
Abstract
The methanogenic degradation of oil hydrocarbons can proceed through syntrophic partnerships of hydrocarbon-degrading bacteria and methanogenic archaea1,2,3. However, recent culture-independent studies have suggested that the archaeon ‘Candidatus Methanoliparum’ alone can combine the degradation of long-chain alkanes with methanogenesis4,5. Here we cultured Ca. Methanoliparum from a subsurface oil reservoir. Molecular analyses revealed that Ca. Methanoliparum contains and overexpresses genes encoding alkyl-coenzyme M reductases and methyl-coenzyme M reductases, the marker genes for archaeal multicarbon alkane and methane metabolism. Incubation experiments with different substrates and mass spectrometric detection of coenzyme-M-bound intermediates confirm that Ca. Methanoliparum thrives not only on a variety of long-chain alkanes, but also on n-alkylcyclohexanes and n-alkylbenzenes with long n-alkyl (C≥13) moieties. By contrast, short-chain alkanes (such as ethane to octane) or aromatics with short alkyl chains (C≤12) were not consumed. The wide distribution of Ca. Methanoliparum4,5,6 in oil-rich environments indicates that this alkylotrophic methanogen may have a crucial role in the transformation of hydrocarbons into methane.
Similar content being viewed by others

Main
In subsurface oil reservoirs and marine oil seep sediments, microorganisms use hydrocarbons as a source of energy and carbon7,8. The microorganisms preferentially consume alkanes, cyclic and aromatic compounds, leaving an unresolved complex mixture as residue and thereby altering the quality of the oil7,8. In the absence of sulfate, microorganisms couple anaerobic hydrocarbon degradation to methane formation1,9,10. This reaction was originally demonstrated by Zengler et al2 as methanogenic ‘microbial alkane cracking’, and alarge number of studies have shown that it can be performed in syntrophic interactions of bacteria and archaea11. In this syntrophy, the bacteria ferment the oil to acetate, carbon dioxide and hydrogen, while hydrogenotrophic and/or acetotrophic methanogenic archaea use the products for methanogenesis1,2,11.
Diverse anaerobic hydrocarbon activation mechanisms exist, including the well-studied fumarate addition pathway catalysed by glycyl radical enzymes12. This mechanism is widespread among bacteria that thrive on alkanes of various chain lengths and other hydrocarbons12,13. By contrast, several archaeal lineages activate gaseous alkanes with the help of a specific type of methyl-coenzyme M reductase (MCR), an enzyme that was originally described to catalyse the reduction of methyl-coenzyme M (methyl-CoM) to methane in methanogens14. Anaerobic methanotrophic archaea use canonical MCRs to activate methane into methyl-CoM, which is then oxidized to CO2. Short-chain alkane-oxidizing archaea contain divergent variants of this enzyme, which are known as alkyl-CoM reductases (ACRs). Analogous to the methane-activating MCR, ACRs activate multicarbon alkanes to form CoM-bound alkyl units15,16,17. The cultured alkane-oxidizing archaea oxidize short-chain alkanes such as ethane, propane or butane to CO2 through the Wood–Ljungdahl and/or the beta-oxidation pathways15,16,17. These archaea require syntrophic partner bacteria that receive the reducing equivalents released during alkane oxidation for sulfate reduction15,16,17. However, many uncultured archaeal lineages harbour acr genes, indicating that hydrocarbon-degrading archaea are far more diverse than pictured by the few cultured representatives5,18,19,20,21,22. The recently described metagenome assembled genomes (MAGs) of ‘Candidatus Methanoliparia’4,5 encode both a canonical MCR and an ACR. This unique MCR–ACR combination, combined with additional genomic features such as membrane-bound methylcobalamin: CoM methyltransferases (Mtr), suggested that these archaea combine the degradation of alkanes and methane formation without the need for syntrophic partners4,5. However, these claims lacked direct physiological evidence.
Methanogenesis in the oily sludge
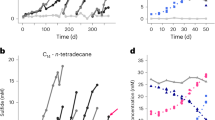
To verify the proposed alternative route of methane formation from oil in archaea, we incubated an anoxic oily sludge derived from subsurface oil reservoirs (1,000–2,000 m below the surface) of the Shengli oilfield (Dong Ying, China)—samples in which Ca. Methanoliparia represented approximately 49% of all archaea (Supplementary Fig. 1a) according to 16S rRNA gene sequencing. Incubation of the oily sludge with sulfate-free anoxic mineral medium resulted in obvious microbial growth at 35, 45 and 55 °C, with a methane production rate of 12.3, 35.8 and 30.1 µmol methane per g of oily sludge per day, and a methane accumulation doubling time of 23.3, 11.3 and 10.1 days, respectively (Fig. 1a, Extended Data Fig. 1a and Supplementary Table 1). These three active cultures consumed most of the n-alkanes with chain lengths of between 13 and 38 carbon atoms, and alkyl substituted cyclohexanes and n-alkylbenzenes, but only if their side alkyl chains contained at least 13 carbon atoms (Fig. 1b and Extended Data Fig. 1b). Previous studies investigating microbial crude oil degradation observed similar preferential consumption of long-chain alkyl-substituted hydrocarbons6,23,24. Analyses based on 16S rRNA sequencing with general primers for archaea revealed that our cultures contained high numbers of the archaeon Ca. Methanoliparum, accounting for 64% to 78% of all archaea (Extended Data Fig. 1c). In situ hybridization with probes specific for Ca. Methanoliparum revealed that this archaeon appears as single coccus-shaped cells attached to oil droplets (Fig. 1c–e and Extended Data Fig. 2). As described previously for marine Ca. Methanoliparia relatives4, Ca. Methanoliparum does not form specific associations with other microorganisms (Fig. 1c–e and Extended Data Fig. 2).

a, Accumulation of CH4 in oily sludge cultures that were incubated with sulfate-free anoxic mineral medium at 55 °C (orange) compared with sterilized control cultures (grey). b, The relative abundance of n-alkanes, n-alkylcyclohexanes and n-alkylbenzenes classified by carbon number in oily sludge cultures incubated at 55 °C (orange) for 301 days compared with sterilized control cultures (grey). c–e, Selection of epifluorescence micrographs of the oil-degrading culture. Cells of Ca. Methanoliparum are targeted with probe DC06-660Mlp in green and bacterial cells are targeted with probe EUB388 I-III in red. Scale bars, 10 µm. Representative images of n = 3 independent samples from one culture are displayed. c, Ca. Methanoliparia and a few bacterial cells accumulate around an oil droplet that is visible in faded green–brown owing to autofluorescence. d, Accumulation of cells of Ca. Methanoliparia with only a few bacterial cells. e, Cells of Ca. Methanoliparia and rare bacterial cells associate with an oil droplet that is visible by red–brown autofluorescence. Additional micrographs are shown in Extended Data Fig. 2.
Metabolic activity of Ca. Methanoliparum
To study the specific turnover of n-alkanes, the most abundant compound class of oil, aliquots of the 55 °C oil-degrading culture grown at stationary phase were transferred into sterilized vials without addition of fresh medium. The cultures were supplemented with 1,2-13C-labelled or unlabelled n-hexadecane (Fig. 2). Within 100 days of incubation, both compounds were quantitatively converted into methane and carbon dioxide (Fig. 2a–c). In the 13C-labelling experiment, around 0.46 mmol of 13CH4 and around 0.15 mmol of 13CO2 were produced, which was equal to 85% to 92% of the stoichiometric conversion of the supplemented labelled hexadecane according to

a, b, The accumulation of CH4 (a) and CO2 (b) in culture aliquots that were incubated with unlabelled hexadecane (Hex.; black) and 1,2-13C labelled n-hexadecane (orange), and controls (grey) lacking the additional substrates. c, The fraction of 13C (atom%) in CH4 (triangles) and CO2 (circles) produced in 1,2-13C n-hexadecane cultures. In cultures amended with unlabelled hexadecane (black) or no substrate (control in grey, not visible), the 13C contents remained stable (~1%). d, The total amount of 13CH4 (triangles) and 13CO2 (circles) produced in the cultures amended with labelled hexadecane (black), and the percentage (orange) of these values compared with theoretical amount of 13CH4 (0.52 mmol) and 13CO2 (0.16 mmol) produced from labelled hexadecane (0.34 mmol) (Methods). Data are mean ± s.d. n = 3 (oily sludge cultures and control cultures) and n = 2 (cultures with unlabelled and labelled hexadecane addition) biologically independent replicates. Where not visible, the error bars are smaller than symbols. e, Archaeal community composition based on the 16S rRNA gene amplicon sequencing. Mba, Methanobacteria; Mlp, Ca. Methanoliparum; Mme, ‘Candidatus Methanomethylicus’. f, The relative abundance of taxonomic groups based on the recruitment of metagenomic reads to 479 dereplicated MAGs (Supplementary Table 4). The names of groups with relative abundances of ≥1% are shown in e and f. ‘Other’ indicates the sum of all groups with a relative abundance of <1% (details of the bacterial community structure are provided in Supplementary Fig. 2). For e, data are mean ± s.d. n = 3 biologically independent replicates, except for day 55 of hexadecane culture (n = 1).
(Fig. 2d). This suggests that only a small portion of the labelled hexadecane was transformed into biomass. Such low carbon assimilation efficiencies have been also reported for other methanogenic hydrocarbon-degrading cultures13,25.
We examined the functioning of Ca. Methanoliparum in the hexadecane-degrading culture using amplicon sequencing, metagenomics and metatranscriptomics (Supplementary Tables 2 and 3). In the archaeal domain, the relative abundance of Ca. Methanoliparum in the hexadecane-degrading cultures comprised up to 75% of the total abundance according to analysis of archaeal 16S rRNA genes (Fig. 2e). Furthermore, Ca. Methanoliparum accounted for approximately 34–40% of the total microbial community as determined by metagenomic read recruitment estimation (Fig. 2f).
