
Abstract
Type A γ-aminobutyric acid receptors (GABAARs) are pentameric ligand-gated chloride channels that mediate fast inhibitory signalling in neural circuits1,2 and can be modulated by essential medicines including general anaesthetics and benzodiazepines3. Human GABAAR subunits are encoded by 19 paralogous genes that can, in theory, give rise to 495,235 receptor types. However, the principles that govern the formation of pentamers, the permutational landscape of receptors that may emerge from a subunit set and the effect that this has on GABAergic signalling remain largely unknown. Here we use cryogenic electron microscopy to determine the structures of extrasynaptic GABAARs assembled from α4, β3 and δ subunits, and their counterparts incorporating γ2 instead of δ subunits. In each case, we identified two receptor subtypes with distinct stoichiometries and arrangements, all four differing from those previously observed for synaptic, α1-containing receptors4,5,6,7. This, in turn, affects receptor responses to physiological and synthetic modulators by creating or eliminating ligand-binding sites at subunit interfaces. We provide structural and functional evidence that selected GABAAR arrangements can act as coincidence detectors, simultaneously responding to two neurotransmitters: GABA and histamine. Using assembly simulations and single-cell RNA sequencing data8,9, we calculated the upper bounds for receptor diversity in recombinant systems and in vivo. We propose that differential assembly is a pervasive mechanism for regulating the physiology and pharmacology of GABAARs.
Similar content being viewed by others


Main
Human GABAARs assemble from a pool of 19 subunits (α1–6, β1–3, γ1–3, δ, ε, π, ρ1–3 and θ) with cell-type-specific expression patterns1,2,10. Their homopentameric and heteropentameric combinations give rise to receptors with different localization and functions1,2,10,11,12,13,14. For example, in the central nervous system, receptors containing the γ-subunit localize both synaptically and extrasynaptically, bind to GABA with lower affinity and desensitize more rapidly than δ-subunit-containing receptors14,15. By contrast, in the predominantly extrasynaptic δ-containing receptors, GABA binds with higher affinity and has lower efficacy than in the γ-containing receptors14.
The stoichiometry and arrangement of subunits in a pentamer are fundamental determinants of receptor signalling because interfaces between subunits have binding sites for agonists, antagonists and allosteric modulators. Therefore, different subunit arrangements within a receptor pentamer can result in the emergence or disappearance of ligand-binding sites16. Synaptic GABAARs are thought to assemble into pentamers with invariant arrangements and stoichiometry (βαγβα, counter-clockwise), which is supported by cryogenic electron microscopy (cryo-EM) structures of receptors containing the α1, β1–3 and γ2 subunits, including human α1β3γ2 bound to the Ro15-4513 benzodiazepine reported in this paper4,5,6,7 (Fig. 1a). However, the arrangements and stoichiometries of extrasynaptic receptors, and of synaptic receptors assembled from different subunit pools, are uncertain17,18,19,20,21. Moreover, the overall landscape of possible receptor subtypes and the extent to which they can diversify GABAAR physiology and pharmacology remain unknown. Given the plethora of pharmacological agents with anti-convulsant, anti-anxiety, analgesic, sedative and anaesthetic properties that target GABAARs3, mechanistic insights into the interplay between stoichiometry and function are important not only to understand GABAergic signalling better but also to provide new opportunities for drug development.

a–e, Structures, subunit arrangements and ligand-binding pockets of α1β3γ2 (a), α4β3δ (b), β3δ solved from the same dataset as b (c), α4β3γ22 (d) and β3γ2 GABAAR solved from the same dataset as d (e). TMD, transmembrane domain.
Structural evidence for GABAAR heterogeneity
To explore the diversity of possible receptor subtypes, we first sought to solve the structure of human α4β3δ, an extrasynaptic GABAAR. Previous work has suggested that, when co-expressed, these subunits form multiple receptor populations depending on transfection ratios and expression systems19,20,22. Thus, we generated two mammalian cell lines by varying the relative amounts of complementary DNA encoding individual subunits (Methods).
Unexpectedly, identical α4β3δ GABAAR structures were solved from both cell lines (Fig. 1b, Extended Data Fig. 1a, c, e, Supplementary Fig. 1). These structures revealed that the receptor contains one α4 subunit, three β3 subunits and one δ-subunit, in a βαδββ arrangement. From the same samples, we classified a second population of receptors, the di-heteromeric β3δ subtype containing four β3 subunits and one δ-subunit (Fig. 1c, Extended Data Fig. 1b, d). Although the α4β3δ GABAAR contains a single putative GABA-binding site at the β3+/α4– interface, it also has two putative histamine-binding pockets at the β3+/β3– interfaces (principal and complementary faces of the interface are denoted as + and −, respectively)23,24. Therefore, both neurotransmitters could bind to these receptors. By contrast, a prototypical synaptic receptor, such as α1β3γ2 (arranged as βαγβα), has GABA-binding pockets at its two β3+/α1– interfaces but no known binding sites for histamine, as the pentameric arrangement of subunits does not present a β3+/β3– interface6,7 (Fig. 1a). Conversely, the β3δ subtype has putative histamine-binding pockets at the three β3+/β3– interfaces.
To investigate whether the stoichiometry observed in the α4β3δ GABAAR is driven by the δ-subunit, we solved the structure of the α4β3γ2 receptor (Fig. 1d, Extended Data Fig. 1f). To our surprise, we found yet another subunit arrangement compatible with a GABA–histamine ‘crosstalk’, albeit different to the one observed in α4β3δ receptors. The α4β3γ2 assembly contains one α4, one γ2 and three β3 subunits, arranged as ββγβα, which has putative binding sites for GABA and histamine at the β3+/α4– and β3+/β3– interfaces, respectively (Extended Data Fig. 2c). From the same dataset, we also solved the structure of a β3γ2 subtype (Fig. 1e, Extended Data Fig. 1g). In contrast to the β3δ receptor, the β3γ2 receptor incorporates two γ2 subunits at non-adjacent positions, in agreement with previous stoichiometry estimates for the β2γ2 receptor25. Both the β2γ2 and β3γ2 subtypes have been shown to form functional, GABA-gated channels25,26. We did not observe receptors with the same subunit composition but with different stoichiometries, such as β3δ with two δ-subunits or β3γ2 with a single γ2 subunit. Therefore, we conclude that cells expressing α4, β3 and δ or α4, β3 and γ2 subunits assemble multiple receptor populations in a differential (that is, context-dependent) but non-random manner.
Differential assembly diversifies signalling
To examine whether and how the observed subunit arrangements may diversify receptor function, we focused on the putative GABA–histamine interplay at the α4β3δ receptor. Histamine modulation has previously been observed for several GABAAR subtypes, including the α1β2γ2 (ref. 24), α1β3δ, α4β3γ2 and α4β3δ (ref. 27). The joint presence of α4 and β3 subunits showed the strongest enhancement of GABA currents and an allosteric mechanism for histamine action was proposed27. We solved the structure of the α4β3δ receptor (βαδββ arrangement) simultaneously bound to both ligands (Fig. 2a–c, Extended Data Fig. 1c). The β3+/α4– agonist pocket is occupied by a GABA molecule, whereas the equivalent pockets at the two β3+/β3– interfaces bind to histamine (Fig. 2a–c, Extended Data Figs. 2, 3). All three pockets adopt compact conformations, with loop C closed and ligands coordinated in each corresponding ‘aromatic cage’ (Extended Data Fig. 3, Supplementary Fig. 1). Although a low-affinity GABA-binding site has previously been proposed at the β3+/δ– interface28, we did not observe any ligand density in this pocket, which is supported by our electrophysiological recordings (Extended Data Fig. 4a–c). The ion channel is desensitized, as previously described for synaptic and homomeric β3 receptors6,7,23,29. From the same dataset, we classified a subpopulation of the α4β3δ subtype (βαδββ arrangement) where all three pockets mentioned above are occupied by histamine molecules (Extended Data Figs. 1e, 2b, 3, Supplementary Fig. 2). Although, in this case, the extracellular domain adopts an activated conformation, the ion channel is closed, illustrating a pre-open ‘flip’ state consistent with the partial agonism of histamine at receptors containing β3+/–β3– interfaces30 (Extended Data Fig. 2f). Moreover, the same dataset also contains the β3δ subtype, devoid of α4 subunits and thus is unable to bind GABA (Extended Data Figs. 1d, 2b, 4b). In this map, histamine molecules occupy the agonist pockets at the three β3+/β3– interfaces, and the ion channel is desensitized (Extended Data Fig. 2f). In the context of a heterogeneous GABAAR population, the relative concentration of agonists and their binding properties will ultimately determine the identity of ligands that occupy individual pockets and their signalling impact.

