEK3 a His6 tag (18 bp, 65% efficiency), a Flag epitope tag (24 bp, 18% efficiency), and an extended Cre recombinase loxP site (44 bp, 23% efficiency) with 3.0–5.9% indels (Fig. 5g). We anticipate that the ability to efficiently and precisely insert new DNA sequences into target sites in living cells will enable many biotechnological and therapeutic applications.
Collectively, the prime editing experiments described here performed 19 insertions up to 44 bp, 23 deletions up to 80 bp, 119 point mutations including 83 transversions, and 18 combination edits at 12 endogenous loci in the human and mouse genomes at locations ranging from 3 bp upstream to 29 bp downstream of a PAM without making explicit DSBs. These results establish prime editing as a remarkably versatile genome editing method. Because 85–99% of insertions, deletions, indels, and duplications in ClinVar are 30 bp in length or smaller (Extended Data Fig. 11), in principle prime editing could correct up to about 89% of the 75,122 pathogenic human genetic variants in ClinVar (Fig. 1a).
Prime editing offers many possible choices of pegRNA-induced nick locations, sgRNA-induced second nick locations, PBS lengths, RT template lengths, and which strand to edit first. This flexibility, which contrasts with more limited options typically available for other precision editing methods11,15,16, allows editing efficiency, product purity, DNA specificity, and other parameters to be optimized to suit a given application (Extended Data Fig. 9).
Much additional research is needed to further understand and improve prime editing in a broad range of cell types and organisms, to assess off-target prime editing in a genome-wide manner, and to further characterize the extent to which prime editors might affect cells. Interfacing prime editing with additional in vitro and in vivo delivery strategies is essential for exploring the potential of prime editing to enable applications, including the study and treatment of genetic diseases. By enabling precise targeted transitions, transversions, insertions, and deletions in the genomes of mammalian cells without requiring DSBs, donor DNA templates, or HDR, however, prime editing provides a new search-and-replace capability that substantially expands the scope of genome editing.
Methods
General methods
DNA amplification was conducted by PCR using Phusion U Green Multiplex PCR Master Mix (ThermoFisher Scientific) or Q5 Hot Start High-Fidelity 2× Master Mix (New England BioLabs) unless otherwise noted. DNA oligonucleotides, including Cy5-labelled DNA oligonucleotides, dCas9 protein, and Cas9(H840A) protein were obtained from Integrated DNA Technologies. Yeast reporter plasmids were derived from previously described plasmids39 and cloned by the Gibson assembly method. All mammalian editor plasmids used in this work were assembled using the USER cloning method as previously described40. Plasmids expressing sgRNAs were constructed by ligation of annealed oligonucleotides into BsmBI-digested acceptor vector (Addgene plasmid no. 65777). Plasmids expressing pegRNAs were constructed by Gibson assembly or Golden Gate assembly using a custom acceptor plasmid (see Supplementary Note 3). Sequences of sgRNA and pegRNA constructs used in this work are listed in Supplementary Tables 2 and 3. All vectors for mammalian cell experiments were purified using Plasmid Plus Midiprep kits (Qiagen) or PureYield plasmid miniprep kits (Promega), which include endotoxin removal steps. All experiments using live animals were approved by the Broad Institute Institutional and Animal Care and Use Committees. Wild-type C57BL/6 mice were obtained from Charles River (#027). No statistical methods were used to predetermine sample size. The experiments were not randomized, and investigators were not blinded to allocation during experiments and outcome assessment.
In vitro biochemical assays
pegRNAs and sgRNAs were transcribed in vitro using the HiScribe T7 in vitro transcription kit (New England Biolabs) from PCR-amplified templates containing a T7 promoter sequence. RNA was purified by denaturing urea PAGE and quality-confirmed by an analytical gel before use. 5′-Cy5-labelled DNA duplex substrates were annealed using two oligonucleotides (Cy5-AVA024 and AVA025; 1:1.1 ratio) for the non-nicked substrate or three oligonucleotides (Cy5-AVA023, AVA025 and AVA026; 1:1.1:1.1) for the pre-nicked substrate by heating to 95 °C for 3 min followed by slowly cooling to room temperature (Supplementary Table 2). Cas9 cleavage and reverse transcription reactions were carried out in 1× cleavage buffer41 supplemented with dNTPs (20 mM HEPES-K, pH 7.5; 100 mM KCl; 5% glycerol; 0.2 mM EDTA, pH 8.0; 3 mM MgCl2; 0.5 mM dNTP mix; 5 mM DTT). dCas9 or Cas9(H840A) (5 µM final) and the sgRNA or pegRNA (5 µM final) were pre-incubated at room temperature in a 5-µl reaction mixture for 10 min before the addition of 0.5 μl of 4 μM duplex DNA substrate (400 nM final), followed by the addition of 0.2 μl of Superscript III reverse transcriptase (ThermoFisher Scientific), an undisclosed M-MLV RT variant, when applicable. Reactions were carried out at 37 °C for 1 h, then diluted to a volume of 10 µl with water, treated with 0.2 µl of proteinase K solution (20 mg/ml, ThermoFisher Scientific), and incubated at room temperature for 30 min. Following heat inactivation at 95 °C for 10 min, reaction products were combined with 2× formamide gel loading buffer (90% formamide; 10% glycerol; 0.01% bromophenol blue), denatured at 95 °C for 5 min, and separated by denaturing urea PAGE gel (15% TBE-urea, 55 °C, 200 V). DNA products were visualized by Cy5 fluorescence signal using a Typhoon FLA 7000 biomolecular imager.
Electrophoretic mobility shift assays were carried out in 1× binding buffer (1× cleavage buffer with 10 µg/ml heparin) using pre-incubated dCas9–sgRNA or dCas9–pegRNA complexes (concentration between 5 nM and 1 µM final) and Cy5-labelled duplex DNA (Cy5-AVA024 and AVA025; 20 nM final). After 15 min of incubation at 37 °C, the samples were analysed by native PAGE gel (10% TBE) and imaged for Cy5 fluorescence.
For DNA sequencing of reverse transcription products, fluorescent bands were excised and purified from urea PAGE gels, then 3′ tailed with terminal transferase (TdT; New England Biolabs) in the presence of dGTP or dATP according to the manufacturer’s protocol. Tailed DNA products were diluted tenfold with binding buffer (40% saturated aqueous guanidinium chloride and 60% isopropanol) and purified by QIAquick spin column (Qiagen), then used as templates for primer extension by Klenow fragment (New England Biolabs) using primer AVA134 (A-tailed products) or AVA135 (G-tailed products) (Supplementary Table 2). Extensions were amplified by PCR for 10 cycles using primers AVA110 and AVA122, then sequenced with AVA037 using the Sanger method (Supplementary Table 2).
