
Abstract
Aortic calcification is an important independent predictor of future cardiovascular events. We performed a genome-wide association meta-analysis to determine SNPs associated with the extent of abdominal aortic calcification (n = 9,417) or descending thoracic aortic calcification (n = 8,422). Two genetic loci, HDAC9 and RAP1GAP, were associated with abdominal aortic calcification at a genome-wide level (P < 5.0 × 10−8). No SNPs were associated with thoracic aortic calcification at the genome-wide threshold. Increased expression of HDAC9 in human aortic smooth muscle cells promoted calcification and reduced contractility, while inhibition of HDAC9 in human aortic smooth muscle cells inhibited calcification and enhanced cell contractility. In matrix Gla protein–deficient mice, a model of human vascular calcification, mice lacking HDAC9 had a 40% reduction in aortic calcification and improved survival. This translational genomic study identifies the first genetic risk locus associated with calcification of the abdominal aorta and describes a previously unknown role for HDAC9 in the development of vascular calcification.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout




Similar content being viewed by others


Data availability
The summary statistics for the cohorts used in the meta-analysis and validation have been deposited with the database of Genotypes and Phenotypes (accession no. phs000930), according to the policy of the CHARGE consortium40. The remaining data that support the findings of this study are available from the corresponding author upon request.
References
Greenland, P., LaBree, L., Azen, S. P., Doherty, T. M. & Detrano, R. C. Coronary artery calcium score combined with Framingham score for risk prediction in asymptomatic individuals. JAMA 291, 210–215 (2004).
Budoff, M. J. et al. Assessment of coronary artery disease by cardiac computed tomography: a scientific statement from the American Heart Association Committee on Cardiovascular Imaging and Intervention, Council on Cardiovascular Radiology and Intervention, and Committee on Cardiac Imaging, Council on Clinical Cardiology. Circulation 114, 1761–1791 (2006).
O’Donnell, C. J. et al. Genome-wide association study for coronary artery calcification with follow-up in myocardial infarction. Circulation 124, 2855–2864 (2011).
Natarajan, P. et al. Multiethnic exome-wide association study of subclinical atherosclerosis. Circ. Cardiovasc. Genet. 9, 511–520 (2016).
Thanassoulis, G. et al. Genetic associations with valvular calcification and aortic stenosis. N. Engl. J. Med. 368, 503–512 (2013).
Post, W. et al. Determinants of coronary artery and aortic calcification in the Old Order Amish. Circulation 115, 717–724 (2007).
Criqui, M. H. et al. Abdominal aortic calcium, coronary artery calcium, and cardiovascular morbidity and mortality in the Multi-Ethnic Study of Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 34, 1574–1579 (2014).
Budoff, M. J. et al. Thoracic aortic calcification and coronary heart disease events: the multi-ethnic study of atherosclerosis (MESA). Atherosclerosis 215, 196–202 (2011).
Bastos Gonçalves, F. et al. Calcification of the abdominal aorta as an independent predictor of cardiovascular events: a meta-analysis. Heart 98, 988–994 (2012).
Wilson, P. W. et al. Abdominal aortic calcific deposits are an important predictor of vascular morbidity and mortality. Circulation 103, 1529–1534 (2001).
Erbel, R. et al. 2014 ESC Guidelines on the diagnosis and treatment of aortic diseases: document covering acute and chronic aortic diseases of the thoracic and abdominal aorta of the adult. The Task Force for the Diagnosis and Treatment of Aortic Diseases of the European Society of Cardiology (ESC). Eur. Heart J. 35, 2873–2926 (2014).
Guerin, A. P., London, G. M., Marchais, S. J. & Metivier, F. Arterial stiffening and vascular calcifications in end-stage renal disease. Nephrol. Dial. Transplant. 15, 1014–1021 (2000).
London, G. M. Mechanisms of arterial calcifications and consequences for cardiovascular function. Kidney Int. Suppl. (2011) 3, 442–445 (2013).