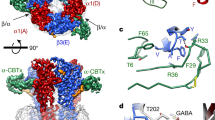
a, b, Histamine (HSM) bound in the two β3+/β3− agonist pockets. c, GABA bound in the β3+/α4− agonist pocket. d, In α4β3δ cells, an equimolar mix of GABA and histamine (10 mM each) gates more receptors than each ligand applied alone. Superimposed, normalized whole-cell current-averaged traces obtained with a two-pulse protocol described in the Methods section. GABA gated 15.0 ± 6.0% (n = 7 cells), histamine gated 50.2 ± 11.0% (n = 6 cells) and histamine + GABA together gated 99.0 ± 18.9% (n = 8 cells) of total receptors gated by 10 mM GABA + 30 μM etomidate. Standard deviations were propagated. Statistical analysis is shown in Extended Data Fig. 4g. Data are mean ± s.d. e, f, Representative deactivating current traces obtained from the same cell by application of a 200-ms pulse of GABA alone or in the presence of 100 μM histamine (the same current traces normalized as described in the Methods section are shown in f). Pre-application of a low concentration of histamine, in which its agonistic action was weak, caused a decrease in peak current amplitude of the deactivating currents by 74.5 ± 11.9% (n = 4 cells) (e), and the deactivation rate became faster, with the current reaching 50% of its original value in 1.0 ± 0.4 s (n = 4) instead of 2.7 ± 0.7 s (n = 4 cells; P = 0.002, two-tailed paired Student’s t-test) in the absence of histamine (f). Statistical analysis of deactivation time constants (τ) and amplitudes is shown in Extended Data Fig. 4h–k. ‘GABA pre’ is the first control GABA pulse before HSM application. ‘GABA post’ is the second control GABA pulse, after GABA and HSM application. Putative hydrogen bonds are indicated by yellow dashed lines.
We investigated the functional consequences of GABA and histamine crosstalk at the α4β3δ receptor by performing whole-cell patch clamp electrophysiology on the cell line used for structural analysis. Both GABA (half-maximal effective concentration (EC50) of 69 nM) and histamine (EC50 of 821 μM) are agonists, with the latter being threefold more efficacious (Fig. 2d, Extended Data Fig. 4d–f), consistent with previous studies in α4β3δ and α4β3γ2 receptors27. Co-application of these ligands results in a cumulative enhancement of current amplitude (Fig. 2d, Extended Data Fig. 4g). However, proving that the observed enhancement is a consequence of the action of GABA and histamine at the α4β3δ receptors is complicated by the presence of other subtypes that respond to either one of the two ligands (such as the β3δ subtype), as demonstrated by structural studies. One indication of crosstalk at the level of a single α4β3δ receptor is that, in the continuous presence of a low concentration of histamine (100 μM), deactivation of currents following a brief pulse of 10 μM GABA is accelerated (Fig. 2e, f). A decrease in peak current amplitude is also observed under these experimental conditions (Fig. 2e, f, Extended Data Fig. 4h–k).
To further deconvolve responses from different receptor subtypes present in the same cell, we first established that GABA at 100 nM robustly activated currents in α4β3δ cells, barely in α4β3 and not in β3 or β3δ cells (Extended Data Fig. 5a–d). Similarly, histamine (300 μM) robustly enhanced GABA currents only in α4β3δ cells. Only modest responses to co-application of 100 nM GABA and 300 μM histamine were observed in α4β3, β3δ and β3 cells, attributable to histamine currents alone (Extended Data Fig. 5e). Together, our results indicate that histamine has dual and opposing actions on α4β3δ receptors: it is an agonist itself, and it also accelerates closure of GABA-activated receptors. Thus, differential assembly of GABAARs in a single cell diversifies signalling by enabling activation and/or modulation of receptor ensembles by multiple neurotransmitters, such as GABA and histamine. The timing, order, strength and duration of neurotransmitter exposure can affect the outcome of signalling through α4β3δ receptors, and the overall output of each cell is a summed response of all receptor subtypes that respond to the particular ligands.
Differential assembly affects drug responses
The binding and functional impact of synthetic GABAAR modulators may also be altered by combinatorial expression of subunits and their assembly permutations and result in off-target effects or complete loss of ligand activity. We illustrate this phenomenon with two drug candidate molecules: 4,5,6,7-tetrahydroisoxazolo[5,4-c]pyridin-3-ol (THIP; also known as gaboxadol), a synthetic agonist of α4β3δ receptors that has recently been investigated as a treatment for insomnia15,31,32, and Ro15-4513, a partial inverse agonist benzodiazepine that is thought to bind to both γ-containing and δ-containing receptors (for example, α1β3γ2 and α4β3δ)33,34. Ro15-4513 has been reported to reverse low-dose alcohol potentiation of GABAARs, and thus ethanol inebriation, by acting specifically on extrasynaptic α4/6β3δ subtypes (also dubbed the ‘one glass of wine’ receptors35), although these findings have been challenged36.
We solved the structure of α4β3δ (βαδββ arrangement) bound to THIP and histamine (Extended Data Fig. 1h). In agreement with our GABA + histamine structure and previous work37, we found THIP bound in the β3+/α4– agonist pocket, and histamine at the two β3+/β3– interfaces (Fig. 3a, Extended Data Figs. 2d, 3, Supplementary Fig. 2). We also found THIP density in the equivalent pocket at the δ+/β3– interface (Fig. 3b), consistent with previous studies28. Binding of this agonist to two distinct sites provides a structural explanation for previous observations that THIP has higher potency and supramaximal efficacy at the α4β3δ subtype than at other receptors subtypes37,38. Furthermore, from the same dataset, we also solved the structure of a β3δ receptor (Extended Data Fig. 1i). Here histamine occupies the agonist sites at the three β3+/β3– interfaces, whereas THIP binds only to the δ+/β3– pocket. These observations directly illustrate why functional measurements for THIP (or any molecule active at GABAARs) represent an integrated response of all receptor subtypes that are present and capable of binding that compound, and that targeting a specific receptor arrangement with unique ligand-binding sites might yield drugs with better specificity and fewer side effects39.

a, Inset showing THIP bound in the β3+/α4− agonist pocket of an α4β3δ GABAAR. b, THIP coordination in the second binding site, the δ+/β3– ‘agonist pocket’, of an α4β3δ GABAAR. The same binding mode is observed in the second receptor subtype solved from the same dataset, the β3δ receptor. c, Ro15-4513 binding mode in the α1+/γ2− pocket of the α1β3γ2 receptor. d, Radioligand assay measuring competition of Ro15-4513 with pre-bound [3H]Ro15-4513 shows that the ligand binds to cells expressing α1, β3, γ2 and α4, β3, γ2 subunits, but not the cells expressing α4, β3, δ or α4, β3 subunits (n = 3 technical repeats for each measurement). Data are mean ± s.d. Putative hydrogen bonds are indicated by yellow dashed lines.
To investigate the mechanism of action of Ro15-4513 as an alcohol antagonist, we first explored its interaction with the α1β3γ2 receptor (βαγβα arrangement; Extended Data Fig. 1j). The ligand unambiguously occupies the α1+/γ2– benzodiazepine pocket in the extracellular domain (Fig. 3c, Supplementary Fig. 1). Attempts to solve the structure of α4β3δ bound to Ro15-4513 did not reveal any density for the drug. Radioligand binding assays confirmed that Ro15-4513 binds to membranes from the α1β3γ2 cell line as well as the α4β3γ2 cell pool, but not those from the α4β3δ or the α4β3 cell lines (Fig. 3d). Furthermore, whole-cell electrophysiology recordings demonstrate that Ro15-4513 has little effect on GABA currents in the α4β3δ cell line (Supplementary Fig. 1). For the α4β3γ2 cell pool, it remains unclear whether Ro15-4513 binds to a non-canonical interface (for example, the β3+/γ2– interface) or whether receptor subtypes containing an α4+/γ2– interface may also be present. The superposition of α1+/γ2– and α4+/δ– pockets shows that, among multiple potentially clashing residues, H92 on the δ– side of the interface would prevent the binding of Ro15-4513 in the mode seen in α1β3γ2 (Extended Data Fig. 6a–d). The structural similarity of Ro15-4513 to all other imidazo-benzodiazepines, and previous knowledge that ‘classical’ benzodiazepines do not bind to α4-containing and α6-containing receptors due to the presence of R135 (α4+ numbering)7,40, help to rationalize why most (if not all) benzodiazepines do not bind to the α4+/δ– and, by extension, α6+/δ– extracellular domain interfaces. Therefore, the identity of subunits and their particular arrangement within pentameric receptors dictate the binding and functional effects of both physiological and synthetic ligands.