Yeast fluorescent reporter assays
Dual fluorescent reporter plasmids containing an in-frame stop codon, a +1 frameshift, or a −1 frameshift were subjected to 5′-extended pegRNA or 3′-extended pegRNA prime editing reactions in vitro as described above using 100 ng of plasmid substrate. Following incubation at 37 °C for 1 h, the reactions were diluted with water and plasmid DNA was precipitated with 0.3 M sodium acetate and 70% ethanol. Resuspended DNA was transformed into Saccharomyces cerevisiae by electroporation as previously described42 and plated on synthetic complete medium without leucine (SC(glucose), L−). GFP and mCherry fluorescence signals were visualized from colonies with the Typhoon FLA 7000 biomolecular imager.
General mammalian cell culture conditions
HEK293T (ATCC CRL-3216), U2OS (ATTC HTB-96), K562 (CCL-243), and HeLa (CCL-2) cells were purchased from ATCC and cultured and passaged in Dulbecco’s modified Eagle’s medium (DMEM) plus GlutaMAX (ThermoFisher Scientific), McCoy’s 5A medium (Gibco), RPMI medium 1640 plus GlutaMAX (Gibco), or Eagle’s minimal essential medium (EMEM, ATCC), respectively, each supplemented with 10% (v/v) fetal bovine serum (Gibco, qualified) and 1× penicillin streptomycin (Corning). All cell types were incubated, maintained, and cultured at 37 °C with 5% CO2. Cell lines were authenticated by their respective suppliers and tested negative for mycoplasma.
HEK293T tissue culture transfection protocol and genomic DNA preparation
HEK293T cells were seeded on 48-well poly-d-lysine coated plates (Corning). Between 16 and 24 h after seeding, cells were transfected at approximately 60% confluency with 1 µl lipofectamine 2000 (Thermo Fisher Scientific) according to the manufacturer’s protocols and 750 ng PE plasmid, 250 ng pegRNA plasmid, and 83 ng sgRNA plasmid (for PE3 and PE3b). Unless otherwise stated, cells were cultured for 3 days following transfection, after which the medium was removed, the cells were washed with 1× PBS solution (Thermo Fisher Scientific), and genomic DNA was extracted by the addition of 150 µl of freshly prepared lysis buffer (10 mM Tris-HCl, pH 7.5; 0.05% SDS; 25 µg/ml proteinase K (ThermoFisher Scientific)) directly into each well of the tissue culture plate. The genomic DNA mixture was incubated at 37 °C for 1–2 h, followed by an 80 °C enzyme inactivation step for 30 min. Primers used for mammalian cell genomic DNA amplification are listed in Supplementary Table 4. For HDR experiments in HEK293T cells, 231 ng Cas9 nuclease-expression plasmid, 69 ng sgRNA-expression plasmid and 50 ng (1.51 pmol) of 100-nt ssDNA donor template (PAGE-purified; Integrated DNA Technologies) was lipofected using 1.4 µl lipofectamine 2000 (ThermoFisher) per well. Genomic DNA from all HDR experiments was purified using the Agencourt DNAdvance Kit (Beckman Coulter), according to the manufacturer’s protocol.
High-throughput DNA sequencing of genomic DNA samples
Genomic sites of interest were amplified from genomic DNA samples and sequenced on an Illumina MiSeq as previously described with the following modifications17,18. In brief, amplification primers containing Illumina forward and reverse adapters (Supplementary Table 4) were used for a first round of PCR (PCR 1) to amplify the genomic region of interest. PCR 1 reactions (25 µl) were performed with 0.5 µM of each forward and reverse primer, 1 µl genomic DNA extract and 12.5 µl Phusion U Green Multiplex PCR Master Mix. PCR reactions were carried out as follows: 98 °C for 2 min, then 30 cycles of [98 °C for 10 s, 61 °C for 20 s, and 72 °C for 30 s], followed by a final 72 °C extension for 2 min. Unique Illumina barcoding primer pairs were added to each sample in a secondary PCR reaction (PCR 2). Specifically, 25 µl of a given PCR 2 reaction contained 0.5 µM of each unique forward and reverse Illumina barcoding primer pair, 1 µl unpurified PCR 1 reaction mixture, and 12.5 µl Phusion U Green Multiplex PCR 2× Master Mix. The barcoding PCR 2 reactions were carried out as follows: 98 °C for 2 min, then 12 cycles of [98 °C for 10 s, 61 °C for 20 s, and 72 °C for 30 s], followed by a final 72 °C extension for 2 min. PCR products were evaluated analytically by electrophoresis in a 1.5% agarose gel. PCR 2 products (pooled by common amplicons) were purified by electrophoresis with a 1.5% agarose gel using a QIAquick Gel Extraction Kit (Qiagen), eluting with 40 µl water. DNA concentration was measured by fluorometric quantification (Qubit, ThermoFisher Scientific) or qPCR (KAPA Library Quantification Kit-Illumina, KAPA Biosystems) and sequenced on an Illumina MiSeq instrument according to the manufacturer’s protocols.
Sequencing reads were demultiplexed using MiSeq Reporter (Illumina). Alignment of amplicon sequences to a reference sequence was performed using CRISPResso243. For all prime editing yield quantification, prime editing efficiency was calculated as: percentage of (number of reads with the desired edit that do not contain indels)/(number of total reads). For quantification of point mutation editing, CRISPResso2 was run in standard mode with “discard_indel_reads” on. Prime editing for installation of point mutations was then explicitly calculated as: (frequency of specified point mutation in non-discarded reads) × (number of non-discarded reads)/(total reads). For insertion or deletion edits, CRISPResso2 was run in HDR mode using the desired allele as the expected allele (e flag), and with “discard_indel_reads” on. Editing yield was calculated as: (number of HDR-aligned reads)/(total reads). For all experiments, indel yields were calculated as: (number of indel-containing reads)/(total reads).
Nucleofection of U2OS, K562, and HeLa cells
Nucleofection was used for transfection in all experiments using K562, HeLa, and U2OS cells. For PE conditions in these cell types, 800 ng prime editor expression plasmid, 200 ng pegRNA expression plasmid, and 83 ng nicking sgRNA expression plasmid was nucleofected in a final volume of 20 μl in a 16-well nucleocuvette strip (Lonza). For HDR conditions in these three cell types, 350 ng Cas9 nuclease expression plasmid, 150 ng sgRNA expression plasmid and 200 pmol (6.6 μg) 100-nt ssDNA donor template (PAGE-purified; Integrated DNA Technologies) was nucleofected in a final volume of 20 µl per sample in a 16-well Nucleocuvette strip (Lonza). K562 cells were nucleofected using the SF Cell Line 4D-Nucleofector X Kit (Lonza) with 5 × 105 cells per sample (program FF-120), according to the manufacturer’s protocol. U2OS cells were nucleofected using the SE Cell Line 4D-Nucleofector X Kit (Lonza) with 3–4 × 105 cells per sample (program DN-100), according to the manufacturer’s protocol. HeLa cells were nucleofected using the SE Cell Line 4D-Nucleofector X Kit (Lonza) with 2 × 105 cells per sample (program CN-114), according to the manufacturer’s protocol. Cells were harvested 72 h after nucleofection for genomic DNA extraction.