In total, the 47 medium-to-high-quality MAGs of Ca. Methanoliparum retrieved from our cultures grouped into four species-level clusters based on phylogenetic inferences and the evaluation of gene- and genome-level identities (Extended Data Figs. 3 and 4 and Supplementary Tables 4 and 5). Cluster 1 includes 15 MAGs that are highly similar to the recently described ‘Candidatus Methanoliparum thermophilum’5. The other species were named ‘Candidatus Methanoliparum widdelii’ (cluster 2, 13 MAGs), ‘Candidatus Methanoliparum whitmanii’ (cluster 3, 10 MGAs) and ‘Candidatus Methanoliparum zhangii’ (cluster 4, 9 MAGs) (Supplementary Table 5). Nineteen MAGs of Ca. Methanoliparum encode ACR and MCR, whereas the 10 MAGs of Ca. M. whitmanii encode only MCR (Fig. 3a, Extended Data Figs. 4e and 5 and Supplementary Table 5). All Ca. Methanoliparum species encode a beta-oxidation pathway with multiple copies of medium- and long-chain acyl-CoA synthases, a Wood–Ljungdahl pathway and a complete methanogenesis pathway with a canonical MCR similar to those of class I/II methanogens (Fig. 3a, Extended Data Fig. 5 and Supplementary Table 6).

a, The metabolic steps and the corresponding gene expression patterns during alkane degradation in Ca. Methanoliparum. The steps are colour coded as follows: orange (substrate activation), blue (beta-oxidation), red (Wood–Ljungdahl and methanogenesis pathways). Energy conservation is indicated in green (Supplementary Table 6). The non-confirmed steps for the oxidation of hexadecanol to hexadecanoate are indicated in grey. Cluster squares indicate the gene expression corresponding to the different ‘Ca. Methanoliparia’ species. Fdred, reduced ferredoxins; Fdox, oxidized ferredoxins. b, Rank of ACR/MCR transcription (filled symbols) of all transcribed genes from the whole microbial community based on log2-transformed fragments per kilobase of transcript per million mapped reads (FPKM) values in cultures incubated with hexadecane and without amendment (Supplementary Table 7). c, Rank of transcript abundances of Ca. Methanoliparum MAGs among all 479 dereplicated MAGs (Supplementary Table 8). The top 100 MAGs recruited most of the transcripts shown here. The positions of the top 1% and 2% are indicated by dashed lines in b and c.
We analysed the gene expression patterns of Ca. Methanoliparum during methanogenic hexadecane degradation (Fig. 3; details are provided in Extended Data Fig. 6 and Supplementary Table 7). The genes encoding the methanogenic hexadecane degradation pathway ranked among the top 10% to 25% of all Ca. M. thermophilum transcribed genes (Fig. 3a). Moreover, genes of Ca. M. thermophilum encoding ACR and MCR ranked among the top 2% of all transcribed genes within the whole community (Fig. 3b). The MAGs of Ca. M. thermophilum also showed the highest transcription among all described MAGs (Fig. 3c and Supplementary Table 8). These analyses indicate that Ca. M. thermophilum performs both the degradation of hexadecane and the formation of methane.
Detection of CoM derivatives
To probe the microbial activation of long-chain alkanes as alkyl-CoM15,16,17, we searched the cell extracts of the hexadecane-degrading cultures for hexadecyl-CoM formation using Q-Exactive Plus Orbitrap mass spectrometry. The unlabelled hexadecane culture contained a prominent mass peak of m/z = 365.21868 that matches the mass produced by synthesized authentic standard of hexadecyl-CoM (Fig. 4a, b). Fragmentation of both peaks yielded hexadecyl-thiol (m/z = 257.23080, C16H33S−), ethenesulfonate (m/z = 106.98074, C2H3SO3−) and bisulfite (m/z = 80.96510, HSO3−) (Fig. 4c). Liquid chromatography analysis revealed that this peak and the authentic hexadecyl standard had identical retention times (Extended Data Fig. 7). Moreover, cultures supplied with 1,2-13C-hexadecane produced a peak at m/z = 367.22524 for 1,2-13C-hexadecyl-CoM and the fragment 259.23721 for 1,2-13C-hexadecyl-thiol, with a mass shift of 2 units compared with the unlabelled group (Fig. 4d, e). These analyses confirmed the activation of n-hexadecane as hexadecyl-CoM.

a, Schematic of ACR-catalysed formation of hexadecyl-CoM thioether (C16H33-SC2H4SO3−) and its expected mass fragments. The dashed arrows and numbers above indicate the positions of chemical bonds splitting. The expected mass change for one of the fragments of 13C-labelled hexadecyl-CoM is shown (red). b, MS analysis of extracts of culture with unlabelled hexadecane showed a peak at m/z = 365.21868 (in blue), which matches the authentic hexadecyl-CoM standard (m/z = 365.21874, dark blue). c, Fragmentation of the isolated m/z range of 365.0–365.4 yields bisulfite (HSO3−, m/z = 80.96510), ethenesulfonate (C2H3SO3−, m/z = 106.98074) and hexadecylthiol (C16H33S−, m/z = 257.23080). These peaks are also produced by hexadecyl-CoM standards. d, MS analysis of extracts of culture with 1,2-13C-labelled hexadecane yielded a peak at m/z = 367.22524. e, Fragmentation of the isolated m/z range of 367.0–367.4 yielded 13C-labelled hexadecylthiol (C16H33S−, m/z = 259.23721), non-labelled ethenesulfonate (C2H3SO3−, m/z = 106.98074) and bisulfite (HSO3−, m/z = 80.96510) (e, from left to right). The mass errors for all mass peaks are <5 ppm.
After the initial incubation of oily sludge, culture aliquots were incubated with n-tetradecane, n-pentadecane, n-hexadecane, n-eicosane, or with a mixture of n-docosane, n-hexadecylcyclohexane and n-hexadecylbenzene. All incubations showed immediate methane formation, and produced the corresponding CoM derivatives from these substrates (Extended Data Figs. 8 and 9 and Supplementary Tables 1 and 9). By contrast, culture aliquots incubated with mixtures of short-chain alkanes (ethane to octane) neither formed the corresponding alkyl-CoMs nor produced more methane than the controls without additional substrates (Supplementary Fig. 3 and Supplementary Table 9). These results showed that the ACR of Ca. Methanoliparum activates diverse hydrocarbons at the long-chain alkyl units. Such a large substrate spectrum in an MCR- or ACR-like enzyme is unprecedented as previous studies suggested that there are high substrate specificities between MCR and ACR and short-chain alkanes such as methane or ethane14,15,17.
On the basis of the observed CoM-type activation of multiple substrate classes, we performed semi-continuous cultivation of Ca. Methanoliparum 55 °C, cultured with a substrate mixture of n-docosane, n-hexadecyl cyclohexane and n-hexadecyl benzene (Extended Data Fig. 9a–c). In each dilution step, 30% to 50% of the culture was transferred and supplemented with fresh mineral medium and hydrocarbons. After dilution, the methanogenic activity recovered with a methane-based doubling time of 10 to 20 days, which is similar to the original oil-degrading cultures (Supplementary Table 1). The abundance of Ca. Methanoliparum recovered and even increased to values of 5 × 108 copies of 16S rRNA gene per ml culture. At the same time, the proportion of the hydrogenotrophic methanogen Methanothermobacter declined (Extended Data Fig. 9d). Moreover, the relative abundance of alkylsuccinate synthases containing bacteria such as ‘Candidatus Caldatribacteriota’26, Actinobacteria27 and Smithella spp2 decreased from 4% in the initial incubation to <0.1% in the sixth transfer (Supplementary Fig. 4a). Similarly, the ratio of assA to acrA transcripts declined from 0.05–0.2 in the original inoculate to <0.005 in the sixth transfer of the culture (Supplementary Fig. 4b). In these cultures, alkylsuccinates—the activation products formed by ASS enzymes—were not detected. In summary, this shows that, in our culture, bacterial hydrocarbon degradation has, at most, a small role.
In yet unresolved reactions, Ca. Methanoliparum transforms the CoM derivatives to CoA-bound acyl units. A previous study proposed that Ca. Methanoliparia cleaves off the CoM group and forms free alcohols as the next intermediate. The alcohols would be sequentially oxidized to aldehydes and fatty acids and then ligated to CoA4. We searched the cell extracts of the 13C-labelled hexadecane experiment for such metabolites, but we could not detect labelled or non-labelled hexadecanol, hexadecanal or palmitic acid. This questions the oxidation of hexadecyl-units as free intermediates. The formed acyl-CoA (hexadecanoyl-CoA) would be split into acetyl units in the beta-oxidation pathway. By contrast, a stepwise degradation of CoM-bound alkylbenzenes would leave benzoyl-CoA residues that cannot directly undergo canonical fatty acid degradation. A previousstudy described a benzoyl-CoA reductase (BCR) gene cluster in Ca. Methanoliparia MAGs4. Indeed, Ca. M. thermophilum also contains a BCR cluster (Supplementary Fig. 5 and Supplementary Table 10). Its bcr genes are flanked by other genes that resemble those of the aromatic degraders Thauera aromatica28 and Rhodopseudomonas palustris29, in which these genes encode a pathway for the degradation of benzoyl-CoA to acetyl CoA (Extended Data Fig. 10).
The known anaerobic alkane-oxidizing archaea completely oxidize the acetyl-CoA through the Wood–Ljungdahl pathway to CO2 and transfer the reducing equivalents to the partner bacteria for sulfate reduction15,16,17. By contrast, Ca. Methanoliparum contains and expresses genes that encode a methyl-H4MPT: CoM methyltransferase and the canonical MCR, features that are absent in alkane-oxidizing archaea that thrive with sulfate-reducing partners15,16,17. These enzymes are used to transfer the methyl groups from H4MPT to CoM, followed by the reduction to methane (Fig. 3a and Extended Data Fig. 6). These steps would degrade the model compound n-hexadecane according to
The excess of reducing equivalents produced in these reactions in form of reduced electron carriers such as ferredoxin or F420, could be balanced by the reduction of CO2 to methane according to
All of the genes of this methanogenesis pathway are present and expressed (Fig. 3a).
