Abstract
Cryo-electron microscopy is a popular method for the determination of protein structures; however, identifying a sufficient number of particles for analysis can take months of manual effort. Current computational approaches find many false positives and require ad hoc postprocessing, especially for unusually shaped particles. To address these shortcomings, we develop Topaz, an efficient and accurate particle-picking pipeline using neural networks trained with a general-purpose positive-unlabeled learning method. This framework enables particle detection models to be trained with few sparsely labeled particles and no labeled negatives. Topaz retrieves many more real particles than conventional picking methods while maintaining low false-positive rates, is capable of picking challenging unusually shaped proteins (for example, small, non-globular and asymmetric particles), produces more representative particle sets and does not require post hoc curation. We demonstrate the performance of Topaz on two difficult datasets and three conventional datasets. Topaz is modular, standalone, free and open source (http://topaz.csail.mit.edu).
Similar content being viewed by others


Main
Single-particle cryo-electron microscopy (cryo-EM) is a method capable of resolving high-resolution structures of proteins in near-native states. Cryo-EM projection images (micrographs) can contain hundreds or thousands of individual protein projections (particles). Given a sufficient number of particles, the three-dimensional (3D) structure of the protein can be determined1. However, owing to the low signal-to-noise ratio (SNR) of cryo-EM images, large numbers of observations are required for accurate reconstruction. Studies show a log–linear relationship between the number of particles included and the inverse resolution of the reconstruction2,3. The concentration of protein on EM grids, efficiency of data collection and completeness and accuracy of particle identification are factors determining the total number of particles available for downstream reconstruction and, hence, the achievable resolution. In particular, particle identification (particle picking) is a major bottleneck, often taking weeks or even months with current workflows for small or non-globular particles owing to variability in particle shapes and structured noise in micrographs.
A variety of methods have been developed for particle picking automation. The most widely used are difference of Gaussians (DoG) and template-based approaches4,5,6,7,8. However, these methods are unable to detect unusually shaped particles and suffer from high false-positive rates necessitating post-picking curation. Most commonly, researchers use iterative 2D–3D classification and discard poor subsets by eye. These picking methods and downstream curation introduce bias into the final particle set, potentially removing rare particle views and conformations9,10,11. Newer methods based on convolutional neural networks (CNNs) have been proposed12,13,14, which use positive- and negative-labeled micrograph regions to train CNN classifiers, which then predict labels for the remaining regions. However, owing to factors like low SNR, structured background and the distribution of particle morphologies, researchers must label a large number of regions for training—a non-trivial and time-consuming task. Moreover, the diverse characteristics of negative data make it difficult to manually label a representative set of negative examples, and, hence, the number of labeled negatives must be an order of magnitude larger than the number of positives to achieve acceptable performance15. This has limited adoption by the cryo-EM community and hand-labeling remains the gold standard.
To overcome the challenges inherent to current automatic particle-picking methods, we frame particle picking as a positive-unlabeled (PU) learning problem. In PU learning, we seek to learn a classifier of positives and negatives given a small number of labeled positive regions and the remaining unlabeled regions. This has proved to be an effective paradigm when working with partially labeled data in other domains (for example, document classification16, time series classification17 and anomaly detection18). Recent work has explored general-purpose PU learning for neural network models based on estimating the true positive–negative risk, but overfitting remains a challenge for PU learning19. Therefore, we instead approach PU learning as a constrained optimization problem in which we wish to find classifier parameters to minimize classification errors on the labeled data subject to a constraint on the expectation over the unlabeled data. By imposing this constraint softly with a novel generalized expectation (GE) criteria20, we are able to mitigate overfitting and train high-accuracy particle classifiers using very few labeled data points. Furthermore, by combining our PU learning method with autoencoder-based regularization, we can further reduce the amount of labeled data required for high performance.
Here we present Topaz, a pipeline for particle picking using CNNs with PU learning. Topaz retrieves many more particles than alternative methods, while maintaining a low false-positive rate. It substantially reduces the need for particle curation, removes systematic bias in particle picking introduced by conventional pickers and 2D–3D classification procedures and allows for robust and representative particle analysis and classification. Furthermore, Topaz is capable of reliably picking previously challenging particles (for example, small, non-globular and asymmetric particles) and avoiding aggregation, grid substrate and other background objects, while requiring minimal example particles.
We first demonstrate the capabilities of Topaz on a novel protein dataset for the Toll receptor—a ~105-kDa non-globular asymmetric particle. Despite aggregation and sparse labeling in the dataset, Topaz enables a 3.7-Å reconstruction and resolves secondary structures that could not be resolved with other methods. Topaz also decreases anisotropy by better detecting conventionally difficult particle views. Additionally, on three publicly available datasets, we find that by using Topaz with only 1,000 labeled training examples, we are able to retrieve many more real particles than were included in the published particle sets. This enables us to solve 3D structures of equal or greater quality to those found using the published particles. Remarkably, the Topaz results do not require any of the ad hoc postprocessing that is typically required for high-resolution structures; we feed Topaz particles directly into alignment and reconstruction. Finally, we compare our GE-based PU learning method against other off-the-shelf PU learning approaches and find that our method offers improvements over the current state-of-the-art methods when applied to training particle-detection models. Topaz was a critical component in determining the single particle behavior of an elongated clustered protocadherin21.
Topaz source code is freely available (https://github.com/tbepler/topaz) and can be installed through Anaconda, pip, Docker, Singularity and SBGrid22. Topaz is designed to be modular, has been integrated into Appion23, is being integrated into Relion24, CryoSparc25, EMAN25, Scipion26 and Focus27, and can easily be integrated into other cryo-EM software suites in the future. Topaz runs efficiently on a single GPU computer and includes a standalone graphical user interface (GUI)28 to assist with particle labeling.
Results
Topaz pipeline
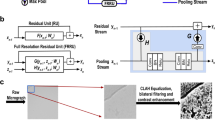
The Topaz particle-picking pipeline is composed of the following main steps (Fig. 1): (1) whole micrograph preprocessing with an optional mixture model newly designed to capture micrograph statistics (Methods; Supplementary Figs. 1, 2 and 3), (2) neural network classifier training with our PU learning framework, and (3) sliding window classification of micrographs and extraction of particle coordinates by non-maximum suppression.

a, Given a set of labeled particles, a CNN is trained to classify positive and negative regions using particle locations as positive regions and all other regions as unlabeled. Labeled particles from EMPIAR-10096 are indicated by blue circles and a few positive and unlabeled regions are depicted. b, Once the CNN classifier is trained, particles are predicted in two steps. First, the classifier is applied to each micrograph region to give per region predictions. Second, coordinates are extracted from the region predictions using non-maximum suppression. The left image shows a raw micrograph from EMPIAR-10096. The middle image depicts the micrograph with overlaid region predictions (blue indicates low confidence and red indicates high confidence). The right image indicates predicted particles after using non-maximum suppression on the region predictions.
