
Abstract
Zika virus (ZIKV) infection during pregnancy can cause microcephaly in newborns, yet the underlying mechanisms remain largely unexplored. Here, we reveal extensive and large-scale metabolic reprogramming events in ZIKV-infected mouse brains by performing a multi-omics study comprising transcriptomics, proteomics, phosphoproteomics and metabolomics approaches. Our proteomics and metabolomics analyses uncover dramatic alteration of nicotinamide adenine dinucleotide (NAD+)-related metabolic pathways, including oxidative phosphorylation, TCA cycle and tryptophan metabolism. Phosphoproteomics analysis indicates that MAPK and cyclic GMP–protein kinase G signaling may be associated with ZIKV-induced microcephaly. Notably, we demonstrate the utility of our rich multi-omics datasets with follow-up in vivo experiments, which confirm that boosting NAD+ by NAD+ or nicotinamide riboside supplementation alleviates cell death and increases cortex thickness in ZIKV-infected mouse brains. Nicotinamide riboside supplementation increases the brain and body weight as well as improves the survival in ZIKV-infected mice. Our study provides a comprehensive resource of biological data to support future investigations of ZIKV-induced microcephaly and demonstrates that metabolic alterations can be potentially exploited for developing therapeutic strategies.
Similar content being viewed by others


Main
ZIKV, a mosquito-transmitted flavivirus, has imposed tremendous global public health challenges in recent years1,2. ZIKV infection during pregnancy has been confirmed to cause severe neurological symptoms, including microcephaly in newborns3. We and others have established mouse models for viral infection and ZIKV-induced microcephaly and demonstrated that ZIKV infection can induce cell death in brains4,5. Previous studies have characterized the mechanisms underlying ZIKV infection and ZIKV-induced microcephaly6,7, but given the lack of effective treatments, it is critical to expand our understanding of ZIKV-induced microcephaly pathogenesis to facilitate the development of new therapies.
Viruses have evolved numerous strategies to reprogram the metabolism of infected host cells to achieve rapid viral replication and to escape host immune responses8,9. The reprogramming of metabolic pathways, including the urea cycle, lipid metabolism and arginine metabolism, has been reported to facilitate replication of viruses and/or modulate host cell responses10,11,12,13. Identifying viral infection-induced metabolic reprogramming in host cells not only contributes to a better understanding of virus–host interactions but also helps to develop potential antiviral strategies by targeting metabolic alterations. For example, 2-deoxy-d-glucose (2-DG), an antimetabolite that inhibits phosphoglucose isomerase, has been previously reported to effectively inhibit the replication of several viruses, such as severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and influenza virus14,15. Our previous studies also show that correcting the dysregulated arginine metabolism that is associated with immune dysfunctions during severe fever with thrombocytopenia syndrome virus and SARS-CoV-2 infection could be exploited as potential therapeutic strategies10,16. However, little is known from in vivo studies about the impacts of ZIKV infection on host metabolism and the consequential pathogenesis of microcephaly.
Multi-omics analysis has allowed for a comprehensive understanding of the molecular mechanisms underlying complex diseases, such as viral infection13,17, cancer18 and diabetes19. We and others have focused on transcriptomics studies in cell lines and/or mouse models and revealed that ZIKV infection causes strong immune responses and aberrant neurodevelopment5,20,21. However, we are unaware of attempts to perform integrative analysis of ZIKV-induced microcephaly in in vivo models by incorporating multi-omics techniques.
To fill this gap, here we performed an extensive and systematic multi-omics characterization of ZIKV-infected mouse brains. Specifically, we analyzed transcriptomics, proteomics, phosphoproteomics and metabolomics data from mock- and ZIKV-infected mouse brains. We thereby detected the anticipated strong immune response as well as previously underexplored ZIKV biological impacts such as profound dysregulation of NAD+ metabolism and activation of MAPK signaling. This crucial role of NAD+ in ZIKV-induced microcephaly was confirmed with our in vivo demonstration that boosting NAD+ by NAD+ or nicotinamide riboside (NR) supplementation alleviated cell death and increased cortex thickness in ZIKV-infected mouse brains; and NR supplementation even more systemically increased brain and body weight as well as improved survival in ZIKV-infected mice. Thus, our study not only introduces a high-quality multi-omics resource but also demonstrates regulatory mechanisms and potential therapeutic targets for ZIKV-induced microcephaly. More generally, the research strategies and data analysis techniques applied in our work on ZIKV-induced microcephaly illustrate principles for effectively using a multi-omics approach to gain biological insights and potential therapeutic strategies for other virus-induced diseases.
Results
Overview of the multi-omics landscape
To obtain a comprehensive molecular understanding of ZIKV-induced microcephaly, we infected mice with ZIKV at E15.5 (embryonic day 15.5) and collected brains at P3 (postnatal day 3) for multi-omics characterization (Fig. 1a and Extended Data Fig. 1a; Methods). Our previous work has revealed transcriptomic alterations relating to immune responses and brain development in ZIKV-infected mouse brains5,22. Here, we further performed proteomics, phosphoproteomics and metabolomics studies to gain systematic biological information from multiple perspectives. Our liquid chromatography–mass spectrometry (LC–MS)-based global proteomics identified a total of 5,080 proteins with an average of 4,294 per sample (Extended Data Fig. 1b). Phosphoproteomics analysis identified a total of 5,935 reliable phosphopeptides (localization prob ≥0.75, score ≥40, δ score ≥17) from 1,927 phosphoproteins, with an average of 2,366 phosphopeptides per sample (Extended Data Fig. 1c). The data from transcriptomics, proteomics and phosphoproteomics were of high quality as evaluated using standard measures (Extended Data Fig. 1b–h). Further, by using a targeted metabolomics approach, we identified 190 metabolites across mouse brain samples from a total of 260 monitored metabolites. The high quality of our metabolic data was revealed by the evaluation presented in Extended Data Fig. 1i. Based on these comprehensive multi-omics datasets, we performed integrative pathway enrichment analysis to improve the system-level understanding of the cellular processes and pathways involved in ZIKV-induced microcephaly and revealed profound alterations in apoptotic process, immune system process, MAPK signaling and metabolic pathways (Fig. 1b).

a, Experimental design and sample collection in the study. ZIKV was injected into one side of the lateral ventricle (LV) of embryonic mouse brains on E15.5. Neonatal pups were born on P0 (postnatal day 0). Pup brains were collected on P3 for transcriptomics, proteomics, phosphoproteomics and metabolomics analyses. b, Summary of representative dysregulated biological processes revealed by multi-omics analysis. Indicated representative pathways are mapped for mRNAs, proteins and metabolites with the greatest variation between ZIKV-infected and mock-infected brains.
Transcriptomics and proteomics analyses of ZIKV-infected brains
In silico deconvolution has been widely used to dissect cell compositions of complex tissues using bulk RNA sequencing (RNA-seq) data23,24. Here, we performed in silico deconvolution to identify cell subpopulations based on our bulk RNA-seq data22 using known marker genes for various cell types in brain tissue. The inferred number of microglia (the innate immune cells of the brain) was increased in the ZIKV infection group, indicating an upregulated immune response induced by ZIKV (Fig. 2a). The inferred number of neural stem cells was increased and numbers of predicted neurons, neuroblasts and astrocytes were decreased in ZIKV-infected mouse brains (Fig. 2a), implying potential defects in differentiation of neural stem cells. These findings were consistent with our previous experimental observations that ZIKV infection leads to S phase arrest and differentiation defects of neural progenitor cells, defects in the development of neurons, activation of microglia and decreased cell numbers of astrocytes and oligodendrocyte progenitor cells5,25.

a, Cell-type enrichment scores (ESs) calculated by GSVA using RNA-seq data of ZIKV-infected samples and mock-infected controls. ESs demonstrating relative abundance of distinct cell types are shown in orange (increased) and green (decreased). b, Volcano plot of protein alterations between mock- and ZIKV-infected brains. Significantly different proteins (FDR < 0.05 and fold change >2) are colored in red (upregulated) and blue (downregulated); others are colored in gray. c, Significantly altered KEGG pathways between mock- and ZIKV-infected brains. Enriched KEGG pathways based on differential proteins in ZIKV-infected brains (FDR < 0.05 and fold change >2) are shown. d, Schema of cholesterol metabolism with upregulated proteins in ZIKV-infected brains (FDR < 0.05 and fold change >2). HDL, high-density lipoprotein; IDL, intermediate-density lipoprotein; VLDL, very-low-density lipoprotein; CM, chylomicrons. e, Schema of phagosome with upregulated proteins in ZIKV-infected brains (FDR < 0.05 and fold change >2, n = 8 per group). f, Integrative GO analysis of proteomics and transcriptomics data. GSEA of upregulated and downregulated pathways in ZIKV-infected brains at mRNA (x axis) and protein (y axis) levels based on GO subset in AmiGO database. Normalized enrichment scores (NES) of GO terms are plotted.
To further characterize host responses induced by ZIKV infection at the protein level, we next conducted an LC–MS-based global proteomics study. Principal-component analysis (PCA) discriminated mock-infected controls from ZIKV-infected mouse brains based on abundance of detected proteins (Extended Data Fig. 2a). In total, expression levels of 577 proteins were identified as significantly different between these two groups (false discovery rate (FDR) < 0.05 and fold change >2), with 299 downregulated and 278 upregulated proteins in the ZIKV-infected group (Fig. 2b).