Here we demonstrate the activation of different hydrocarbon classes by ACRs of Ca. Methanoliparum, expanding the substrate range of this enzyme to an unforeseen number of compounds. Ca. Methanoliparum couples the degradation of long-chain alkanes and alkyl-substituted hydrocarbons to methane formation, proposed as alkylotrophy. Its metabolic pathways represent an additional mode of methanogenesis, adding to CO2 reduction, methylotrophy, methyl reduction, acetate fermentation30,31,32,33 and the recently reported methoxydotrophy34. Ca. Methanoliparum grows in a wide temperature range, at least between 35 and 55 °C, covering the temperature range of most biodegraded oil reservoirs. Indeed, sequences of Ca. Methanoliparum are present in various anoxic hydrocarbon-rich environments worldwide4,5,19 (Supplementary Table 11). Thus, the demonstration of the unique features of Ca. Methanoliparum in hydrocarbon conversion may fundamentally change our view of crude oil transformation and biogeochemical processes in subsurface oil reservoirs. Future studies with Ca. Methanoliparia cultures will resolve the biochemical mechanisms of methanogenic hydrocarbon degradation in archaea, and will be helpful for the application of microbial-enhanced energy recovery from depleted oil reservoirs35.
Methods
Data reporting
No statistical methods were used to predetermine sample size. The experiments were not randomized, and investigators were not blinded to allocation during experiments and outcome assessment.
Cultivation of methanogenic hydrocarbon-degrading organisms
The oily sludge used as inoculum for the incubations was sampled from an oil tank of the Shengli oilfield in eastern China (37° 54′ N, 118° 33′ E). The crude oil in the tank was derived from multiple oil wells of subsurface oil reservoirs with a depth range of 1,000–2,000 m. The oily sludge is a mixture of water (27–46%, w/w), crude oil (35–59%, w/w) and sands (13–19%, w/w). The sludge was stored anoxically in bottles at 4 °C. The microbial community structures and abundances of bacteria and archaea in the oily sludge are shown in Supplementary Fig. 1a–c. The anoxic medium was prepared as described previously36 using 9 g l−1 NaCl, 3 g l−1 MgCl2·6H2O, 0.15 g l−1 CaCl2·2H2O, 0.3 g l−1 NH4Cl, 0.2 g l−1 KH2PO4, 0.5 g l−1 KCl, 0.5 g l−1 cysteine-HCl, 1 ml l−1 resazurin solution and 2 ml l−1 trace element solution37. The medium was autoclaved at 121 °C for 30 min. After cooling to room temperature under N2 (99.999%), sterile Na2S·9H2O (0.25 g l−1), vitamin mixture (2 ml l−1), vitamin B12 (2 ml l−1) and vitamin B1 (2 ml l−1) solutions were added, and the pH of this prereduced medium (PRM) was adjusted to 6.8–7.0 by addition of HCl or NaOH solution (1 M). The pH of the active cultures was repeatedly controlled using a portable pH meter (Laqua Twin, AS-712, Horiba).
The initial incubations were set up under N2 (99.999%) atmosphere with 5 g of oily sludge and 20 ml PRM medium in 60 ml pressure vials (Yaobo). Vials were sealed with butyl rubber stoppers (Bellco Glass) and aluminium caps. Replicate cultures were incubated statically in the dark at temperatures between 25 °C and 75 °C, with steps of 10 °C. The sterilized controls were set up by autoclaving the cultures at 121 °C for 30 min, and then incubated at 25, 55 or 75 °C. Four parallel cultures supplemented with100–200 g oily sludge and 1 l PRM were also set up in 2 l serum bottles (Shuniu) and incubated at 55 °C in the dark without shaking for physiological experiments (for example, labelling substrate experiments, see below).
For the 13C-labelled hexadecane incubations, the headspace gas of sterilized 300 ml serum bottles (Shuniu) containing 5 g sterile glass beads (Qinghua) was purged with N2 (99.999%) for 15 min. An aliquot of 100 ml of the oil-degrading cultures was then directly distributed into each bottle, with the addition of 100 µl of unlabelled or 1,2-13C-labelled hexadecane (Sigma-Aldrich). Control cultures received no additional substrates. To further verify the growth of Ca. Methanoliparum and the methanogenic activity of the cultures, we set up semi-continuous incubations as follows: the 150 ml oil-degrading cultures were inoculated into the 600 ml serum bottle with 150 ml PRM, and amended with a mixture of n-docosane (0.15 g, Sigma-Aldrich), n-hexadecylcyclohexane (150 µl, TCI) and n-hexadecylbenzene (150 µl, TCI) (C22 mix). After each incubation reached the late logarithmic methane-production phase, a total of 50–70% (v/v) of the growing cultures was removed with a syringe, and then the same amount of fresh PRM and C22 mix was supplemented in each transfer incubation.
To test the utilization of short- and medium-chain alkanes, we set up two treatments with 10 ml PRM and 10 ml oily-sludge-degrading cultures in 100 ml serum bottles (Shuniu) and added substrates as follows: (1) a mixture of n-ethane (11 ml), n-propane (17 ml), n-butane (10 ml) and n-pentane (2 ml); (2) a mixture (10 µl of each) of n-hexane, n-heptane and n-octane (Kelong). The gaseous alkane mixture was produced by the Southwest Research & Design Institute of the Chemical Industry. All of the cultures described above were incubated at 55 °C in the dark without shaking.
Calculation of the methane production
Methane production data of previous studies were digitized from scanned plots of methane production curves using Plot Digitizer38. The data from the logarithmic phase of methane accumulation curve were used for the calculation of the maximum specific methane production rate (μmax) and methane doubling time (G) as follows:39
where t1 is the first time point at which methane data were used; t2 is the second time point at which methane data were used; y1 is the amount of methane at the sampling time t1; y2 is the amount of methane at the sampling time t2. t1 and t2 were sampled from the early logarithmic methane-production phase.
The methane production rate of the oily-sludge-degrading cultures was calculated as follows:
where T1 is the time from the date of culture to 5% of the maximum methane production (Y1); T2 is the time from the date of culture to 90% of the maximum methane production (Y2); W is the weight (in grams) of oily sludge.
Chemical analyses
Gas samples (200 µl) were taken from the headspace of the vials or serum bottles at different time points using a pressure lock syringe (Vici). Concentrations of CH4 and CO2 were measured using the Agilent GC 7820A gas chromatography system equipped with a Porapak Q column (length, 3 m; inner diameter, 0.32 mm) and a thermal conductivity detector40. The column, oven and detector temperatures were set to 65 °C, 120 °C and 130 °C, respectively. The carrier gas was hydrogen (99.999%) at a flow rate of 27 ml min−1. The gas pressure in the headspace of vials or serum bottles was determined using a barometer at room temperature (Ashcroft). The carbon isotopic compositions of formed CH4 and CO2 were analysed by gas chromatography coupled via combustion to isotope ratio mass spectrometry as described previously41. CH4 and CO2 were separated by gas chromatography (Agilent GC 7890B), using the HP-PLOT/Q column (30 m; inner diameter, 0.32 mm; film thickness, 20 µm) and helium as the carrier (99.999%; flow rate, 2.514 ml min−1). The oven and injector temperatures were set to 60 °C and 105 °C, respectively. After in line combustion of CH4 to CO2 on an IsoPrime GC5 combustion interface (IsoPrime), the isotopic composition of CO2 was determined using the IsoPrime100 isotope ratio mass spectrometer.
The amounts of CH4 and CO2(g) in the headspace were calculated according to Avogadro’s law40. Trace amounts of methane dissolved in the liquid phase were not considered. Total dissolved CO2 (TDC) in the liquid medium was calculated according to Henry’s Law with the equilibrium constants of the dissociation of CO2 as described by Stumm et al42. In brief, equilibrium values (25 °C, temperature during gas measuring) of the following equations were retrieved:42
Here [H2CO3*] = [CO2(aq)] + [H2CO3], which could be calculated by the following equation based on the headspace CO2 concentration measured:42
Then, the TDC and total generated CO2 (t) were calculated on the basis of equations (11) and (12).
where CO2(t) is the total CO2 measured from the sum of liquid and gaseous CO2; CO2(g) is the CO2 in the headspace of vials; and CO2(aq) is the gaseous CO2 in the liquid medium. K1, K2, K3 are the equilibrium constants listed in equations 7–9.
To calculate rates of conversion of hexadecane to CH4 and CO2 (Fig. 2d), the isotopic compositions of 13CH4 and 13CO2 were used to avoid the effect of oil carryover from inoculum. The amounts of 13C gas (13CH4 and 13CO2) produced from 1,2-13C-hexadecane (Fig. 2d) were calculated according to equations (13) and (14) (see below). The total amount of 13C gas in the labelled cultures and control cultures was calculated on the basis of equation (13) and the amount of 13C gas derived from 1,2-13C-hexadecane in the labelled cultures was calculated on the basis of equation (14) by subtracting total amount of 13C gas in control cultures from the values of the labelled cultures. The isotope fractionation of CO2 dissolving in water was neglected43. According to equation (1) in the main text, the complete degradation of added 1,2-13C-hexadecane (100 µl, 0.34 mmol) would theoretically produce 0.52 mmol 13CH4 and 0.16 mmol 13CO2. The conversion ratio of hexadecane during the incubation was then calculated by dividing the labelled hexadecane-derived 13CH4 and 13CO2 by the theoretical yield (equation (15)).
‘Gas’ in equations (13)–(15) represents CH4 or CO2.
Fractionation and GC–MS analysis of petroleum hydrocarbons
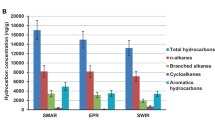
Oil samples (30–50 mg) were retrieved from the incubation vials after 301 days of incubation via glass capillaries, and separated into four fractions (saturates, aromatics, resins and asphaltenes) as previously described23. In brief, both saturated and aromatic hydrocarbon fractions were analysed using gas chromatography coupled with mass spectrometry (GC–MS) (Agilent 6890-5975i) equipped with a HP-5MS-fused silica column (length, 60 m; inner diameter, 0.25 mm; thickness, 0.25 µm)23. For later quantifications, we added pristane, phytane and 17a(H), 21b(H)-hopane (for saturates), and methylbiphenyl, 3-methylbiphenyl and chrysene (for aromatics), which are not biodegraded under anoxic conditions. Relative quantification of the target compounds in saturated and aromatic fractions was obtained by comparing their mass chromatogram peak areas to those of reference standards. Figure 1b and Extended Data Figure 1b display the amounts of n-alkanes (m/z = 85), n-alkylcyclohexanes (m/z = 82), and n-alkylbenzene (m/z = 92) relative to pristane23 (equation (16)).