Sigrist, M. K., Taal, M. W., Bungay, P. & McIntyre, C. W. Progressive vascular calcification over 2 years is associated with arterial stiffening and increased mortality in patients with stages 4 and 5 chronic kidney disease. Clin. J. Am. Soc. Nephrol. 2, 1241–1248 (2007).
Psaty, B. M. et al. Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) Consortium: design of prospective meta-analyses of genome-wide association studies from 5 cohorts. Circ. Cardiovasc. Genet. 2, 73–80 (2009).
Yang, J. et al. Genomic inflation factors under polygenic inheritance. Eur. J. Hum. Genet. 19, 807–812 (2011).
Nikpay, M. et al. A comprehensive 1,000 Genomes-based genome-wide association meta-analysis of coronary artery disease. Nat. Genet. 47, 1121–1130 (2015).
Markus, H. S. et al. Evidence HDAC9 genetic variant associated with ischemic stroke increases risk via promoting carotid atherosclerosis. Stroke 44, 1220–1225 (2013).
Franceschini, N. et al. GWAS and colocalization analyses implicate carotid intima-media thickness and carotid plaque loci in cardiovascular outcomes. Nat. Commun. 9, 5141 (2018).
Bellenguez, C. et al. Genome-wide association study identifies a variant in HDAC9 associated with large vessel ischemic stroke. Nat. Genet. 44, 328–333 (2012).
Foroud, T. et al. Genome-wide association study of intracranial aneurysm identifies a new association on chromosome 7. Stroke 45, 3194–3199 (2014).
Ramos, E. M. et al. Phenotype-Genotype Integrator (PheGenI): synthesizing genome-wide association study (GWAS) data with existing genomic resources. Eur. J. Hum. Genet. 22, 144–147 (2014).
Grunstein, M. Histone acetylation in chromatin structure and transcription. Nature 389, 349–352 (1997).
Haberland, M., Montgomery, R. L. & Olson, E. N. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat. Rev. Genet. 10, 32–42 (2009).
Chang, S. et al. Histone deacetylases 5 and 9 govern responsiveness of the heart to a subset of stress signals and play redundant roles in heart development. Mol. Cell. Biol. 24, 8467–8476 (2004).
Lobera, M. et al. Selective class IIa histone deacetylase inhibition via a nonchelating zinc-binding group. Nat. Chem. Biol. 9, 319–325 (2013).
Rusanescu, G., Weissleder, R. & Aikawa, E. Notch signaling in cardiovascular disease and calcification. Curr. Cardiol. Rev. 4, 148–156 (2008).
Leopold, J. A. Vascular calcification: mechanisms of vascular smooth muscle cell calcification. Trends Cardiovasc. Med. 25, 267–274 (2015).
Ngo, P., Ramalingam, P., Phillips, J. A. & Furuta, G. T. Collagen gel contraction assay. Methods Mol. Biol. 341, 103–109 (2006).
Luo, G. et al. Spontaneous calcification of arteries and cartilage in mice lacking matrix GLA protein. Nature 386, 78–81 (1997).
Kato, N. et al. Trans-ancestry genome-wide association study identifies 12 genetic loci influencing blood pressure and implicates a role for DNA methylation. Nat. Genet. 47, 1282–1293 (2015).
McEniery, C. M. et al. Aortic calcification is associated with aortic stiffness and isolated systolic hypertension in healthy individuals. Hypertension 53, 524–531 (2009).
Paultre, F. & Mosca, L. Association of blood pressure indices and stroke mortality in isolated systolic hypertension. Stroke 36, 1288–1290 (2005).
Schurgers, L. J., Uitto, J. & Reutelingsperger, C. P. Vitamin K-dependent carboxylation of matrix Gla-protein: a crucial switch to control ectopic mineralization. Trends Mol. Med. 19, 217–226 (2013).
Nigwekar, S. U. et al. Vitamin K-dependent carboxylation of matrix Gla protein influences the risk of calciphylaxis. J. Am. Soc. Nephrol. 28, 1717–1722 (2017).