To identify the biological processes and pathways impacted by ZIKV infection, enrichment analyses were performed based on differential proteins. Gene Ontology (GO) enrichment analysis for biological processes indicated upregulation of immune response and inflammatory process and downregulation of neuron and axon development in ZIKV-infected mouse brains (Extended Data Fig. 2b–e). Notably, inflammatory cytokines, such as CSF1 and CXCL10, were upregulated in ZIKV-infected mouse brains at both protein (Extended Data Fig. 2d) and messenger RNA (Extended Data Fig. 2f) levels, which were also found to be significantly increased after SARS-CoV-2 infection26,27, suggesting the similar inflammatory responses induced by ZIKV and SARS-CoV-2. Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis revealed significant upregulation of cholesterol metabolism-, phagosome- and lysosome-related pathways and downregulation of synapse-related pathways (Fig. 2c). Previous studies have shown that cholesterol metabolism can facilitate virus infection28. Thus, enhanced cholesterol metabolism may contribute to ZIKV entry via upregulating components involved in extracellular cholesterol transport, for example, APOA1, APOA4, APOC3, APOE, APOH and PLTP and intracellular cholesterol transport, NPC2 (Fig. 2d). Additionally, ZIKV infection has been shown to induce activation of microglia25. Upregulation of phagosomes (Fig. 2e) may be attributed to the activated phagocytosis function of microglia induced by ZIKV, which possibly contributes to neuronal cell death29.
In silico deconvolution was also performed on a proteomics dataset based on cell-type-specific markers. Notably, deconvolution of our protein dataset revealed a similar cell-type change to that in the RNA-seq dataset (Extended Data Fig. 2g), which provided an orthogonal cross-validation between protein and mRNA levels. We next integrated proteomics and transcriptomics datasets to gain a deep insight into the dysregulation of cellular processes induced by ZIKV infection. By adopting gene set enrichment analysis (GSEA) pathway enrichment, we found several upregulated pathways, including processes of immune response, apoptosis and MAPK signaling, as well as downregulated metabolic pathways, such as the TCA cycle, at both protein and mRNA levels (Fig. 2f).
Proteomics reveals aberrant metabolism in ZIKV-infected brains
Given the above-mentioned findings that several metabolic processes were affected by ZIKV (Fig. 2f), we performed GSEA of KEGG metabolic pathways in mock- and ZIKV-infected mouse brains on the basis of our proteomics data (Fig. 3a). We found that enriched upregulated pathways in ZIKV-infected brains were frequently related to amino acid and purine and pyrimidine metabolism, evidenced with upregulated proteins (FDR < 0.05), such as GATM, PNP and NT5C3 (Fig. 3b), whereas downregulated pathways included the TCA cycle, oxidative phosphorylation (OXPHOS) and pyruvate metabolism, evidenced with downregulated proteins (FDR < 0.05), such as IDH1, NDUFA4 and DLAT (Fig. 3c). Considering the metabolic heterogeneity among different neural cell types30, we then dissected in silico the proteomics data using cell-specific markers and protein–protein interaction networks to identify cell-specific metabolic changes (Extended Data Fig. 3a). We found that the downregulated OXPHOS and TCA cycle occur in neurons and neuroblast cells, suggesting a correlation between mitochondrial dysfunction and ZIKV-induced neural cell death (Extended Data Figs. 2g and 3a).

a, GSEA of KEGG metabolic pathways enriched in mock- and ZIKV-infected brains. Upregulated and downregulated pathways in ZIKV-infected brains are shown in red and blue, respectively. b,c, Heat map and quantitative analysis of altered proteins (FDR < 0.05) in upregulated (b) and downregulated pathways (c) in ZIKV-infected brains. d, Reprogrammed metabolic pathways in ZIKV-infected brains. Protein and mRNA changes of ZIKV-infected brains are indicated in comparison with mock-infected controls.
To precisely investigate the system-level changes in the altered metabolic pathways, we next re-analyzed our transcriptomics and proteomics data. The downregulation of OXPHOS (NDUFS3, NDUFA4, NDUFA5, COX4I1, COX6C and ATP8) and TCA cycle (DLAT, PDHB, ACLY, IDH1 and IDH3A) components that we observed at mRNA and/or protein levels indicated the potential dysregulated mitochondria functions and bioenergy metabolism (Fig. 3d), both of which are associated with neural cell death29,31. In addition, an independent experiment at a different time point of E18.5 (infected on E13.5 and analyzed on E18.5) showed similar alterations in OXPHOS and the TCA cycle induced by ZIKV infection (Extended Data Fig. 3b). Additionally, downregulated purine and pyrimidine metabolism toward RNA and DNA syntheses at the protein level implied a low proliferation state for the cells of ZIKV-infected mouse brains (Fig. 3d). Thus, our data suggest that ZIKV infection induces aberrant metabolic reprogramming, bioenergy deficiency and mitochondrial dysfunction.
Metabolomics reveals NAD+ depletion in ZIKV-infected brains
Given that transcriptomics and proteomics integrative analysis clearly indicated the strong connection between metabolic reprogramming and ZIKV-induced microcephaly, we next performed metabolomics analysis on mouse brains from mock- and ZIKV-infected groups. Visualization of detected metabolites distinguished between these two groups by PCA and hierarchical clustering analysis (Fig. 4a and Extended Data Fig. 4a). To identify key metabolic pathways altered by ZIKV infection, 121 differential metabolites were selected (FDR < 0.05) and subjected to KEGG pathway enrichment analysis (Fig. 4b). Further multi-omics analysis of metabolic pathways showed similar alterations at mRNA, protein and metabolite levels, including the TCA cycle, OXPHOS, purine metabolism and pyrimidine metabolism (Fig. 4c and Extended Data Fig. 4b,c). Our analysis revealed downregulation of the TCA cycle in ZIKV-infected mouse brains, evidenced with significantly declined levels of metabolites (such as fumarate and malate) and key metabolic enzymes (such as DLAT, ACLY, IDH1 and IDH3A) (Fig. 4c). Moreover, subunits of mitochondrial respiration chain complexes, such as NDUFS3, COX6C and ATP8, along with metabolites, including NAD+ and fumarate, were significantly reduced (Fig. 4c), suggesting downregulation of OXPHOS induced by ZIKV infection.

a, PCA of metabolomics data in mock- (n = 8) and ZIKV-infected (n = 8) brains. b, Altered KEGG metabolic pathways in ZIKV-infected brains compared with mock-infected brains enriched by significantly altered metabolites (FDR < 0.05). c, Transcriptomic, proteomic and metabolic analyses of OXPHOS and TCA cycle metabolic pathways. Metabolites with significantly increased or decreased abundance in ZIKV-infected brains are marked in red or blue, respectively (FDR < 0.05). d, Differential metabolites between mock- and ZIKV-infected brains (FDR < 0.05). Top 35 of the most upregulated metabolites and top 20 of the most downregulated metabolites are shown. e, Relative abundance of representative metabolites related with NAD+ and arginine metabolism (n = 8 mice per group). a.u., arbitrary unit. To calculate relative abundance, the peak area of each metabolite was divided by the sum peak areas of all detected metabolites in the same sample and the resulting ratio value was used as the relative abundance of each metabolite. Data are shown as mean ± s.e.m. Two-tailed unpaired Student’s t-tests followed by a Benjamini–Hochberg multiple comparison test were used for statistical analysis. Exact FDR values are indicated. f, Transcriptomic, proteomic and metabolic analyses of NAD+ salvage pathway. Metabolites upregulated and downregulated in ZIKV-infected mouse brains are marked in red or blue, respectively (FDR < 0.05). g, Western blotting analysis of key enzymes (NAMPT and NMNAT2) in NAD+ salvage pathway (n = 3 mice per group). Data are representative of three independent experiments. Data are shown as mean ± s.e.m. A two-tailed unpaired Student’s t-test was used for statistical analysis. Exact P values are indicated.
To gain a deep insight into the significantly altered metabolites and metabolic pathways, we examined the differential metabolites (FDR < 0.05) and found several prominently upregulated and downregulated metabolites (Fig. 4d). Of note, a significantly decreased level of NAD+ and increased levels of its precursors from de novo and salvage pathways, including tryptophan, kynurenine, NR and nicotinamide mononucleotide (NMN), were observed (Fig. 4d,e), suggesting dysregulation of NAD+ metabolism and tryptophan–kynurenine metabolism in ZIKV-infected mouse brains. Enhanced tryptophan–kynurenine metabolism is known to contribute to both immune suppression and neuronal excitotoxicity32. The significantly elevated levels of tryptophan and kynurenine in ZIKV-infected mouse brains (Fig. 4e) suggest that ZIKV infection may lead to neuronal excitotoxic and/or immunosuppressant effects via disturbing tryptophan–kynurenine metabolism. Moreover, in ZIKV-infected mouse brains, we observed a significantly increased level of arginine (Fig. 4e), which has been reported as a regulator of antiviral adaptive immunity10,11, indicating the potential role of altered arginine metabolism in the adaptive immune response induced by ZIKV infection. Of note, the metabolic changes in E18.5 model showed high similarities with those observed in the P3 model (Extended Data Fig. 5a,b). In particular, NAD+ metabolism and the TCA cycle showed strong consistency between the two time points (Extended Data Fig. 5c), including upregulated NAD+ precursors (tryptophan, kynurenine and NAM), downregulated NAD+ and intermediates in the TCA cycle (fumarate and malate).