Estimation of GABAAR diversity
Prompted by the observation that a cell line expressing three GABAAR subunit genes gives rise to at least two distinct receptor arrangements, we sought to investigate the possible subtype diversity in the brain. Because cryo-EM reconstructions are biased, that is, limited to particles one can purify and classify, it is possible that the receptor heterogeneity in engineered cell lines, as well as in GABAAR-expressing neurons, might be even greater. We analysed single-cell RNA sequencing (scRNA-seq) data from the human cortex8,9 and found that mRNAs of up to 14 different GABAAR subunits can be simultaneously present in individual cell types (Methods; Extended Data Fig. 7a, b). Although we acknowledge that mRNA abundance may not be a reliable predictor of protein levels, the specific pattern of 14 co-expressed subunits observed in the cortex can theoretically produce up to 62,847 distinct receptor subtypes (Supplementary Methods, Supplementary Table 1).
To overcome current cryo-EM limitations and estimate the potential for GABAAR diversity, we simulated the equilibrium distribution of pentameric receptors assembled from a pool of three distinct monomeric subunits, denoted as α, β and δ/γ (Methods, Fig. 4a, Supplementary Discussion). With the simulation, we sought to calculate the distribution of receptor subtypes given two sets of parameters: subunit abundances and relative interface likelihoods. We simulated the distributions over a large range of relative subunit abundance and interface likelihoods and searched for mutually consistent conditions that mimic the experimentally observed subtype distributions (Methods, Supplementary Discussion). In such conditions, we consistently found additional receptor subtypes that may exist and contribute to the overall signalling response of a cell (Fig. 4b). For example, αβ-heterodimeric receptors represent a major population across many conditions in the α4β3δ simulation, in agreement with previous observations that a large fraction of α4-containing GABAA receptors isolated from the rat brain do not contain γ-subunits or δ-subunits41. Of note, α4β3δ and α4β3γ2 receptors with two α4 subunits are predicted to exist, but we are unable to experimentally identify them due to the lack of specific nanobodies for their inter-subunit interfaces. Together, the scRNA-seq data and computational simulations suggest that the diversity of subtypes, in our cell lines and in the brain, is probably greater than what we observe by cryo-EM and may also include less abundant subtypes with distinct signalling properties.

a, Schematic diagram of the simulation process. b, Expression of selected GABAAR subtypes across different simulated conditions. Out of all conditions that favour the expression patterns observed in experimental data from the α4β3δ (top) or the α4β3γ2 (bottom) cell line (Methods), 20 examples were randomly chosen for display. Here x denotes δ (top) or γ (bottom). Each row represents one simulated condition. The colour scale indicates subtype abundance in each condition. Receptor subtypes expressed across different simulated conditions are shown between the two panels. Experimentally observed subtypes are denoted with dashed boxes (red for α4β3δ and blue for α4β3γ2).
Discussion
It has been recognized in the past that co-expression of multiple GABAAR paralogue genes could increase the diversity of receptor subtypes and responses to GABA42. Our study provides a direct, structural demonstration that differential GABAAR assembly gives rise to an ensemble of receptors with distinct signalling properties. Several lines of evidence support the hypothesis that similar diversity occurs in vivo. For example, three distinct populations of extrasynaptic receptors were identified based on conductance measured by single-channel electrophysiology in cerebellar granule cells43. Multiple receptor subtypes were also observed by native pulldowns with subunit-specific antibodies44 or by immunofluorescence45,46. More recently, cerebellar granule cells have been found to assemble distinct populations of α1α6βγ2 receptors, in which either the α1 or α6 subunit is at the principal side of the α+/γ– interface16. Structures of native receptors and a detailed characterization of assembly pathways are needed for a more complete understanding of the GABAAR signalling pathways. The potential physiological implications of simultaneous binding of GABA and histamine to GABAARs are discussed in Supplementary Information 1.2.
Our simulations and analysis of scRNA-seq data suggest that, by controlling relative subunit abundance and by modulating interface affinities, perhaps through assembly factors or chaperones, it is possible to generate a large ensemble of receptors. Because these parameters are regulated in vivo, differential assembly of GABAARs may be a mechanism to rapidly adapt cellular responses to specific signalling needs by enabling diversification of input recognition and enhanced capacity to finely tune the summed output39. Individual neurons or synapses may also assemble distinct receptor subtypes across spatial locations, developmental stages, and physiological or disease states47,48,49,50. Such flexibility may have enabled the establishment of intricate developmental programmes and facilitated the evolution of complex neuronal circuits and behaviours in animals.
Methods
Protein production and purification
Generation of the α4β3δ cell lines
Stable tetracycline-inducible HEK293S TetR54 cell lines expressing full-length human α4 subunits, β3 subunits and δ-subunits under antibiotic selection (zeocin, hygromycin and geneticin (also known as G418), respectively) were prepared as previously described21. The δ-subunit was modified to include an N-terminal FLAG tag and a C-terminal linker (GGS)3GK followed by the 1D4 tag (TETSQVAPA). To investigate stoichiometric variability of α4β3δ GABAARs, we generated two stable cell lines using different transfection ratios. One cell line was transfected with molar ratios of α4:β3:δ = 2:1:0.25, predicted to yield receptors with a subunit composition of two α4 subunits, two β3 subunits and one δ-subunit19. The other cell line was transfected with about three times less β3 subunit relative to the first one (α4:β3:δ = 2:0.3:0.25 molar ratios), to minimize β3 homo-oligomerization.
α4β3δ protein production
Suspension cultures were grown at 37 °C at 160 rpm in 8% CO2, in FreeStyle 293 expression medium (Gibco), supplemented with 1% fetal bovine serum (Invitrogen), 2 mM l-glutamine, 1% non-essential amino acids and antibiotics: 200 μg ml−1 geneticin, 50 μg ml−1 hygromycin-B, 250 μg ml−1 zeocin, 5 μg ml−1 blasticidin and 10,000 units per ml penicillin–streptomycin (zeocin, hygromycin and blasticidin from Thermo Fisher Scientific; the penicillin–streptomycin mix was prepared in-house). Once cell density reached 2.5 × 106 cells ml−1, expression was induced with 2 μg ml−1 doxycycline (Sigma) in the presence of 5 mM sodium butyrate and 1 mg l−1 I α-mannosidase inhibitor kifunensine (Toronto Research Chemicals). After 24 h, cells were collected by centrifugation at 300g and snap-frozen in liquid nitrogen.
Generation of the α4β3γ2 cell pool and protein production
Full-length human γ2L subunit codon-optimized for expression in human cells was cloned into the pHR vector55. A (GGS)3GK linker followed by the 1D4 tag was added to the C terminus of the γ2L subunit for purification purposes. Lentiviral particles containing the γ2L subunit cDNA were prepared as previously described55, and were used to infect a stable, tetracycline-inducible HEK293S TetR cell line expressing full-length human α4 and β3 under antibiotic selection (zeocin and hygromycin, respectively)21. Protein production in suspension cultures proceeded as described above for the α4β3δ cell lines.
Production of the α1β3γ2 receptor
The cell line and protocols used to produce the α1β3γ2 receptor were previously published6,7,56.
GABAA receptor purification and nanodisc reconstitution
Frozen cell pellets were resuspended on ice in buffer A (50 mM HEPES pH 7.5, and 300 mM NaCl) supplemented with 1% (v/v) mammalian protease inhibitor cocktail (Sigma-Aldrich). Cells were lysed by 1% (w/v) lauryl maltose neopentyl glycol (LMNG; Anatrace) for 1 h at 4 °C then centrifuged for 30 min at 10,000g (4 °C)6. The supernatant was incubated with 1D4 affinity resin rotating slowly for 1 h at 4 °C29. The 1D4 affinity resin was generated in-house using the anti-Rho-1D4 antibody from the University of British Columbia. The resin was recovered by centrifugation (300g for 5 min) then washed with buffer B (buffer A supplemented with 0.1% (w/v) LMNG). For the α4β3δ/β3δ + HEPES and α4β3δ/β3δ + histamine + GABA samples, the wash buffer also contained 0.01% BBE (w/v). While attached to 1D4 resin, receptors were incubated with phosphatidylcholine (POPC; Avanti) and bovine brain lipid (BBL) extract (type I, Folch fraction I; Sigma-Aldrich) mixture (POPC:BBL = 85:15) for 30 min at 4 °C. Excess lipids were removed by pipetting after allowing the beads to settle, then samples were mixed with 100 μl (5 mg ml−1) of MSP 2N2 and incubated for 30 min at 4 °C7. The detergent was removed by incubating the resin with 20 mg Bio-Beads for 90 min at 4 °C, followed by washing with 20–30 bed volumes of buffer A. Receptor samples were eluted with buffer C (12.5 mM HEPES pH 7.5, and 125 mM NaCl) supplemented with 2 mM 1D4 peptide (TETSQVAPA).