Genomic DNA extraction for HDR experiments
Genomic DNA from all HDR comparison experiments in HEK293T, HEK293T HBB(E6V), K562, U2OS, and HeLa cells was purified using the Agencourt DNAdvance Kit (Beckman Coulter), according to the manufacturer’s protocol.
Comparison between PE2, PE3, BE2, BE4max, ABEdmax, and ABEmax
HEK293T cells were seeded on 48-well poly-d-lysine coated plates (Corning). After 16–24 h, cells were transfected at approximately 60% confluency. For base editing with CBE or ABE constructs, cells were transfected with 750 ng base editor plasmid, 250 ng sgRNA expression plasmid, and 1 µl of lipofectamine 2000 (Thermo Fisher Scientific). PE transfections were performed as described above. Genomic DNA extraction for PE and BE was performed as described above.
Determination of PE3 activity at known Cas9 off-target sites
To evaluate PE3 off-target editing activity at known Cas9 off-target sites, genomic DNA extracted from HEK293T cells 3 days after transfection with PE3 was used as template for PCR amplification of 16 previously reported Cas9 off-target genomic sites32,33 (the top four off-target sites each for the HEK3, EMX1, FANCF, and HEK4 spacers; primer sequences are listed in Supplementary Table 4). These genomic DNA samples were identical to those used for quantifying on-target PE3 editing activities shown in Fig. 4 or Extended Data Fig. 5d, e; pegRNA and nicking sgRNA sequences are listed in Supplementary Table 3. Following PCR amplification of off-target sites, amplicons were sequenced on the Illumina MiSeq platform as described above (see ‘High-throughput DNA sequencing of genomic DNA samples’ section). To determine the on-target and off-target editing activity of Cas9 nuclease, Cas9(H840A) nickase, dCas9, and PE2-dRT, we transfected HEK293T cells with 750 ng editor plasmid (Cas9 nuclease, Cas9(H840A) nickase, dCas9, or PE2-dRT), 250 ng pegRNA or sgRNA plasmid, and 1 μl lipofectamine 2000. Genomic DNA was isolated from cells 3 days after transfection as described above. On-target and off-target genomic loci were amplified by PCR using the primer sequences in Supplementary Table 4 and sequenced on an Illumina MiSeq.
High-throughput sequencing (HTS) data analysis was performed using CRISPResso243. The editing efficiencies of Cas9 nuclease, Cas9 H840A nickase, and dCas9 were quantified as the percentage of total sequencing reads containing indels. For quantification of PE3 and PE3-dRT off-targets, aligned sequencing reads were examined for point mutations, insertions, or deletions that were consistent with the anticipated product of pegRNA reverse transcription initiated at the Cas9 nick site. Single nucleotide variations occurring at <0.1% overall frequency among total reads within a sample were excluded from analysis. For reads containing single nucleotide variations that both occurred at frequencies ≥0.1% and were partially consistent with the pegRNA-encoded edit, t-tests (unpaired, one-tailed, α = 0.5) were used to determine whether the variants occurred at significantly higher levels compared to samples treated with pegRNAs that contained the same spacer but encoded different edits. To avoid differences in sequencing errors, comparisons were made between samples that were sequenced simultaneously within the same MiSeq run. Variants that did not meet the criteria of P > 0.05 were excluded. Off-target PE3 editing activity was then calculated as the percentage of total sequencing reads that met the above criteria.
Generation of a HEK293T cell line containing HBB(E6V) using Cas9-initiated HDR
HEK293T cells were seeded in a 48-well plate and transfected at approximately 60% confluency with 1.5 µl lipofectamine 2000, 300 ng Cas9(D10A) nickase plasmid, 100 ng sgRNA plasmid, and 200 ng 100-mer ssDNA donor template (Supplementary Table 5). Three days after transfection, the medium was exchanged for fresh medium. Four days after transfection, cells were dissociated using 30 µl TrypLE solution and suspended in 1.5 ml medium. Single cells were isolated into individual wells of two 96-well plates by fluorescence-activated cell sorting (FACS) (Beckman-Coulter Astrios). See Supplementary Note 1 for representative FACS sorting examples. Cells were expanded for 14 days before genomic DNA sequencing as described above. Of the isolated clonal populations, none was found to be homozygous for the HBB allele encoding the E6V mutation, so a second round of editing by lipofection, sorting, and outgrowth was repeated in a partially edited cell line to yield a cell line homozygous for the E6V-encoding allele.
Generation of a HEK293T cell line containing HBB(E6V) using PE3
HEK293T cells (2.5 × 104) were seeded on 48-well poly-d-lysine coated plates (Corning). Between 16 and 24 h after seeding, cells were transfected at approximately 70% confluency with 1 µl lipofectamine 2000 (Thermo Fisher Scientific) according to the manufacturer’s protocols and 750 ng PE2-P2A-GFP plasmid, 250 ng pegRNA plasmid, and 83 ng sgRNA plasmid. After 3 days, cells were washed with 1× PBS (Gibco) and dissociated using TrypLE Express (Gibco). Cells were then diluted with DMEM plus GlutaMax (Thermo Fisher Scientific) supplemented with 10% (v/v) FBS (Gibco) and passed through a 35-µm cell strainer (Corning) before sorting. Flow cytometry was carried out on a LE-MA900 cell sorter (Sony). Cells were treated with 3 nM DAPI (BioLegend) 15 min before sorting. After gating for doublet exclusion, single DAPI-negative cells with GFP fluorescence above that of a GFP-negative control cell population were sorted into 96-well flat-bottom cell culture plates (Corning) filled with pre-chilled DMEM with GlutaMax supplemented with 10% FBS. See Supplementary Note 1 for representative FACS sorting examples and allele tables. Cells were cultured for 10 days before genomic DNA extraction and characterization by HTS, as described above. A total of six clonal cell lines were identified that are homozygous for the E6V-encoding mutation in HBB.
Generation of a HEK293T cell line containing the HEXA 1278+TATC insertion using PE3
HEK293T cells containing the HEXA1278+TATC allele were generated following the protocol described above for creation of the HBB(E6V) cell line; pegRNA and sgRNA sequences are listed in Supplementary Table 3 under the Fig. 5 subheading. After transfection and sorting, cells were cultured for 10 days before genomic DNA was extracted and characterized by HTS, as described above. We recovered two heterozygous cell lines that contained 50% HEXA1278+TATC alleles and two homozygous cell lines containing 100% HEXA1278+TATC alleles.