NAD+ is an essential component of both bioenergy metabolism and signal transduction in cells33, whereas bioenergy depletion is known to cause neuronal cell death29. Given the significantly declined level of NAD+ (Fig. 4e) and downregulated OXPHOS and TCA cycle metabolism as described above (Fig. 4c), it is reasonable to speculate that NAD+ depletion caused bioenergy deficiency and thereby cell death as we previously demonstrated in ZIKV-induced microcephaly5. To further elucidate possible causes of NAD+ reduction in ZIKV-infected mouse brains, we systematically examined the NAD+ salvage pathway, the major synthesis pathway of NAD+ in brains34, at the metabolite, protein and mRNA levels. Our data showed that NAMPT, which converts NAM to NMN, was significantly upregulated. However, NMNAT2, which converts NMN to NAD+, was downregulated at both protein and mRNA levels (Fig. 4f,g). Conversely, most of the Parp and Sirt genes, encoding the major NAD+-consuming enzymes, were significantly upregulated at the mRNA level in ZIKV-infected brains (Fig. 4f). Of note, NAMPT, PARP9, PARP10, PARP12 and PARP14 were reportedly upregulated after SARS-CoV-2 infection35, suggesting the reprogramming and potential role of host NAD+ metabolism in both ZIKV and SARS-CoV-2 infection. Thus, our data highlight the significantly altered NAD+ salvage metabolism in ZIKV-infected mouse brains, possibly resulting from dysregulation of metabolic enzymes in the pathway.
Phosphoproteomics reveals disturbed signaling pathways in ZIKV-infected brains
Phosphorylation is a common post-translational modification of proteins that is known to be essential for host cell responses to infection by a variety of viruses36. Given that both transcriptomics and proteomics analyses clearly indicated the strong connection between MAPK and ZIKV infection, we therefore examined relative abundance of both phosphopeptides and putative phosphosites in mock- and ZIKV-infected mouse brains to identify impacts of phosphorylation on ZIKV-induced microcephaly. Among the detected 5,935 phosphopeptides, 50 were significantly upregulated (FDR < 0.05 and fold change >2) and 15 were significantly downregulated (FDR < 0.05 and fold change >2) in ZIKV-infected mouse brains (Fig. 5a). Enrichment analysis of these differential phosphoproteins allowed us to identify specific phosphorylation events occurring throughout a given cellular pathway, which focused our attention on the apparently strong upregulation for the cyclic GMP (cGMP)–protein kinase G (PKG) and MAPK signaling pathways and cell adhesion molecules (Fig. 5b). Previous studies have reported that calcium signaling, cGMP–PKG and cAMP can stimulate MAPK signaling37,38,39. We therefore examined the altered phosphoproteins involved in MAPK, calcium, cGMP–PKG and cAMP signaling pathways and found several upregulated phosphoproteins (P < 0.05 and fold change >2), for example, CACNA1E, SLC8A1, ADCY9, RASGRP2 and MAP4K4 (Fig. 5c). Notably, we detected significant upregulation of phosphoproteins and phosphosites in the MAPK pathway in ZIKV-infected mouse brains after normalization to individual protein levels, such as CACNA1B, CACNA1E, RASGRP2, MAP4K4, MAPT and MEF2C (Fig. 5d and Extended Data Fig. 6a), suggesting the potential role of MAPK signaling in ZIKV-induced microcephaly. To fully validate activation of MAPK signaling, we examined expression levels of phosphorylated proteins in MAPK signaling, including pERK, pJNK and p-p38. Our results showed significant activation of phosphorylated ERK in ZIKV-infected mouse brains (Fig. 5e), but not p-p38 and pJNK (Extended Data Fig. 6b). To nominate potential therapeutic targets for ZIKV-induced microcephaly, we screened the US Food and Drug Administration (FDA)-approved drugs that target hyperphosphorylated proteins in MAPK, cGMP–PKG, cAMP and calcium signaling pathways (P < 0.05 and fold change >2). Our results showed that drugs, such as bepridil, dronedarone, ivabradine and trametinib, may have potential therapeutic effects against ZIKV-induced microcephaly (Extended Data Fig. 6c).

a, Volcano plot indicating expression of differential phosphopeptides in mock- and ZIKV-infected brains. Upregulated and downregulated phosphopeptides with FDR < 0.05 and fold change >2 are colored in red and blue, respectively; others are colored in gray. b, Enriched KEGG pathways based on significantly upregulated (red) and downregulated (blue) phosphopeptides in ZIKV-infected brains (FDR < 0.05 and fold change >2). c, Cellular localization and expression levels of altered phosphopeptides in MAPK, calcium, cGMP and cAMP signaling pathways in ZIKV-infected brains (P < 0.05 and fold change >2). Color of protein node indicates the measured level (log2(fold change)) of the phosphorylation site of individual protein with the strongest alteration between mock- and ZIKV-infected mouse brains. Individual phosphorylation sites are shown next to the corresponding molecules. d, Normalized levels of phosphoproteins that were significantly upregulated in MAPK pathway in ZIKV-infected mouse brains (P < 0.05 and fold change >2, n = 8 per group). e, Expression of phosphorylated ERK in mock- and ZIKV-infected mouse brains (n = 3 per group). Data are representative of three independent experiments. Data are shown as mean ± s.e.m. (d and e). Two-tailed unpaired Student’s t-tests were used for statistical analysis (d,e). Exact P values are indicated.
Kinases are commonly used as drug targets40, therefore phosphoproteomics data could be mined for identifying kinases with therapeutic potentials24. Notably, our phosphoproteomics data experimentally detected several differential kinases (P < 0.05 and fold change >2), for example, AAK1, CAMK2B, DCLK1, MAP4K4, PRKCE, were upregulated in ZIKV-infected mouse brains compared to mock-infected controls (Extended Data Fig. 6d), providing us with potential drug targets for treating ZIKV-induced microcephaly. Furthermore, potential drug targets can be predicted by inferring the activity of a kinase based on the variation of its substrate expression41. Here we used pLogo42 and NetPhos 3.1 Server43 to predictably identify activity-altered kinases as potential therapeutic targets based on our data for significantly altered phosphopeptides in ZIKV-infected mouse brains (FDR < 0.05 and fold change >2). Our analysis predicted several upregulated kinases in ZIKV-infected mouse brains with high reliability, such as GSK3, p38MAPK, PKA and PKC, as well as several downregulated kinases, such as ATM (Extended Data Fig. 6e). Indeed, consistent with predicted results, the levels of these predicted kinases showed a difference between mock- and ZIKV-infected mouse brains in our experimental data, such as GSK3B, PRKCE and PRKAR2B (P < 0.05 and fold change >2) (Extended Data Fig. 6f). Thus, our phosphoproteomics study provides a data-rich resource for identifying differential kinases that could be further explored as drug targets for the treatment of ZIKV-induced microcephaly.
NAD+ supplementation suppresses ZIKV-induced cell death
On the basis of the observation that the level of NAD+ was dramatically declined in ZIKV-infected mouse brains (Fig. 4e) and the previous reports that supplementation of NAD+ or its precursors (NAM and NR) played a protective role in brain and nervous system in vivo44,45,46, we were prompted to test whether locally boosting NAD+ can confer protective effects against ZIKV-induced cell death in vivo. To this end, we treated pups with NAD+ or one of its precursors, NAM (nicotinamide) via λ point injection once a day from postnatal day 6 (P6) to P8 after ZIKV infection on P0 and killed the pups on P9 (Fig. 6a).

a, Experimental overview of NAD+ and NAM supplementation in ZIKV-infected mouse brains. ZIKV was injected into the λ point on P0. Six days later on P6, NAD+, NAM or vehicle (Veh.) (RPMI medium 1640 basic + 2% FBS) was injected into the λ point of ZIKV- or mock-infected mouse brains once a day for three consecutive days on P6–8. Mice were killed on P9 and brains were collected for immunocytochemistry analysis. b,c, Immunostaining images of ZIKV (green), DAPI (blue) and cleaved Cas3 (red) on mock- and ZIKV-infected brains. HIP, hippocampus; CTX, cortex. Scale bars, 500 μm (b) and 50 μm (c). d,e, ZIKV intensity (d) and cell death (e) in cortex (left) and hippocampus (right) respectively. n = 3 brains for each group, three slices for each brain. f, Quantification of cortex thickness in mock- and ZIKV-infected brains. n = 3 brains for Ctrl + Veh., Ctrl + NAD+ and ZIKV + NAD+ groups, n = 2 brains for ZIKV + Veh. group, six slices for each brain. g, Immunostaining images of Iba-1 (red), CD68 (green) and DAPI (blue) on ZIKV-infected brains with or without NAD+ supplementation. Scale bar, 50 μm. h,i, Cell numbers with Iba-1+, CD68+ and CD68+/Iba-1+ ratio in hippocampus (h) and cortex (i) of ZIKV-infected brains with or without NAD+ supplementation. n = 3 brains for the ZIKV + NAD+ group, n = 2 brains for the ZIKV + Veh. group, three slices for each brain. Data in b,c,g are representative of two independent experiments. Data are shown as mean ± s.e.m. (d–f,h,i). Two-tailed unpaired Student’s t-test was used for statistical analysis (d–f,h,i). Exact P values are indicated.