Cryo-EM sample preparation
Before freezing, all samples were deglycosylated with 0.01 mg ml−1 endoglycosidase F1 for 1 h at room temperature. Samples were incubated for 30 min with 5 μM Nb25 (ref. 57) and 1.7 μM Mb192 (ref. 58) to facilitate particle alignment and improve orientation distribution, respectively. During this incubation, ligands were also added at the following concentrations: 0.2 mM GABA, 1 mM histamine, 1 mM THIP and 10 μM Ro15-4513, for the respective samples. A 3.5-μl volume of sample was applied to glow-discharged (PELCO easiGlow, 30 mA for 30 s) gold R1.2/1.3 300 mesh UltraAuFoil grids59 (Quantifoil) and incubated between 0 and 30 s at 14 °C. The excess liquid was blotted for 4.0–4.5 s before plunge-freezing into liquid ethane using a Leica EM GP2 plunger (Leica Microsystems; 95% humidity, 14 °C). Grids were stored in liquid nitrogen before data collection.
Cryo-EM data collection
Cryo-EM datasets were collected on Titan Krios G3 microscopes at the MRC LMB or the Department of Biochemistry EM facility (BiocEM, University of Cambridge) in electron counting mode at 300 kV. Both microscopes were equipped with Gatan K3 cameras and Gatan BioQuantum energy filters. Before data acquisition, twofold astigmatism was corrected and beam tilt was adjusted to the coma-free axis using the autoCTF function (EPU v2.00–2.11, Thermo Fisher Scientific). All datasets were acquired automatically using EPU software (Thermo Fisher Scientific, version 2.0–2.11). Detailed data acquisition parameters for all datasets are given in Extended Data Table 1.
Cryo-EM image processing
A typical image processing pipeline is shown in Extended Data Fig. 8. Gain-uncorrected K3 super-resolution movies in TIFF format were motion and gain corrected using RELION’s implementation of the MotionCor2 algorithm60, with frames grouped to yield a total fluence corresponding to approximately 1 e− Å−2 per frame and binned by 2. Contrast transfer function (CTF) estimation was performed with CTFFIND-4.1.13 (ref. 61) using the sums of power spectra from combined fractions corresponding to an accumulated fluence of 4 e− Å−2. Micrographs whose estimated resolution from CTFFIND was worse than 5 Å were removed. Particles were picked using a re-trained BoxNet2D neural network in Warp v1.0.7 (ref. 62) and then re-extracted in RELION with a pixel size of approximately 1.1 Å and (246 pixels)2 box size. All initial data cleaning procedures were performed in cryoSPARC (from v2.15 to v3.2.0)63. First, particles were imported into cryoSPARC and subjected to 2D classification, then good classes were selected to generate an ab initio model using stochastic gradient descent with at least two seeds. After homogeneously refining the ab initio model, all picked particles were included in one or more rounds of heterogeneous refinement in cryoSPARC using three or more classes and the refined model as reference. Aiming to retain as many particles as possible, only particles belonging to classes displaying features of structural damage (for example, incomplete extracellular domain (ECD) or transmembrane domain (TMD), or collapsed TMD) were excluded and the rest were converted into STAR format using csparc2star from the UCSF PyEM v0.5 suite64. Particles were then re-imported into RELION v3.1 (refs. 65,66) for a standard 3D auto-refinement. Refined maps were visually inspected and an optional 3D classification step without alignment was performed if the maps displayed structural damage features. Particles belonging to the best class were re-refined, followed by three steps of CTF refinement: first refining magnification anisotropy, then refining optical aberrations (up to the fourth order), and finally refining per-particle defocus67. Next, 3D auto-refinement was performed, followed by Bayesian polishing to optimize per-particle beam-induced motion tracks68, and another round of auto-refinement. During the polishing step, target particle box size was approximately (270 Å)2. CTF refinement was then repeated for optical aberration correction, magnification anisotropy, per-particle defocus and per-micrograph astigmatism, followed by auto-refinement. For the highest resolution α4β3δ/β3δ dataset (+HEPES), additional steps at this stage included a second round of Bayesian polishing with trained parameters, auto-refinement, CTF refinement as in previous step, followed by auto-refinement. To separate α4β3δ and β3δ receptors, a soft mask surrounding only the Nb25 at all five possible symmetry-related positions was created by simulating Nb25 density from a previously published model (Protein Data Bank (PDB) ID: 7A5V) with UCSF Chimera v1.0 (ref. 69) molmap function, and low-pass filtered to 15 Å. To separate α4β3γ2 and β3γ2 receptors, a soft mask surrounding only the vestibule glycan and vestibule-lining protein residues was created by simulating density from a previously published model (PDB ID 6HUG) with UCSF Chimera molmap function, which was then low-pass filtered to 15 Å. These masks were used during 3D classification without alignment and regularization parameter T = 32 or T = 64. In some instances, classification on Nb25 alone did not provide sufficient separation of α4β3δ and β3δ particles. To overcome this, we focused the classification simultaneously on the vestibule glycan of the α4 subunit and the N80 glycan of the β3 subunit, with T = 128. After selecting classes corresponding to α4β3δ, β3δ, α4β3γ2 or β3γ2 receptors, a final round of 3D auto-refinement with local signal-to-noise filtering using SIDESPLITTER70 implemented in RELION was followed by standard post-processing procedures in RELION. Local resolution plots were generated with Resmap (version 1.1.4)71. Orientation distributions were analysed by cryoEF v1.2 (ref. 72). Renderings of maps and models were done in ChimeraX-1.1.1 (ref. 73) or PyMOL v1.8.4.
Atomic model building and refinement
The initial models used were PDB IDs 7A5V (for the β3 subunit)23 and 6HUG (for the α2 and γ2 subunits)7. Starting models for the α4 and δ subunits were generated in SWISS-MODEL74. Restrains for small molecules were generated by the Grade webserver (Global Phasing Ltd) using SMILES strings75 from ChemDraw JS v2.0.0.9 (PerkinElmer). Iterative rounds of model building and refinement were performed in Coot v0.9.4 (ref. 76), REFMAC v5.8.0258 (ref. 51) and Phenix v1.19.2 (ref. 53). Secondary structure restraints from ProSMART v0.859 were used during the initial stages of refinement77. Models were validated using MolProbity v4.2 (ref. 52). Model building and refinement parameters and statistics are provided in Extended Data Table 2.
Electrophysiology
Electrophysiology measurements were performed on the α4β3δ cell line described above (α4β3N-FLAG-δ-C-L3-1D4, subunit cDNA transfection ratio α4:β3:δ = 2:0.3:0.25), an α4β3 cell line21, and a β3 cell line (Supplementary Methods) transiently transfected with N-Flag-δ-C-L3-1D4 pCMV as indicated in the figures. Cells were seeded on glass coverslips and GABAA gene expression was induced with tetracycline (2 μg ml−1) for 28–32 h. GABAA receptor-mediated chloride currents were recorded using whole-cell patch-clamp electrophysiology at room temperature (20–22 °C). The recording chamber was continuously perfused with the bath solution: 145 mM NaCl, 5 mM KCl, 10 mM HEPES, 2 mM CaCl2, 1 mM MgCl2 and 10 mM glucose, pH 7.4 (pH adjusted with NaOH). The pipette solution for whole-cell recordings contained: 140 mM KCl, 10 mM HEPES, 1 mM EGTA, 2 mM MgCl2, and 2 mM Mg-ATP at pH 7.3 (pH adjusted with KOH). Open pipette resistances ranged from 2 to 2.3 MΩ. Series resistance ranged from 0.5 to 2.8 MΩ and was monitored before and after recordings. Cells whose series resistances changed by 10% or more during recordings were not analysed. Cell capacitances ranged from 4 to 16 pF. The membrane capacitance and series resistance were compensated electronically by more than 85% with a lag of 10 μs. Cells were voltage clamped at –50 mV using a patch-clamp amplifier (Axopatch 200A or Axopatch 200B, Molecular Devices Corp.). GABAARs were activated with agonists delivered via a quad-channel superfusion pipette coupled to a piezoelectric element that switched the superfusion solution in less than 1 ms78. Cells were washed with bath solution alone for at least 1 min between each pulse of agonist application to allow the receptors to recover from desensitization. In some cases, more than one pulse was delivered 1 min apart and the traces acquired were averaged for analysis. Data were manually leak subtracted before analysis and low-pass filtered offline with a Gaussian filter at 250 Hz for presentation.