Cell viability assays
HEK293T cells were seeded in 48-well plates and transfected at approximately 70% confluency with 750 ng editor plasmid (PE2, PE2(R110S/K103L), Cas9(H840A) nickase, or dCas9), 250 ng HEK3-targeting pegRNA plasmid, and 1 μl lipofectamine 2000, as described above. Cell viability was measured every 24 h post-transfection for 3 days using the CellTiter-Glo 2.0 assay (Promega) according to the manufacturer’s protocol. Luminescence was measured in 96-well flat-bottomed polystyrene microplates (Corning) using a M1000 Pro microplate reader (Tecan) with a 1-s integration time.
Lentivirus production
Lentivirus was produced as previously described44. T-75 flasks of rapidly dividing HEK293T cells (ATCC; Manassas, VA, USA) were transfected with lentivirus production helper plasmids pVSV-G and psPAX2 in combination with modified lentiCRISPRv2 genomes carrying intein-split PE2 editor using FuGENE HD (Promega, Madison, WI, USA) according to the manufacturer's protocol. Four split-intein editor constructs were designed: 1) a viral genome encoding a U6-pegRNA expression cassette and the N-terminal portion (1–573) of Cas9(H840A) nickase fused to the Npu N-intein, a self-cleaving P2A peptide, and GFP-KASH; 2) a viral genome encoding the Npu C-intein fused to the C-terminal remainder of PE2; 3) a viral genome encoding the Npu C-intein fused to the C-terminal remainder of Cas9 for the Cas9 control; and 4) a nicking sgRNA for DNMT1 (derived from Addgene plasmid no. 52963). The split-intein45 mediates trans splicing to join the two halves of PE2 or Cas9, while the P2A GFP-KASH enables co-translational production of a nuclear membrane-localized GFP. After 48 h, supernatant was collected, centrifuged at 500g for 5 min to remove cellular debris, and filtered using a 0.45-µm filter. Filtered supernatant was concentrated using the PEG-it Virus Precipitation Solution (System Biosciences, Palo Alto, CA, USA) according to the manufacturer’s directions. The resulting pellet was resuspended in Opti-MEM (Thermo Fisher Scientific, Waltham, MA, USA) using 1% of the original medium volume. Resuspended pellet was flash-frozen and stored at −80 °C until use.
Mouse primary cortical neuron dissection and culture
E18.5 dissociated cortical cultures were taken from timed-pregnant C57BL/6 mice (Charles River). Embryos were removed from pregnant mice after euthanasia by CO2 followed by decapitation. Cortical caps were dissected in ice-cold Hibernate-E supplemented with penicillin/streptomycin (Life Technologies). Following a rinse with ice-cold Hibernate-E, tissue was digested at 37 °C for 8 min in papain/DNase (Worthington/Sigma). Tissue was triturated in NBActiv4 (BrainBits) supplemented with DNase. Cells were counted and plated in 24-well plates at 100,000 cells per well. Half of the medium was changed twice per week.
Prime editing in primary neurons and nucleus isolation
At days in vitro (DIV) 1, 15 µl lentivirus was added at a 10:10:1 ratio of N-terminal:C-terminal:nicking sgRNA. At DIV 14, neuronal nuclei were isolated using the EZ-PREP buffer (Sigma D8938) following the manufacturer’s protocol. All steps were performed on ice or at 4 °C. Medium was removed from dissociated cultures, and cultures were washed with ice-cold PBS. PBS was aspirated and replaced with 200 µl EZ-PREP solution. Following a 5-min incubation on ice, EZ-PREP was pipetted across the surface of the well to dislodge remaining cells. The sample was centrifuged at 500g for 5 min, and the supernatant removed. Samples were washed with 200 µl EZ-PREP and centrifuged again at 500g for 5 min. Samples were resuspended with gentle pipetting in 200 µl ice-cold Nuclei Suspension Buffer (NSB) consisting of 100 µg/ml BSA and 3.33 µM Vybrant DyeCycle Ruby (Thermo Fisher) in 1×PBS, then centrifuged at 500g for 5 min. The supernatant was removed and nuclei were resuspended in 100 µl NSB and sorted into 100 µl Agencourt DNAdvance lysis buffer using a MoFlo Astrios (Beckman Coulter) at the Broad Institute flow cytometry facility. Genomic DNA was purified according to the manufacturer’s Agencourt DNAdvance instructions.
RNA-seq and data analysis
HEK293T cells were co-transfected with PRNP-targeting or HEXA-targeting pegRNAs and PE2, PE2-dRT, or Cas9(H840A) nickase. Seventy-two hours after transfection, total RNA was harvested from cells using TRIzol reagent (Thermo Fisher) and purified with RNeasy Mini kit (Qiagen) including on-column DNaseI treatment. Ribosomes were depleted from total RNA using the rRNA removal protocol of the TruSeq Stranded Total RNA library prep kit (Illumina) and subsequently washed with RNAClean XP beads (Beckman Coulter). Sequencing libraries were prepared using ribo-depleted RNA on a SMARTer PrepX Apollo NGS library prep system (Takara) following the manufacturer’s protocol. The resulting libraries were visualized on a 2200 TapeStation (Agilent Technologies), normalized using a Qubit dsDNA HS assay (Thermo Fisher), and sequenced on a NextSeq 550 using high output v2 flow cell (Illumina) as 75-bp paired-end reads. Fastq files were generated with bcl2fastq2 version 2.20 and trimmed using TrimGalore version 0.6.2 (https://github.com/FelixKrueger/TrimGalore) to remove low-quality bases, unpaired sequences, and adaptor sequences. Trimmed reads were aligned to a Homo sapiens genome assembly GRCh38 with a custom Cas9(H840A) gene entry using RSEM version 1.3.146. The limma-voom47 package was used to normalize gene expression levels and perform differential expression analysis with batch effect correction. Differentially expressed genes were called with FDR-corrected P < 0.05 and fold change > 2 cutoffs, and results were visualized in R.
ClinVar analysis
The ClinVar variant summary was downloaded from NCBI (accessed July 15, 2019), and the information contained therein was used for all downstream analysis. The list of all reported variants was filtered by allele ID in order to remove duplicates and by clinical significance in order to restrict the analysis to pathogenic variants. The list of pathogenic variants was filtered sequentially by variant type in order to calculate the fraction of pathogenic variants that are insertions, deletions, and so on. Single nucleotide variants (SNVs) were separated into two categories (transitions and transversions) on the basis of the reported reference and alternate alleles. SNVs that did not report reference or alternate alleles were excluded from the analysis.