We inspected ZIKV density in brains via immunostaining with monoclonal antibody ZK2B10 (ref. 47). Comparable levels of ZIKV in the hippocampus and higher levels of ZIKV in the cortex were detected in the NAD+ treatment group compared to the vehicle treatment group (Fig. 6b–d), indicating that NAD+ did not significantly inhibit replication of ZIKV in mouse brains. Cell death is considered to be a major cause of microcephaly in ZIKV-infected brains48. We observed that ZIKV infection led to massive cell death as shown by the activation of caspase-3 (cleaved Cas3) in both the cortex and hippocampus (Fig. 6b,c,e). Notably, NAD+ treatment suppressed cell death and significantly rescued cortex thinning induced by ZIKV infection (Fig. 6e,f), although it did not increase brain and body weight (Extended Data Fig. 7a,b). ZIKV infection is known to cause progressive activation of microglia and excessive immune responses, both of which are known to promote cell death25,49. We stained ZIKV-infected brain slices with antibodies for Iba-1, a marker for microglia and CD68, a marker for phagolysosome and activated microglia. As expected, many cells stained positive for both Iba-1 and CD68 were detected in the cortex and hippocampus of ZIKV-infected mouse brains (Fig. 6g). Notably, in brains that received NAD+ supplementation, the number of CD68+ cells was significantly reduced in the hippocampus (Fig. 6h), although no such reduction was observed for the cortex (Fig. 6i), suggesting that NAD+ supplementation can prevent progressive activation of microglia induced by ZIKV infection. However, NAM supplementation did not inhibit the replication of ZIKV (Extended Data Fig. 7c), alleviate cell death in the cortex and hippocampus in ZIKV-infected mice (Extended Data Fig. 7d,e) or increase brain and body weight (Extended Data Fig. 7f,g).
We next sought to investigate how NAD+ levels change in the brain after treatment with NAD+/NAM. Notably, NAD+ injection led to a marked increase in NAD+ levels in the homogenous extract of cortex and hippocampus at 6 h after NAD+ injection (Extended Data Fig. 8a), whereas NAM injection lowered NAD+ levels at 2 h after NAM injection, although a significant increase in NAM levels was observed (Extended Data Fig. 8b). Therefore, the beneficial effects of NAD+ supplementation may possibly be attributed to elevated levels of NAD+ in brains. In addition, by culturing primary isolated neurons with 1 mM NAD+ for 0.5 h to 12 h, we found that the level of NAD+ was significantly increased in cells (Extended Data Fig. 8c) and levels of its precursors (NR, NMN and NAM) were dramatically increased in both cells and culture medium, concomitantly with a decreasing trend in levels of NAD+ in the culture medium (Extended Data Fig. 8c,d), suggesting that elevated levels of NAD+ in cells were most likely achieved via NAD+-derived precursors that can enter cells to resynthesize NAD+ intracellularly; however, we could not exclude the possibility that a small proportion of intact NAD+ might have entered the neurons. Collectively, these data establish that supplementation with NAD+ does elevate its level and confer protective effects against ZIKV-induced cell death in mouse brains.
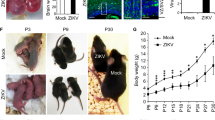
NR supplementation protects brains and improves survival in ZIKV-infected mice
NR, a widely utilized NAD+ precursor, has been demonstrated to elevate NAD+ levels in tissues in mice and humans50,51,52,53. To explore the effects of NR against ZIKV-induced microcephaly in a manner that better mimics the pathology and treatment strategy in humans52, we treated pregnant mice with NR in drinking water from E13.5 (2 d before infecting embryos with ZIKV on E15.5) to P0 and thereafter, their neonates with NR intraperitoneally once a day starting on P0 (Fig. 7a). Our data revealed that although NR supplementation did not affect replication of ZIKV (Fig. 7b,c), it significantly reduced cell death induced by ZIKV infection in mouse brains (Fig. 7b,d). More notably, NR treatment significantly increased cortex thickness (Fig. 7e), brain weight (Fig. 7f) and body weight (Fig. 7g), as well as improved survival of ZIKV-infected mice (Fig. 7h). Collectively, our results demonstrate that NR confers protective effects against ZIKV-induced microcephaly at both molecular and systemic levels.

a, Overview for the study of NR supplementation. Pregnant mice were supplemented with (ZIKV + NR) or without (ZIKV + Veh.) NR in drinking water (12 mM) starting on E13.5. Two days later on E15.5, embryos received a ZIKV injection. Pregnant mice were continuously supplemented with or without NR in drinking water until the day of birth (P0). Born littermate neonates were continuously treated with (ZIKV + NR) or without (ZIKV + Veh.) NR (200 mg kg−1) once a day from P0. For mice grouped for monitoring of lifespan, pups received treatment until death. For mice grouped for checking brain impairment, pups were killed on P6 and brains were collected, weighed and examined for immunostaining. Body weights of pups were measured from P0 to the day before being killed (P5). i.p., intraperitoneal. b, Immunostaining images of ZIKV (green), DAPI (blue) and cleaved Cas3 (red) on ZIKV-infected and mock-infected brains. Scale bar, 50 μm. Data are representative of two independent experiments. c,d, Relative ZIKV (c) and cleaved Cas3 (d) intensity in cortex. e, Quantification of thickness of cortices in mock- and ZIKV-infected brains. n = 5 for Ctrl + NR and ZIKV + Veh. groups, n = 4 for Ctrl + Veh. and ZIKV + NR groups; three slices of each mouse brain were used (c–e). f, Brain weight of Veh.- (n = 5) and NR-treated (n = 4) pups. g, Body weight of Veh.- (n = 36) and NR-treated (n = 39) pups. h, Survival of Veh.- (n = 31) and NR-treated (n = 35) pups. All values are presented as mean ± s.e.m. Two-tailed unpaired Student’s t-test (c–g) and log-rank test (h) were used for statistical analysis. Exact P values are indicated.
We then set out to assess the metabolic alterations in mouse brains upon NR supplementation. Heat map plotting showed that metabolic profiles of ZIKV-infected mouse brains were different from those of mock-infected mouse brains (Extended Data Fig. 9a). ZIKV-infected mouse brains from vehicle and NR-treated groups were separated by partial least squares discriminant analysis (PLS-DA) (Extended Data Fig. 9b). Differential metabolites (variable importance in projection (VIP) > 1) between vehicle- and NR-treated groups were enriched to nicotinate and nicotinamide metabolism, purine metabolism, pyrimidine metabolism and several metabolic pathways related to amino acid metabolism (Extended Data Fig. 9c). Furthermore, we performed absolute quantifications of NAD+ and its precursors involved in nicotinate and nicotinamide metabolism in mock- and ZIKV-infected mouse brains. Our results showed that NAD+ and its precursors (NAM and NMN) were significantly upregulated after NR supplementation (Extended Data Fig. 9d–g). Notably, the dramatic decline in NAD+ levels in ZIKV-infected mouse brains was significantly elevated after NR supplementation (Extended Data Fig. 9d). In addition, perturbations of several other metabolites induced by ZIKV infection were attenuated with NR supplementation. More specifically, several upregulated metabolites in ZIKV-infected mouse brains, such as choline, guanosine, 2ʹ-deoxycytidine and 2ʹ-deoxyadenosine, were reduced by NR supplementation (Extended Data Fig. 9h), whereas downregulated metabolites, such as fructose 1,6-biphosphate, ADP, γ-aminobutyric acid and serotonin, were elevated upon NR treatment (Extended Data Fig. 9i). Taken together, NR supplementation rescues the decline in NAD+ levels, corrects metabolic perturbations in ZIKV-infected mouse brains and confers protective effects against ZIKV-induced microcephaly.
Discussion
Since 2016, ZIKV-induced microcephaly has emerged as a major threat to newborns2; however, the mechanisms underlying ZIKV-induced microcephaly are largely unknown, highlighting an urgent need to expand our knowledge of its pathogenesis. This study systematically investigates ZIKV-induced microcephaly in vivo based on integrative analysis of transcriptomics, proteomics, phosphoproteomics and metabolomics data. Although ideally all omics data should be derived from the same sample, this was not feasible due to the limited amount of sample and incompatible sample preparation protocols for RNA, protein, phosphoproteins and metabolites. Therefore, we performed proteomics, phosphoproteomics and metabolomics on the same sample, but employed transcriptomics data generated from a published study of ours22,54 that was performed under exactly the same conditions as those of the current study. Thus, the four omics data in our study are completely comparable. Our multi-omics analysis implicated various biological events in mice with ZIKV-induced microcephaly, highlighting, for example, the activation of key signaling pathways and extensive metabolic reprogramming.