The GABA EC50 was determined by exposing the cells to three 8-s pulses: (1) 10 mM GABA; (2) varying concentrations of GABA (1 nM to 10 μM), and (3) 10 mM GABA. Peak current amplitudes obtained with the second pulse were normalized to the average peak amplitudes obtained in the first and third pulses. The histamine EC50 was determined by a two-pulse protocol: (1) a 4-s pulse of various concentrations of histamine (0.03–10 mM), and (2) a 1-s pulse of 10 mM GABA. Peak current amplitudes obtained in the first pulse were normalized to those obtained in the second pulse. All experimental pulses were separated by a 6-s wash.
GABA and histamine efficacy were determined in whole-cell configuration using a two-pulse protocol. The first pulse was 2 s of either GABA (10 mM), histamine (10 mM) or equimolar GABA + histamine (10 mM). After a wash of 6 s, the second pulse was 1 s of 10 mM GABA + 30 μM etomidate, which was assumed to open the maximum number of receptors. Current traces shown in Fig. 2d and peak amplitudes were normalized to the peak amplitude of this second pulse. Current deactivation was studied in the whole-cell configuration because the receptor amounts were too low for measurable currents in outside-out macro patches. Unlike GABA, histamine gates α4β3δ receptors in the same concentration range as β3 receptors21,24,28 (Extended Data Fig. 4d, e). Therefore, we utilized the specificity of GABA in the 1–10 μM range to selectively gate α4β3δ receptors. A small fraction of the β3δ and α4β3 receptors (if present) may be activated by 10 μM GABA28. Low concentration of histamine (100 μM) was used to modulate the gating equilibrium. Deactivating currents were elicited from the same cell by a 200-ms pulse of 10 μM GABA alone and data were acquired for 5 s. For each cell, the three traces separated by 60-s washes were: (1) GABA alone, (2) histamine (100 μM) present for 60 s before the GABA pulse and present throughout deactivation, and (3) GABA alone. Current traces were normalized to their own peak amplitudes for better visual comparison of deactivation rates shown in Fig. 2f.
Electrophysiology data acquisition and analysis
Electrophysiology data were acquired using Clampex version 8.1 (Molecular Devices), digitized at 5 kHz or 10 kHz depending on the length of the pulse. Data were low-pass filtered at either 5 kHz or 10 kHz. Deactivating phases of the currents were fit with a one-term or two-term exponential equation, as determined by an F-test, using the Levenberg–Marquardt algorithm in Clampfit 9.0 (Molecular Devices). The low relative amplitude of the fast phase of deactivation means that its fitted parameters are less accurate than those of the slow phase. Statistical analysis was done using Prism 6 (GraphPad Software). Concentration–response curves were fitted to a Hill equation in the following form:
where Inorm is the normalized peak current amplitude in the presence of the agonist and the EC50 is the agonist concentration that gives a response halfway to the maximum. Figures were prepared in Origin 6 (OriginLab).
Radioactive ligand-binding assays
Radioactive ligand-binding assays were carried out as previously described56.
Simulations of receptor subtype distributions
Here we describe the implementation details of the simulations for the particular case of three subunits as used in the paper. For the general case description of the computational method for simulating the subtype distribution, simulation aims, assumptions, limitations and alternative models of assembly, please refer to the Supplementary Discussion 1.4. Derivation of the equation for calculating the theoretical number of receptor subtypes is also presented in the Supplementary Information.
Model parameters
In the simulation, we define three distinct monomeric subunits, arbitrarily denoted as α, β and δ/γ. Two parameter sets are initiated at the start of each run: subunit abundances (an 3D vector) and affinities (a 3 × 3 matrix of pairwise affinity coefficients, where [m,n] denotes the relative probability of forming an m+–n− interface). The subunit abundance vector was normalized to the unit sum of its components.
Computational setup
We iterated over a range of relative subunit abundances and affinity coefficients and simulate M = 1,000 receptors for each of these cases. We sampled a range of relative abundances of any pair of subunits from 1:64 to 64:1 and increased by a factor of 2. Each of the coefficients in the 3 × 3 affinity matrix loops over a discrete set of values 100, 101, 102, 104 and 105, with the exception of the αα coefficient, which is kept 0. Together, we generated a total of 117,100,607 simulated conditions. For every condition, each of the 1,000 receptors was assigned to one of the 51 unique subtypes, and the distribution saved together with the parameters that generated it. Custom scripts79 were written in Python v3.6–3.8.
Identification of conditions that favour experimentally observed subtype distributions
To identify simulated conditions that favour subtype distributions observed in our cryo-EM experiments, we searched among all simulated conditions and respective subtype distributions for those that satisfy the following criteria that are conservatively derived from our cryo-EM observations: (1) of all receptors produced under a given condition, at least 50% incorporate δ-subunit (containing the purification tag), (2) of all purifiable receptors (that is, those from condition 1), at least 50% are either α4β3δ or β3δ, (3) both α4β3δ and β3δ receptor populations should be abundant and above the noise level, and (4) all other purifiable and solvable receptor subtypes (that is, those containing the purification tag and the β3+/β3− interface to which the Nb25 nanobody binds) are below the noise level (estimated as \(\sqrt{M}\)). Because our estimations of the constraints from cryo-EM are imperfect, we confirmed that the general observations are robust to changes in constraint 1 (tested down to 10%). Using analogous constraints, we searched for parameters that favour the receptor distribution observed from the α4β3γ2 cell lines. In addition, given that both of these cell lines express identical α4 and β3 subunit constructs and the cells themselves are identical, we used an additional constraint that all 2 × 2 pairwise affinity coefficients between these two subunits must be identical between the conditions identified for the α4β3δ and the α4β3γ2 cell lines (Supplementary Fig. 5a, Supplementary Discussion). This allowed us to narrow down the range of plausible conditions. All analyses were performed using custom written scripts79 in R v3.5.2 and RStudio v1.4.1106.
Analysis of GABAA receptor gene expression
scRNA-seq data were obtained from the Allen Brain Atlas on 25 July 2019 at 13:00 GMT and 18 December 2019 at 23:00 GMT (dataset ‘Human Multiple Cortical Areas SMART-seq’; download link: https://portal.brain-map.org/atlases-and-data/rnaseq/human-multiple-cortical-areas-smart-seq)8,9. Handling of raw data was performed using the rhdf5 2.26.2 R package. For binarized expression data in Extended Data Fig. 7a, a subunit was considered expressed if its precalculated trimmed mean number of counts was greater than zero. Trimmed means are provided by the Allen Brain Institute and are generated by first taking the log2 of the summed intron and exon counts of a particular gene across all sequenced cells from a particular cluster (that is, cell type), then calculating the average number of counts for the middle 50% of the data (that is, excluding the 25% highest and lowest values). Visualization was done using the UpsetR package in R. A heatmap of trimmed mean counts was generated using the pheatmap package in R (v1.0.12).
Biological materials availability
Cell lines, cell pools and cDNA constructs generated for the purpose of this study are available from A.R.A. on reasonable request.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Data availability
Atomic coordinates for the GABAAR models reported here have been deposited to the PDB with accession codes 7QN5-9 and 7QNA-E, and the cryo-EM density maps have been deposited in the Electron Microscopy Data Bank with accession codes EMD-14067–EMD-14076. Raw movies of all datasets have been deposited in the Electron Microscopy Public Image Archive with accession codes EMPIAR-10909–EMPIAR-10914. Coordinate files used during model building can be found in the PDB under the accession codes 7A5V and 6HUG. scRNA-seq data used in the study are available from Allen Brain Map: https://portal.brain-map.org/atlases-and-data/rnaseq/human-multiple-cortical-areas-smart-seq.Source data are provided with this paper.
Code availability
The scripts for scRNA-seq analysis and for running and analysing the receptor assembly simulations are available at www.github.com/andrijasente/GABA-differential-assembly.
References
Sigel, E. & Steinmann, M. E. Structure, function, and modulation of GABAA receptors. J. Biol. Chem. 287, 40224–40231 (2012).
Sieghart, W. & Sperk, G. Subunit composition, distribution and function of GABAA receptor subtypes. Curr. Top. Med. Chem. 2, 795–816 (2002).