Classifier training from positive and unlabeled data
We frame particle picking as a PU learning problem in which we seek to learn a classifier that discriminates between particle and non-particle micrograph regions given a small number of labeled particles and many unlabeled micrograph regions. CNN classifiers are trained using minibatched stochastic gradient descent with a novel objective function, GE-binomial (Methods), which explicitly models the sampling statistics of minibatches to regularize the posterior of the classifier over the unlabeled data. Combining this with an optional autoencoder module allows high-accuracy classifiers to be trained using very few positive examples. This approach allows us to overcome overfitting problems associated with recent PU learning methods developed for neural networks in domains other than cryo-EM analysis, and to effectively pick particles in challenging cryo-EM datasets.
Micrograph region classification and particle extraction
Given a trained CNN particle classifier, we extract predicted particle coordinates and their associated predicted probabilities. First, we calculate the per pixel predicted probabilities by applying the classifier to each micrograph region in a sliding window. Then, to extract coordinates from these dense predictions, we use the well-known non-maximum suppression algorithm which iteratively selects high-scoring pixels and removes their neighbors from consideration as particle centers. This yields a list of predicted particle coordinates and their associated model scores for each micrograph.
Topaz picks challenging particles and orientations
We explore the ability of Topaz to detect challenging particles of a small, asymmetric, non-globular and aggregated protein, a Toll receptor. To this end, we compared particles picked by Topaz (trained with 686 labeled particles) with particles picked using several other methods (DoG7 and template picking followed by 2D class averaging and manual filtering, and the CNN-based methods crYOLO29 and DeepPicker12) (Methods). The CNN-based methods were all trained following the software instructions with default settings and identical labeled particles.
After four rounds of 2D classification and filtering, DoG found 770,263 good particles from an initial stack of 1,599,638 and template picking found 627,533 good particles from an initial stack of 1,265,564. Using Topaz, after one round of 2D classification, we were left with 1,006,089 of an initial 1,010,937 particles, indicating that Topaz gives a remarkably low false-positive rate of only 0.5% on this data. We then compared the quality of the picked particles by taking each particle set through reconstruction (Fig. 2a–c). We found that particles picked using Topaz yield a structure with 0.731 sphericity at Fourier shell correlation (FSC)0.143 = 3.70 Å resolution, as compared to 0.706 sphericity at 3.92 Å for template-picked particles and 0.652 sphericity at 3.86 Å for particles picked using DoG. Furthermore, only the density map based on Topaz particles was of high enough quality to reliably resolve secondary structure (β-strands) and allow for model building. Other CNN-based picking methods, crYOLO and DeepPicker, were unable to find sufficient numbers of good particles for high-resolution reconstruction. crYOLO found 131,300 particles resulting in a 6.8 Å structure while DeepPicker failed to find any meaningful particles in this dataset (Supplementary Figs. 4–7).

Template and DoG particles were filtered through multiple rounds of 2D classification before analysis. Topaz particles were not filtered. a, Density map using particles picked with Topaz. The global resolution is 3.70 Å at FSC0.143 with a sphericity of 0.731. Scale bar, 5 nm. b, Density map using particles picked using template picking. The global resolution is 3.92 Å at FSC0.143 with a sphericity of 0.706. c, Density map using particles picked using DoG. The global resolution is 3.86 Å at FSC0.143 with a sphericity of 0.652. d, Quantification of picked particles for each protein view on the basis of 2D classification. e, Example micrograph (representative of >100 micrographs examined) showing Topaz picks (red circles) and protein aggregation (outlined in green).
We next quantified the ability of these methods to detect different particle views. The Toll receptor is strongly asymmetric and non-globular, thus it is important for picking methods to retrieve the full spectrum of view angles. By counting the number of particles assigned to each view in 2D class averages, we found that Topaz retrieved a much larger fraction of oblique, side and top views of the Toll receptor as compared to DoG and template-based methods (Fig. 2d). In addition, we note that these micrographs are challenging, containing junk and protein aggregation, yet Topaz is uniquely able to avoid these micrograph regions while picking only good particles (Fig. 2e and Supplementary Fig. 4).
Topaz enables high-resolution reconstruction with no postprocessing
We next evaluated the full Topaz particle-picking pipeline by generating reconstructions for three cryo-EM datasets containing T20S proteasome (EMPIAR-10025), 80S ribosome (EMPIAR-10028) and rabbit muscle aldolase (EMPIAR-10215). Each of these datasets already had a curated set of particles yielding high-quality reconstructions, which we compared with particles predicted by Topaz (trained with 1,000 positives on the basis of reconstruction quality; Methods). We standardized the reconstruction procedure by using cryoSPARC homogeneous refinement on the raw Topaz particle sets (that is, no postprocessing was applied) and published particle sets with identical settings for each dataset. By considering the reconstruction resolution at decreasing probability thresholds (increasing numbers of particles) predicted by Topaz, we selected the particle set that optimized the resolution for each dataset.
We found that Topaz was able to retrieve substantially more good particles than were present in the curated particle sets, finding 3.22, 1.72 and 3.68 times more particles in EMPIAR-10025, EMPIAR-10028 and EMPIAR-10215, respectively. Furthermore, reconstructions from the Topaz particle sets were of equal or higher quality as compared to those given by the curated particles (Fig. 3). Topaz maps reached roughly equivalent resolution to the published structures for the 80S ribosome and rabbit muscle aldolase while improving the resolution by ~0.15 Å over the published structures for the T20S proteasome. Remarkably, this was achieved using only 1,000 labeled examples and no filtering of the particle set (for example, particle filtering with 2D or 3D class averaging or iterative reconstructions removing poor particles). We note that even though these labeled training particles are extremely sparse, PU learning enabled Topaz to pick with high precision as seen in example micrographs (Supplementary Figs. 8, 9 and 10). We verified that the additional particles found by Topaz were good particles by performing reconstructions using only the newly picked particles and find nearly identical structures (Fig. 3). For aldolase, although Topaz found many more particles than were in the published dataset, the Topaz, curated and the Topaz minus curated particle sets achieved the same reconstruction resolution (2.63 Å at FSC0.143), suggesting that the ~200,000 particles in the published set is already sufficient to reach the resolution limit of the data given standard reconstruction methods.

Published particles are on the left, Topaz particles in the middle, and Topaz particles with published particles removed are on the right. Below each reconstruction is the corresponding 3D FSC plot. a, T20S proteasome (EMPIAR-10025) using the provided aligned and dose-weighted micrographs. b, 80S ribosome (EMPIAR-10028). c, Rabbit muscle aldolase (EMPIAR-10215). Scale bars, 3 nm.
Topaz particle predictions are well-ranked and contain few false positives
We next quantified the quality of the particles predicted by Topaz over varying predicted probability thresholds by calculating the reconstruction resolution and estimating the number of false-positive particles on the basis of 2D class averaging. For each dataset, reconstructions were calculated using particles predicted by Topaz at decreasing probability cutoffs (Fig. 4a). The resolution of Topaz structures increased as we included more good particles and then dropped once the threshold became small and too many false positives were included, as demonstrated by the dip in resolution for the last threshold of EMPIAR-10025. Furthermore, we compared these curves with those obtained by randomly subsampling the published particle sets and found that Topaz particles quickly matched the resolution of the published particles for the proteasome and ribosome datasets. For the aldolase dataset, we saw that more Topaz particles were required to match and then exceed the resolution of the curated particle set. This could be because Topaz did not find enough side views of the particle until the probability was sufficiently lowered whereas the curated dataset had been filtered to be enriched for these views (Supplementary Fig. 11).