We and others have reported ZIKV-related alteration of immune responses5,54 and our current study identified upregulated immune responses in ZIKV-infected mouse brains through transcriptomics and proteomics, underscoring the utility of the multi-omics dataset. Viruses are known to reprogram host cell metabolism to support rapid viral replication and such reprogramming can lead to bioenergetic stress and apoptosis for host cells7,12,15. More specifically, bioenergy depletion caused by mitochondrial dysfunction has been reported to contribute to neuronal cell death29,31. Notably, our integrated pathway analysis of proteomics and metabolomics data revealed dramatic alterations of metabolic pathways, including significantly downregulated OXPHOS and TCA cycle, indicating mitochondrial dysfunction in ZIKV-infected brains. A recent study revealed that ZIKV infection can lead to mitochondrial damage and oxidative-stress-induced apoptosis in astrocytes, cells known to play important roles in neural development55. Thus, our data suggest the possibility that alterations of mitochondrial metabolic pathways may cause ZIKV-induced microcephaly in vivo through promoting neural cell apoptosis. Dysregulated host cholesterol metabolism has been known to facilitate viral entry, virus particle assembly and release and to modulate immune response during virus infection56,57,58. Another recent study has revealed that the SARS-CoV-2 S protein binds to cholesterol and high-density lipoprotein (HDL) components to enhance SARS-CoV-2 attachment and entry59. Notably, in our study, we identified significant upregulation of components in cholesterol metabolism, such as APOA1, APOA4, APOC3, APOE and NPC2 upon ZIKV infection. Moreover, our previous work has demonstrated that 25-hydroxycholesterol, an intermediate of cholesterol metabolism, exhibited protective effects against microcephaly caused by ZIKV infection60. These findings suggest that cholesterol metabolism reprogramming may be a potential therapeutic target for ZIKV infection.
NAD+ serves as a critical coenzyme or a co-substrate for enzymes involved in various metabolic reactions, such as bioenergy homeostasis61. Targeted manipulation of NAD+ metabolism has been confirmed as a successful therapeutic approach for treating several diseases, including cancer62, aging63, and neurodegeneration61,64. Notably, our multi-omics analysis revealed a significant alteration in metabolite levels involved in NAD+ metabolism in ZIKV-infected brains. More specifically, we observed that the level of NAD+ was dramatically decreased, possibly owing to downregulation of the key enzyme NMNAT2 in the NAD+ salvage pathway. Considering that PARP isozymes require NAD+ as a substrate65, upregulation of PARPs in ZIKV-infected brains may also contribute to decreases in NAD+ level. A recent study revealed that SARS-CoV-2 infection upregulates PARP expression and reprograms NAD+ metabolism, suggesting the potential of an NAD+-boosting strategy in activating innate immunity to coronaviruses35. The increased NAD+ level enhances mitochondrial function in different model organisms33,66, which is consistent with the observations in our study, with significantly reduced levels of NAD+ and downregulated OXPHOS and TCA cycle metabolism. These results emphasize the possibility that NAD+ depletion caused bioenergy deficiency and that mitochondrial dysfunction may contribute to cell death in ZIKV-induced microcephaly.
Emerging evidence supporting the critical role of NAD+ metabolism in aging and neurodegenerative has triggered an interest in approaches to boost NAD+ levels for treatment63,66,67,68,69. Notably, NAD+ supplementation has shown a protective role in the brain and nervous system in vivo44,70,71. In addition, considering the neuroprotective effects of NR and NAM in Alzheimer’s disease model mice45,46, we introduced NAD+ and its precursors, NAM and NR, as NAD+-boosting strategies in our study. NAD+ supplementation significantly increased NAD+ level and suppressed ZIKV-induced cell death in the cortex and hippocampus, although higher levels of ZIKV in the cortex were observed, which might be attributed to improved survival of neural cells that provide a more-parasitic basis for ZIKV replication. In addition, NAD+ supplementation prevents progressive activation of microglia in the hippocampus. Microglia, the brain’s specialized phagocytes, can remove dead, dying, damaged or infected neurons and synapses during brain development and inflammation72. Thus, the observed reduction of microglia activation may be attributed to amelioration of ZIKV-induced cell death upon NAD+ supplementation; however, NAM supplementation showed no protective effects in ZIKV-infected brains, possibly because it failed to increase levels of NAD+ in brains after NAM injection, which is in line with previous findings that NAM treatment depresses NAM salvage and does not produce a net boost in tissue NAD+ levels73. Our and others’ results underline the complex pharmacological effects of NAM acting as both an NAD+ precursor and sirtuin inhibitor.
Of note, NR supplementation elevated the level of NAD+ in brains, corrected metabolic perturbations, protected brains and improved survival in ZIKV-infected mice. Nmrk2, encoding nicotinate riboside kinase (NRK), which converts NR to NMN, was dramatically upregulated by nearly 150-fold at the mRNA level after ZIKV infection (Extended Data Fig. 9j). In contrast, expression of Nampt, encoding the NAMPT enzyme that converts NAM to NMN, was upregulated by merely twofold after ZIKV infection (Extended Data Fig. 9j). This gene expression pattern (dramatic upregulation of Nmrk2 and slight upregulation of Nampt) may suggest that ZIKV-infected mouse brains were more inclined to mobilize and utilize NR as an NAD+ precursor rather than increasing NAM usage, thus leading to differential effects of NR and NAM supplementation on ZIKV-induced microcephaly, although we could not exclude the possibility of other unexplored underlying mechanisms.
Previous studies have identified that several signaling pathways are altered in ZIKV infection, such as AKT–mTOR, AMPK, MAPK–ERK and ATM–ATR6,7,74. Here, our phosphoproteomics analysis identified that MAPK signaling is activated in ZIKV-induced microcephaly. The increase in p38/MAPK activity has been reported to regulate proinflammatory cytokine production and viral replication in influenza virus and SARS-CoV-2 infections75,76. Notably, MAPK signaling was also found to regulate NAD+ metabolism via NMNAT2, thereby promoting axon degeneration77,78. Given the findings of downregulated levels of NAD+ and NMNAT2, we speculate that the MAPK–NMNAT2–NAD+ axis may have a crucial neuron-specific impact in the pathogenesis of ZIKV-induced microcephaly, thus its functional contributions and therapeutic potential is worthy of further investigation in future studies.
In summary, our study strongly underscores the value of multi-omics approaches for studying diseases and represents a major step forward toward a deep mechanistic understanding of the cellular and molecular basis of ZIKV-induced microcephaly. Our work also indicated multiple attractive therapeutic targets. To illustrate how the rich datasets we obtained can facilitate hypothesis-driven applications, we tested our hypothesis that considers NAD+ metabolic reprogramming as a pathogenic driver of ZIKV-induced microcephaly by conducting in vivo experiments that confirmed the beneficial effects of boosting NAD+ on reducing ZIKV-induced cell death and tissue impairment and improving survival. Our study thus uncovered fundamental insights related to both viral infection and pathogenesis of ZIKV-induced microcephaly and thereby provided potential therapeutic targets.
Methods
Mice
In vivo studies were performed in pregnant ICR mice (8–10 weeks old) and their pups (including males and females) after birth. Mice were obtained from the Beijing Vital River Laboratory Animal Technology. Pregnant mice were kept in separate cages and maintained on a 12-h light–dark cycle throughout the experiment at 21–23 °C and humidity of 30–60%. Experimental procedures were performed according to protocols approved by the Institutional Animal Care and Use Committee (issue no. AP2020021) at Institute of Genetics and Developmental Biology, Chinese Academy of Sciences.
Virus strains
ZIKV, SZ-SMGC-1 (ref. 79) and SZ01 (GenBank accession no. KU866423), were obtained from Y. Shi (Institute of Microbiology) and C. Qin (Beijing Institute of Microbiology and Epidemiology), respectively.
Cell experiments
Neurons were prepared from the cortex and hippocampus of fetal (E18.5) mice as previously described80. Cells were cultured in six-well plates (seeding density, 1.0 × 105 cells per well) in Neurobasal medium (Gibco, cat. no. 21103049) with 2% B-27 (Gibco, cat. no. 17504001) and 1% Glutamax supplement (Gibco, cat. no. 35050079). The medium was exchanged by 50% with fresh culture medium every 3 d. Experiments were conducted when neurons were 15 d in vitro at 80% confluence.
After incubation in culture medium supplemented with 1 mM NAD+ for 0, 0.5, 1, 2, 6 and 12 h, cells were collected and extracted with cold 80% methanol in water (containing D4-NAM as an internal standard), medium was collected and extracted by adding cold methanol (containing D4-NAM as an internal standard) to achieve a final concentration of 80% methanol, followed by vortexing and centrifugation. The supernatant was evaporated to dryness. NAD+ and its precursors (NAM, NR and NMN) were quantified by LC–MS/MS-based targeted metabolomics analysis.
Mouse procedures
For proteomics, phosphoproteomics and metabolomics analysis with the P3 model (infected on E15.5 and analyzed on P3), 650 plaque-forming units (p.f.u.) of ZIKV SZ-SMGC-1 virus stock (2 × 106 p.f.u. ml−1)81 or culture medium (RPMI medium 1640 basic + 2% FBS) was injected into one side of the LV of embryonic mouse brains on E15.5 as described previously5. Mock- and ZIKV-infected mice were killed on P3 and the injected-side brains were subjected to proteomics, phosphoproteomics and metabolomics analyses. Transcriptomics experiment was described in our previously published paper22.