CARD-FISH analysis
Catalysed reported deposition-fluorescence in situ hybridization (CARD-FISH) analyses were performed as described by Laso-Pérez et al4. In brief, an aliquot (2–3 ml) of the 55 °C oily sludge cultures grown in exponential phase was fixed with 2% formaldehyde at 25 °C for 3 h. Fixed samples were washed and stored in phosphate-buffered saline (PBS, pH 7.4)-ethanol (1:1, v/v) and sent to the Max Planck Institute for Marine Microbiology. Aliquots of 10–20 µl of the stored sample were filtered using GTTP polycarbonate filters (0.2 µm pore size; Millipore). The CARD-FISH reaction was performed as described previously44 with the following modifications. Cells were permeabilized with lysozyme (PBS pH 7.4, 0.005 M EDTA pH 8.0, 0.02 M Tris-HCl pH 8.0, 10 mg ml−1 lysozyme; Sigma-Aldrich) at 37 °C for 30 min, and with proteinase K (PBS pH 7.4, 0.005 M EDTA pH 8.0, 0.02 M Tris-HCl pH 8.0, 15 µg ml−1 proteinase K; Merck) at room temperature for 3 min. Endogenous peroxidases were inactivated by incubation with 0.15% H2O2 in methanol for 30 min at room temperature. The 16S rRNA gene was targeted with the specific oligonucleotide probes EUB-338 I-III45,46, ARCH-915 (refs.47,48) and a newly designed probe for members of the Ca. Methanoliparum genus named DC06-660Mlp. All of the probes were purchased from biomers.net. The sequence of the DC06-660Mlp probe is 5′-GTACCTCTGGTCTCTCCT-3′, a modification of probe DC06-660 (ref.4) that enables specific detection of the genus Ca. Methanoliparum. The stringency of all probes was tested with a formamide concentration of between 10% to 50% in the hybridization buffer. A 35% formamide was selected for the EUB-338 and ARCH-915 probes, whereas 20% formamide was applied for the DC06-660Mlp probe. Double hybridization was performed after inactivation of peroxidases from the first hybridization by incubation in a solution of 0.30% H2O2 in methanol for 30 min at room temperature in the dark. For signal amplification, the fluorochromes Alexa Fluor 488 and Alexa Fluor 594 were used. Filters were counterstained with 4′,6′-diamino-2-phenylindole and analysed using epifluorescence microscopy (Axiophot II imaging).
Extraction of genomic DNA, 16S rRNA gene amplicon sequencing and data analysis
For amplicon sequencing, 2 ml of growing culture was pelleted by centrifugation (17,000g for 5 min at 4 °C) during incubations at different time points. DNA was extracted by using a bead-beating method as previously described6. Bacterial and archaeal 16S rRNA genes were amplified using the primer sets 341F (5′-CCTAYGGGRBGCASCAG-3′) and 806R (5′-GGACTACNNGGGTATCTAAT-3′)49, and Arch519F (5′-CAGCCGCCGCGGTAA-3′) and Arch915R (5′-GTGCTCCCCCGCCAATTCCT-3′)50, respectively, with 34 thermal cycles of 94 °C for 1 min; 57 °C for 45 s; and 72 °C for 1 min. Amplicon sequences were generated using a NovaSeq 6000 sequencer (Illumina) with paired-end 250 bp mode (PE250) at Novogene Bioinformatics Technology. Raw reads were filtered as previously described51,52. Quality-filtered reads were loaded into the Qiime2 pipeline53. Operational taxonomic units (OTUs) were defined with a cut-off value of 97% and were then taxonomically classified by using the Naive Bayes method implemented in Qiime2, with the Silva NR99 database (release 138) as the reference. 54.
Metagenome sequencing, reads processing, assembly, genome binning and annotation
For metagenomic sequencing, 10–15 ml cultures were collected at different time points, and total DNA was extracted using the beat-beating method as described above. Sequencing libraries were prepared by Novogene using their in-house pipelines (https://en.novogene.com) and metagenomic sequencing data were generated using a NovaSeq 6000 instrument with PE150 at Novogene. The raw reads were quality-trimmed using Trimmomatic55 with the default parameters. For each sample, the quality-trimmed reads were de novo assembled using metaSPAdes (v.3.12.0)56 with the following parameter: -k 21, 33, 55, 77. Genome binning of the assembled contigs was performed using MetaBAT (v.2.12.1) using the coverage information of the contigs57. The quality (completeness, contamination and strain heterogeneity) of recovered MAGs was evaluated using CheckM (v.1.0.11)58. Taxonomic classification of MAGs was performed with GTDB-Tk (v.1.3.0)59 based on the GTDB database60 (release 95.0, July 2020). In total, we obtained 2,179 MAGs, including 47 medium-to-high quality MAGs belonging to the genus Ca. Methanoliparum. Annotations of the 47 MAGs were carried out by the DRAM pipeline (https://github.com/shafferm/DRAM)61, which implements annotations from Kyoto Encyclopedia of Genes and Genomes (KEGG), UniRef90 and PFAM databases. Moreover, annotations of predicted open reading frames were also compared and evaluated by using the KEGG server (BlastKOALA) and eggNOG-mapper62,63. To determine the similarity of the 47 MAGs of Ca. Methanoliparum, the 16S rRNA gene sequence identity, the average amino acid identity (AAI), the average nucleotide identity (ANI) and the percentage of conserved proteins (POCP) were calculated. The 16S rRNA gene sequences were retrieved using barrnap (https://github.com/tseemann/barrnap, v0.9). The AAI was calculated using CompareM (v.0.0.22) (https://github.com/dparks1134/CompareM) using the ‘aai_wf’ command with the default parameters. The ANI between genomes was calculated using the Orthologous Average Nucleotide Identity Tool64. The POCP was calculated using a script by Qin et al65.
Identification of genes encoding enzymes involved in hydrocarbon activation
To identify genes involved in the anaerobic degradation of hydrocarbons, a custom hidden Markov model (HMM) profile was constructed based on the protein sequences of known genes involved in the anaerobic hydrocarbon oxidation through fumarate addition. Sequences from hydrocarbon-degrading enzymes that are present in the AnHyDeg database (https://github.com/AnaerobesRock/AnHyDeg) were combined with recently published protein sequences of alkyl-succinate synthases26,27. These sequences were the alpha subunits from alkyl-succinate synthase/(1-methylalkyl)succinate synthase (AssA/MasD), benzylsuccinate synthase (BssA), naphtylmethylsuccinate synthase (NmsA) and archaeal type alkylsuccinate synthases (PflD) (Supplementary Fig. 6). To identify alkyl CoM reductase sequences, HMM profiles for the methyl CoM reductase alpha subunit (PF02249 for MCR_alpha and PF02745 for MCR_alpha_N) from PFAM database were used. Scaffolds with length > 500 bp from all metagenomic data were kept and coding sequences were predicted with Prodigal (v.2.6.3)66 using the ‘-p meta’ parameter. Putative marker genes were screened using the profiles described above with hmmsearch (HMMER v.3.1b2)67 and validated by phylogenetic analysis (as described below). The acrA and assA-related genes were dereplicated with 99% nucleic acid identity using CD-hit68 and used as references for metatranscriptomic reads mapping.
Phylogenomic analyses of MAGs and phylogenetic analyses of functional genes
For the phylogenomic analysis, reference genomes of related archaea were downloaded from the NCBI database (https://www.ncbi.nlm.nih.gov/). A list of the reference genomes is provided in Supplementary Table 12. To construct a phylogenomic tree, CheckM was used to extract 16 ribosomal proteins (ribosomal proteins L2-L6, L14-L16, L18, L22, L24, S3, S8, S10, S17 and S19) from the MAGs of this study and the downloaded reference genomes. Then, genes were aligned independently using MUSCLE69 as described previously70. The concatenated amino acid alignment was used to construct a phylogenomic tree with IQ-TREE (v.1.6.1)71 using the WAG model, with the option ‘-bb 1000’. Moreover, a 16S rRNA gene phylogenetic tree was constructed. For this, 16S rRNA gene sequences retrieved from Ca. Methanoliparum MAGs were aligned with the SINA aligner72. Alignments were imported into the SILVA ribosomal RNA database (NR99, release 138)54 using ARB (v.6.1)73. 16S rRNA gene sequences of Ca. Methanoliparia were inserted (with quick add options) into phylogenetic tree using maximum parsimony methods.
For the phylogenetic analysis of AcrA/McrA, reference sequences were retrieved from Annotree (based on GTDB release version 95)74 based on the HMM profiles for the methyl-CoM reductase alpha subunit (PF02249, MCR_alpha and PF02745, MCR_alpha_N) from the PFAM database. AcrA sequences from a recent publication were also included19. For AssA, sequences used for the AssA custom HMM profile construction (see above) were used as references. Reference sequences of the BcrB and BcrC amino acid sequences were also downloaded by searching for TIGR02260 and TIGR02263 from Annotree74 with a cut-off value of 1 × 10−10. BcrB/BcrC sequences from Thauera, Rhodopseudomonas, ‘Ca. Methanolliviera hydrocarbonicum’, Ca. M. thermophilum and GoM_asphalt were also included as refs. 4,5. The amino acid sequences of AcrA/McrA, BcrB and BcrC were retrieved from 47 Ca. Methanoliparum using hmmsearch (HMMER v.3.1b2)67 as described above. The amino acid sequences of AcrA/McrA, AssA related, BcrB and BcrC proteins were aligned using MUSCLE, respectively69. Phylogenetic trees for AcrA/McrA, AssA-related, BcrB and BcrC protein sequences were constructed using IQ-TREE (v.1.6.1) using the WAG model with the option ‘-bb 1000’. All trees were visualized in iTOL75.