a, Number of particles versus reconstruction resolution for Topaz particles (increasing number of particles corresponds to decreasing log-likelihood threshold) and randomly sampled subsets of the published particle set. Resolution is as reported by cryoSPARC. For the published particle sets the mean of three replicates is marked with s.d. shaded in gray. b, Stacked bar plots show the quantification of the number of true and false positives at each threshold on the basis of 2D class averages. The decreasing threshold corresponds to an increasing number of predicted particles. True and false positives are shown in blue and orange, respectively. c, 2D class averages obtained at each score threshold for the T20S proteasome (EMPIAR-10025). Number of particles (ptcls) and effective sample size (ess) for each class are reported by cryoSPARC. NaN is reported for classes without any particles assigned. Classes determined to be false positives are marked with orange boxes. Several classes which appear to be false positives at high score thresholds do not contain any particles and, therefore, are not highlighted.
We also classified the particle sets at each threshold into ten classes and manually examined the class averages to determine whether each class represented true particles or false positives. As expected, we found that as the probability threshold was decreased, the fraction of false positives increased (Fig. 4b), yet remained remarkably low even at relaxed thresholds. Furthermore, particles appear to be well-ranked, in that noisy or unusual particle classes only start to appear at low thresholds. For example, the T20S proteasome dataset was contaminated with gold particles, which appeared as dark spots in the micrographs. Particles in close proximity to gold were only selected when the probability threshold was decreased (Fig. 4). Similar trends can be observed in the ribosome (Supplementary Fig. 12) and aldolase (Supplementary Fig. 11) class averages. This can also be seen in the precision-recall curves for these datasets (Supplementary Figs. 13), in which Topaz maintains remarkably high precision even at high recall levels.
GE-criteria-based PU learning method outperforms other general-purpose PU learning approaches
Comparison of PU learning methods
We considered two GE-based approaches to PU learning, GE-KL and GE-binomial (Methods), and evaluated their effectiveness by benchmarking against the recent non-negative risk estimator approach of Kiryu et al.19 (NNPU) and the naive approach in which unlabeled data are considered as negative for classifier training (the naive approach is hereafter referred to as PN) on two additional cryo-EM datasets. This is important to keep the development of our PU learning methods separate from the full Topaz evaluation above. The first dataset, EMPIAR-10096, is a publicly available dataset containing influenza hemagglutinin trimer particles and the second, EMPIAR-10234 (clustered protocadherin), is a challenging dataset provided by the Shapiro lab containing a stick-like particle with low SNR (Supplementary Fig. 14). For purposes of comparison, we simulated positively labeled datasets of varying sizes by randomly subsampling the set of all positive examples within the training set of each dataset.
We found that across all experiments, classifiers trained with our GE-criteria-based objective functions dramatically outperformed those trained with the NNPU or PN methods. Generally, GE-binomial and GE-KL classifiers displayed similar performance with a few important exceptions where GE-binomial gave better results. For the dataset with more compact particles, EMPIAR-10096, GE-binomial gave significantly (P < 0.05, Student’s paired t test) better test set average-precision scores than GE-KL when the number of data points was tiny (ten positive examples; Fig. 5a). At larger numbers of positives, both methods were statistically equivalent. On the challenging EMPIAR-10234 dataset, GE-binomial significantly outperformed GE-KL at 1,000 labeled examples (P < 0.05) whereas GE-KL gave better results (P < 0.05) within the 50–250 range of labeled examples. These results indicate that our GE-based PU learning approaches dramatically outperform previous PU learning methods, enabling particle picking despite few labeled positives on the challenging EMPIAR-10234 dataset and substantially improving picking quality on the easier EMPIAR-10096 dataset. Although GE-binomial and GE-KL performed similarly in this experiment, we did find that GE-binomial outperformed GE-KL in the two important cases of ten easy particles and 1,000 difficult particles.

a, Mean ± s.d. of the average-precision score for predicting positive regions in the EMPIAR-10096 and EMPIAR-10234 test set micrographs for models trained using the naive PN, NNPU, GE-KL or GE-binomial objective functions. Each number of labeled positives was sampled ten times independently. Asterisks indicate experiments in which GE-binomial achieved higher average-precision than GE-KL with P < 0.05. Daggers indicate experiments in which GE-KL achieved higher average-precision than GE-binomial with P < 0.05 according to a two-sided dependent t test. b, Mean ± s.d. of the average-precision score for models trained jointly with autoencoders with different reconstruction loss weights (γ). γ = 0 corresponds to training the classifier without the autoencoder. γ = 10/N means the reconstruction loss is weighted by ten divided by the number of labeled positives used to train the model.
Augmentation with autoencoders
We next considered whether classifier performance could be improved when few labeled data points are available by introducing a generator network with a corresponding reconstruction error term in the objective to form a hybrid classifier with autoencoder network (Methods). We hypothesized that including this reconstruction component would improve the generalizability of the classifier when few labeled data points are available by requiring that the feature vectors given by the encoder network be descriptive of the input—acting as a sort of machine learning technique known as regularization.
We evaluated this hypothesis by training classifiers on the EMPIAR-10096 and EMPIAR-10234 datasets with different settings of the autoencoder weight, γ, and varying numbers of labeled data points, N (Methods). We found that including the decoder network with a reconstruction error term in the objective (γ = 1 and \(\gamma = \frac{{10}}{N}\)) improved classifier performance in the regime with few labeled data points (Fig. 5b). As the number of data points increased, the benefit of using the autoencoder decreased and then hurt classifier performance owing to over-regularization. Our results from both datasets suggest that using the autoencoder with \({\mathrm{\gamma = }}\frac{{{\mathrm{10}}}}{N}\) gives best results when N ≤ 250
Discussion
As our work originally appeared in RECOMB 201830 and as an arXiv preprint, other works have followed on bioRxiv that propose alternative CNN-based particle-picking methods29,31. However, these methods follow the supervised learning paradigm (that is, some variant of PN learning) and are limited by the associated assumptions. In the future, it may also be possible to provide particle-detection models pretrained on many publicly available datasets; however, we note that fully labeled ground-truth datasets are presently unavailable and that these models are unlikely to generalize to new datasets with conventionally difficult particles, which we focus on here. While it may seem difficult to provide labeled data upfront, in practice we find that explicitly relaxing the requirement to completely label micrographs eases this burden, and is a major advantage of Topaz over other CNN-based methods. Users may also ‘bootstrap’ the labeling procedure using existing picking and curation methods, while remaining cautious against reintroducing bias. We note that there may be some difference between randomly sampling from a curated particle set and particles that would be labeled by a user. However, the Toll receptor and clustered protocadherin training sets were both provided by hand labeling and demonstrate that labeling a small representative set of particles is easily achievable even for conventionally difficult datasets.