For proteomics, phosphoproteomics and metabolomics experiments with E18.5 model (infected on E13.5 and analyzed on E18.5), 650 p.f.u. of ZIKV SZ-SMGC-1 virus stock (2 × 106 p.f.u. ml−1) or culture medium (RPMI medium 1640 basic + 2% FBS) was injected into one side of the LV of embryonic mouse brains on E13.5. Mock- and ZIKV-infected mice were killed on E18.5 and injected-side brains were subjected to proteomics, phosphoproteomics and metabolomics analyses.
For NAD+ and NAM supplementation experiments, 500 p.f.u. of ZIKV SZ-SMGC-1 virus stock (2 × 106 p.f.u. ml−1) or culture medium (mock) were injected into the λ point of pup brains on P0. Six days later (P6), we injected NAD+ (500 μM, 2 μl, Sigma-Aldrich, cat. no. N7004) or NAM (500 μM, 2 μl, Sigma-Aldrich, cat. no. 72340) or culture medium (mock) into the λ point of mock- or ZIKV-infected mice once a day for three consecutive days (P6–8). Mock- and ZIKV-infected mice were weighed and killed on P9 and brains were weighed and subjected to immunocytochemistry analysis.
For assessment of time-dependent alterations after NAD+ or NAM supplementation, 500 p.f.u. of ZIKV SZ-SMGC-1 virus stock (2 × 106 p.f.u. ml−1) or culture medium (mock) were injected into the λ point of ICR pup brains on P0. Nine days later (P9), we injected NAD+ or NAM or culture medium (mock) into of the λ point of mock- or ZIKV-infected mice 0, 1, 2, 6 and 12 h before the end point, when mice were killed and brains were subjected to NAD+ and NAM quantification analyses.
For the NR supplementation experiment, pregnant mice (n = 4 per group) were supplemented with or without NR (Selleck, cat. no. S2935) in drinking water (12 mM) starting on E13.5. Two days later on E15.5, embryos received a ZIKV injection as follows: 500 p.f.u. of ZIKV SZ-SMGC-1 virus stock (2 × 106 p.f.u. ml−1)81 or culture medium (mock) was injected into one side of the LV of the embryonic mouse brains on E15.5 as described previously5. Pregnant mice were continuously supplemented with or without NR in drinking water until the day of birth (P0). The born littermate neonates were continuously treated with vehicle (PBS) or NR (200 mg kg−1) i.p. once a day from P0. For mice grouped for monitoring of lifespan, pups received treatment until death. For mice grouped for checking brain impairment and metabolomics analyses, pups were killed 6 h after the last dosing of NR on P6 and brains were collected, weighed and examined for immunostaining and NAD+-related metabolite quantification, as well as metabolomics. Body weights of pups were measured from P0 to the day before being killed (P5).
Sample preparation for metabolomics, proteomics and phosphoproteomics
Metabolite extraction
Each mouse brain sample was accurately weighed and homogenized in 80% methanol in water (50 mg tissue ml−1 solution) and vortexed for 1 min. Then, 200 µl of homogenate was transferred into a tube pre-added with 800 µl of ice-cold 80% methanol in water, vortexed for 1 min, centrifuged at 14,000 r.p.m. for 15 min. Then, 900 µl of metabolite-containing supernatant was transferred into a new tube and 500 µl of 80% methanol was added to the pellet, followed by vortexing for 1 min and centrifugation at 14,000 r.p.m. for 15 min. Then, 500 µl of the metabolite-containing supernatant was transferred to combine with the previous extract in the tube. Supernatant was evaporated to dryness under a speed vacuum concentrator. Dried metabolite pellets were kept at −80 °C until analysis and remaining precipitates were used for proteomics study.
Protein extraction and trypsin digestion
Protein was precipitated by 80% methanol as per metabolome methods and lysed in lysis buffers (1% sodium deoxycholate, 10 mM 2-carboxyethyl phosphine, 40 mM 2-chloroacetamide and 100 mM 2-carboxyethyl, pH 8.5) containing protease and phosphatase inhibitors (Thermo Scientific) followed by sufficiently vortexing and heating at 95 °C for 5 min. Centrifugation was performed at 16,000g for 10 min and the supernatant was transferred to a new centrifuge tube. Protein concentration was determined by Nanodrop One. The supernatant was mixed with trypsin in a ratio of protein to trypsin (50:1) and digested at 37 °C for 4 h. Finally, this lysate was added to formic acid to 1% and centrifuged at 16,000g for 10 min to remove the white precipitate. Supernatant desalination was accomplished using a homemade C18 tip. Peptide mixtures were concentrated by a speed vacuum concentrator at 60 °C.
Enrichment of phosphopeptides
Ten percent of the peptide mixtures was used for determination of total proteome and remaining peptide mixtures (90%, ~100 μg) were processed for phosphopeptide enrichment. Binding buffer (80% acetonitrile, 5% trifluoroacetic acid (Sigma-Aldrich) and 1 M lactic acid (Sigma-Aldrich)) was added to peptide mixtures followed by vortexing and centrifugation at 16,000g for 10 min. Supernatant was transferred to a new centrifuge tube with addition of 3 mg TiO2 beads. Incubation was processed at room temperature for 30 min on the rotor with a middle speed. Then TiO2 beads were collected by centrifugation at 2,000g for 2 min and supernatant phosphopeptide enrichment was performed twice with new TiO2 beads. TiO2 beads were collected and washed in a clear tube with wash buffer (50% acetonitrile and 5% trifluoroacetic acid (Sigma-Aldrich)) five times. Afterward, beads were transferred into a homemade C18 tip (0.2-ml StageTip with two pieces of C18) and followed by washing twice. Phosphorylated peptides were eluted five times with elution buffer (30% acetonitrile and 18% aqua ammonia). Peptide samples were concentrated by a speed vacuum concentrator at 60 °C.
Metabolomics
Separation was achieved on an RP-UPLC column (HSS T3, 2.1 mm × 150 mm, 1.8 μm, Waters) using a Nexera ultra-high performance liquid chromatograph system (Shimadzu Corporation). The mobile phases employed were 0.03% formic acid in water (A) and 0.03% formic acid in acetonitrile (B). The gradient program was as follows: 0–3 min 99% mobile phase A; 3–15 min, 99–1% A; 15–17 min, 1% A; 17–17.1 min, 1–99% A; 17.1–20 min, 99% A. The column was maintained at 35 °C and samples were kept in the autosampler at 4 °C. The flow rate was 0.25 ml min−1 and injection volume was 10 µl.
The mass spectrometer was an SCIEX QTRAP 6500+ (SCIEX) with electrospray ionization source in multiple reaction monitoring (MRM) mode. Sample analysis was performed in positive/negative switching mode. The MRM transition (m/z), declustering potential (DP) and collision energy of each metabolite were optimized using the standard compound. The MS detector conditions were set as follows: curtain gas 30 psi; ion spray voltages 4,500 V (positive) and −3,500 V (negative); temperature 550 °C; ion source gas 1 50 psi; ion source gas 2 50 psi; entrance potential 10 V (positive) and −10 V (negative). In total, around 260 endogenous metabolites were monitored. MRM data were acquired using Analyst v.1.6.3 software (SCIEX).
Quantification of NAD+, NAM, NR and NMN
To quantitatively measure concentrations of NAD+, NAM, NR and NMN in primary neurons, culture medium and mouse brains, 5 pmol D4-nicotinamide (on-column amounts) (CIL, cat. no. 6883) was used as an internal standard. Chromatographic separation and mass spectrometric detection were the same as described in the Metabolomics section above. The concentrations of analytes were calculated by peak area ratios of the analytes to internal standard using standard curves generated with serial concentrations of each authentic standard prepared in water.
Proteomics and phosphoproteomics
Peptides were resuspended in 10 µl buffer containing 0.1% formic acid (10% for phosphorylated peptides) for LC–MS/MS analysis. Profiling and phosphorylated peptide samples were detected by Orbitrap Fusion Lumos (Thermo Fisher Scientific) mass spectrometer coupled to an Easy-nLC1000 liquid chromatography system (Thermo Fisher Scientific). A 50 °C thermostatic column incubator was used. Then, 5 µl of peptide solution was eluted from a homemade 100-μm ID × 2 cm C18 trapping column and separated on a homemade 150-μm ID × 30 cm column (C18, 1.9 μm, 120 Å, Dr. Maisch GmbH) with a 150-min linear 5–40% acetonitrile gradient at 600 nl min−1. The dynamic exclusion of previously acquired precursor ions was enabled at 25 s. For one full scan, the automatic gain control target was 5 × 105 ions. For MS/MS scans, the automatic gain control target was 5 × 103 ions and max injection time was 35 ms.
Immunohistochemistry
For immunostaining of brains, mice were perfused with PBS followed by 4% paraformaldehyde and brains were post-fixed in 4% paraformaldehyde at 4 °C overnight. Brains were dehydrated in 30% sucrose for 2 d and frozen in tissue-freezing medium. Sections (thickness 40 μm) were blocked with blocking buffer (PBS + 10% FBS + 3% BSA + 0.2% TritonX100) at room temperature for 1 h and incubated with the first antibody at 4 °C overnight. After washing with PBST for 10 min three times, brain sections were incubated with fluorescence-conjugated secondary antibodies at room temperature for 1 h, followed by PBST washing for 10 min three times. The antibodies used in immunostaining were as follows: cleaved Cas-3 (CST, cat. no. 9664s, 1:500 dilution), ZK2B10 (ref. 47) (1:2,000 dilution), Iba-1 (Abcam, cat. no. ab5076, 1:1,000 dilution), CD68 (Abcam, cat. no. 125212, 1:1,000 dilution), DAPI (CST, cat. no. 4083s). Immunostaining slices were imaged on LSM 700 (Carl Zeiss) confocal microscope and analyzed with ZEN2010 and ImageJ, as described previously5,49.