The lengths of reported insertions, deletions, and duplications were calculated using reference/alternate alleles, variant start/stop positions, or appropriate identifying information in the variant name. Variants that did not report any of the above information were excluded from the analysis. The lengths of reported indels (single variants that include both insertions and deletions relative to the reference genome) were calculated by determining the number of mismatches or gaps in the best pairwise alignment between the reference and alternate alleles. Frequency distributions of variant lengths were calculated using GraphPad Prism 8.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Data availability
High-throughput sequencing data have been deposited to the NCBI Sequence Read Archive database under accession PRJNA565979. Plasmids encoding PE1, PE2 (same as PE3), and pegRNA expression vectors are available from Addgene. Previously described plasmids expressing sgRNAs are also available from Addgene, such as Addgene plasmid no. 65777.
Code availability
The script used to quantify pegRNA scaffold insertion is provided as Supplementary Note 4.
References
Landrum, M. J. et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 44, D862–D868 (2016).
Jinek, M. et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821 (2012).
Cong, L. et al. Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823 (2013).
Mali, P. et al. RNA-guided human genome engineering via Cas9. Science 339, 823–826 (2013).
Kosicki, M., Tomberg, K. & Bradley, A. Repair of double-strand breaks induced by CRISPR–Cas9 leads to large deletions and complex rearrangements. Nat. Biotechnol. 36, 765–771 (2018).
Haapaniemi, E., Botla, S., Persson, J., Schmierer, B. & Taipale, J. CRISPR–Cas9 genome editing induces a p53-mediated DNA damage response. Nat. Med. 24, 927–930 (2018).
Ihry, R. J. et al. p53 inhibits CRISPR–Cas9 engineering in human pluripotent stem cells. Nat. Med. 24, 939–946 (2018).
Rouet, P., Smih, F. & Jasin, M. Expression of a site-specific endonuclease stimulates homologous recombination in mammalian cells. Proc. Natl Acad. Sci. USA 91, 6064–6068 (1994).
Chapman, J. R., Taylor, M. R. G. & Boulton, S. J. Playing the end game: DNA double-strand break repair pathway choice. Mol. Cell 47, 497–510 (2012).
Cox, D. B. T., Platt, R. J. & Zhang, F. Therapeutic genome editing: prospects and challenges. Nat. Med. 21, 121–131 (2015).
Paquet, D. et al. Efficient introduction of specific homozygous and heterozygous mutations using CRISPR/Cas9. Nature 533, 125–129 (2016).
Chu, V. T. et al. Increasing the efficiency of homology-directed repair for CRISPR–Cas9-induced precise gene editing in mammalian cells. Nat. Biotechnol. 33, 543–548 (2015).
Maruyama, T. et al. Increasing the efficiency of precise genome editing with CRISPR–Cas9 by inhibition of nonhomologous end joining. Nat. Biotechnol. 33, 538–542 (2015).
Rees, H. A., Yeh, W.-H. & Liu, D. R. Development of hRad51-Cas9 nickase fusions that mediate HDR without double-stranded breaks. Nat. Commun. 10, 2212 (2019).
Shen, M. W. et al. Predictable and precise template-free CRISPR editing of pathogenic variants. Nature 563, 646–651 (2018).
Rees, H. A. & Liu, D. R. Base editing: precision chemistry on the genome and transcriptome of living cells. Nat. Rev. Genet. 19, 770–788 (2018).
Komor, A. C., Kim, Y. B., Packer, M. S., Zuris, J. A. & Liu, D. R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 533, 420–424 (2016).
Gaudelli, N. M. et al. Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature 551, 464–471 (2017).
Gao, X. et al. Treatment of autosomal dominant hearing loss by in vivo delivery of genome editing agents. Nature 553, 217–221 (2018).
Marraffini, L. A. & Sontheimer, E. J. CRISPR interference limits horizontal gene transfer in staphylococci by targeting DNA. Science 322, 1843–1845 (2008).
Barrangou, R. et al. CRISPR provides acquired resistance against viruses in prokaryotes. Science 315, 1709–1712 (2007).
Liu, Y., Kao, H.-I. & Bambara, R. A. Flap endonuclease 1: a central component of DNA metabolism. Annu. Rev. Biochem. 73, 589–615 (2004).
Keijzers, G., Bohr, V. A. & Rasmussen, L. J. Human exonuclease 1 (EXO1) activity characterization and its function on flap structures. Biosci. Rep. 35, e00206 (2015).
Baranauskas, A. et al. Generation and characterization of new highly thermostable and processive M-MuLV reverse transcriptase variants. Protein Eng. Des. Sel. 25, 657–668 (2012).
Gerard, G. F. et al. The role of template-primer in protection of reverse transcriptase from thermal inactivation. Nucleic Acids Res. 30, 3118–3129 (2002).
Arezi, B. & Hogrefe, H. Novel mutations in Moloney murine leukemia virus reverse transcriptase increase thermostability through tighter binding to template-primer. Nucleic Acids Res. 37, 473–481 (2009).
Kotewicz, M. L., Sampson, C. M., D’Alessio, J. M. & Gerard, G. F. Isolation of cloned Moloney murine leukemia virus reverse transcriptase lacking ribonuclease H activity. Nucleic Acids Res. 16, 265–277 (1988).
Nishimasu, H. et al. Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell 156, 935–949 (2014).
Thuronyi, B. W. et al. Continuous evolution of base editors with expanded target compatibility and improved activity. Nat. Biotechnol. 37, 1070–1079 (2019).
Kim, Y. B. et al. Increasing the genome-targeting scope and precision of base editing with engineered Cas9-cytidine deaminase fusions. Nat. Biotechnol. 35, 371–376 (2017).
Koblan, L. W. et al. Improving cytidine and adenine base editors by expression optimization and ancestral reconstruction. Nat. Biotechnol. 36, 843–846 (2018).
Tsai, S. Q. et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat. Biotechnol. 33, 187–197 (2015).
Kleinstiver, B. P. et al. High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature 529, 490–495 (2016).
Bannert, N. & Kurth, R. Retroelements and the human genome: new perspectives on an old relation. Proc. Natl Acad. Sci. USA 101 (Suppl. 2), 14572–14579 (2004).
Halvas, E. K., Svarovskaia, E. S. & Pathak, V. K. Role of murine leukemia virus reverse transcriptase deoxyribonucleoside triphosphate-binding site in retroviral replication and in vivo fidelity. J. Virol. 74, 10349–10358 (2000).
Mead, S. et al. A novel protective prion protein variant that colocalizes with kuru exposure. N. Engl. J. Med. 361, 2056–2065 (2009).
Asante, E. A. et al. A naturally occurring variant of the human prion protein completely prevents prion disease. Nature 522, 478–481 (2015).
Kügler, S., Kilic, E. & Bähr, M. Human synapsin 1 gene promoter confers highly neuron-specific long-term transgene expression from an adenoviral vector in the adult rat brain depending on the transduced area. Gene Ther. 10, 337–347 (2003).
Anzalone, A. V., Lin, A. J., Zairis, S., Rabadan, R. & Cornish, V. W. Reprogramming eukaryotic translation with ligand-responsive synthetic RNA switches. Nat. Methods 13, 453–458 (2016).
