
Abstract
Loci discovered by genome-wide association studies predominantly map outside protein-coding genes. The interpretation of the functional consequences of non-coding variants can be greatly enhanced by catalogs of regulatory genomic regions in cell lines and primary tissues. However, robust and readily applicable methods are still lacking by which to systematically evaluate the contribution of these regions to genetic variation implicated in diseases or quantitative traits. Here we propose a novel approach that leverages genome-wide association studies’ findings with regulatory or functional annotations to classify features relevant to a phenotype of interest. Within our framework, we account for major sources of confounding not offered by current methods. We further assess enrichment of genome-wide association studies for 19 traits within Encyclopedia of DNA Elements- and Roadmap-derived regulatory regions. We characterize unique enrichment patterns for traits and annotations driving novel biological insights. The method is implemented in standalone software and an R package, to facilitate its application by the research community.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout





Similar content being viewed by others

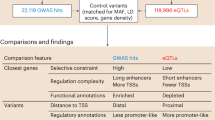
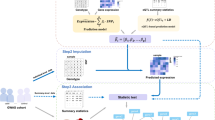
Code availability
Custom codes can be found at http://www.ebi.ac.uk/birney-srv/GARFIELD/.
Data availability
Web links for publicly available GWAS datasets and regulatory information databases are included in the URLs section. Restriction of availability applies to blood cell indices GWAS from van der Harst et al.33 and Gieger et al.34, which have been obtained through the manuscripts’ authors. Any other data that support the findings of this study are available from the corresponding authors upon reasonable request.
References
Visscher, P. M., Brown, M. A., McCarthy, M. I. & Yang, J. Five years of GWAS discovery. Am. J. Hum. Genet. 90, 7–24 (2012).
Hindorff, L. A. et al. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc. Natl. Acad. Sci. USA 106, 9362–9367 (2009).
ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012).
Thurman, R. E. et al. The accessible chromatin landscape of the human genome. Nature 489, 75–82 (2012).
Bernstein, B. E. et al. The NIH roadmap epigenomics mapping consortium. Nat. Biotechnol. 28, 1045–1048 (2010).
Adams, D. et al. BLUEPRINT to decode the epigenetic signature written in blood. Nat. Biotechnol. 30, 224–226 (2012).
1000 Genomes Project Consortium et al. An integrated map of genetic variation from 1,092 human genomes. Nature 491, 56–65 (2012).
Shen, H. et al. Comprehensive characterization of human genome variation by high coverage whole-genome sequencing of forty-four Caucasians. PLoS ONE 8, e59494 (2013).
Chung, D., Yang, C., Li, C., Gelernter, J. & Zhao, H. GPA: a statistical approach to prioritizing GWAS results by integrating pleiotropy and annotation. PLoS. Genet. 10, e1004787 (2014).
Finucane, H. K. et al. Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat. Genet. 47, 1228–1235 (2015).
Maurano, M. T. et al. Systematic localization of common disease-associated variation in regulatory DNA. Science 337, 1190–1195 (2012).
Schork, A. J. et al. All SNPs are not created equal: genome-wide association studies reveal a consistent pattern of enrichment among functionally annotated SNPs. PLoS. Genet. 9, e1003449 (2013).
Pickrell, J. K. Joint analysis of functional genomic data and genome-wide association studies of 18 human traits. Am. J. Hum. Genet. 94, 559–573 (2014).
Trynka, G. et al. Disentangling the effects of colocalizing genomic annotations to functionally prioritize non-coding variants within complex-trait loci. Am. J. Hum. Genet. 97, 139–152 (2015).
Roadmap Epigenomics Consortium et al. Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330 (2015).
Schmidt, E. M. et al. GREGOR: evaluating global enrichment of trait-associated variants in epigenomic features using a systematic, data-driven approach. Bioinformatics 31, 2601–2606 (2015).
Galwey, N. W. A new measure of the effective number of tests, a practical tool for comparing families of non-independent significance tests. Genet. Epidemiol. 33, 559–568 (2009).
Dunham, I., Kulesha, E., Iotchkova, V., Morganella, S. & Birney, E. FORGE: a tool to discover cell specific enrichments of GWAS associated SNPs in regulatory regions. F1000Res. https://doi.org/10.12688/f1000research.6032.1 (2015).
Astle, W. J. et al. The allelic landscape of human blood cell trait variation and links to common complex disease. Cell 167, 1415–1429.e19 (2016).
Harrow, J. et al. GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res. 22, 1760–1774 (2012).
Speliotes, E. K. et al. Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nat. Genet. 42, 937–948 (2010).
Lango Allen, H. et al. Hundreds of variants clustered in genomic loci and biological pathways affect human height. Nature 467, 832–838 (2010).