Quantification of 16S rRNA genes
Bacteria and Ca. Methanoliparum in cultures were assessed using quantitative PCR. The primers 519F/907R (5′-CAGCMGCCGCGGTAANWC-3′/ 5′-CCGTCAATTCMTTTRAGTT-3′)76 and Mlp1F/ Mlp1R (5′-GGGAATTCGACTAAGCCATGCAA-3′/ 5′-CCCGGCCCTTTCTATTAGGTG-3′) were used for bacteria and Ca. Methanoliparum, respectively. Triplicate amplifications were conducted in a 10 μl reaction system containing 5 μl of SsoFast EvaGreen Super Mix (BIORAD), 3.25 μl of sterilized distilled H2O, 0.25 μl of each primer (10 μM), 0.25 μl of 5 mg ml−1 bovine serum albumin and 1 μl of template DNA. The thermal cycling steps were performed using the Bio-Rad qPCR instrument (Bio-Rad CFX96), which consisted of an initial denaturation step at 95 °C for 3 min, 45 cycles of 95 °C for 15 s, 65 °C for 15 s and 72 °C for 10 s with plate reading, and a final extension step at 72 °C for 10 min. After the main program, melt curve analysis was performed from 65 °C to 95 °C, with an increment of 0.5 °C and 0.5 s plate reading at each step. The sample DNA was diluted for about 10–100 times when necessary. The standard curve was generated with tenfold serially diluted linear plasmids containing a single copy of the bacterial or Ca. Methaoliparum 16S rRNA gene77.
Metatranscriptomics workflow
For metatranscriptomics, about 15–20 ml of active growing culture was collected by centrifugation (17,000g, 5 min, 4 °C; Beckman Coulter) (Supplementary Table 3). Cells pellets were transferred immediately to liquid N2 and stored at −80 °C. Cells were lysed by using bead-beating methods and total RNA was extracted using an acid phenol chloroform isoamyl alcohol-based protocol as described above78, and sent to Novogene on dry ice. Quality checking, genomic DNA digestion, ribosome RNA removal, cDNA synthesis and library construction were performed by Novogene with their in-house pipelines (https://en.novogene.com). Metatranscriptomic sequencing data were generated using the NovaSeq 6000 instrument with PE150 at Novogene. The raw reads were first quality-trimmed using Trimmomatic55 with the default parameters and the mRNA sequences were obtained by removing tRNA and rRNA sequences with SortMeRNA79.
Evaluation of the relative abundance and activity of Ca. Methanoliparum
In addition to 16S rRNA gene amplicon sequencing, the relative abundance of Ca. Methanoliparum was estimated by mapping the metagenomic reads to the 16S rRNA genes (whole prokaryotic community) and to all binned MAGs. Sequences of the 16S rRNA gene extracted from three quality-filtered metagenomic datasets obtained from cultures with and without hexadecane were taxonomically assigned by Kraken2 (ref. 80). For that, a custom database was built based on Silva (NR99 database, release 138)54 and the reads from each sample were searched against this custom database. After the best-hit processing, classification of reads at different taxonomic levels was obtained. A total of 479 medium-to-high-quality MAGs were obtained after dereplicationwith dRep81 with an ANI cut-off of 97% (Supplementary Table 4). The abundance of dereplicated MAGs was determined by mapping the quality-filtered metagenomic reads to the contigs of the MAGs using Bowtie2 (v.2.2.8)82. The resulting SAM mapping files were converted to BAM files using SAMtools83. The read coverage in MAG contigs was calculated using BEDTools84. The number of reads was normalized to the length of MAGs. The relative abundance of each MAG is the number of normalized reads mapped to individual MAG divided by the total number of normalized metagenomic reads mapped to all dereplicated MAGs.
To estimate the expression of Ca. Methanoliparum in hexadecane-degrading cultures, the abundance of transcripts was calculated at both the genome and gene level. At the genome level, the relative transcript abundance of Ca. Methanoliparum was calculated similarly to the relative abundance of Ca. Methanoliparum in the metagenomic data, but mapping the metatranscriptomic reads (Fig. 3c). The transcription of key genes encoding enzymes that are involved in alkane degradation in the four Ca. Methanoliparum species was calculated by mapping high-quality non-rRNA reads to all of the annotated genes using Burrows–Wheeler Aligner (BWA) with the default settings85. The resulting SAM mapping files were converted to BAM files using SAMtools83. The read coverage in MAG contigs was calculated using BEDTools84. We used the FPKM method to normalize the expression level. All annotated genes were ranked according to FPKM values (Fig. 3a). Similarly, FPKM expression values of all the annotated genes from assembled contigs were calculated for the culture with hexadecane addition. All of the transcribed genes were ranked based on the log2[FPKM] values (Fig. 3b).
Moreover, the expression activities of acrA and assA-related genes were calculated by mapping high-quality non-rRNA reads to all dereplicated acrA and assA-related gene sequences (see above) using BWA with the default settings85. The relative transcript abundances of acrA and assA-related genes were calculated by dividing the FPKM value of individual gene by the sum of FPKM values of all acrA and assA-related genes (Supplementary Fig. 4b).
Synthesis of authentic alkyl-CoM standards
To synthesize hexadecyl-CoM and eicosyl-CoM standards, 0.125 g of sodium 2-mercaptoethanesulfonate (CoM, purity ≥ 98%; Aladdin) and 470 µl of 1-iodohexadecane or 1-iodoeicosane (purity ≥ 97%; Aladdin) were dissolved in 2 ml of 30% ammonium hydroxide solution in a sealed serum bottle. After magnetic stirring (~500 rpm) for 14 h at 55 °C, the solution was bubbled with nitrogen gas (99.999%) for 30 min at room temperature. To obtain the pure hexadecyl-CoM and eicosyl-CoM standards, the mixture was passed through a HC-C18 SPE Cartridge (100 mg, 1 ml, ANPEL Lab Technologies) with 0.8 ml 80% acetonitrile water solution as an eluent. The standards were kept at −20 °C until analysis.
Metabolite extraction
To prepare metabolite extracts, cells were collected from 15–25 ml active growing cultures by centrifugation (12,000g for 8 min at 4 °C) and washed twice with 5 ml of 7 mM PBS solution. The cell pellets were transferred into 2 ml microtubes (SARSTEDT) containing 1 ml of a mixture of acetonitrile:methanol:water (40:40:20, v/v/v) and 0.3 g sterile glass beads (0.1 mm diameter; Sigma-Aldrich). Cells were lysed by shaking at 6 m s−1 for 50 s with a homogenizer (FastPrep-24, MP). Glass beads and cell debris were removed by centrifugation (12,000g for 10 min at 4 °C). The cell extracts were diluted in 10 ml deionized water and further purified with an HC-C18 SPE Cartridge as described above. The cell extracts were stored at −20 °C until analysis.
MS analysis of synthesized standards and cell extracts
Authentic standards and cell extract samples were analysed using the Q-Exactive Plus Orbitrap mass spectrometer (QE Plus Orbitrap, Thermo Fisher Scientific). To detect hexadecyl-CoM, heated electrospray ionization (HESI) ion monitoring (SIM) MS was performed in the negative ionization mode with an m/z scan range of 365–366 Da. The parameters for the HESI source were set as follows: a CE-Inject Voltage of 3.6 kV, a de-measure voltage of 450 V, a de-inject voltage of 40 V, a Z-lens 3 voltage of 230 V, a lens 6 voltage of 680 V, CLT pull voltage of 270 V, CLT push voltage of 120 V, CLT offset voltage of 1.6 kV, a spray voltage of 3.2 kV, sheath gas flow rate of 12 p.s.i., auxiliary-gas flow rate of 2 p.s.i., capillary temperature of 320 °C, S-lens level of 55 and a probe heater temperature of 50 °C. The injection volume was 1–5 μl. The formation of an even-electron fragment HSO3− from bisulfite is favoured in the presence of a beta H atom and SO3−•, which was also produced after fragmentation, as described by Laso-Pérez et al16. and the references within. Fragments with masses of HSO3−, C2H3SO3− and C16H33S− were indicative of hexadecyl-CoM in both of the standard and the cell extracts. The mass errors of less than 5 ppm were applied in data processing accordingly and all data were acquired and processed using Xcalibur 3.0 (Thermo Fisher Scientific).
The hexadecyl-CoM in cell extracts was further confirmed by high performance liquid chromatography–tandem MS using a triple quadrupole mass spectrometer (HPLC–MS/MS, AB4500, SCIEX). The hexadecyl-CoM was detected by multiple-reaction monitoring mode (MRM). All three hexadecyl-CoM transitions (m/z = 365.2– 80.9, m/z = 365.2–107.0 and m/z = 365.2–257.2) were initially optimized by direct infusion of standard solution into the mass spectrometer. For comparison of retention times of peaks of interest and standard, the mass spectrometer was coupled to a HPLC system (LC-30AD, Shimadzu) equipped with a reversed-phase C18 column (1.7 μm particle size; length, 100 mm; inner diameter, 2.1 mm; Thermo Fisher Scientific), and run with a binary gradient (15% to 90% acetonitrile in water) at a flow rate of 0.3 ml min−1 and with an oven temperature of 40 °C. For eachanalysis, 1–10 μl standards or samples were injected into the HPLC. The retention time and the presence of both mass transitions as compared to the standards were used as quality criteria.
Screening of public datasets for Ca. Methanoliparum
The 16S rRNA gene sequences longer than 700 bp retrieved from all Ca. Methanoliparum MAGs were searched against the April 2019 NCBI Sequence Read Archive (422,880 runs) using the Integrated Microbial Next Generation Sequencing (IMNGS) platform (https://www.imngs.org/). SRA reads were considered to be Ca. Methanoliparum, if they matched this 16S rRNA gene at a minimum of 97% identity over at least 200 bp. Information of latitude, longitude and origin sources was obtained from the metadata of each BioSample with significant hits. Output SRA environment categories were combined into ‘marine water’, ‘marine sediment’, ‘microbial mat’, ‘oil field’, ‘anaerobic digester’, ‘sludge’ or ‘soils’. The relative abundance refers to the number of reads of Ca. Methanoliparum normalized to the total number of reads for the sample (Supplementary Table 11).