Sieghart, W. & Savic, M. M. International Union of Basic and Clinical Pharmacology. CVI: GABAA receptor subtype- and function-selective ligands: key issues in translation to humans. Pharmacol. Rev. 70, 836–878 (2018).
Zhu, S. et al. Structure of a human synaptic GABAA receptor. Nature 559, 67–72 (2018).
Phulera, S. et al. Cryo-EM structure of the benzodiazepine-sensitive α1β1γ2S tri-heteromeric GABAA receptor in complex with GABA. eLife 7, e39383 (2018).
Laverty, D. et al. Cryo-EM structure of the human α1β3γ2 GABAA receptor in a lipid bilayer. Nature 565, 516–520 (2019).
Masiulis, S. et al. GABAA receptor signalling mechanisms revealed by structural pharmacology. Nature 565, 454–459 (2019).
Tasic, B. et al. Shared and distinct transcriptomic cell types across neocortical areas. Nature 563, 72–78 (2018).
Hodge, R. D. et al. Conserved cell types with divergent features in human versus mouse cortex. Nature 573, 61–68 (2019).
Wisden, W., Laurie, D. J., Monyer, H. & Seeburg, P. H. The distribution of 13 GABAA receptor subunit mRNAs in the rat brain. I. Telencephalon, diencephalon, mesencephalon. J. Neurosci. 12, 1040–1062 (1992).
Nusser, Z., Sieghart, W. & Somogyi, P. Segregation of different GABAA receptors to synaptic and extrasynaptic membranes of cerebellar granule cells. J. Neurosci. 18, 1693–1703 (1998).
Pearce, R. A. Physiological evidence for two distinct GABAA responses in rat hippocampus. Neuron 10, 189–200 (1993).
Baude, A., Sequier, J. M., McKernan, R. M., Olivier, K. R. & Somogyi, P. Differential subcellular distribution of the alpha 6 subunit versus the alpha 1 and beta 2/3 subunits of the GABAA/benzodiazepine receptor complex in granule cells of the cerebellar cortex. Neuroscience 51, 739–748 (1992).
Farrant, M. & Nusser, Z. Variations on an inhibitory theme: phasic and tonic activation of GABAA receptors. Nat. Rev. Neurosci. 6, 215–229 (2005).
Brickley, S. G. & Mody, I. Extrasynaptic GABAA receptors: their function in the CNS and implications for disease. Neuron 73, 23–34 (2012).
Scholze, P. et al. Two distinct populations of α1α6-containing GABAA-receptors in rat cerebellum. Front. Synaptic Neurosci. 12, 591129 (2020).
Barrera, N. P. et al. Atomic force microscopy reveals the stoichiometry and subunit arrangement of the α4β3δ GABAA receptor. Mol. Pharmacol. 73, 960–967 (2008).
Patel, B., Mortensen, M. & Smart, T. G. Stoichiometry of delta subunit containing GABAA receptors. Br. J. Pharmacol. 171, 985–994 (2014).
Wagoner, K. R. & Czajkowski, C. Stoichiometry of expressed α4β2δ γ-aminobutyric acid type A receptors depends on the ratio of subunit cDNA transfected. J. Biol. Chem. 285, 14187–14194 (2010).
Botzolakis, E. J. et al. Comparison of gamma-aminobutyric acid, type A (GABAA), receptor αβγ and αβδ expression using flow cytometry and electrophysiology: evidence for alternative subunit stoichiometries and arrangements. J. Biol. Chem. 291, 20440–20461 (2016).
Zhou, X. et al. High-level production and purification in a functional state of an extrasynaptic gamma-aminobutyric acid type A receptor containing α4β3δ subunits. PLoS ONE 13, e0191583 (2018).
Eaton, M. M. et al. γ-Aminobutyric acid type A α4, β2, and δ subunits assemble to produce more than one functionally distinct receptor type. Mol. Pharmacol. 86, 647–656 (2014).
Nakane, T. et al. Single-particle cryo-EM at atomic resolution. Nature 587, 152–156 (2020).
Saras, A. et al. Histamine action on vertebrate GABAA receptors: direct channel gating and potentiation of GABA responses. J. Biol. Chem. 283, 10470–10475 (2008).
Wongsamitkul, N. et al. α Subunits in GABAA receptors are dispensable for GABA and diazepam action. Sci. Rep. 7, 15498 (2017).
Taylor, P. M. et al. Identification of amino acid residues within GABAA receptor beta subunits that mediate both homomeric and heteromeric receptor expression. J. Neurosci. 19, 6360–6371 (1999).
Bianchi, M. T., Clark, A. G. & Fisher, J. L. The wake-promoting transmitter histamine preferentially enhances alpha-4 subunit-containing GABAA receptors. Neuropharmacology 61, 747–752 (2011).
Lee, H. J. et al. A pharmacological characterization of GABA, THIP and DS2 at binary α4β3 and β3δ receptors: GABA activates β3δ receptors via the β3+δ− interface. Brain Res. 1644, 222–230 (2016).
Miller, P. S. & Aricescu, A. R. Crystal structure of a human GABAA receptor. Nature 512, 270–275 (2014).
Hoerbelt, P. et al. Mutagenesis and computational docking studies support the existence of a histamine binding site at the extracellular β3+ β3− interface of homooligomeric β3 GABAA receptors. Neuropharmacology 108, 252–263 (2016).
Falch, E. & Krogsgaard-Larsen, P. The binding of the GABA agonist [3H]THIP to rat brain synaptic membranes. J. Neurochem. 38, 1123–1129 (1982).
Wafford, K. A. & Ebert, B. Gaboxadol—a new awakening in sleep. Curr. Opin. Pharmacol. 6, 30–36 (2006).
Hanchar, H. J. et al. Ethanol potently and competitively inhibits binding of the alcohol antagonist Ro15-4513 to α4/6β3δ GABAA receptors. Proc. Natl Acad. Sci. USA 103, 8546–8551 (2006).
Wallner, M., Hanchar, H. J. & Olsen, R. W. Low-dose alcohol actions on α4β3δ GABAA receptors are reversed by the behavioral alcohol antagonist Ro15-4513. Proc. Natl Acad. Sci. USA 103, 8540–8545 (2006).
Olsen, R. W., Hanchar, H. J., Meera, P. & Wallner, M. GABAA receptor subtypes: the “one glass of wine” receptors. Alcohol 41, 201–209 (2007).
Linden, A. M. et al. Ro 15-4513 antagonizes alcohol-induced sedation in mice through αβγ2-type GABAA receptors. Front. Neurosci. 5, 3 (2011).
Storustovu, S. I. & Ebert, B. Pharmacological characterization of agonists at delta-containing GABAA receptors: functional selectivity for extrasynaptic receptors is dependent on the absence of γ2. J. Pharmacol. Exp. Ther. 316, 1351–1359 (2006).
Brown, N., Kerby, J., Bonnert, T. P., Whiting, P. J. & Wafford, K. A. Pharmacological characterization of a novel cell line expressing human α4β3δ GABAA receptors. Br. J. Pharmacol. 136, 965–974 (2002).
Marti-Solano, M. et al. Combinatorial expression of GPCR isoforms affects signalling and drug responses. Nature 587, 650–656 (2020).
Rudolph, U. & Knoflach, F. Beyond classical benzodiazepines: novel therapeutic potential of GABAA receptor subtypes. Nat. Rev. Drug Discov. 10, 685–697 (2011).
Bencsits, E., Ebert, V., Tretter, V. & Sieghart, W. A significant part of native γ-aminobutyric acidA receptors containing α4 subunits do not contain γ or δ subunits. J. Biol. Chem. 274, 19613–19616 (1999).
Levitan, E. S. et al. Structural and functional basis for GABAA receptor heterogeneity. Nature 335, 76–79 (1988).
Brickley, S. G., Cull-Candy, S. G. & Farrant, M. Single-channel properties of synaptic and extrasynaptic GABAA receptors suggest differential targeting of receptor subtypes. J. Neurosci. 19, 2960–2973 (1999).
Mertens, S., Benke, D. & Mohler, H. GABAA receptor populations with novel subunit combinations and drug binding profiles identified in brain by α5- and δ-subunit-specific immunopurification. J. Biol. Chem. 268, 5965–5973 (1993).
Fritschy, J. M. et al. Five subtypes of type A gamma-aminobutyric acid receptors identified in neurons by double and triple immunofluorescence staining with subunit-specific antibodies. Proc. Natl Acad. Sci. USA 89, 6726–6730 (1992).