Heid, I. M. et al. Meta-analysis identifies 13 new loci associated with waist-hip ratio and reveals sexual dimorphism in the genetic basis of fat distribution. Nat. Genet. 42, 949–960 (2010).
Saxena, R. et al. Genetic variation in GIPR influences the glucose and insulin responses to an oral glucose challenge. Nat. Genet. 42, 142–148 (2010).
Dupuis, J. et al. New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk. Nat. Genet. 42, 105–116 (2010).
Strawbridge, R. J. et al. Genome-wide association identifies nine common variants associated with fasting proinsulin levels and provides new insights into the pathophysiology of type 2 diabetes. Diabetes 60, 2624–2634 (2011).
Soranzo, N. et al. Common variants at 10 genomic loci influence hemoglobin A1(C) levels via glycemic and nonglycemic pathways. Diabetes 59, 3229–3239 (2010).
Teslovich, T. M. et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature 466, 707–713 (2010).
Liu, J. Z. et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat. Genet. 47, 979–986 (2015).
International Consortium for Blood Pressure Genome-Wide Association Studies et al. Genetic variants in novel pathways influence blood pressure and cardiovascular disease risk. Nature 478, 103–109 (2011).
Morris, A. P. et al. Large-scale association analysis provides insights into the genetic architecture and pathophysiology of type 2 diabetes. Nat. Genet. 44, 981–990 (2012).
Schizophrenia Working Group of the Psychiatric Genomics Consortium. Biological insights from 108 schizophrenia-associated genetic loci. Nature 511, 421–427 (2014).
Van der Harst, P. et al. Seventy-five genetic loci influencing the human red blood cell. Nature 492, 369–375 (2012).
Gieger, C. et al. New gene functions in megakaryopoiesis and platelet formation. Nature 480, 201–208 (2011).
Purcell, S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007).
UK10K Consortium. et al. The UK10K project identifies rare variants in health and disease. Nature 526, 82–90 (2015).
Acknowledgements
This study made use of data generated by the UK10K Consortium. A full list of the investigators who contributed to the generation of the data is available from http://www.UK10K.org/. Funding for UK10K was provided by the Wellcome Trust under award no. WT091310. Research by N.S. is supported by the Wellcome Trust (grants WT098051 and WT091310). N.J.T. is supported as a Wellcome Trust Investigator (no. 202802/Z/16/Z), is supported as the principal investigator of the Avon Longitudinal Study of Parents and Children (no. MRC & WT 102215/2/13/2), is supported by the University of Bristol NIHR Biomedical Research Centre (no. BRC-1215-20011) and the MRC Integrative Epidemiology Unit (no. MC_UU_12013/3), and works within the CRUK Integrative Cancer Epidemiology Programme (no. C18281/A19169). G.R.S.R. and V.I. are supported by European Molecular Biology Laboratory-Wellcome Trust Sanger Institute postdoctoral fellowships.
Author information
Authors and Affiliations
Consortia
Contributions
G.R.S.R., J.L.M., K.W., N.J.T., I.D. and N.S. contributed data or materials. E.B., G.R.S.R., I.D., J.L.M., N.S. and V.I. developed the method. V.I. analyzed the data. E.B., I.D., N.J.T., N.S. and V.I. provided critical interpretation of the results. M.G. and S.M. designed the tools. E.B., N.S. and V.I. wrote the manuscript. E.B., G.R.S.R., I.D., J.L.M., K.W., M.G., N.J.T., N.S., S.M. and V.I. evaluated the manuscript. E.B. and N.S. designed and managed the project.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Integrated supplementary information
Supplementary Fig. 1 GARFIELD method assessment.
a, GARFIELD-estimated false-positive rate (FPR) from 29 real data GWAS and n = 1,000 independent simulated annotations. The black horizontal line denotes the 5% FPR threshold. Traits are shown on the x axis. Colored bars denote the GARFIELD threshold of T < 10–8 (red) and the GARFIELD threshold of T < 10–5 (gray). Error bars denote standard errors. b,c, Effect of feature correction for each of 29 GWAS at the T < 10–8 significance threshold when analyzing 424 DHS cell types. The figure shows the proportion of significant annotations with respect to feature correction, where N denotes the number of LD proxies and T distance to the nearest TSS. The y axis shows the corresponding values when no feature correction is employed. d, Difference in proportion of significant enrichments between a model not accounting for any feature to a model accounting for any combination of the features, respectively, applied to 424 DHS cell types.
Supplementary Fig. 2 GARFIELD enrichment wheel plots.