Badran, A. H. et al. Continuous evolution of Bacillus thuringiensis toxins overcomes insect resistance. Nature 533, 58–63 (2016).
Anders, C. & Jinek, M. in Methods in Enzymology (eds. Doudna, J. A. & Sontheimer, E. J.) 546, 1–20 (Academic, 2014).
Pirakitikulr, N., Ostrov, N., Peralta-Yahya, P. & Cornish, V. W. PCRless library mutagenesis via oligonucleotide recombination in yeast. Protein Sci. 19, 2336–2346 (2010).
Clement, K. et al. CRISPResso2 provides accurate and rapid genome editing sequence analysis. Nat. Biotechnol. 37, 224–226 (2019).
Levy, J. M. & Nicoll, R. A. Membrane-associated guanylate kinase dynamics reveal regional and developmental specificity of synapse stability. J. Physiol. (Lond.) 595, 1699–1709 (2017).
Zettler, J., Schütz, V. & Mootz, H. D. The naturally split Npu DnaE intein exhibits an extraordinarily high rate in the protein trans-splicing reaction. FEBS Lett. 583, 909–914 (2009).
Li, B. & Dewey, C. N. RSEM: accurate transcript quantification from RNA-seq data with or without a reference genome. BMC Bioinformatics 12, 323 (2011).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015).
Acknowledgements
We thank J. M. Madison for neuron cell culture advice. This work was supported by the Merkin Institute of Transformative Technologies in Healthcare, US NIH grants U01AI142756, RM1HG009490, R01EB022376, and R35GM118062, and the HHMI. A.V.A. acknowledges a Jane Coffin Childs postdoctoral fellowship. P.B.R. and A.R. acknowledge NIH T32 GM095450. A.A.S. acknowledges NIH T32 GM007726. P.J.C. and A.R. acknowledge NSF graduate fellowships. C.W. acknowledges a Damon Runyon Cancer Research Foundation fellowship (DRG-2343-18). G.A.N acknowledges a Helen Hay Whitney postdoctoral fellowship.
Author information
Authors and Affiliations
Contributions
A.V.A. designed the research, performed experiments, analysed data, and wrote the manuscript. P.B.R., J.R.D., A.A.S., and G.A.N. performed human cell experiments and analysed data. L.W.K. and J.M.L. performed neuron experiments. P.J.C. and C.W. performed and analysed RNA-seq experiments. A.R. analysed ClinVar data. D.R.L designed and supervised the research and wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
Authors through the Broad Institute have filed patent applications on prime editing. D.R.L. is a consultant and co-founder of Prime Medicine, Beam Therapeutics, Pairwise Plants, and Editas Medicine, companies that use genome editing.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Peer review information Nature thanks Guangping Gao, Randall Platt and Fyodor Urnov for their contribution to the peer review of this work.
Extended data figures and tables
Extended Data Fig. 1 In vitro prime editing validation studies with fluorescently labelled DNA substrates.
a, Electrophoretic mobility shift assays with dCas9, 5′-extended pegRNAs and 5′-Cy5-labelled DNA substrates. pegRNAs 1–5 contain a 15-nt linker sequence (linker A for pegRNA 1, linker B for pegRNAs 2–5) between the spacer and the PBS, a 5-nt PBS sequence, and RT templates of 7 nt (pegRNAs 1 and 2), 8 nt (pegRNA 3), 15 nt (pegRNA 4), and 22 nt (pegRNA 5). pegRNAs are those used in e and f; full sequences are listed in Supplementary Table 2. b, In vitro nicking assays of Cas9(H840A) using 5′-extended and 3′-extended pegRNAs. Data in a, b are representative of n = 2 independent replicates. c, Cas9-mediated indel formation in HEK293T cells at HEK3 using 5′-extended and 3′-extended pegRNAs. Mean ± s.d. of n = 3 independent biological replicates. d, Overview of prime editing in vitro biochemical assays. 5′-Cy5-labelled pre-nicked and non-nicked dsDNA substrates were tested. sgRNAs, 5′-extended pegRNAs, or 3′-extended pegRNAs were pre-complexed with dCas9 or Cas9(H840A) nickase, then combined with dsDNA substrate, Superscript III M-MLV RT, and dNTPs. Reactions were allowed to proceed at 37 °C for 1 h before separation by denaturing urea PAGE and visualization by Cy5 fluorescence. e, Primer extension reactions using 5′-extended pegRNAs, pre-nicked DNA substrates, and dCas9 lead to substantial conversion to RT products. f, Primer extension reactions using 5′-extended pegRNAs as in b with non-nicked DNA substrate and Cas9(H840A) nickase. Product yields are greatly reduced by comparison to pre-nicked substrate. g, An in vitro primer extension reaction using a 3′-pegRNA generates a single apparent product by denaturing urea PAGE. The RT product band was excised, eluted from the gel, then subjected to homopolymer tailing with terminal transferase (TdT) using either dGTP or dATP. Tailed products were extended using poly-T or poly-C primers, and the resulting DNA was sequenced. Sanger traces indicate that three nucleotides derived from the pegRNA scaffold were reverse-transcribed (added as the final 3′ nucleotides to the DNA product). Note that pegRNA scaffold insertion is much rarer in mammalian cell prime editing experiments than in vitro (Extended Data Fig. 6), potentially owing to the inability of the tethered RT to access the Cas9-bound guide RNA scaffold, and/or cellular excision of mismatched 3′ ends of 3′ flaps containing pegRNA scaffold sequences. Data in e–g are representative of n = 2 independent replicates. For gel source data, see Supplementary Fig. 1.
Extended Data Fig. 2 Cellular repair in yeast of 3′ DNA flaps from in vitro prime editing reactions.
a, Dual fluorescent protein reporter plasmids contain GFP and mCherry open reading frames separated by a target site encoding an in-frame stop codon, a +1 frameshift, or a −1 frameshift. Prime editing reactions were carried out in vitro with Cas9(H840A) nickase, pegRNA, dNTPs, and M-MLV RT, then transformed into yeast. Colonies that contain unedited plasmids produce GFP but not mCherry. Yeast colonies containing edited plasmids produce both GFP and mCherry as a fusion protein. b, Overlay of GFP and mCherry fluorescence for yeast colonies transformed with reporter plasmids containing a stop codon between GFP and mCherry (unedited negative control, top), or containing no stop codon or frameshift between GFP and mCherry (pre-edited positive control, bottom). c–f, Visualization of mCherry and GFP fluorescence from yeast colonies transformed with in vitro prime editing reaction products. c, d, Stop codon correction via T•A-to-A•T transversion using a 3′-extended pegRNA (c) or a 5′-extended pegRNA (d). e, +1 frameshift correction via a 1-bp deletion using a 3′-extended pegRNA. f, −1 frameshift correction via a 1-bp insertion using a 3′-extended pegRNA. g, Sanger DNA sequencing traces from plasmids isolated from GFP-only colonies in b and GFP and mCherry double-positive colonies in c. Data in b–g are representative of n = 2 independent replicates.