Although we use a simple CNN architecture with reasonable default hyperparameters, and show that it performs well on these datasets, any model architecture that can be trained with gradient descent can use our GE criteria objective functions to learn from positive and unlabeled data. Furthermore, additional hyperparameter tuning, such as L2 or dropout regularization, can improve model performance. The only hyperparameters introduced by our objective function is the unknown positive class prior, π, and the constraint strength, λ. Although the positive class prior could also be chosen by cross validation, we observed that our results were relatively insensitive to its choice (Supplementary Fig. 15). Furthermore, we do not find that λ needs to be changed from the default setting. Our proposed GE-binomial PU learning method could also have widespread utility for object detection in other domains where positive labels are frequently incomplete, for example, in light microscopy or medical imaging. Additionally, although we proposed GE-binomial for positive-unlabeled learning, it is straightforward to extend to the typical semisupervised case (where some labeled negative regions are provided) by taking the expectation of the loss over all labeled data in the first term.
Topaz particle probability thresholding allows particles to be included iteratively until the reconstruction resolution stops improving. It is possible for reconstruction algorithms to explicitly take these probabilities into account when determining 3D structures in the future.
Topaz requires researchers to label very few particles to achieve high quality predictions. It performs well independently of particle shape, opening automated picking to a wide selection of proteins previously too difficult to locate computationally. In addition, our pipeline is computationally efficient—training in a few hours on a single GPU and producing predictions for hundreds of micrographs in only minutes. Furthermore, once a model is trained for a specific particle, it can be applied to new imaging runs of the same particle. Topaz greatly expedites structure determination by cryo-EM, enabling particle picking for previously difficult datasets, reducing the manual effort required to achieve high-resolution structures, and thus increasing the efficiency of cryo-EM workflows and the completeness of particle analytics.
Methods
Dataset description
Aligned and summed micrographs and star files containing published particle sets were retrieved from the Electron Microscopy Public Image Archive (EMPIAR) for datasets EMPIAR-10025 (ref. 32), EMPIAR-10028 (ref. 33) and EMPIAR-10096 (ref. 34). Aligned and summed micrographs and hand-labeled particle coordinates were provided by the Shapiro lab for the EMPIAR-10234 dataset. Aligned and summed micrographs and a curated in-house particle set were provided by the New York Structural Biology Center for the EMPIAR-10215 dataset. Micrographs for each dataset were downsampled to the resolution specified in Table 1 and normalized as described in the following section. Each dataset was then split into training and test sets at the micrograph level. The number of micrographs and labeled particles in each split are also reported in Table 1. To demonstrate the utility of our Gaussian mixture model (GMM) normalization method, we also retrieved micrographs for EMPIAR-10261 (ref. 35) from EMPIAR.
Micrograph normalization
Images were normalized using a per-image scaled two component GMM. Given K images, each pixel is modeled as being drawn from a two component GMM, parameterized by ρ, the mixing parameter and μ0, σ0, μ1 and σ1, the means and standard deviations of the Gaussian distributions, with a scalar multiplier for each image, α1…K. Let xi,j,k be the value of the pixel at position i,j in image k, it is distributed according to
where \(z_{i,j,k}\) is a random variable denoting the component membership of the pixel. The maximum likelihood values of the parameters ρ, μ0, σ0, μ1, σ1 and α1…K are found by expectation-maximization for each dataset. Then, the pixels are normalized by first dividing by the image scaling factor and then standardizing to the dominant mixture component. Let μ′,σ′ be μ0,σ0 if ρ < 0.5 and μ1,σ1 otherwise, then the normalized pixel values \(x^{\prime}_{i,j,k}\) are given by
We positively contrasted this normalization with standard affine normalization of micrographs (Supplementary Figs. 1, 2, and 3). In affine normalization, micrographs are transformed by subtracting the mean and dividing by the s.d. of all pixel values in each micrograph.
PU learning baselines
Let P be the set of labeled positive micrograph regions (centered on a particle), and U be the set of unlabeled micrograph regions where π is the fraction of positive examples within U. Then, the task is to learn a classifier (g) that discriminates between positive and negative regions given P and U. When π is small, treating the unlabeled examples as negatives for the purposes of classifier training with the following standard loss minimization objective, given cost function L, can be effective (referred to as PN)
However, in general, this approach suffers from overfitting owing to poor specification of the classification objective—it is minimized when positives are perfectly separated from unlabeled data points. To address this, Kiryo et al.19 recently proposed an unbiased estimator of the true positive–negative classification objective for positive and unlabeled data with known π and a non-negative estimator (PU), which is shown to reduce overfitting still present in the unbiased estimator.
PU learning with generalized expectation criteria
Here we adopt an alternative approach to positive-unlabeled learning that is not based on estimating the PN misclassification risk. Instead, we observe that unlabeled data with known π can be used to constrain a classifier such that it minimizes the classification loss on the labeled data and matches the expectation (π) over the unlabeled data. In other words, we wish to find the classifier, g, that minimizes Ex~P[L(g(x),1)] subject to the constraint Ex~U[g(x)] = π. This constraint can be imposed ‘softly’ through a regularization term in the objective function with weight λ (referred to as GE-KL)
In this objective function, we impose a constraint through the KL-divergence between the expectation of the classifier over the unlabeled data and the known fraction of positives, which is minimized when these terms are equal. This approach is an instance of a general class of posterior regularization called GE criteria, as specifically proposed by Mann and McCallum20. However, because we wish for our classifier to be a neural network and to optimize the objective using minibatched stochastic gradient descent, the gradient of the objective must be approximated using samples from the data. Estimates of the gradient of the GE-KL objective from samples are biased, which could cause stochastic gradient descent to find a suboptimal solution.
To address this issue, we propose an alternative GE criteria, GE-binomial, defined so as to minimize the difference between the distribution over the number of positives in the minibatch and the binomial distribution parameterized by π. The number of positive data points, k, in a minibatch of N samples from U follows the binomial distribution with parameter π. Furthermore, the classifier g also describes a distribution over the number of positives in the minibatch as
where x is a micrograph region, y is an indicator vector (\(y_i \in \left\{ {0,1} \right\}\)) denoting which data points are positive (yi = 1) and negative (yi = 1) and Y(k) is the set of all such vectors summing to k. This allows us to define the new GE criteria as the cross entropy between these two distributions \(\mathop {\sum }\limits_{k = 1}^N q\left( k \right){\mathrm{logp}}\left( k \right)\) giving the full GE-binomial objective function
In practice, because computing exact q(k) is slow, we make a Gaussian approximation with mean \(\mathop {\sum }\limits_{i = 1}^N g\left( {x_i} \right)\) and variance \(\mathop {\sum }\limits_{i = 1}^N g\left( {x_i} \right)\left( {1 - g\left( {x_i} \right)} \right)\) and substitute the Gaussian probability density function with these parameters for q in the above equation.
Autoencoder-based classifier regularization
When including the autoencoder component, we break our classifier network into the following two components: an encoder network composed of all layers except the final linear layer and the linear classifier layer. We denote these networks as f and c, respectively, with the full network, g, being given by g(x) = c(f(x)). Furthermore, we introduce a deconvolutional (also called transposed convolutional; see next section) decoder network, d, which takes the output of the feature extractor network and returns a reconstruction of the input image, x′ = d(f(x)). The objective function is then modified to include a term penalizing the expected reconstruction error over all images in the dataset, D, with weight γ
This forms the full GE-binomial objective function with autoencoder component used in Topaz.