Enrichment of genome-wide association analysis P-values in DNase I–hypersensitive sites (hotspots) for 27 disease/quantitative traits. Radial lines show OR values at eight GWAS –log10 P-value thresholds (T) for n = 424 ENCODE and Roadmap Epigenomics DHS cell lines, sorted by tissue on the outer circle. Dots on the outer side of the circle denote significant enrichment (if present) at T < 10–5 (outermost) to T < 10–8 (innermost).
Supplementary Fig. 3 Comparison between real data results for 29 real GWAS and 424 open chromatin annotations at the T < 10–8 and T < 10–5 GWAS P-value thresholds.
Left, −log10 P-value comparison for all trait annotation pairs (n = 29 × 424 points). Horizontal and vertical lines denote the threshold for detecting enrichment after multiple-testing correction. The numbers in each corner denote the number of points in it. Right, odds ratio (OR) comparison for trait annotation pairs with significant enrichment in both the T < 10–8 and T < 10–5 GWAS P-value thresholds.
Supplementary Fig. 4 Multiple-annotation enrichment of genome-wide association analysis P-values in DNase I–hypersensitive sites (hotspots) for 15 disease or quantitative GWAS traits.
Cell types/tissues remaining after a heuristic multiple-annotation approach are shown on the y axis for each trait. Odds ratios (on log scale) are represented as dots and 95% CI with lines. The multiple-annotation model estimates are represented in red and the marginal effects of analysis of each annotation on its own are represented in black. Only phenotypes with at least a single detected enrichment are shown.
Supplementary Fig. 5 Enrichment levels (log OR) and extent of sharing between traits for 25-state chromatin segmentations of the NIH Roadmap and ENCODE projects at the T < 10–8 GWAS significance threshold.
a, Distribution of significant OR values across the 29 traits considered, split by segmentation state and colored to highlight predicted functional elements by Roadmap Epigenomics (see Supplementary Table 9). Number of points n is shown on the x axis below each category. b, Distribution of the pairwise difference between ORs from all enhancer, promoter and transcriptional enhancers and transcriptional regulatory states tested (‘state 1’) to ORs from transcription states for significant enrichments only (‘state 2’; for example, measuring ORc,tEnhA1 − ORc,tTx for all cell types c and traits t for which P-valuec,tEnhA1 and P-valuec,tTx are both significant). Number of points n is shown on the x axis below each category. c, Sharing of significantly enriched/depleted annotations across 27 phenotypes (excluding CD and UC). The bar plot displays the number of cell types where an annotation is uniquely enriched in a trait or shared among multiple traits.
Supplementary information
Supplementary Text and Figures
Supplementary Figures 1–5 and Supplementary Note
Supplementary Table 1
Summary of available enrichment analysis approaches
Supplementary Table 2
Summary of overlap of UK10K sequence variants with DNase I–hypersensitive sites
Supplementary Table 3
Summary of the number of variants per disease/quantitative trait
Supplementary Table 4
GARFIELD enrichment of 29 publicly available GWAS studies in DNase I–hypersensitive sites from 424 ENCODE and Roadmap Epigenomics cell types
Supplementary Table 5
Enrichment P values (–log10) from five methods of 21 publicly available GWAS studies in DNase I–hypersensitive sites from 424 ENCODE and Roadmap Epigenomics cell types; in H3K27ac peaks from 127 ENCODE and Roadmap Epigenomics cell types; and in H3K4me3 peaks from 127 ENCODE and Roadmap Epigenomics cell types
Supplementary Table 6
Method running time and memory usage for 21 traits and 424 DNase I–hypersensitive site annotations
Supplementary Table 7
Roadmap epigenomics and ENCODE cell lines used for segmentations
Supplementary Table 8
GARFIELD enrichment of 29 publicly available GWAS studies in 25 state genome segmentations in 127 cell types
Supplementary Table 9
Segmentation state summary
Rights and permissions
About this article

Cite this article
Iotchkova, V., Ritchie, G.R.S., Geihs, M. et al. GARFIELD classifies disease-relevant genomic features through integration of functional annotations with association signals. Nat Genet 51, 343–353 (2019). https://doi.org/10.1038/s41588-018-0322-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41588-018-0322-6
This article is cited by
-
Genetics of chronic respiratory disease
Nature Reviews Genetics (2024)
-
Multi-ancestry genome-wide association study of kidney cancer identifies 63 susceptibility regions
Nature Genetics (2024)
-
Integration of epigenetic and genetic profiles identifies multiple sclerosis disease-critical cell types and genes
Communications Biology (2023)
-
Functional genomics identify causal variant underlying the protective CTSH locus for Alzheimer’s disease
Neuropsychopharmacology (2023)
-
Genetic variation in cis-regulatory domains suggests cell type-specific regulatory mechanisms in immunity
Communications Biology (2023)