Extended Data Fig. 3 Prime editing of genomic DNA in human cells by PE1.
a, pegRNAs contain a spacer sequence, an sgRNA scaffold, and a 3′ extension containing an RT template (purple), which contains the edited base(s) (red), and a primer-binding site (PBS, green). The primer-binding site hybridizes to the nicked target DNA strand. The RT template is homologous to the DNA sequence downstream of the nick, with the exception of the encoded edited base(s). b, Installation of a T•A-to-A•T transversion at the HEK3 site in HEK293T cells using Cas9(H840A) nickase fused to wild-type M-MLV RT (PE1) and pegRNAs with varying PBS lengths. c, T•A-to-A•T transversion editing efficiency and indel generation by PE1 at the +1 position of HEK3 using pegRNAs containing 10-nt RT templates and PBS sequences ranging from 8 to 17 nt. d, G•C-to-T•A transversion editing efficiency and indel generation by PE1 at the +5 position of EMX1 using pegRNAs containing 13-nt RT templates and PBS sequences ranging from 9 to 17 nt. e, G•C-to-T•A transversion editing efficiency and indel generation by PE1 at the +5 position of FANCF using pegRNAs containing 17-nt RT templates and PBS sequences ranging from 8 to 17 nt. f, C•G-to-A•T transversion editing efficiency and indel generation by PE1 at the +1 position of RNF2 using pegRNAs containing 11-nt RT templates and PBS sequences ranging from 9 to 17 nt. g, G•C-to-T•A transversion editing efficiency and indel generation by PE1 at the +2 position of HEK4 using pegRNAs containing 13-nt RT templates and PBS sequences ranging from 7 to 15 nt. h, PE1-mediated +1 T deletion, +1 A insertion, and +1 CTT insertion at the HEK3 site using a 13-nt PBS and a 10-nt RT template. Sequences of pegRNAs are as in Fig. 2a (Supplementary Table 3). Editing efficiencies reflect sequencing reads that contain the intended edit and do not contain indels among all treated cells, with no sorting. Mean ± s.d. of n = 3 independent biological replicates.
Extended Data Fig. 4 Evaluation of M-MLV RT variants for prime editing.
a, Abbreviations for prime editor variants used in this figure. b, Targeted insertion and deletion edits with PE1 at the HEK3 locus. c–h, Comparison of 18 prime editor constructs containing M-MLV RT variants for their ability to install a +2 G•C-to-C•G transversion edit at HEK3 (c), a 24-bp Flag insertion at the +1 position of HEK3 (d), a +1 C•G-to-A•T transversion edit at RNF2 (e), a +1 G•C-to-C•G transversion edit at EMX1 (f), a +2 T•A-to-A•T transversion edit at HBB (g), and a +1 G•C-to-C•G transversion edit at FANCF (h). i–n, Comparison of four prime editor constructs containing M-MLV variants for their ability to install the edits shown in c–h in a second round of independent experiments. o–s, PE2 editing efficiency at five genomic loci with varying PBS lengths. o, +1 T•A-to-A•T at HEK3. p, +5 G•C-to-T•A at EMX1. q, +5 G•C-to-T•A at FANCF. r, +1 C•G-to-A•T at RNF2. s, +2 G•C-to-T•A at HEK4. Editing efficiencies reflect sequencing reads that contain the intended edit and do not contain indels among all treated cells, with no sorting. Mean ± s.d. of n = 3 independent biological replicates.
Extended Data Fig. 5 Design features of pegRNA PBS and RT template sequences, and additional editing examples with PE3.
a, PE2-mediated +5 G•C-to-T•A transversion editing efficiency (blue line) at VEGFA in HEK293T cells as a function of RT template length. Indels (grey line) are plotted for comparison. The sequence below the graph shows the last nucleotide templated for synthesis by the pegRNA. G nucleotides (templated by a C in the pegRNA) are highlighted in red; RT templates that end in C should be avoided during pegRNA design to maximize prime editing efficiency. b, +5 G•C-to-T•A transversion editing and indels for DNMT1 as in a. c, +5 G•C-to-T•A transversion editing and indels for RUNX1 as in a. d–f, PE3-mediated transition and transversion edits at the specified positions for FANCF (d), EMX1 (e), and DNMT1 (f). Mean ± s.d. of n = 3 independent biological replicates.
Extended Data Fig. 6 Comparison of prime editing and base editing, and off-target editing by Cas9 and prime editors at known Cas9 off-target sites.
a, C•G-to-T•A editing efficiency at the same target nucleotides for PE2, PE3, BE2max, and BE4max at endogenous HEK3, FANCF, and EMX1 sites in HEK293T cells. b, Indel frequency from treatments in a. c, Editing efficiency of precise C•G-to-T•A edits (without bystander edits or indels) at HEK3, FANCF, and EMX1. d, Total A•T-to-G•C editing efficiency for PE2, PE3, ABEdmax, and ABEmax at HEK3 and FANCF. e, Precise A•T-to-G•C editing efficiency without bystander edits or indels at HEK3 and FANCF. f, Indel frequency from treatments in d. g, Average triplicate Cas9 nuclease editing efficiencies (indel frequencies) in HEK293T cells at four endogenous on-target sites and their 16 known top off-target sites32,33. For each on-target site, Cas9 was paired with an sgRNA or with each of four pegRNAs that recognize the same protospacer. h, Average triplicate on-target and off-target editing efficiencies and indel efficiencies (below in parentheses) in HEK293T cells for PE2 or PE3 paired with each pegRNA in g. Editing efficiencies reflect sequencing reads that contain the intended edit and do not contain indels among all treated cells, with no sorting. Off-target editing efficiencies in h reflect off-target locus modification consistent with prime editing. Mean ± s.d. of n = 3 independent biological replicates.
Extended Data Fig. 7 Incorporation of pegRNA scaffold sequence into target loci.
HTS data were analysed for pegRNA scaffold sequence insertion as described in Supplementary Note 4. a, Analysis for the EMX1 locus. Shown is the percentage of total sequencing reads containing one or more pegRNA scaffold sequence nucleotides within an insertion adjacent to the RT template (left); the percentage of total sequencing reads containing a pegRNA scaffold sequence insertion of the specified length (middle); and the cumulative total percentage of pegRNA insertion up to and including the length specified on the x-axis. b, As in a for FANCF. c, As in a for HEK3. d, As in a for RNF2. Mean ± s.d. of n = 3 independent biological replicates.
Extended Data Fig. 8 Effects of PE2, PE2-dRT, Cas9(H840A) nickase, and dCas9 on cell viability and on transcriptome-wide RNA abundance.