Taxonomic description of new Ca. Methanoliparum species
Ca. Methanoliparum thermophilum was proposed and described by Borrel and colleagues5.
Ca. Methanoliparum widdelii (wid.del’i.i. N.L. gen. n. widdelii, of, Widdel, named after F. Widdel), an alkylotrophic methanogen of genus Ca. Methanoliparum named in honour of F. Widdel for his work on anaerobic hydrocarbon transformation. The type material is the genome designated HX_O_T65_bin.11 representing Ca. M. widdelii. The genome HX_O_T65_bin.11 represents a MAG comprising 1.54 Mbp in 35 contigs with an estimated completeness of 92.13%, an estimated contamination of 1.96%, a 23S rRNA gene, a 5S rRNA gene and 44 tRNAs. The MAG was recovered from oily sludge of Shengli oilfield incubated anaerobically with crude oil as substrate and has a G + C content of 36.07% (further details are provided in Supplementary Table 5).
Ca. Methanoliparum whitmanii (whit.man’i.i. N.L. gen. n. whitmanii, of, Whitman, named after W. B. Whitman), a novel methanogen of genus Ca. Methanoliparum named in honour of W. B. Whitman for his work on methanogens. The type material is the genome designated XY_O_T55_M2_bin.61 representing Candidatus M. whitmanii. The genome XY_O_T55_M2_bin.61 represents a MAG comprising 1.41 Mbp in 60 contigs with an estimated completeness of 90.5%, an estimated contamination of 1.31%, a 5S rRNA gene and 38 tRNAs. The MAG was recovered from oily sludge of Shengli oilfield incubated anaerobically with crude oil as substrate and has a G + C content of 36.49% (further details are provided in Supplementary Table 5).
Ca. Methanoliparum zhangii (zhang’i.i. N.L. gen. n. zhangii, of, Zhang, named after H. Zhang), an alkylotrophic methanogen of genus Ca. Methanoliparum named in honour of H. Zhang for his work on methanogens and petroleum microbiology. The type material is the genome designated GD_Cm_T35_P3_bin.32 representing Ca. M. zhangii. The genome GD_Cm_T35_P3_bin.32 represents a MAG comprising 1.62 Mbp in 74 contigs with an estimated completeness of 91.8%, an estimated contamination of 1.96%, a 23S rRNA gene, a 16S rRNA gene, a 5S rRNA gene and 43 tRNAs. The MAG was recovered from oily sludge of Shengli oilfield incubated anaerobically with a mixture of n-docosane, hexadecylcyclohexane and hexadecylbenzene as substrate, and has a G + C content of 37.2% (further details are provided in Supplementary Table 5).
Figure generation
The manuscript figures were generated mainly using ggplot2 (ref. 86), pheatmap (https://github.com/raivokolde/pheatmap), gggenes and custom R87 scripts in Rstudio (https://rstudio.com/), ARB73 and Adobe Illustrator (http://www.adobe.com/au/products/illustrator.html).
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Data availability
The 16S rRNA gene amplicon sequences, metagenomic and metatranscriptomic data generated in current study are available in the NODE database (http://www.biosino.org/node/project/detail/OEP001282). The data of dereplicated MAGs analysed during the current study are available in the NODE database under the accession numbers OEZ006960 and OEZ007009–OEZ007026. Further details are provided in Supplementary Table 13. All other data are available in the main text or the Supplementary Information.
Code availability
The sources of the code and programs used for analyses are mentioned in the Methods, and are also available at GitHub (https://github.com/liupfskygre/Methanoliparum_MS_code/tree/main).
References
Jones, D. M. et al. Crude-oil biodegradation via methanogenesis in subsurface petroleum reservoirs. Nature 451, 176–180 (2008).
Zengler, K., Richnow, H. H., Rossello-Mora, R., Michaelis, W. & Widdel, F. Methane formation from long-chain alkanes by anaerobic microorganisms. Nature 401, 266–269 (1999).
Dolfing, J., Larter, S. R. & Head, I. M. Thermodynamic constraints on methanogenic crude oil biodegradation. ISME J. 2, 442–452 (2008).
Laso Pérez, R. et al. Anaerobic degradation of non-methane alkanes by “Candidatus Methanoliparia” in hydrocarbon seeps of the Gulf of Mexico. mBio 10, e01814-19 (2019).
Borrel, G. et al. Wide diversity of methane and short-chain alkane metabolisms in uncultured archaea. Nat. Microbiol. 4, 603–613 (2019).
Cheng, L. et al. Progressive degradation of crude oil n-alkanes coupled to methane production under mesophilic and thermophilic conditions. PLoS ONE 9, e113253 (2014).
Head, I. M., Jones, D. M. & Röling, W. F. M. Marine microorganisms make a meal of oil. Nat. Rev. Microbiol. 4, 173–182 (2006).
Van Hamme, J. D., Singh, A. & Ward, O. P. Recent advances in petroleum microbiology. Microbiol. Mol. Biol. Rev. 67, 503–549 (2003).
Aitken, C. M., Jones, D. M. & Larter, S. R. Anaerobic hydrocarbon biodegradation in deep subsurface oil reservoirs. Nature 431, 291–294 (2004).
Head, I. M., Jones, D. M. & Larter, S. R. Biological activity in the deep subsurface and the origin of heavy oil. Nature 426, 344–352 (2003).
Gieg, L. M., Fowler, S. J. & Berdugo-Clavijo, C. Syntrophic biodegradation of hydrocarbon contaminants. Curr. Opin. Biotechnol. 27, 21–29 (2014).
Rabus, R. et al. Anaerobic microbial degradation of hydrocarbons: from enzymatic reactions to the environment. J. Mol. Microbiol. Biotechnol. 26, 5–28 (2016).
Fowler, S. J., Dong, X., Sensen, C. W., Suflita, J. M. & Gieg, L. M. Methanogenic toluene metabolism: community structure and intermediates. Environ. Microbiol. 14, 754–764 (2012).
Thauer, R. K. Methyl (alkyl)-coenzyme M reductases: nickel F-430-containing enzymes involved in anaerobic methane formation and in anaerobic oxidation of methane or of short chain alkanes. Biochemistry 58, 5198–5220 (2019).
Hahn, C. J. et al. “Candidatus Ethanoperedens”, a thermophilic genus of Archaea mediating the anaerobic oxidation of ethane. mBio 11, e00600-20 (2020).
Laso-Pérez, R. et al. Thermophilic archaea activate butane via alkyl-coenzyme M formation. Nature 539, 396–401 (2016).
Chen, S.-C. et al. Anaerobic oxidation of ethane by archaea from a marine hydrocarbon seep. Nature 568, 108–111 (2019).
Wang, Y., Wegener, G., Hou, J., Wang, F. & Xiao, X. Expanding anaerobic alkane metabolism in the domain of Archaea. Nat. Microbiol. 4, 595–602 (2019).
Wang, Y., Wegener, G., Ruff, S. E. & Wang, F. Methyl/alkyl-coenzyme M reductase-based anaerobic alkane oxidation in Archaea. Environ. Microbiol. 23, 530–541 (2020).
Boyd, J. A. et al. Divergent methyl-coenzyme M reductase genes in a deep-subseafloor Archaeoglobi. ISME J. 13, 1269–1279 (2019).
Baker, B. J. et al. Diversity, ecology and evolution of Archaea. Nat. Microbiol. 5, 887–900 (2020).
Seitz, K. W. et al. Asgard archaea capable of anaerobic hydrocarbon cycling. Nat. Commun. 10, 1822 (2019).
Cheng, L. et al. Preferential degradation of long-chain alkyl substituted hydrocarbons in heavy oil under methanogenic conditions. Org. Geochem. 138, 103927 (2019).
Oldenburg, T. B. P. et al. The controls on the composition of biodegraded oils in the deep subsurface—part 4. Destruction and production of high molecular weight non-hydrocarbon species and destruction of aromatic hydrocarbons during progressive in-reservoir biodegradation. Org. Geochem. 114, 57–80 (2017).
Cheng, L. et al. DNA-SIP reveals that Syntrophaceae play an important role in methanogenic hexadecane degradation. PLoS ONE 8, e66784 (2013).
Liu, Y.-F. et al. Anaerobic hydrocarbon degradation in candidate phylum ‘Atribacteria’ (JS1) inferred from genomics. ISME J. 13, 2377–2390 (2019).
Liu, Y.-F. et al. Anaerobic degradation of paraffins by thermophilic Actinobacteria under methanogenic conditions. Environ. Sci. Technol. 54, 10610–10620 (2020).
Breese, K., Boll, M., Alt‐Mörbe, J., Schägger, H. & Fuchs, G. Genes coding for the benzoyl‐CoA pathway of anaerobic aromatic metabolism in the bacterium Thauera aromatica. Eur. J. Biochem. 256, 148–154 (1998).
Egland, P. G., Pelletier, D. A., Dispensa, M., Gibson, J. & Harwood, C. S. A cluster of bacterial genes for anaerobic benzene ring biodegradation. Proc. Natl Acad. Sci. USA 94, 6484–6489 (1997).
Borrel, G. et al. Comparative genomics highlights the unique biology of Methanomassiliicoccales, a Thermoplasmatales-related seventh order of methanogenic archaea that encodes pyrrolysine. BMC Genom. 15, 679 (2014).
Lyu, Z., Shao, N., Akinyemi, T. & Whitman, W. B. Methanogenesis. Curr. Biol. 28, R727–R732 (2018).
Ferry, J. G. & Lessner, D. J. Methanogenesis in marine sediments. Ann. N. Y. Acad. Sci. 1125, 147–157 (2008).
Thauer, R. K., Kaster, A.-K., Seedorf, H., Buckel, W. & Hedderich, R. Methanogenic archaea: ecologically relevant differences in energy conservation. Nat. Rev. Microbiol. 6, 579–591 (2008).
Mayumi, D. et al. Methane production from coal by a single methanogen. Science 354, 222–225 (2016).
Suflita, J. M., Davidova, I. A., Gieg, L. M., Nanny, M. & Prince, R. C. in Studies in Surface Science and Catalysis Vol. 151 (eds Vazquez-Duhalt, R. & Quintero-Ramirez, R.) 283–305 (Elsevier, 2004).