Classifier and autoencoder architectures and hyperparameters
We use a simple three-layer convolutional neural network with striding, batch normalization36 and parametric rectified linear units (PReLU) as the classifier in this work. The model is organized as 32 conv7×7 filters with batch normalization and PReLU, stride by 2, 64 conv5×5 filters with batch normalization and PReLU, stride by 2, 128 conv5×5 filters with batch normalization and PReLU, and a final fully connected layer with a single output. We use sigmoid activation on this output to convert it into the predicted probability of a region being from the positive class (that is, the output is interpreted as the log-likelihood ratio between positive and negative classes).
When augmenting with an autoencoder, we use a decoder structure similar to that of DCGAN37. The d-dimensional representation that is output by the final convolutional layer of the classifier network is projected to a representation with small spatial dimensions but large feature dimensions. This is repeatedly projected into representations with larger spatial dimensions and smaller feature dimensions until the final output is of the original input image size. Specifically, this model is structured as repeated transpose convolutions with batch normalization and leaky ReLU activations. Let z be the representation output by the final convolutional layer of the classifier and X′ be the image reconstruction given by the decoder, the decoder structure is z → transpose conv4×4 128-d, batch normalization, leaky ReLU → transpose conv4×4 64-d, stride 2, batch normalization, leaky ReLU → transpose conv4×4 32-d, stride 2, batch normalization, leaky ReLU → transpose conv3×3 1-d, stride 2 → X′.
PU learning benchmarking
To compare classifiers trained with the different objective functions, we simulated hand-labeling with various amounts of effort by randomly sampling varying numbers of particles from the training sets to treat as the positive examples. All other particles were considered unlabeled. We used cross entropy loss for the labeled particles. The values of π used for training are specified in Table 1. For GE-KL we set the GE criteria weight, λ, to 10 as recommended by Mann and McCallum20. For GE-binomial, we set this parameter to 1. The classifier was then trained with those positives and evaluated by average-precision score (see next section for description of classifier evaluation) on the test set micrographs. This was repeated with ten independent samples of particles for each number of positives. Statistical significance of performance differences between methods at each number of labeled positive examples was assessed using a two-sided t test.
We also evaluated classifiers trained with autoencoder components and input reconstruction weight, γ, and varying numbers of labeled data points, N. We compared models trained with γ = 0 (no autoencoder), γ = 0, and \(\gamma = \frac{{10}}{N}\). For each setting of γ and N, we trained ten models with different sets of N randomly sampled positives and calculated the average-precision score for each model on the test split of each dataset.
Classifier evaluation
Classifiers were evaluated by average-precision score. This score is a measure of how well ranked the micrograph regions were when ordered by the predicted probability of containing a particle, and corresponds to the area under the precision-recall curve. It is calculated as the sum over the ranked micrograph regions of the precision at k elements times the change in recall
where precision (Pr) is the fraction of predictions that are correct and recall (Re) is the fraction of labeled particles that are retrieved in the top k predictions. Let TP(k) be the number of true positives in the top k predictions, then Pr and Re are given by
This measure is commonly used in information retrieval.
Non-maximum suppression algorithm for extracting particle coordinates
Non-maximum suppression chooses coordinates and the corresponding predicted probabilities of being a particle greedily starting from the highest scoring region. To prevent nearby pixels from also being considered particle candidates, all pixels within a second user-defined radius are excluded when a coordinate is selected. We set this radius to be the half major-axis length of the particle; however, smaller radii may give better results for closely packed, irregularly shaped particles.
Micrograph preprocessing
For EMPIAR-10025 and EMPIAR-10096, the aligned and summed micrographs along with contrast transfer function (CTF) estimates were taken directly from the public data release on EMPIAR. For EMPIAR-10028 and EMPIAR-10261, frames were aligned and summed without dose compensation using MotionCor238. Whole micrograph CTF estimates provided with the public release were used for this dataset.
For the clustered protocadherin dataset (EMPIAR-10234), single particle micrographs were collected on a Titan Krios electron microscope (Thermo Fisher Scientific) equipped with a K2 counting camera (Gatan); the microscope was operated at 300 kV with a calibrated pixel size of 1.061 Å. Ten-second exposures were collected (40 frames per micrograph) for a total dose of 68 e− Å−2 with a defocus range of 1–4 µm. A total of 896 micrographs were collected using Leginon39. Frames were aligned using MotionCor238. A total of 1,540 particles were picked manually using Appion Manual Picker23 from 87 micrographs and used as a training dataset for Topaz.
The rabbit muscle aldolase dataset (EMPIAR-10215) was collected on a Titan Krios electron microscope (Thermo Fisher Scientific) equipped with a K2 counting camera (Gatan) in super-resolution mode; the microscope was operated at 300 kV with a calibrated super-resolution pixel size of 0.416 Å. Six-second exposures were collected (30 frames per micrograph) for a total dose of 70.32 e− Å−2 with a defocus range of 1–2 µm. A total of 1,052 micrographs were collected using Leginon39. Frames were aligned, Fourier binned by a factor of 2 and dose compensated using MotionCor238. Whole-image CTF estimation was performed using CTFFIND440.
The Toll receptor dataset was collected on a Titan Krios electron microscope (Thermo Fisher Scientific) equipped with a K2 counting camera (Gatan); the microscope was operated at 300 kV with a calibrated pixel size of 0.832 Å. Six-second exposures were collected (40 frames per micrograph) for a total dose of 73.48 e− Å−2 with a defocus range of 1.5–2.0 µm. A total of 9,323 micrographs were collected using Leginon. Frames were aligned using MotionCor238. Whole-image CTF estimation was performed using CTFFIND440.
3D reconstruction procedure
Reconstruction was performed using cryoSPARC25. For each particle set, we first generated an ab initio structure with a single class. These structures were then refined using the homogeneous refinement option of cryoSPARC with symmetry specified depending on the dataset (T20S proteasome, D7; 80S ribosome, C1; and aldolase, D2). For the aldolase dataset, we used C2 symmetry for ab initio structure determination. Otherwise, all other parameters were left in the default setting. When evaluating the quality of Topaz particle sets for decreasing score thresholds, each particle set was selected by taking all particles predicted by the Topaz model with scores greater than or equal to the given threshold. Reconstructions were calculated for each of these sets independently as described above.
Removal of overlapping particles
To evaluate the quality of the extra particles predicted by Topaz, we removed particles from the Topaz particle set that were also included in the published particle set. This was done by removing all Topaz particles with centers within the particle radius of a particle center in the published particle set.
2D class averages (EMPIAR-10025, EMPIAR-10028 and EMPIAR-10215)
Class averages were calculated using the cryoSPARC 2D classification option. All settings were left as default except the number of 2D classes, which was set to ten for every particle set.
3D structure analysis (EMPIAR-10025, EMPIAR-10028 and EMPIAR-10215)
The final 3D reconstructions were analyzed visually in UCSF Chimera41 and 3DFSC34. In Chimera, the previous 3D reconstruction was first loaded (with the fitted Protein Data Bank structure, if available), the newly processed 3D reconstruction was then aligned to the previous reconstruction. The structures were visually compared and representative areas were chosen for display in Fig. 4. The 3DFSC reconstructions were calculated using the public server (https://3dfsc.salk.edu), which compares Fourier shell components for several solid angles to determine the range of resolutions and the amount of anisotropy in the reconstruction.