Steitz, S. A. et al. Smooth muscle cell phenotypic transition associated with calcification: upregulation of Cbfa1 and downregulation of smooth muscle lineage markers. Circ. Res. 89, 1147–1154 (2001).
Sun, Y. et al. Smooth muscle cell-specific runx2 deficiency inhibits vascular calcification. Circ. Res. 111, 543–552 (2012).
Brozovich, F. V. et al. Mechanisms of vascular smooth muscle contraction and the basis for pharmacologic treatment of smooth muscle disorders. Pharm. Rev. 68, 476–532 (2016).
Lino Cardenas, C. L. et al. An HDAC9-MALAT1-BRG1 complex mediates smooth muscle dysfunction in thoracic aortic aneurysm. Nat. Commun. 9, 1009 (2018).
Rich, S. S. et al. Rapid evaluation of phenotypes, SNPs and results through the dbGaP CHARGE Summary Results site. Nat. Genet. 48, 702–703 (2016).
Agatston, A. S. et al. Quantification of coronary artery calcium using ultrafast computed tomography. J. Am. Coll. Cardiol. 15, 827–832 (1990).
Rumberger, J. A. & Kaufman, L. A rosetta stone for coronary calcium risk stratification: agatston, volume, and mass scores in 11,490 individuals. AJR Am. J. Roentgenol. 181, 743–748 (2003).
Budoff, M. J. et al. Effect of scanner type on the reproducibility of extracoronary measures of calcification: the multi-ethnic study of atherosclerosis. Acad. Radiol. 14, 1043–1049 (2007).
Budoff, M. J. et al. Reproducibility of CT measurements of aortic valve calcification, mitral annulus calcification, and aortic wall calcification in the multi-ethnic study of atherosclerosis. Acad. Radiol. 13, 166–172 (2006).
Chuang, M. L. et al. Distribution of abdominal aortic calcium by computed tomography: impact of analysis method on quantitative calcium score. Acad. Radiol. 20, 1422–1428 (2013).
Willer, C. J., Li, Y. & Abecasis, G. R. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26, 2190–2191 (2010).
Zhang, X. et al. Identification of common genetic variants controlling transcript isoform variation in human whole blood. Nat. Genet. 47, 345–352 (2015).
Ongen, H. & Dermitzakis, E. T. Alternative splicing QTLs in European and African populations. Am. J. Hum. Genet. 97, 567–575 (2015).
Zhang, C. L. et al. Class II histone deacetylases act as signal-responsive repressors of cardiac hypertrophy. Cell 110, 479–488 (2002).
O’Rourke, C. Calcification of vascular smooth muscle cells and imaging of aortic calcification and inflammation. J. Vis. Exp. https://doi.org/10.3791/54017 (2016).
Kang, H. et al. Bone morphogenetic protein 4 promotes vascular smooth muscle contractility by activating microRNA-21 (miR-21), which down-regulates expression of family of dedicator of cytokinesis (DOCK) proteins. J. Biol. Chem. 287, 3976–3986 (2012).
Aikawa, E. et al. Osteogenesis associates with inflammation in early-stage atherosclerosis evaluated by molecular imaging in vivo. Circulation 116, 2841–2850 (2007).
Aikawa, E. et al. Multimodality molecular imaging identifies proteolytic and osteogenic activities in early aortic valve disease. Circulation 115, 377–386 (2007).
Malhotra, R. et al. Inhibition of bone morphogenetic protein signal transduction prevents the medial vascular calcification associated with matrix Gla protein deficiency. PLoS ONE 10, e0117098 (2015).
Malhotra, R. et al. Hepcidin deficiency protects against atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 39, 178–187 (2019).
Derwall, M. et al. Inhibition of bone morphogenetic protein signaling reduces vascular calcification and atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 32, 613–622 (2012).