HEK293T cells were transiently transfected with plasmids encoding PE2, PE2(R110S/K103L), Cas9(H840A) nickase, or dCas9, together with a HEK3-targeting pegRNA plasmid. Cell viability was measured for the bulk cellular population every 24 h after transfection for 3 days using the CellTiter-Glo 2.0 assay (Promega). a, Viability, as measured by luminescence, at 1, 2, or 3 days after transfection. Mean ± s.e.m. of n = 3 independent biological replicates, each performed in technical triplicate. b, Percentage editing and indels for PE2, PE2(R110S/K103L), Cas9(H840A) nickase, or dCas9, together with a HEK3-targeting pegRNA plasmid that encodes a +5 G-to-A edit. Editing efficiencies were measured on day 3 after transfection from cells treated alongside those used for assaying viability in a. Mean ± s.d. of n = 3 independent biological replicates. c–k, Analysis of cellular RNA, depleted for ribosomal RNA, isolated from HEK293T cells expressing PE2, PE2-dRT, or Cas9(H840A) nickase and a PRNP-targeting or HEXA-targeting pegRNA. RNAs corresponding to 14,410 genes and 14,368 genes were detected in PRNP and HEXA samples, respectively. c–h, Volcano plot displaying the −log10 FDR-adjusted P value versus log2-fold change in transcript abundance for each RNA, comparing PE2 versus PE2-dRT with PRNP-targeting pegRNA (c), PE2 versus Cas9(H840A) with PRNP-targeting pegRNA (d), PE2-dRT versus Cas9(H840A) with PRNP-targeting pegRNA (e), PE2 versus PE2-dRT with HEXA-targeting pegRNA(f), PE2 versus Cas9(H840A) with HEXA-targeting pegRNA (g), PE2-dRT versus Cas9(H840A) with HEXA-targeting pegRNA (h). Red dots indicate genes that show twofold or more changes in relative abundance that are statistically significant (FDR-adjusted P < 0.05). i–k, Venn diagrams of upregulated and downregulated transcripts (twofold change or more) comparing PRNP and HEXA samples for PE2 versus PE2-dRT (i), PE2 versus Cas9(H840A) (j), and PE2-dRT versus Cas9(H840A) (k). Values for each RNA-seq condition reflect the mean of n = 5 biological replicates. Differential expression was assessed using a two-sided t-test with empirical Bayesian variance estimation.
Extended Data Fig. 9 PE3-mediated correction of E6V-encoding HBB mutation and HEXA1278+TATC by various pegRNAs.
a, Screen of 14 pegRNAs for correction of the HBB E6V-encoding allele in HEK293T cells with PE3. All pegRNAs evaluated convert the mutant HBB allele back to wild-type HBB without the introduction of any silent PAM mutation. b, Screen of 41 pegRNAs for correction of the HEXA1278+TATC allele in HEK293T cells with PE3 or PE3b. Those pegRNAs labelled HEXAs correct the pathogenic allele by a shifted 4-bp deletion that disrupts the PAM and leaves a silent mutation. Those pegRNAs labelled HEXA correct the pathogenic allele back to wild-type. Entries ending in b use an edit-specific nicking sgRNA in combination with the pegRNA (the PE3b system). Mean ± s.d. of n = 3 independent biological replicates.
Extended Data Fig. 10 PE3 activity in human cell lines and comparison of PE3 and Cas9-initiated HDR.
a, Prime editing in K562 (leukaemic bone marrow), U2OS (osteosarcoma), and HeLa (cervical cancer) cells. b–e, Efficiency of generating the correct edit (without indels) and indel frequency for PE3 and Cas9-initiated HDR in HEK293T cells (b), K562 cells (c), U2OS cells (d), and HeLa cells (e). Each bracketed editing comparison installs identical edits with PE3 and Cas9-initiated HDR. Non-targeting controls are PE3 and a pegRNA that targets a non-target locus. (f) Control experiments with non-targeting pegRNA + PE3, and with dCas9 + sgRNA, compared with wild-type Cas9 HDR experiments confirming that ssDNA donor HDR template, a common contaminant that artificially elevates apparent HDR efficiencies, does not contribute to the HDR measurements in a–d. g, Example HEK3 site allele tables from genomic DNA samples isolated from K562 cells after editing with PE3 or with Cas9-initiated HDR. Alleles were sequenced on an Illumina MiSeq and analysed using CRISPResso243. The reference HEK3 sequence from this region is at the top. Allele tables are shown for a non-targeting pegRNA negative control, a +1 CTT insertion at HEK3 using PE3, and a +1 CTT insertion at HEK3 using Cas9-initiated HDR. Allele frequencies and corresponding Illumina sequencing read counts are shown for each allele. All alleles observed with frequency ≥0.20% are shown. Mean ± s.d. of n = 3 independent biological replicates.
Extended Data Fig. 11 Distribution by length of pathogenic insertions, duplications, deletions, and indels in the ClinVar database.
The ClinVar variant summary was downloaded from NCBI on 15 July 2019. The lengths of reported insertions, deletions, and duplications were calculated using reference and alternate alleles, variant start and stop positions, or appropriate identifying information in the variant name. Variants that did not report any of the above information were excluded from the analysis. The lengths of reported indels (single variants that include both insertions and deletions relative to the reference genome) were calculated by determining the number of mismatches or gaps in the best pairwise alignment between the reference and alternate alleles. a, Length distribution of insertions. b, Length distribution of duplications. c, Length distribution of deletions. d, Length distribution of indels.
Supplementary information
Supplementary Information
The Supplementary Information file contains: a Supplementary Discussion that provides additional background and descriptions of the data; Supplementary Tables 1-5 listing activity data and short DNA and RNA sequences; Supplementary Sequences 1-3 that contain DNA and protein sequences of key constructs; Supplementary Notes 1-4 that contain FACS information, cloning methods, and scripts; Supplementary Figure 1 that contains original gel image data, and Supplementary References
Rights and permissions
About this article

Cite this article
Anzalone, A.V., Randolph, P.B., Davis, J.R. et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 576, 149–157 (2019). https://doi.org/10.1038/s41586-019-1711-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-019-1711-4
This article is cited by
-
scSNV-seq: high-throughput phenotyping of single nucleotide variants by coupled single-cell genotyping and transcriptomics
Genome Biology (2024)
-
Eukaryotic-driven directed evolution of Cas9 nucleases
Genome Biology (2024)
-
Decoding the complexity of on-target integration: characterizing DNA insertions at the CRISPR-Cas9 targeted locus using nanopore sequencing
BMC Genomics (2024)
-
Plant genome editing to achieve food and nutrient security
BMC Methods (2024)
-
HiHo-AID2: boosting homozygous knock-in efficiency enables robust generation of human auxin-inducible degron cells
Genome Biology (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