Toll receptor particle picking
A total of 1,599,638 particles were picked using DoG Picker 2 (ref. 7) from 8,974 micrographs and imported into cryoSPARC for all subsequent processing. After particle curation using 2D classification described below, the particle picks from 44 micrographs were visually inspected. Picks in areas of obvious particle aggregation were removed, and lower SNR particles corresponding to views typically missed by DoG Picker were selected. The resulting 1,048 particles were split into 686 training and 362 testing particles at the micrograph level. Topaz was then trained on the training particles and applied with the default score threshold of 0 for particle prediction. The ‘oblique’, ‘side’, and ‘top’ 2D classes (Fig. 3d) were lowpass filtered to 15 Å and used for template correlation with FindEM42 implemented in the Appion23 software package.
The crYOLO29 network was trained on the complete set of 1,048 labeled particles with 20% held out for validation by default. Micrographs were filtered and training was performed as described in the crYOLO tutorial. Picking was performed at the default threshold of 0.3.
The DeepPicker12 network was also trained on the complete set of 1,048 particles. Though no micrograph processing is required in the DeepPicker tutorial, micrographs were binned in Fourier space and lowpass filtered to 10 Å using EMAN25. Even with a threshold of 0, no particles were predicted by DeepPicker.
Toll receptor 3D reconstruction
All reconstructions were performed using cryoSPARC25. For all particle-picking approaches, we performed 2D classification with default parameters and 100 2D classes, then removed obvious non-particles. For the DoG dataset, four rounds of 2D classification yielded 770,263 particles from an initial stack of 1,599,638. For the template dataset, four rounds of 2D classification yielded 627,533 particles from an initial stack of 1,265,564. For the Topaz dataset, one round of 2D classification yielded 1,006,089 particles from an initial stack of 1,010,937. For the crYOLO dataset, one round of 2D classification yielded 131,300 particles from an initial stack of 133,644. For all datasets, ab initio reconstruction was used to generate an initial model, and the structures were further refined using homogeneous refinement with C1 symmetry, followed by non-uniform refinement. All parameters were left in their default setting. Unfiltered half maps and masks were used to calculate 3DFSC reconstructions using the public server (https://3dfsc.salk.edu).
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
Single-particle half maps, full sharpened maps and masks for T20S proteasome, 80S ribosome, rabbit muscle aldolase and the Toll receptor (DoG, template and Topaz picks) have been deposited in the Electron Microscopy Data Bank (EMDB) under accessions EMD-9194, EMD-9201, EMD-9202, EMD-9206, EMD-9207, EMD-9208, EMD-9209, EMD-9210, EMD-9211, EMD-20529, EMD-20531 and EMD-20532. The full rabbit muscle aldolase dataset has been deposited in the Electron Microscopy Pilot Image Archive (EMPIAR) under accession EMPIAR-10215.
Code availability
Source code for Topaz is publicly available via Code Ocean43 and on GitHub at https://github.com/tbepler/topaz. Updates to Topaz will be posted at http://topaz.csail.mit.edu. Topaz is licensed under the GNU General Public License v.3.0.
Change history
11 October 2019
In the version of this article originally published, scale bars were missing from Supplementary Figs 1–3, 8–10 and 14. This has now been amended and the Supplementary Information file has been updated.
References
Cheng, Y., Grigorieff, N., Penczek, P. A. & Walz, T. A primer to single-particle cryo-electron microscopy. Cell 161, 438–449 (2015).
Stagg, S. M., Noble, A. J., Spilman, M. & Chapman, M. S. ResLog plots as an empirical metric of the quality of cryo-EM reconstructions. J. Struct. Biol. 185, 41–426 (2014).
Rosenthal, P. B. & Henderson, R. Optimal determination of particle orientation, absolute hand, and contrast loss in single-particle electron cryomicroscopy. J. Mol. Bio. 333, 721–745 (2003).
Scheres, S. H. W. Semi-automated selection of cryo-EM particles in RELION-1.3. J. Struct. Biol. 189, 114–122 (2015).
Tang, G. et al. EMAN2: an extensible image processing suite for electron microscopy. J. Struct. Biol. 157, 38–46 (2007).
Roseman, A. M. Particle finding in electron micrographs using a fast local correlation algorithm. Ultramicroscopy 94, 225–236 (2003).
Voss, N. R., Yoshioka, C. K., Radermacher, M., Potter, C. S. & Carragher, B. DoG Picker and TiltPicker: software tools to facilitate particle selection in single particle electron microscopy. J. Struct. Biol. 166, 205–213 (2009).
Zhang, K., Li, M. & Sun, F. Gautomatch: an efficient and convenient gpu-based automatic particle selection program. https://www.mrc-lmb.cam.ac.uk/kzhang/ (2011).
Henderson, R. Avoiding the pitfalls of single particle cryo-electron microscopy: Einstein from noise. Proc. Natl Acad. Sci. USA 110, 18037–18041 (2013).
Subramaniam, S. Structure of trimeric HIV-1 envelope glycoproteins. Proc. Natl Acad. Sci. USA 110, E4172–E4174 (2013).
van Heel, M. Finding trimeric HIV-1 envelope glycoproteins in random noise. Proc. Natl Acad. Sci. USA 110, E4175–E4177 (2013).
Wang, F. et al. DeepPicker: a deep learning approach for fully automated particle picking in cryo-EM. J. Struct. Biol. 195, 325–336 (2016).
Zhu, Y., Ouyang, Q. & Mao, Y. A deep convolutional neural network approach to single-particle recognition in cryo-electron microscopy. BMC Bioinformatics 18, 348 (2017).
Xiao, Y. & Yang, G. A fast method for particle picking in cryo-electron micrographs based on fast R-CNN. AIP Conf. Proc. 1836, 020080 (2017).
Chen, M. et al. Convolutional neural networks for automated annotation of cellular cryo-electron tomograms. Nat. Methods 14, 983–985 (2017).
Li, X.-L. & Liu, B. in Machine Learning: ECML 2005 (eds Gama, J. et al.) 218–229 (Springer, 2005).
Nguyen, M. N., Li, X.-L. & Ng, S.-K. Positive unlabeled learning for time series classification. IJCAI 11, 1421–1426 (2011).
Zhang, J., Wang, Z., Yuan, J. & Tan, Y.-P. Positive and unlabeled learning for anomaly detection with multi-features. in Proc. 2017 ACM on Multimedia Conference 854–862 (ACM, 2017).
Kiryo, R., Niu, G., du Plessis, M. C. & Sugiyama, M. in Advances in Neural Information Processing Systems 30 (eds Guyon, I. et al.) 1675–1685 (Curran Associates, 2017).
Mann, G. S. & McCallum, A. Generalized expectation criteria for semi-supervisedl earning with weakly labeled data. J. Mach. Learn. Res. 11, 955–984 (2010).
Brasch, J. et al. Visualization of clustered protocadherin neuronal self-recognition complexes. Nature 569, 280–283 (2019).