Rong, J. X., Shapiro, M., Trogan, E. & Fisher, E. A. Transdifferentiation of mouse aortic smooth muscle cells to a macrophage-like state after cholesterol loading. Proc. Natl Acad. Sci. USA 100, 13531–13536 (2003).
Chomczynski, P. & Sacchi, N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem. 162, 156–159 (1987).
Acknowledgements
We acknowledge the essential role of the CHARGE Consortium in the development and support of this manuscript. The views expressed in this manuscript are those of the authors and do not necessarily represent the views of the National Heart, Lung, and Blood Institute (NHLBI), the NIH or the U. S. Department of Health and Human Services. The Framingham Heart Study was supported by the NHLBI (contract no. N01-HC-25195) and its contract with Affymetrix for genotyping services (contract No. N02-HL-6-4278). R.M. was supported by the NHLBI (grant nos. K08HL111210 and R01HL142809), the American Heart Association (grant no. 18TPA34230025), the Wild Family Foundation and the Hassenfeld Scholar Award. F.W. was supported by Deutsche Forschungsgemeinschaft (DFG; no. Wu 841/1-1). A.B. was supported by the Department of Defense Peer Reviewed Medical Research Program/Discovery Award (no. W81XWH-17-1-0058). S.U.N. was supported by the American Heart Association (grant no. 15FTF25980003). C.Song was supported by an international postdoctoral fellowship from the Swedish research council (no. 2016-00598). U.H. received research support unrelated to the current project: research grants on behalf of the institution from Kowa, MedImmune, HeartFlow, Duke University (Abbott), Oregon Health & Science University (American Hospital Association; no. 13FTF16450001), Columbia University (NIH; no. 5R01-HL109711) and NIH/NHLBI grant nos. 5K24HL113128, 5T32HL076136 and 5U01HL123339. M.E.L. was supported by the Fredman Fellowship, the Toomey Fund for Aortic Dissection Research and grant no. R01 HL130113. D.B.B. was supported by the Leducq Foundation and the National Institutes of Diabetes and Digestive and Kidney Diseases (no. 5R01DK082971). The Age, Gene/Environment Susceptibility-Reykjavik Study has been funded by National Institute on Aging (NIA) contract no. N01-AG-12100 with contributions from the National Eye Institute, National Institute on Deafness and Other Communication Disorders, NHLBI, NIA Intramural Research Program, Hjartavernd (the Icelandic Heart Association) and Althingi (Icelandic Parliament). MESA and the MESA SHARe project are conducted and supported by the NHLBI in collaboration with MESA investigators. MESA was supported by grant nos. R01 HL071739 and R01 HL72403, and by contract nos. HHSN268201500003I, N01-HC-95159, N01-HC-95160, N01-HC-95161, N01-HC-95162, N01-HC-95163, N01-HC-95164, N01-HC-95165, N01-HC-95166, N01-HC-95167, N01-HC-95168, N01-HC-95169, UL1-TR-000040, UL1-TR-001079 and UL1-TR-001420. Funding for the SHARe genotyping was provided by NHLBI contract no. N02-HL-64278. Genotyping was performed at Affymetrix and the Broad Institute of Harvard and MIT using the Affymetrix Genome-Wide Human SNP Array 6.0. This work was also supported in part by the National Center for Advancing Translational Sciences, Clinical Translational Science Institute grant no. UL1TR001881 and the National Institute of Diabetes and Digestive and Kidney Disease (NIDDK) Diabetes Research Center (DRC) grant no. DK063491 to the Southern California Diabetes Endocrinology Research Center. M.B. was supported by the NIH grant no. R01HL071739. We thank the other investigators, staff and participants of the MESA study for their valuable contributions. A full list of participating MESA investigators and institutions can be found at http://www.mesa-nhlbi.org. The FamHS research was supported by NIH grant no. R01-HL-117078 from the NHLBI and grant no. R01-DK-089256 from the NIDDK. We