Western blotting
Brain tissues were prepared in RIPA buffer. Samples were heated at 95 °C for 5 min in loading buffer and separated with SDS–PAGE using a 10% gel and then transferred to a PVDF membrane. After blocking with 5% skim milk in TBS-Tween (TBST), membranes were incubated with primary antibodies, α-Tubulin (1:2,000 dilution, CST, cat. no. 3873s), NAMPT (1:1,000 dilution, a gift from G. Wang), NMNAT2 (1:1,000 dilution, a gift from W. Zeng), ERK (1:1,000 dilution, CST, cat. no. 4695), phospho-ERK (1:1,000 dilution, CST, cat. no. 4370), JNK (1:1,000 dilution, CST, cat. no. 9252), phospho-JNK (1:1,000 dilution, CST, cat. no. 4668), p38 (1:1,000 dilution, CST, cat. no. 8690) and phospho-p38 (1:1,000 dilution, CST, cat. no. 4511), overnight at 4 °C. After washing in TBST, blots were incubated with secondary antibodies for 2 h at room temperature and developed using enhanced chemiluminescence (Thermo Fisher Scientific), according to the manufacturer’s instructions.
Multi-omics analysis
RNA-seq data analysis
For transcriptomics, we revisited datasets published in a paper of ours22,54, which was acquired under the same conditions as those in the current study, including the used virus strain, mouse model, viral dosages and time points of sample collection. Briefly, 1 μl of ZIKV SZ01 (GenBank accession no. KU866423) virus stock (6.5 × 105 p.f.u. ml−1) or culture medium (RPMI medium 1640 basic + 2% FBS) was injected into one side of the LV of E15.5 mouse brains. Half of the P3 brains (injected side) from the mock- and ZIKV-infected mice (n = 3 for each group) were used for global transcriptomics analysis. Sequencing and expression analysis were conducted by Beijing Institute of Genomics, Chinese Academy of Sciences. Total RNA for each sample was sequenced with HiSeq3000. After preprocessing, high-quality reads were compared with the mouse reference genome (GRCm38.p4) using TopHat2 (ref. 82).
Metabolomics data analysis
For metabolomics analysis, half of the E18.5 (n = 8 for control group and n = 7 for ZIKV group) or P3 (n = 8 for each group) brains (injected side) from the mock- and ZIKV-infected mice were used. Chromatogram review and peak area integration were performed using MultiQuant software v.3.0.2 (SCIEX). Each detected metabolite was normalized to the sum of total peak area from all detected metabolites within that sample to correct any variations introduced from sample handling through instrument analysis. The normalized area values were used as variables for multivariate and univariate statistical data analysis.
Proteomics and phosphoproteomics data analysis
For proteomics and phosphoproteomics analyses, half of the E18.5 (n = 3 for each group) or P3 (n = 8 for each group) brain (injected side) for the mock- and ZIKV-infected mouse brains were used. Raw data files were searched against the mouse protein RefSeq database (containing 76,770 entries) added with different protein isoforms in MaxQuant software (v.1.5.3.30) to achieve an FDR < 0.01. The specificity of the enzyme was set to ‘trypsin’ and peptide identification required at least seven amino acids. For proteomics data, oxidation (M) and acetyl (protein-N term) were chosen as variable modifications and carbamidomethyl (C) was set as fixed modification. For phosphoproteomics data, oxidation (M), acetyl (protein-N term) and phospho (STY) were chosen as variable modifications and carbamidomethyl (C) was set as fixed modification. We used MaxQuant LFQ algorithm to quantitate MS signals and protein intensities were represented in iFOT (for each sample, iFOT = iBAQ/∑[iBAQall]).
Principal-component analysis of proteomics and metabolomics data
We performed PCA to illustrate proteomics and metabolomics differences between mock- and ZIKV-infected brains, respectively. The prcomp function under R package stats was implemented on the abundance matrixes of detected proteins and global metabolites. The 95% confidence coverage was represented by colored ellipses, and was calculated based on the mean and covariance of points in each group.
Gene set enrichment analysis
To further explore the biological characteristics of transcriptomics and proteomics datasets in mock- and ZIKV-infected brains, we performed GSEA to recognize altered pathways between two groups. Gene expression data of mRNA and protein levels were used to obtain NES over the active GO biological processes using GSEA software (v.3.0) (http://software.broadinstitute.org/gsea/index.jsp)83. GO terms with unadjusted P < 0.01 at the mRNA level were considered significant and plotted with NES values for mRNA and protein. GO gene sets belonging to Mus musculus used in this article were integrated from AmiGO database (http://amigo.geneontology.org/amigo/landing).
Cell-type-based RNA and protein deconvolution analyses
Cell-type-based RNA and protein deconvolution analyses were performed as follows. First, we confirmed brain cell markers for microglial cells, neural stem cells, astrocytes, neuroblasts and neurons from ‘CellMarker’ database (http://biocc.hrbmu.edu.cn/CellMarker/index.jsp)84. Second, relative RNA or protein abundance of five types of brain cells in mock- and ZIKV-infected brains was assessed by ES using R package GSVA (v.1.34.0)85. GSVA was performed with the following parameters: mx.diff = FALSE, method = ‘gsva’, verbose = TRUE, parallel.sz = 0, min.sz = 1 and max.sz = Inf. ESs were normalized ranging from −1 to 1. Finally, the ES value was used to estimate cell-type abundance from bulk tissue transcriptomes and protein profiles23.
Deconvolution of metabolic protein changes in individual cell types
Deconvolution analysis was also used to determine metabolic changes in the indicated five cell types, including microglial cells, neural stem cells, astrocytes, neuroblasts and neurons, in mock- and ZIKV-infected brains based on our proteomics dataset. First, we extracted markers of various brain cells, including microglial cells, neural stem cells, astrocytes, neuroblasts and neurons, from the ‘CellMarker’ database84 and proteins participated in ‘Metabolism’ pathways from the KEGG pathway database. Second, we calculated a pairwise Spearman correlation coefficient between protein levels of brain cell markers and levels of metabolic proteins. Third, protein pairs with a correlation coefficient >0.8 were used to construct a protein–protein interaction network using Cytoscape (v.3.7.2)86. Finally, enrichment analysis of metabolic proteins participated in metabolism pathways were performed using the R package clusterProfiler (v.3.12.0) in individual cell types. Pathways with P value <0.05 were considered significant. A similar approach was adopted in previous studies and protein–protein interaction correlation studies proved the effectiveness of this type of approach87.
Differential analysis and pathway enrichment analysis
Two-tailed unpaired Student’s t-tests followed by Benjamini–Hochberg FDR correction was performed to compare ZIKV-infected mouse brains versus mock-infected samples. Differential mRNAs, proteins, phosphoproteins between mock- and ZIKV-infected mouse brains were filtered with the restricted condition: FDR < 0.05 and fold change >2 (ref. 88). Subsequently, KEGG pathways and GO biological processes enriched in ZIKV-infected and mock-infected brains were calculated by R package clusterProfiler (v.3.12.0)89. Pathways were considered significant with FDR < 0.05 in proteins and P < 0.05 in phosphoproteins. Metabolites with significant differences (FDR < 0.05) between two groups were used for KEGG enrichment analysis using MetaboAnalyst v.4.0 (http://www.metaboanalyst.ca/). Metabolic pathways with P < 0.05 were considered significant.
Kinase activity analysis
Phosphorylated peptides for unique sequence motifs (±15 amino acids from the phosphorylated site) were normalized by iFOT values. Kinase activities were predicted using NetPhos 3.1 Server (http://www.cbs.dtu.dk/services/NetPhos/) based on differential phosphorylated peptides (FDR < 0.05 and fold change >2).
Drug target analysis
Drug targets approved by the US FDA were retrieved from the Drugbank database (v.5.1.1, released 2018-07-03) (https://www.drugbank.ca/). Target phosphoproteins that were upregulated (P < 0.05 and fold change >2) in ZIKV-infected mouse brains compared to mock-infected brains with potential curative drugs were chosen.
Statistical analyses
Details pertaining to the statistical analysis of omics can be found in ‘Multi-omics analysis’ above. For statistical analysis in mouse and cell experiments, differences between two groups were assessed using two-tailed unpaired Student’s t-tests and differences between more than two groups were assessed using one-way analysis of variance followed by Benjamini–Hochberg post hoc test. GraphPad Prism v.8.3.0 was used for all statistical analyses. P < 0.05 was considered as significant.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The RNA-seq data are published22,54 and can be downloaded from the Sequence Read Archive database, BioProjectID PRJNA358758. The MS proteomics data have been deposited in the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the iProX partner repository86 with dataset identifier PXD026814. Raw metabolomics data are included in Supplementary Data 1. Source data are provided with this paper.
Code availability
Codes for data analysis are available at https://github.com/pang2021/ZIKV_NMet.
References
Rostaing, L. P. & Malvezzi, P. Zika virus and microcephaly. N. Engl. J. Med. 374, 982–984 (2016).