Morin, A. et al. Cutting edge: collaboration gets the most out of software. eLife 2, e01456 (2013).
Lander, G. C. et al. Appion: an integrated, database-driven pipeline to facilitate EM image processing. J. Struct. Biol. 166, 95–102 (2009).
Scheres, S. H. W. RELION: implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol. 180, 519–530 (2012).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
de la, Rosa-Trevín et al. Scipion: a software framework toward integration, reproducibility and validation in 3D electron microscopy. J. Struct. Biol. 195, 93–99 (2016).
Biyani, N. et al. Focus: the interface between data collection and data processing in cryo-EM. J. Struct. Biol. 198, 124–133 (2017).
Dutta, A. & Zisserman, A. The VIA annotation software for images, audio and video. Preprint at https://arxiv.org/abs/1904.10699 (2019).
Wagner, T. et al. SPHIRE-crYOLO: a fast and well-centering automated particle picker for cryo-EM. Comm. Biol. 2, 218 (2019).
Bepler, T. et al. Positive-unlabeled convolutional neural networks for particle picking in cryo-electron micrographs. in Proc. 22nd Annual International Conference on Research in Computational Molecular Biology. (ed. Raphael, B. J.) 245–247 (Springer, 2018).
Tegunov, D. & Cramer, P. Real-time cryo-EM data pre-processing with Warp. Preprint at https://doi.org/10.1101/338558 (2018).
Campbell, M. G., Veesler, D., Cheng, A., Potter, C. S. & Carragher, B. 2.8 Å resolution reconstruction of the Thermoplasma acidophilum 20S proteasome using cryo-electron microscopy. eLife 4, e06380 (2015).
Wong, W. et al. Cryo-EM structure of the Plasmodium falciparum 80S ribosome bound to the anti-protozoan drug emetine. eLife 3, e03080 (2014).
Tan, Y. Z. et al. Addressing preferred specimen orientation in single-particle cryo-EM through tilting. Nat. Methods 14, 793–796 (2017).
Xu, H. et al. Structural basis of Nav1.7 inhibition by a gating-modifier spider toxin. Cell 176, 702–715 (2019).
Ioffe, S. & Szegedy, C. in Proc. 32nd International Conference on Machine Learning (eds Bach, F. & Blei, D.) 448–456 (PMLR, 2015).
Radford, A., Metz, L. & Chintala, S. Unsupervised representation learning with deep convolutional generative adversarial networks. Preprint at https://arxiv.org/abs/1511.06434 (2015).
Zheng, S. Q. et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 (2017).
Carragher, B. et al. Leginon: an automated system for acquisition of images from vitreous ice specimens. J. Struct. Biol. 132, 33–45 (2000).
Rohou, A. & Grigorieff, N. CTFFIND4: fast and accurate defocus estimation from electron micrographs. J. Struct. Biol. 192, 216–221 (2015).
Pettersen, E. F. et al. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Roseman, A. M. FindEM—a fast, efficient program for automatic selection of particles from electron micrographs. J. Struct. Biol. 145, 91–99 (2004).
Bepler, T. et al. Topaz: positive-unlabeled convolutional neural networks for particle picking in cryo-electron micrographs. Code Ocean https://doi.org/10.24433/CO.1911124.v1 (2019).
Acknowledgements
The authors wish to thank Simons Electron Microscopy Center (SEMC) OPs for the aldolase sample preparation and collection, Y. Z. Tan for SPA discussion, and the Electron Microscopy Group at the New York Structural Biology Center (NYSBC) for microscope calibration and assistance. We thank J. Sampson (Columbia University) for expressing the Toll receptor. We would also like to thank T. Jaakkola for his valuable feedback on the machine learning methods. We thank the developers of Relion, cryoSPARC, Appion, EMAN2, Scipion and Focus for their efforts in integrating Topaz. The Topaz GUI is based on VGG Image Annotator (VIA), which is developed and maintained with the support of EPSRC program grant Seebibyte: Visual Search for the Era of Big Data (EP/M013774/1). T.B., A.M. and B.B. were supported by NIH grant R01-GM081871. M.R. was supported by NSF GRFP (DGE-1644869). L.S. was supported by NIH grant R01-MH114817. A.J.N. was supported by a grant from the NIH National Institute of General Medical Sciences (NIGMS) (F32GM128303). The cryo-EM work was performed at the SEMC and National Resource for Automated Molecular Microscopy located at NYSBC, supported by grants from the Simons Foundation (SF349247), NYSTAR and the NIH NIGMS (GM103310) with additional support from the Agouron Institute (F00316) and NIH (OD019994).
Author information
Authors and Affiliations
Contributions
T.B., A.M. and B.B. conceived the project. T.B. developed the PU learning methods and implemented Topaz, processed and analyzed single particle datasets, and carried out the computational experiments under the guidance of B.B. M.R. prepared and collected the Toll receptor dataset. J.B. prepared and collected the clustered protocadherin dataset. A.J.N. analyzed the single particle cryo-EM reconstructions. A.J.N. developed the Topaz GUI based on VIA. T.B., A.M., M.R., J.B., L.S., A.J.N. and B.B. designed the experiments. T.B., M.R., A.J.N. and B.B. wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Additional information
Peer review information Allison Doerr was the primary editor on this article and managed its editorial process and peer review in collaboration with the rest of the editorial team.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Integrated supplementary information
Supplementary Figure 1 Normalization methods on EMPIAR-10261.
Comparison of standard affine normalization and our proposed mixture model normalization on EMPIAR-10261 micrographs downsampled 4x. For affine normalization, micrographs are transformed by subtracting the mean and dividing by the standard deviation of the pixel values. (a) Visualization of three example micrographs with either affine (top) or GMM (bottom) normalization. Affine normalized micrographs are washed out when there are dark grid regions present in the micrographs. (b) Histograms of the pixel intensities of the same three micrographs after affine or GMM normalization. GMM normalization correctly centers the pixel intensities around the high intensity peak. Results are consistent across >20 micrographs examined.
Supplementary Figure 2 Normalization methods on EMPIAR-10234.
Comparison of standard affine normalization and our proposed mixture model normalization on EMPIAR-10234 micrographs downsampled 8x. For affine normalization, micrographs are transformed by subtracting the mean and dividing by the standard deviation of the pixel values. (a) Visualization of three example micrographs with either affine (top) or GMM (bottom) normalization. The left-most micrograph contains light carbon grid that is correctly removed by GMM normalization. (b) Histograms of the pixel intensities of the same three micrographs after affine or GMM normalization. Results are consistent across >20 micrographs examined.
Supplementary Figure 3 Normalization methods on EMPIAR-10096.
Comparison of standard affine normalization and our proposed mixture model normalization on EMPIAR-10096 micrographs downsampled 8x. For affine normalization, micrographs are transformed by subtracting the mean and dividing by the standard deviation of the pixel values. (a) Visualization of three example micrographs with either affine (top) or GMM (bottom) normalization. No grid is present in these micrographs, so affine and GMM normalization given nearly identical results. (b) Histograms of the pixel intensities of the same three micrographs after affine or GMM normalization. Results are consistent across >20 micrographs examined.
Supplementary Figure 4 Toll receptor example micrograph with picks.