are indebted to all the study participants and to the dedicated personnel of both the study center of the Heinz Nixdorf Recall study and the electron beam tomography scanner facilities (D. Grönemeyer and R. Seibel) as well as to the investigative group, in particular to U. Roggenbuck, U. Slomiany, E.M. Beck, A. Öffner, S. Münkel, M. Bauer, S. Schrader, R. Peter and H. Hirche. Scientific advisory board: We thank T. Meinertz (Chair), M. Blettner, C. Bode, P. J. de Feyter, B. Güntert, F. Gutzwiller, H. Heinen, O. Hess (deceased), B. Klein (deceased), H. Löwel, M. Reiser, G. Schmidt (deceased), M. Schwaiger, C. Steinmüller, T. Theorell and S.N. Willich. Criteria and end point committee: C. Bode (Chair), K. Berger, H.R. Figulla, C. Hamm, P. Hanrath, W. Köpcke, C. Weimar, A. Zeiher. Funding: We thank the Heinz Nixdorf Foundation (Chairman: M. Nixdorf; Past Chairman: G. Schmidt (deceased)), the German Ministry of Education and Science and the DFG (project nos. ER 155/6-1 and ER 155/6-2) for the generous support of this study. An additional research grant was received from Imatron, which provided the electron beam computerized tomography scanners, and GE-Imatron after the acquisition of Imatron. We acknowledge the support of the Sarstedt AG & Co. concerning laboratory equipment. We received generous support from the Ministry of Culture and Science of North Rhine-Westphalia and the Faculty of Medicine, University Duisburg-Essen for the genotyping of the Heinz Nixdorf Recall study participants. S.P. was supported by the DFG (project no. PE 2309/2-1). M.M.N. received support from the DFG (German Research Foundation) through the Transregional Collaborative Research Centre grant TRR259 (subproject B07 ”GUARD–Genes Underlying AoRtic valve Disease“; project number 426128588) and is a member of the Excellence Cluster ImmunoSensation2, which is funded by the DFG under Germany’s Excellence Strategy–EXC2151 (project number 390873048). Technical support for the imputation of the Heinz Nixdorf Recall Study data on the Supercomputer Cray XT6m was provided by the Center for Information and Media Services, University of Duisburg-Essen. The AA-DHS was supported by grant nos. R01-DK-071891 from the NIDDK, RO1-HL-67348 from the NHLBI and General Clinical Research Center of Wake Forest School of Medicine M01-RR-07122. N.F. is supported by NIH grant nos. MD012765, DK117445 and HL140385.
Author information
Authors and Affiliations
Contributions
R.M., A.C.M., X.G., J.Y., C.Y.C., W.S.P. and C.J.O. conceived and designed the genome-wide association study. A.V.S., Q.W., S.P., S.-J.H., J.W., L.L., C.Y.C., T.H., M.B., M.H.C., J.I.R., A.D.J., C.Song, N.F., S.D., U.H., H.K., M.M.N., S.S., B.I.F., D.W.B., K.-H.J., S.M., R.E., M.F.F., V.G., G.T., W.S.P. and C.J.O. acquired the cohort-based phenotype and genotype data. R.M., A.C.M., X.G., J.Y., X.Z., W.S.P. and C.J.O. carried out the meta-analysis and sQTL analysis. R.M. conceived and designed the cell-based assays and in vivo studies. R.M., C.L.L.C., F.W., C.J.N., G.S., M.D.B., H.J.B., C.Slocum, H.H.S., C.O., D.K.R., A.B., S.U.N., E.S.B., W.M.Z., M.E.L. and D.B.B. acquired, analyzed or interpreted the data in the in cell-based assays and in vivo studies. R.M., A.C.M. and C.J.O. drafted the manuscript. All authors critically revised the manuscript for important intellectual content and approved the content of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
R.M. serves as a consultant for MyoKardia and Third Pole. A.B. is a consultant for Lungpacer Medical. U.H. is a consultant for Abbott, Duke University (NIH) and ReCor Medical.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
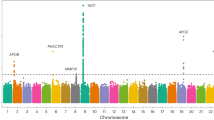
Extended Data Fig. 1 Genome-wide association study does not identify polymorphisms associated with thoracic aortic calcification.