Duggan, M. J., Pollard, S. & Stephenson, F. A. Immunoaffinity purification of GABAA receptor α-subunit iso-oligomers. Demonstration of receptor populations containing α1 α2, α1 α3, and α2 α3 subunit pairs. J. Biol. Chem. 266, 24778–24784 (1991).
Owens, D. F. & Kriegstein, A. R. Is there more to GABA than synaptic inhibition? Nat. Rev. Neurosci. 3, 715–727 (2002).
Datta, D., Arion, D. & Lewis, D. A. Developmental expression patterns of GABAA receptor subunits in layer 3 and 5 pyramidal cells of monkey prefrontal cortex. Cereb. Cortex 25, 2295–2305 (2015).
Fatemi, S. H., Folsom, T. D., Rooney, R. J. & Thuras, P. D. Expression of GABAA α2-, β1- and ε-receptors are altered significantly in the lateral cerebellum of subjects with schizophrenia, major depression and bipolar disorder. Transl. Psychiatry 3, e303 (2013).
Mueller, T. M., Remedies, C. E., Haroutunian, V. & Meador-Woodruff, J. H. Abnormal subcellular localization of GABAA receptor subunits in schizophrenia brain. Transl. Psychiatry 5, e612 (2015).
Murshudov, G. N. et al. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 67, 355–367 (2011).
Chen, V. B. et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 (2010).
Afonine, P. V. et al. Real-space refinement in PHENIX for cryo-EM and crystallography. Acta Crystallogr. D Struct. Biol. 74, 531–544 (2018).
Reeves, P. J., Kim, J. M. & Khorana, H. G. Structure and function in rhodopsin: a tetracycline-inducible system in stable mammalian cell lines for high-level expression of opsin mutants. Proc. Natl Acad. Sci. USA 99, 13413–13418 (2002).
Elegheert, J. et al. Lentiviral transduction of mammalian cells for fast, scalable and high-level production of soluble and membrane proteins. Nat. Protoc. 13, 2991–3017 (2018).
Dostalova, Z. et al. Human α1β3γ2L γ-aminobutyric acid type A receptors: high-level production and purification in a functional state. Protein Sci. 23, 157–166 (2014).
Miller, P. S. et al. Structural basis for GABAA receptor potentiation by neurosteroids. Nat. Struct. Mol. Biol. 24, 986–992 (2017).
Uchanski, T. et al. Megabodies expand the nanobody toolkit for protein structure determination by single-particle cryo-EM. Nat. Methods 18, 60–68 (2021).
Russo, C. J. & Passmore, L. A. Electron microscopy: ultrastable gold substrates for electron cryomicroscopy. Science 346, 1377–1380 (2014).
Zheng, S. Q. et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 (2017).
Rohou, A. & Grigorieff, N. CTFFIND4: fast and accurate defocus estimation from electron micrographs. J. Struct. Biol. 192, 216–221 (2015).
Tegunov, D. & Cramer, P. Real-time cryo-electron microscopy data preprocessing with Warp. Nat. Methods 16, 1146–1152 (2019).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Asarnow, D., Palovcak, E. & Cheng, Y. asarnow/pyem: UCSF pyem v0.5 (Zenodo, 2019).
Scheres, S. H. RELION: implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol. 180, 519–530 (2012).
Zivanov, J. et al. New tools for automated high-resolution cryo-EM structure determination in RELION-3. eLife 7, e42166 (2018).
Zivanov, J., Nakane, T. & Scheres, S. H. W. Estimation of high-order aberrations and anisotropic magnification from cryo-EM data sets in RELION-3.1. IUCrJ 7, 253–267 (2020).
Zivanov, J., Nakane, T. & Scheres, S. H. W. A Bayesian approach to beam-induced motion correction in cryo-EM single-particle analysis. IUCrJ 6, 5–17 (2019).
Pettersen, E. F. et al. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Ramlaul, K., Palmer, C. M., Nakane, T. & Aylett, C. H. S. Mitigating local over-fitting during single particle reconstruction with SIDESPLITTER. J. Struct. Biol. 211, 107545 (2020).
Kucukelbir, A., Sigworth, F. J. & Tagare, H. D. Quantifying the local resolution of cryo-EM density maps. Nat. Methods 11, 63–65 (2014).
Naydenova, K. & Russo, C. J. Measuring the effects of particle orientation to improve the efficiency of electron cryomicroscopy. Nat. Commun. 8, 629 (2017).
Goddard, T. D. et al. UCSF ChimeraX: meeting modern challenges in visualization and analysis. Protein Sci. 27, 14–25 (2018).
Waterhouse, A. et al. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 46, W296–W303 (2018).
Weininger, D. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J. Chem. Inf. Comput. Sci. 28, 31–36 (1988).
Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 (2004).
Nicholls, R. A., Fischer, M., McNicholas, S. & Murshudov, G. N. Conformation-independent structural comparison of macromolecules with ProSMART. Acta Crystallogr. D Biol. Crystallogr. 70, 2487–2499 (2014).
Forman, S. A. A hydrophobic photolabel inhibits nicotinic acetylcholine receptors via open-channel block following a slow step. Biochemistry 38, 14559–14564 (1999).
Sente, A. & Naydenova, K. Scripts for differential assembly diversifies GABAA receptor structures and signaling (v1.0.1). Zenodo https://doi.org/10.5281/zenodo.5884871 (2022).
Acknowledgements
We thank M. Madan Babu, D. Barford, T. Blundell, R. Calinescu, T. Flock, A. B. Kleist, A. Murzin, D. Malinverni, C. J. Russo, N. Unwin and the members of Aricescu laboratory for helpful discussions and comments on this work; V. B. Kasaragod for help with model building; T. Nakane and K. Yamashita for help with data processing; J. Grimmett and T. Darling for support with scientific computing; S. Chen, G. Cannone, G. Sharov, A. Yeates and B. Ahsan for electron microscopy support; T. Uchanski, E. Pardon and J. Steyaert for the Mb192 megabody. Cryo-EM datasets were collected at the MRC-LMB and Cambridge University Department of Biochemistry EM (BiocEM) facilities. We acknowledge funding from the UK Medical Research Council (MR/L009609/1 and MC_UP_1201/15 to A.R.A.), National Institute for General Medical Sciences (1R01-GM135550 to K.W.M. and A.R.A.), the Department of Anesthesia, Critical Care & Pain Medicine at Massachusetts General Hospital (to K.W.M.), MRC-LMB and Cambridge Trust (LMB Cambridge Scholarship to A.S. and K.N.), School of Clinical Medicine, University of Cambridge (MB/PhD fellowship to A.S.), and Boehringer Ingelheim Fonds (PhD Fellowship to J.M.). The cryo-EM facility at the Department of Biochemistry is funded by the Wellcome Trust (206171/Z/17/Z and 202905/Z/16/Z) and the University of Cambridge.
Author information
Authors and Affiliations
Contributions
A.S., K.W.M. and A.R.A. conceived the project. A.S., Y.J. and R.D. generated and characterized cell lines. A.S. and S.M. purified proteins and prepared the cryo-EM samples. A.S., S.W.H. and D.Y.C. collected the cryo-EM data. A.S., K.N. and S.M. processed the cryo-EM data. A.S., T.M., J.M. and A.R.A. built and refined the atomic models. R.D. and K.W.M. designed, performed and analysed the electrophysiology experiments. X.Z. and K.W.M. designed, performed and analysed the radioligand binding assays. A.S. designed and performed the bioinformatics analyses. A.S. and K.N. designed, performed and analysed the simulations for receptor assembly. A.S., K.N., R.D., K.W.M. and A.R.A. wrote the manuscript, with input from all authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature thanks Margot Ernst and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Quality of cryo-EM maps and models.
Each panel contains masked, unmasked, phase-randomised and map vs model Fourier shell correlation (FSC) curves (top), particle orientation distribution (middle) and local resolution-coloured maps for each structure. a, α4β3δ "apo" (+HEPES). b, β3δ "apo" (+HEPES). c, α4β3δ + GABA + histamine. d, β3δ + histamine. e, α4β3δ + histamine. f, α4β3γ2 + GABA. g, β3γ2 + GABA. h, α4β3δ + THIP + histamine. i, β3δ + THIP + histamine. j, α1β3γ2 + Ro15-4513. The probability density function for a certain orientation ranges between 0 (blue, low) and 0.0001 (red, high).