Example micrograph for the Toll receptor dataset with the unlabeled micrograph (top left), training picks (top right), DoG Picker picks (middle left), Topaz picks (middle right), FindEM picks (bottom left), and crYOLO picks (bottom right). This result is consistent over >100 micrographs examined.
Supplementary Figure 5 Toll receptor 3DFSC curves and crYOLO reconstruction.
Toll receptor 3DFSC plots and 3D reconstruction of the Toll receptor using 131,300 particles picked using crYOLO. (a) 3DFSC plots for Toll receptor structures solved using particles from Topaz, DoG, Template, and crYOLO. (b) Density map of the crYOLO structure. The crYOLO structure reaches a resolution of 6.83 Å at FSC0.143 with a sphericity of 0.734.
Supplementary Figure 6 Sparse picking in aggregates: Topaz vs crYOLO.
Example micrograph with a large amount of aggregate but still a number of real particles, comparing Topaz with crYOLO after following crYOLO’s sparse picking procedure. Topaz (top left) picks 40 particles while avoiding aggregation and ice contamination at the default threshold of 0.0. crYOLO picks 1 particle at its default threshold of 0.3. Lowering the threshold to 0.07 (top right) yields 26 particles with a small cluster of picks in the aggregation near the bottom right hand corner of the image. Further decreasing the threshold to 0.06 (bottom left) yields even more particles, but now it is clear the network is picking a large number of pixels within the aggregation. At a threshold of 0.05 (bottom right), crYOLO is no longer able to avoid aggregation or ice contamination. Note that the particle picking threshold increment in crYOLO is 0.01. We found these results to be consistent across >100 micrographs examined.
Supplementary Figure 7 Sparse picking with low SNR: Topaz vs. crYOLO.
Example micrograph with real particles but thicker ice and thus lower contrast, comparing Topaz with crYOLO after following crYOLO’s sparse picking procedure. Topaz (top left) picks 127 particles at the default threshold of 0.0. crYOLO picks 0 particle at its default threshold of 0.3. Lowering the threshold to 0.07 (top right) yields 37 particles. Further decreasing the threshold to 0.06 (bottom left) yields more particles, but the network is still missing real particles while starting to select some background pixels. At a threshold of 0.05 (bottom right), there are clear artifacts at the edges of the micrograph. We found these results to be consistent across >100 micrographs examined.
Supplementary Figure 8 T20S proteasome example micrographs.
Two example micrographs for the T20S proteasome dataset (EMPIAR-10025) with (top) published particles circled in blue, (middle) training particles sampled from the published particles circled in blue, and (bottom) Topaz particles circled in red. Each column is a different micrograph. The circled training particles illustrates how sparse the Topaz picks are for this dataset. The PU learning framework allows picking to be performed with high accuracy despite the sparsity of examples, as seen by the Topaz picks in red. Furthermore, Topaz recovers many more real particles than are present in the published set. We found this result to be consistent across >100 micrographs examined.
Supplementary Figure 9 80S ribosome example micrographs.
Two example micrographs for the 80S ribosome dataset (EMPIAR-10028) with (top) published particles circled in blue, (middle) training particles sampled from the published particles circled in blue, and (bottom) Topaz particles circled in red. Each column is a different micrograph. The circled training particles illustrates how sparse the Topaz picks are for this dataset. The PU learning framework allows picking to be performed with high accuracy despite the sparsity of examples, as seen by the Topaz picks in red. Furthermore, Topaz recovers many more real particles than are present in the published set. We found this result to be consistent across >100 micrographs examined.
Supplementary Figure 10 Rabbit muscle aldolase example micrographs.
Two example micrographs for the rabbit muscle aldolase dataset (EMPIAR-10215) with (top) published particles circled in blue, (middle) training particles sampled from the published particles circled in blue, and (bottom) Topaz particles circled in red. Each column is a different micrograph. The circled training particles illustrates how sparse the Topaz picks are for this dataset. The PU learning framework allows picking to be performed with high accuracy despite the sparsity of examples, as seen by the Topaz picks in red. Furthermore, Topaz recovers many more real particles than are present in the published set. In this aldolase dataset, the particles are tightly packed, but Topaz correctly identifies and centers the particles. We found this result to be consistent across >100 micrographs examined.
Supplementary Figure 11 EMPIAR-10215 2D class averages.
2D class averages of Topaz particles with decreasing score threshold for the aldolase dataset. Classes identified as false positives for quantification in Fig. 5 are indicated by orange boxes.
Supplementary Figure 12 EMPIAR-10028 2D class averages.
2D class averages of Topaz particles with decreasing score threshold for the 80S ribosome. Classes identified as false positives for quantification in Fig. 5 are indicated by orange boxes.
Supplementary Figure 13 Precision-recall and F1 curves for EMPIAR-10025, EMPIAR-10028, and EMPIAR-10215.
Precision-recall curves and threshold vs precision, recall, and F1 score curves for classifiers trained on the EMPIAR-10025, EMPIAR-10028, and EMPIAR-10215 datasets. Curves were calculated by matching the particles predicted by the Topaz models on the test set micrographs of each dataset to the published particle annotations on those micrographs. We note that the precision and average-precision scores are underestimates of the true precision and true average-precision of the Topaz models due to incompleteness of the published particle set.
Supplementary Figure 14 EMPIAR-10096 and EMPIAR-10234 example micrographs with Topaz picks.
Representative micrographs from (a) the EMPIAR-10096 and (b) the EMPIAR-10234 test sets. For EMPIAR-10096, curated particles from EMPIAR (blue) and particles predicted by Topaz (red) are circled. For EMPIAR-10234, manually selected (blue) and predicted (red) particles are circled. Topaz avoids ice chunks, particles in proximity to the edge of the hole, and particles on carbon and correctly identifies many particles missing from the manually labeled/curated particles sets. We found this result to be consistent over >20 micrographs examined.
Supplementary Figure 15 Sensitivity to π of GE-binomial.
Sensitivity of GE-binomial objective function to the setting of π. For EMPIAR-10096 and the EMPIAR-10234 datasets we report average-precision scores for classifiers trained with 100 and 1,000 labeled particles and values of π varying from 0.5x to 1.5x the values reported in Table 1. We report the mean and standard deviation of 10 runs.
Supplementary information
Supplementary Information
Supplementary Figs. 1–15.
Rights and permissions
About this article

Cite this article
Bepler, T., Morin, A., Rapp, M. et al. Positive-unlabeled convolutional neural networks for particle picking in cryo-electron micrographs. Nat Methods 16, 1153–1160 (2019). https://doi.org/10.1038/s41592-019-0575-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41592-019-0575-8
This article is cited by
-
Structure of the M. tuberculosis DnaK−GrpE complex reveals how key DnaK roles are controlled
Nature Communications (2024)
-
Structural basis for lysophosphatidylserine recognition by GPR34
Nature Communications (2024)
-
Molecular mechanisms underlying the BIRC6-mediated regulation of apoptosis and autophagy
Nature Communications (2024)
-
The PfRCR complex bridges malaria parasite and erythrocyte during invasion
Nature (2024)
-
Cryo-EM structures of PP2A:B55–FAM122A and PP2A:B55–ARPP19
Nature (2024)