Bryant, M. Commentary on the Hungate technique for culture of anaerobic bacteria. Am. J. Clin. Nutr. 25, 1324–1328 (1972).
Friedrich, W., Antje, B. & Ralf, R. in The Prokaryotes: Ecophysiology and Biochemistry Vol. 2 (eds Martin Dworkin et al.) 1028–1049 (Springer, 2006).
Aydin, O. & Yassikaya, M. Y. Validity and reliability analysis of the plotdigitizer software program for data extraction from single-case graphs. Perspect. Behav. Sci. (2021).
Dolfing, J. & Mulder, J.-W. Comparison of methane production rate and coenzyme F420 content of methanogenic consortia in anaerobic granular sludge. Appl. Environ. Microbiol. 49, 1142–1145 (1985).
Cheng, L., Dai, L., Li, X., Zhang, H. & Lu, Y. Isolation and characterization of Methanothermobacter crinale sp. nov, a novel hydrogenotrophic methanogen from the Shengli oil field. Appl. Environ. Microbiol. 77, 5212–5219 (2011).
Ma, T.-T. et al. Coexistence and competition of sulfate-reducing and methanogenic populations in an anaerobic hexadecane-degrading culture. Biotechnol. Biofuels 10, 207 (2017).
Stumm, W. & Morgan, J. J. Aquatic Chemistry: Chemical Equilibria and Rates in Natural Waters (Wiley, 1996).
Deines, P., Langmuir, D. & Harmon, R. S. Stable carbon isotope ratios and the existence of a gas phase in the evolution of carbonate ground waters. Geochim. Cosmochim. Acta 38, 1147–1164 (1974).
Pernthaler, A., Pernthaler, J. & Amann, R. Fluorescence in situ hybridization and catalyzed reporter deposition for the identification of marine bacteria. Appl. Environ. Microbiol. 68, 3094–3101 (2002).
Amann, R. I. et al. Combination of 16S rRNA-targeted oligonucleotide probes with flow cytometry for analyzing mixed microbial populations. Appl. Environ. Microbiol. 56, 1919–1925 (1990).
Daims, H., Brühl, A., Amann, R., Schleifer, K.-H. & Wagner, M. The domain-specific probe EUB338 is insufficient for the detection of all bacteria: development and evaluation of a more comprehensive probe set. Syst. Appl. Microbiol. 22, 434–444 (1999).
Stahl, D. A. in Nucleic Acid Techniques in Bacterial Systematics 205–248 (1991).
Pernthaler, A., Preston, C. M., Pernthaler, J., DeLong, E. F. & Amann, R. Comparison of fluorescently labeled oligonucleotide and polynucleotide probes for the detection of pelagic marine bacteria and archaea. Appl. Environ. Microbiol. 68, 661–667 (2002).
Sofie, T. et al. Comparative evaluation of four bacteria-specific primer pairs for 16S rRNA gene surveys. Front. Microbiol. 8, 494 (2017).
Wei, S. et al. Comparative evaluation of three archaeal primer pairs for exploring archaeal communities in deep-sea sediments and permafrost soils. Extremophiles 23, 747–757 (2019).
Magoč, T. & Salzberg, S. L. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27, 2957–2963 (2011).
Caporaso, J. G. et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336 (2010).
Bolyen, E. et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37, 852–857 (2019).
Quast, C. et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596 (2012).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Nurk, S., Meleshko, D., Korobeynikov, A. & Pevzner, P. A. metaSPAdes: a new versatile metagenomic assembler. Genome Res. 27, 824–834 (2017).
Kang, D. D. et al. MetaBAT 2: an adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ 7, e7359 (2019).
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P. & Tyson, G. W. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055 (2015).
Chaumeil, P.-A., Mussig, A. J., Hugenholtz, P. & Parks, D. H. GTDB-Tk: a toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics 36, 1925–1927 (2019).
Parks, D. H. et al. A complete domain-to-species taxonomy for Bacteria and Archaea. Nat. Biotechnol. 38, 1079–1086 (2020).
Shaffer, M. et al. DRAM for distilling microbial metabolism to automate the curation of microbiome function. Nucleic Acids Res. 48, 8883–8900 (2020).
Kanehisa, M., Sato, Y. & Morishima, K. BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J. Mol. Biol. 428, 726–731 (2016).
Huerta-Cepas, J. et al. Fast genome-wide functional annotation through orthology assignment by eggNOG-mapper. Mol. Biol. Evol. 34, 2115–2122 (2017).
Yoon, S. H., Ha, S. M., Lim, J., Kwon, S. & Chun, J. A large-scale evaluation of algorithms to calculate average nucleotide identity. Antonie Van Leeuwenhoek 110, 1281–1286 (2017).
Qin, Q.-L. et al. A proposed genus boundary for the prokaryotes based on genomic insights. J. Bacteriol. 196, 2210–2215 (2014).
Hyatt, D. et al. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 11, 119 (2010).
Eddy, S. R. A probabilistic model of local sequence alignment that simplifies statistical significance estimation. PLoS Comput. Biol. 4, e1000069 (2008).
Fu, L., Niu, B., Zhu, Z., Wu, S. & Li, W. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152 (2012).
Edgar, R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004).
Hug, L. A. et al. A new view of the tree of life. Nat. Microbiol. 1, 16048 (2016).
Nguyen, L. T., Schmidt, H. A., von Haeseler, A. & Minh, B. Q. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274 (2015).
Pruesse, E., Peplies, J. & Glöckner, F. O. SINA: accurate high-throughput multiple sequence alignment of ribosomal RNA genes. Bioinformatics 28, 1823–1829 (2012).
Ludwig, W. et al. ARB: a software environment for sequence data. Nucleic Acids Res. 32, 1363–1371 (2004).
Mendler, K. et al. AnnoTree: visualization and exploration of a functionally annotated microbial tree of life. Nucleic Acids Res. 47, 4442–4448 (2019).
Letunic, I. & Bork, P. Interactive Tree Of Life (iTOL) v4: recent updates and new developments. Nucleic Acids Res. 47, W256–W259 (2019).
Lane, D. J. 16S/23S rRNA Sequencing 205–248 (John Wiley & Sons, 1991).
Selvaraj, V. A.-O. et al. Development of a duplex droplet digital PCR assay for absolute quantitative detection of "Candidatus Liberibacter asiaticus". PLoS ONE 13, e0197184 (2018).
Peng, J., Lü, Z., Rui, J. & Lu, Y. Dynamics of the methanogenic archaeal community during plant residue decomposition in an anoxic rice field soil. Appl. Environ. Microbiol. 74, 2894–2901 (2008).
Kopylova, E., Noé, L. & Touzet, H. SortMeRNA: fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics 28, 3211–3217 (2012).
Wood, D. E., Lu, J. & Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 20, 257 (2019).
Olm, M. R., Brown, C. T., Brooks, B. & Banfield, J. F. dRep: a tool for fast and accurate genomic comparisons that enables improved genome recovery from metagenomes through de-replication. ISME J. 11, 2864–2868 (2017).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Quinlan, A. R. BEDTools: the Swiss-army tool for genome feature analysis. Curr. Protoc. Bioinform. 47, 11.12.1–11.12.34 (2014).
Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. Preprint at https://arxiv.org/abs/1303.3997 (2013).
Wickham, H. ggplot2. Wiley Interdiscip. Rev. Comput. Stat. 3, 180–185 (2011).
RCore Team R: A Language and Environment for Statistical Computing (R Foundation for Statistical Computing, 2020); http://www.R-project.org/
Acknowledgements
We thank A. Oren (The Hebrew University of Jerusalem) for discussing the naming of the different Ca. Methanoliparum species; R. Conrad and W. B. Whitman for discussing the manuscript; K. Wrighton for providing access to the server Zenith; Q. Yuan, Y. Liu, J. Pan, M.-w. Cai and Y.-n. Tang for assisting in data analysis; L.-r. Dai, D. Zhang and L. Li for assisting in cultivation and experiments; and Z. Zhou for technical support. This study was supported by National Natural Science Foundation of China (nos 92051108, 91851105, 41802179, 31970066, 31570009 and 31970105), Agricultural Science and Technology Innovation Project of the Chinese Academy of Agriculture Science (no. CAAS-ASTIP-2016-BIOMA), the Innovation Team Project of Universities in Guangdong Province (no. 2020KCXTD023) and the Shenzhen Science and Technology Program (no. JCYJ20200109105010363), the Fundamental Research Funds for the Central Universities (LZUJBKY-2021-KB16), the Central Public-interest Scientific Institution Basal Research Fund (Y2021PT02, Y2021XK06). R.L.-P. was supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany’s Excellence Strategy (EXC-2077-390741603) via Excellence Chair Victoria Orphan. G.W. was funded by DFG under Germany’s Excellence Strategy-EXC-2077-390741603 and the Max Planck Society.
Author information
Authors and Affiliations
Contributions
L.C. and M.L. initiated the study. L.C., M.L., G.W. and P.-f.L. designed research. J.-z.L., W.-d.W. and Z.Z. collected the oily sludge samples. Z.Z., J.L., M.Y. and L.C. conducted cultivation experiments. Z.Z. and L.Y. performed oil analysis. C.-j.Z., P.-f.L., Z.Z., R.L.-P. and M.L. performed all bioinformatics analyses. R.L.-P. and L.C. designed CARD-FISH probes, and R.L.-P. performed CARD-FISH and cell visualization. L.F., L.C. and L.-p.B. performed metabolite analyses. P.-f.L., R.L.-P., G.W., M.L. and L.C. analysed data and wrote the manuscript with contributions from all of the co-authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review information
Nature thanks Guillaume Borrel, Rudolph Thauer and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Molecular characterization of the methanogenic oily sludge incubated at different temperatures.
a, Accumulation of methane in the headspace of treatments at different temperatures over an incubation time of 301 days. The estimates of reported methane production rates base on the time interval for the formation of 5% and 90% of the maximum methane formation. b, Mass spectrometric analysis of extracted residual oil for n-alkanes m/z = 85, n-alkylcyclohexanes m/z = 82, n-alkylbenzenes m/z = 92. Exemplary data of the 55 °C culture is presented in Figs. 1b–1d. Data shown are mean ± standard deviation (n = 3 biologically independent replicates). c and d, Archaeal and bacterial community structure revealed by amplicon sequencing in the different temperature treatments after 204 days of incubation, respectively. Only families with relative abundances ≥ 1% are shown. “Other” indicates the sum of groups with relative abundance < 1%. Data shown are mean - standard deviation (n = 3 biologically independent replicates).