Manhattan (left) and Quantile-Quantile (right) plots for the association of thoracic aortic calcification with ~9 million SNPs in the GWAS meta-analysis of 8,422 participants. The hashed line indicates the genome-wide threshold for significance (P < 5 x 10-8).
Extended Data Fig. 2 MGP and HDAC9 deficiency do not result in aortic accumulation of HDAC4 or HDAC7.
Immunofluorescence staining of aortic sections from wild-type, Mgp-/- Hdac9+/+, Mgp-/- Hdac9+/-, and Mgp-/- Hdac9-/- mice. a, Genetic deletion of Mgp and Hdac9 had no influence on aortic HDAC4 expression (red), whereas the vascular smooth muscle contractile marker MYH11 (gold) was more abundant in sections from Mgp-/- Hdac9-/- mice when compared to Mgp-/- Hdac9+/+ mice. b, Immunofluorescence shows no accumulation of HDAC7 (red) in aortas from Mgp-/- Hdac9+/+, Mgp-/- Hdac9+/-, and Mgp-/- Hdac9-/- mice compared to wild-type mice. In contrast, as also shown in Fig. 4f, MMP9 expression was increased in Mgp-/- Hdac9+/+ mice compared to wild-type mice and HDAC9-deficient Mgp-/- mice. Three independent replicates in each group were assessed with representative images shown. Scale bars: 50 μm.
Extended Data Fig. 3 Primary aortic smooth muscle cells from Hdac9-/- mice are protected from calcification.
a, Primary aortic vascular SMCs isolated from Hdac9-/- mice displayed markedly lower mRNA expression of the osteogenic markers Runx2 (left) and tissue non-specific alkaline phosphatase (Tnap, middle) compared to aortic vascular SMCs isolated from wild-type mice. There was also a trend towards decreased expression of Col3a1 (right) in aortic vascular SMCs isolated from Hdac9-/- mice (n = 6 biologically independent samples in each group). b, Hdac9-/- primary aortic vascular SMCs failed to calcify when treated with calcifying media for 10 days compared with vascular SMCs isolated from wild-type mice as determined by Alizarin Red staining. Two independent experiments were performed with representative images shown. Scale bar is 1 cm. c, Hdac9-/- primary aortic vascular SMCs exhibited increased contractility compared with vascular SMCs isolated from wild-type mice (n = 6 biologically independent samples in each group). In a and c, statistical comparisons were made using a two-tailed Student t test. Mean ± s.e.m. is depicted in all plots.
Supplementary information
Supplementary Information
Supplementary Tables 1–6 and Supplementary Note
Supplementary Table 7
SNPs associated with stroke, MI, or CAD were tested for association with AAC and TAC
Rights and permissions
About this article

Cite this article
Malhotra, R., Mauer, A.C., Lino Cardenas, C.L. et al. HDAC9 is implicated in atherosclerotic aortic calcification and affects vascular smooth muscle cell phenotype. Nat Genet 51, 1580–1587 (2019). https://doi.org/10.1038/s41588-019-0514-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41588-019-0514-8
This article is cited by
-
Protein interaction networks in the vasculature prioritize genes and pathways underlying coronary artery disease
Communications Biology (2024)
-
Dysregulated cellular metabolism in atherosclerosis: mediators and therapeutic opportunities
Nature Metabolism (2024)
-
DNA methylation and histone post-translational modifications in atherosclerosis and a novel perspective for epigenetic therapy
Cell Communication and Signaling (2023)
-
Genetics and mechanisms of thoracic aortic disease
Nature Reviews Cardiology (2023)
-
Targeting protein modifications in metabolic diseases: molecular mechanisms and targeted therapies
Signal Transduction and Targeted Therapy (2023)