Extended Data Fig. 2 Survey of ligands bound in agonist pockets in different datasets.
a, α4β3δ and β3δ GABAARs solved in the absence of any known modulators. In both subtypes, HEPES is bound in the β3+/β3− agonist pocket under loop C. b, α4β3δ and β3δ GABAARs solved in the presence of 0.2 mM GABA and 1 mM histamine. c, α4β3γ2 and β3γ2 GABAARs solved in the presence of 0.2 mM GABA. d, α4β3δ and β3δ GABAARs solved in the presence of 1 mM THIP and 1 mM histamine. e, Summary table illustrating ligand vs interface compatibility. Empty spaces are left in cases where insufficient data is available to make a conclusion. For the GABA binding to β3+/γ2− pocket, a question mark indicates variable occupancy. f, Comparison of pore profiles of α4β3δ + GABA + histamine, α4β3δ + histamine, α1β3γ2 + alprazolam (6HUO) and α1β3γ2 + bicuculline (6HUK).
Extended Data Fig. 3 Ligand coordination in the “agonist pockets” under the loops C at various inter-subunit interfaces.
All pockets in which a ligand was observed in our structures are shown with a single representative example. Potential hydrogen bonds are displayed as dashed yellow lines. Histamine and GABA in the β3+/α4− pocket are taken from the α4β3δ + GABA + histamine structure. THIP in the β3+/α4− and δ+/β3− pocket is taken from the α4β3δ + THIP + histamine structure. Histamine in the β3+/β3− pocket is taken from the α4β3δ + histamine structure. HEPES in the β3+/β3− pocket is taken from the "apo" α4β3δ structure. GABA in the β3+/γ2− pocket is taken from the α4β3γ2 + GABA structure. Chain IDs represented in the panels are given as capital letters next to the subunit type.
Extended Data Fig. 4 Characterizing GABA and histamine effects on GABAA receptors composed of α4, β3 and δ subunits.
a–c, Unlike α4β3δ receptors (a), β3δ (b) and β3 (c) receptors are not gated by 200 μM GABA. Representative current traces from three successive pulses separated by 1 min: (1) 4 s pulse of 10 mM GABA plus 30 μM Etomidate (total current, purple); (2) 8 s pulse of 200 μM GABA (green), and (3) repeat of pulse (1) (pink). These pulses are applied to: a, cells co-expressing α4, β3 and δ subunits; b, cells co-expressing β3 and δ subunits; c, cells expressing the β3 subunit alone. d, e, Representative currents normalized to 10 mM GABA currents in the same cell were obtained with varying concentration of (a) GABA (0.001–10 μM, n = 38 cells with 3-8 cells for each concentration) or (b) histamine (0.03–10 mM, n = 31 cells with 3-8 cells for each concentration). In panel e, orange dashed line denotes the baseline. f, Hill plot of peak amplitude obtained with the normalized currents from (a) and (b) plotted against agonist concentration. The EC50 for GABA was 69.3 nM (95% CI 40.5–118.3 nM) and for histamine was 821.2 μM (95% CI 340.5–1981.0 μM) and Hill coefficient of 0.9 (95% CI 0.5–1.3) and 1.1 (95% CI 0.3–2.0) respectively. g, Scatter plot of peak amplitudes of currents shown in Fig. 2d by GABA (10 mM, n = 7 cells) and histamine (10 mM, n = 6 cells) alone or co-applied (n = 8 cells) in the first pulse as a percent of currents elicited by 10 mM GABA + 30 μM Etomidate. One-way ANOVA P < 0.0001. h–k, Scatter plots of the time constants (τ) and amplitudes for the slow and fast phases of deactivation that were obtained by fitting a double exponential to the falling phase of currents such as those in Fig. 2e (n = 4 cells for each experiments). In every paired experiment, the direction of change was consistent. In the presence of histamine fast and slow time constants decreased significantly (P = 0.03, two-tailed paired t test) and the fraction of receptors with slow deactivation increased (P = 0.04, two-tailed paired t test). Significance levels compared to controls were determined by ordinary one–way ANOVA or by the Paired Student’s t test and are denoted as **** P < 0.0001 and * P < 0.05. Data are presented as mean ± SD.
Extended Data Fig. 5 GABA and histamine crosstalk happens at the level of individual α4β3δ receptors.
To address the complexity arising from the presence of multiple receptor subtypes, we investigated the modulation of 100 nM GABA currents by 300 μM histamine in cells expressing different combinations of α4, β3 and δ subunits. We first established that 100 nM GABA robustly activates currents in α4β3δ cells (n = 6 cells) (a), barely in α4β3 (n = 5 cells) (b) and not in β3δ (n = 6 cells) (c) or β3 cells (n = 4 cells) (d). Histamine at 300 μM robustly enhances the GABA current in α4β3δ cells but only modestly in the α4β3, β3δ and β3 cells. The modest enhancement is attributable to histamine currents alone (e.g., from the β3δ or β3-homomeric subtypes). Representative current traces from three successive pulses separated by 1 min: (1) 8 s pulse of 100 nM GABA (dark green); (2) 8 s pulse of 100 nM GABA with a 4 s notch of 300 μM histamine added after two seconds (blue), and (3) 8 s pulse of 100 nM GABA (light green). Finally, to open all receptor isoforms, a normalization pulse of (10 mM GABA + 30 μM etomidate) was applied. e, Histamine enhances GABA current strongly only in α4β3δ receptors (P < 0.0001, one-way ANOVA). Scatter plot shows peak current amplitudes of the blue traces in panels a–d, normalized to the total cell current (10 mM GABA + 30 μM etomidate). Data are presented as mean ± SD. Sample size is the same as in panels a–d.
Extended Data Fig. 6 Ro15-4513 cannot bind to the α4+/δ− interface.
a, b, Two views of Ro15-4513 bound in the α1+/γ2− benzodiazepine pocket of an α1β3γ2 receptor. c, d, Equivalent views of an α4β3δ receptor show that clashes with the side chains of R135 (α4+), E71 and H92 (δ−) prevent Ro15-4513 (and, likely, any benzodiazepine ligand) from binding to the α4+/δ− interface in the same mode as observed in α1β3γ2.
Extended Data Fig. 7 Single-cell expression patterns of GABAAR subunits in the human cortex.
a, Grouping cortical cell types by their GABAAR subunit expression. The majority of cell types (31) expresses a combination of 11 different GABAAR subunits, including α1-5, β1-3, δ and γ2-3. Cell type "Exc L5-6 THEMIS THTPA" expresses the highest number of subunits (14): α1-5, β1-3, δ, ε, γ1-3 and ρ1. Expression in this panel is binarized such that any subunit with a trimmed mean of summed intron and exon counts larger than 0 count per million (CPM) is considered as expressed (Methods). b, Heat map showing trimmed means of CPMs for every GABAAR subunit across individual cell types in the human cortex. Cell types are clustered by their gene expression profiles.
Extended Data Fig. 8 Cryo-EM data processing strategy.
a, A typical cryo-EM data processing workflow. The numbers in this case are derived from the α4β3δ "apo" (+HEPES) dataset. b, Soft mask used to separate receptors with 2 or 3 Nb25 bound, which largely (but not completely) separates α4β3δ and β3δ receptors. The separation is incomplete in all cases because Nb25 may also bind weakly to the β3+/α4− interface. c, To further separate α4β3δ and β3δ receptors, a soft mask around the putative position of the vestibule glycan on the α4 subunit and the putative position of the N149 glycan on the β3 subunit is used. d, Soft mask around the α4 vestibule glycan used to separate α4β3γ2 from β3γ2 receptor subtypes.
Supplementary information
Supplementary Information
This file contains Supplementary Figures; Supplementary Tables; Supplementary Methods; Supplementary Discussion and Supplementary References
Rights and permissions
About this article

Cite this article
Sente, A., Desai, R., Naydenova, K. et al. Differential assembly diversifies GABAA receptor structures and signalling. Nature 604, 190–194 (2022). https://doi.org/10.1038/s41586-022-04517-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-022-04517-3
This article is cited by
-
De novo variants in GABRA4 are associated with a neurological phenotype including developmental delay, behavioral abnormalities and epilepsy
European Journal of Human Genetics (2024)
-
Inhibition of acyl-CoA binding protein (ACBP) by means of a GABAARγ2-derived peptide
Cell Death & Disease (2024)
-
Atypical antipsychotics antagonize GABAA receptors in the ventral tegmental area GABA neurons to relieve psychotic behaviors
Molecular Psychiatry (2023)
-
A pentameric TRPV3 channel with a dilated pore
Nature (2023)
-
The molecular basis of drug selectivity for α5 subunit-containing GABAA receptors
Nature Structural & Molecular Biology (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