Extended Data Fig. 2 Epifluorescence micrographs of different community members of the oily sludge.
a-c, Visualization of archaea (green) and bacteria (red). d-f, Visualization of ‘Ca. Methanoliparum’ (green) and archaea (red). Hybridization of ‘Ca. Methanoliparum’ with the general archaeal probe and the specific DC06-660Mlp probe. The vast majority of archaea hybridized also with the probe for ‘Ca. Methanoliparum’. g-i, Visualization of ‘Ca. Methanoliparum’ (green) and bacteria (red). Oligonucleotide probes were ARCH-915 for archaea, EUB388 I-III for bacteria and DC06-660Mlp for ‘Ca. Methanoliparum’. Three representative recorded images from n = 3 independent samples (a-i, 9 rows of images in total) of one culture are shown. Scale bars in all images are 10 µm.
Extended Data Fig. 3 Phylogenetic analyses of MAGs and 16S rRNA gene sequences of ‘Ca. Methanoliparia’.
a, Phylogenomic analyses of ‘Ca. Methanoliparia’ MAGs based on the concatenated alignments of 16 ribosomal proteins67. Bootstrap values > 0.95 are marked with grey dots, ‘Ca. Bathyarchaeota’ set as outgroup. The maximum-likelihood tree was constructed by using the IQ-TREE software with the parameters ‘-m WAG -bb 1000’. b, Phylogenetic analysis of 16S rRNA gene sequences retrieved from all ‘Ca. Methanoliparia’ MAGs. For MAG- derived sequences source information is given: i.e., T55 indicates temperature of the culture (55 °C) and after the MAGs (bin) number the substrate used is indicated (e.g., n-hexadecane). The asterisk (*) marking ‘Ca. M. whitmanii’ sequence identifiers indicates 16S rRNA genes that were truncated during assembly. In these cases, the longest partial sequence was used for the phylogenetic analyses. The 16S rRNA gene sequences were added to the consensus tree with ‘quick add’ option, thus no bootstrap values are available.
Extended Data Fig. 4 Identities between ‘Ca. Methanoliparum’ clusters and phylogenetic analysis of their AcrA and McrA protein sequences.
a, Identities of the 16S rRNA gene. b, Genome based average Amino Acid Identity (AAI). c, Genome based Average Nucleotide Identity (ANI). d, Identity based on the percentage of conserved proteins (POCP). All matrices consistently showed that all ‘Ca. Methanoliparia’ MAGs from this study grouped into four species-level clusters within the genus ‘Ca. Methanoliparum’. In the box plots the central line represents the median; the lower and upper box limits correspond to the 25th and 75th percentiles, respectively; Numbers represent the times of pairwise comparisons of MAGs between two groups. Cluster 1 (C1): ‘Ca. M. thermophilum’; Clusters 2 (C2): ‘Ca. M. widdelii’; Cluster 3 (C3): ‘Ca. M. whitmanii’; Cluster 4 (C4): ‘Ca. M. zhangii’. Mv indicates the genomes of the sister marine clade ‘Ca. Methanolliviera’. e, Maximum-likelihood tree of the protein sequences of AcrA and McrA present in ‘Ca. Methanoliparum’ MAGs retrieved in the present studied. Different colours indicate the different ‘Ca. Methanoliparum’ species. Numbers in parenthesis indicate the number of acrA/mcrA sequences detected in the different metagenomes. In each MAG, maximum one acr and one mcr were detected. Trees were constructed by using IQ-TREE with the parameters ‘-m WAG, -bb 1000’, with bootstrap values >0.95 shown in grey dots.
Extended Data Fig. 5 Gene clusters associated with the alkane degradation and methanogenesis pathways detected in the representative MAGs of the four ‘Ca. Methanoliparum’ species.
Several copies of fadD and ACADM were detected and only copies with the highest transcript abundances are shown. In orange, alkane activation and conversion to a fatty acid; in blue, beta oxidation pathway and in red, the ACS/CODH complex and the methanogenesis pathway. Details of all copies are included in Supplementary Table 6.
Extended Data Fig. 6 Relative transcript abundances of alkane-degrading and methane-producing pathways coding genes.
The colour code shows the log2(FPKM) values of each gene. For enzymes or subunits with several putative coding genes, only the ones with the highest level of log2(FPKM) are shown here. Two samples were taken for cultures with n-hexadecane addition (Hex.) at day 31 and 55, while sampling at one time point (day 55) with 3 replicates (designated as r1-r3) was performed for control cultures without n-hexadecane amendment (Con.). Grey cells indicate that the corresponding genes were not found in the MAGs. Details of all copies are included in Supplementary Table 7.
Extended Data Fig. 7 Identification of coenzyme M derivatives in cultures by HPLC-MS/MS based on the corresponding retention times.
a and b, hexadecyl-CoM and the corresponding 3 characterized fragments (in blue) in cell extracts from cultures with hexadecane (C16H34) addition. c and d, eicosyl-CoM and 3 characterized fragments (in blue) in cell extracts from cultures with eicosane (C20H42) additions. Standard appears in black primary anions and second anions (produced by fragmentation) detected in hexadecane and eicosane cultures showed the same retention time as the synthetic standards of hexadecyl-CoM and eicosyl-CoM, respectively.
Extended Data Fig. 8 Identification of coenzyme M derivatives in cultures incubated with specific hydrocarbons.
a, Scheme for the activation of long-chain alkanes and alkyl-substituted compounds as CoM thioethers in ACR, and their expected fragmentation patterns. The residual ‘R-’ describes a methyl-, cyclohexane- or aromatic unit with an alkyl chain CnH2n+1 for n ≥ 13. Dash arrows and numbers above indicate the fragmentation positions. b and c, QE Plus-Orbitrap MS analyses of cultures supplemented with eicosane resulted in a mass peak of eicosyl-CoM (C20H41-SC2H4SO3− at m/z = 421.28162 and the fragments eicosyl-thiol (C20H33S-, m/z = 313.29373), ethenesulfonate (C2H3SO3−, m/z = 106.98092) and bisulfite (HSO3−, m/z = 80.96519). All peaks match those of an eicoysl-CoM standard. d-i, QE Plus-Orbitrap MS analyses of cultures supplemented with a mixture of n-docosane (C22H46), n-hexadecyl benzene (C22H38) and n-hexadecyl cyclohexane (C22H44) as substrates, and detection of d and e docosyl-CoM (C24H49S2O3−, m/z = 449.31134) with the fragment C22H45S− (m/z = 341.32495); of f and g n-hexadecyl benzene coenzyme M (C24H41S2O3−, m/z = 441.25064) with the predicted fragment C22H37S− (m/z = 333.26212) and of h and i n-hexadecyl cyclohexane CoM (C24H47S2O3−, m/z = 447.29730) with the fragment C22H43S− (m/z = 339.30939). The mass error for all mass peaks shown here are < 5 p.p.m.
Extended Data Fig. 9 Semi-continuous cultivation of the ‘Ca. Methanoliparum’ cultures at 55 °C.
Microorganisms were cultured using a mixture of n-docosane, n-hexadecyl benzene, n-hexadecyl cyclohexane as substrate. The culture was transferred when 15 to 20 mmol of methane were formed, and 30% to 50% of the culture were transferred. Displayed are transfers 3 to 6. a, Methane formation in the headspace. Grey arrows indicate transfer events. b and c, Abundance of 16S rRNA gene of ‘Ca. Methanoliparum’ and bacteria as determined by qPCR, respectively. d, Relative abundance of main archaeal groups determined by 16S rRNA gene sequencing with archaeal primer set Arch519F/Arch915R.
Extended Data Fig. 10 Proposed metabolic pathway and related gene clusters for benzene-CoA degradation in ‘Ca. Methanoliparum’.
a, Gene clusters found in the four representative MAGs with potential for benzoyl-CoA degradation. Numbers in the gene clusters indicate kilobases. b, Annotations and Locus tag for the corresponding genes shown in panel a that are found in the representative MAG of ‘Ca. M. thermophilum’ (XY_C20_T55_P2_bin.5 of Cluster 1). c, Proposed pathway for the degradation of benzoyl-CoA based on the pairwise comparison of the candidate genes of ‘Ca. Methanoliparum’ (red) with the genes involved in benzoyl-CoA degradation in the model organisms Thauera aromatica (green) and Rhodopseudomonas palustris (blue). The letters for candidate genes of ‘Ca. Methanoliparum’ refer to the letters indicate in the panel a (see Supplementary Table 10 for more details).
Supplementary information
Supplementary Information
Supplementary Figs. 1–6 and the legends for Supplementary Tables 1–13.
Supplementary Tables
Supplementary Tables 1–13.
Rights and permissions
About this article

Cite this article
Zhou, Z., Zhang, Cj., Liu, Pf. et al. Non-syntrophic methanogenic hydrocarbon degradation by an archaeal species. Nature 601, 257–262 (2022). https://doi.org/10.1038/s41586-021-04235-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-021-04235-2
This article is cited by
-
Unraveling the phylogenomic diversity of Methanomassiliicoccales and implications for mitigating ruminant methane emissions
Genome Biology (2024)
-
Methane-dependent complete denitrification by a single Methylomirabilis bacterium
Nature Microbiology (2024)
-
Electronic regulation to achieve efficient anaerobic digestion of organic fraction of municipal solid waste (OFMSW): strategies, challenges and potential solutions
Frontiers of Environmental Science & Engineering (2024)
-
Soil microbial ecology through the lens of metatranscriptomics
Soil Ecology Letters (2024)
-
Variations in the archaeal community and associated methanogenesis in peat profiles of three typical peatland types in China
Environmental Microbiome (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