Lessler, J. et al. Assessing the global threat from Zika virus. Science 353, aaf8160 (2016).
Heymann, D. L. et al. Zika virus and microcephaly: why is this situation a PHEIC? Lancet 387, 719–721 (2016).
Cugola, F. R. et al. The Brazilian Zika virus strain causes birth defects in experimental models. Nature 534, 267–271 (2016).
Li, C. et al. Zika virus disrupts neural progenitor development and leads to microcephaly in mice. Cell Stem Cell 19, 120–126 (2016).
Liang, Q. et al. Zika virus NS4A and NS4B proteins deregulate Akt-mTOR signaling in human fetal neural stem cells to inhibit neurogenesis and induce autophagy. Cell Stem Cell 19, 663–671 (2016).
Thaker, S. K. et al. Differential metabolic reprogramming by Zika virus promotes cell death in human versus mosquito cells. Cell Metab. 29, 1206–1216 (2019).
Olive, A. J. & Sassetti, C. M. Metabolic crosstalk between host and pathogen: sensing, adapting and competing. Nat. Rev. Microbiol. 14, 221–234 (2016).
Wang, A., Luan, H. H. & Medzhitov, R. An evolutionary perspective on immunometabolism. Science https://doi.org/10.1126/science.aar3932 (2019).
Li, X. K. et al. Arginine deficiency is involved in thrombocytopenia and immunosuppression in severe fever with thrombocytopenia syndrome. Sci. Translat. Med. https://doi.org/10.1126/scitranslmed.aat4162 (2018).
Lercher, A. et al. Type I interferon signaling disrupts the hepatic urea cycle and alters systemic metabolism to suppress T cell function. Immunity 51, 1074–1087 (2019).
Tisoncik-Go, J. et al. Integrated omics analysis of pathogenic host responses during pandemic H1N1 influenza virus infection: the crucial role of lipid metabolism. Cell Host Microbe 19, 254–266 (2016).
Shen, B. et al. Proteomic and metabolomic characterization of COVID-19 patient sera. Cell https://doi.org/10.1016/j.cell.2020.05.032 (2020).
Kilbourne, E. D. Inhibition of influenza virus multiplication with a glucose antimetabolite (2-deoxy-D-glucose). Nature 183, 271–272 (1959).
Bojkova, D. et al. Proteomics of SARS-CoV-2-infected host cells reveals therapy targets. Nature https://doi.org/10.1038/s41586-020-2332-7 (2020).
Xiao, N. et al. Integrated cytokine and metabolite analysis reveals immunometabolic reprogramming in COVID-19 patients with therapeutic implications. Nat. Commun. 12, 1618 (2021).
Eisfeld, A. J. et al. Multi-platform ‘omics analysis of human ebola virus disease pathogenesis. Cell Host Microbe 22, 817–829 (2017).
Gao, Q. et al. Integrated proteogenomic characterization of HBV-related hepatocellular carcinoma. Cell 179, 561–577 (2019).
Zhou, W. et al. Longitudinal multi-omics of host-microbe dynamics in prediabetes. Nature 569, 663–671 (2019).
Aid, M. et al. Zika virus persistence in the central nervous system and lymph nodes of rhesus monkeys. Cell 169, 610–620 (2017).
Caires-Junior, L. C. et al. Discordant congenital Zika syndrome twins show differential in vitro viral susceptibility of neural progenitor cells. Nat. Commun. 9, 475 (2018).
Chang, Y. et al. Different gene networks are disturbed by Zika virus infection in a mouse microcephaly model. Genom. Proteom. Bioinform. https://doi.org/10.1016/j.gpb.2019.06.004 (2021).
Newman, A. M. et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat. Biotechnol. 37, 773–782 (2019).
Vasaikar, S. et al. Proteogenomic analysis of human colon cancer reveals new therapeutic opportunities. Cell 177, 1035–1049 (2019).
Li, C. et al. Disruption of glial cell development by Zika virus contributes to severe microcephalic newborn mice. Cell Disco. 4, 43 (2018).
Chua, R. L. et al. COVID-19 severity correlates with airway epithelium-immune cell interactions identified by single-cell analysis. Nat. Biotechnol. 38, 970–979 (2020).
Blanco-Melo, D. et al. Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell https://doi.org/10.1016/j.cell.2020.04.026 (2020).
Osuna-Ramos, J. F., Reyes-Ruiz, J. M. & Del Angel, R. M. The role of host cholesterol during flavivirus infection. Front. Cell. Infect. Microbiol. 8, 388 (2018).
Fricker, M., Tolkovsky, A. M., Borutaite, V., Coleman, M. & Brown, G. C. Neuronal cell death. Physiol. Rev. 98, 813–880 (2018).
Zhu, H. Y. et al. Single-neuron identification of chemical constituents, physiological changes, and metabolism using mass spectrometry. Proc. Natl Acad. Sci. USA 114, 2586–2591 (2017).
Court, F. A. & Coleman, M. P. Mitochondria as a central sensor for axonal degenerative stimuli. Trends Neurosci. 35, 364–372 (2012).
Cervenka, I., Agudelo, L. Z. & Ruas, J. L. Kynurenines: tryptophan’s metabolites in exercise, inflammation, and mental health. Science https://doi.org/10.1126/science.aaf9794 (2017).
Canto, C., Menzies, K. J. & Auwerx, J. NAD(+) metabolism and the control of energy homeostasis: a balancing act between mitochondria and the nucleus. Cell Metab. 22, 31–53 (2015).
Liu, L. et al. Quantitative analysis of NAD synthesis-breakdown fluxes. Cell Metab. 27, 1067–1080 (2018).
Heer, C. D. et al. Coronavirus infection and PARP expression dysregulate the NAD metabolome: an actionable component of innate immunity. J. Biol. Chem. https://doi.org/10.1074/jbc.RA120.015138 (2020).
Wojcechowskyj, J. A. et al. Quantitative phosphoproteomics reveals extensive cellular reprogramming during HIV-1 entry. Cell Host Microbe 13, 613–623 (2013).
Eckel-Mahan, K. L. et al. Circadian oscillation of hippocampal MAPK activity and cAmp: implications for memory persistence. Nat. Neurosci. 11, 1074–1082 (2008).
Chen, H. et al. The bacterial T6SS effector EvpP prevents NLRP3 inflammasome activation by inhibiting the Ca(2+)-dependent MAPK–Jnk pathway. Cell Host Microbe 21, 47–58 (2017).
Etgen, A. M., Gonzalez-Flores, O. & Todd, B. J. The role of insulin-like growth factor-I and growth factor-associated signal transduction pathways in estradiol and progesterone facilitation of female reproductive behaviors. Front. Neuroendocrinol. 27, 363–375 (2006).
Ferguson, F. M. & Gray, N. S. Kinase inhibitors: the road ahead. Nat. Rev. Drug Disco. 17, 353–377 (2018).
Casado, P. et al. Kinase-substrate enrichment analysis provides insights into the heterogeneity of signaling pathway activation in leukemia cells. Sci. Signal 6, rs6 (2013).
O’Shea, J. P. et al. pLogo: a probabilistic approach to visualizing sequence motifs. Nat. Methods 10, 1211–1212 (2013).
Blom, N., Sicheritz-Ponten, T., Gupta, R., Gammeltoft, S. & Brunak, S. Prediction of post-translational glycosylation and phosphorylation of proteins from the amino acid sequence. Proteomics 4, 1633–1649 (2004).
Zhou, M. et al. Neuronal death induced by misfolded prion protein is due to NAD+ depletion and can be relieved in vitro and in vivo by NAD+ replenishment. Brain 138, 992–1008 (2015).
Hou, Y. et al. NAD(+) supplementation normalizes key Alzheimer’s features and DNA damage responses in a new AD mouse model with introduced DNA repair deficiency. Proc. Natl Acad. Sci. USA 115, E1876–E1885 (2018).
Liu, D. et al. Nicotinamide forestalls pathology and cognitive decline in Alzheimer mice: evidence for improved neuronal bioenergetics and autophagy procession. Neurobiol. Aging 34, 1564–1580 (2013).
Li, C. et al. A single injection of human neutralizing antibody protects against Zika virus infection and microcephaly in developing mouse embryos. Cell Rep. 23, 1424–1434 (2018).
Xu, D., Zhang, F., Wang, Y., Sun, Y. & Xu, Z. Microcephaly-associated protein WDR62 regulates neurogenesis through JNK1 in the developing neocortex. Cell Rep. 6, 104–116 (2014).
Zhang, F. et al. American strain of Zika virus causes more severe microcephaly than an old Asian strain in neonatal mice. EBioMedicine 25, 95–105 (2017).
Bieganowski, P. & Brenner, C. Discoveries of nicotinamide riboside as a nutrient and conserved NRK genes establish a Preiss–Handler independent route to NAD+ in fungi and humans. Cell 117, 495–502 (2004).
Belenky, P., Bogan, K. L. & Brenner, C. NAD+ metabolism in health and disease. Trends Biochem. Sci. 32, 12–19 (2007).
Ear, P. H. et al. Maternal nicotinamide riboside enhances postpartum weight loss, juvenile offspring development, and neurogenesis of adult offspring. Cell Rep. 26, 969–983 (2019).
Trammell, S. A. et al. Nicotinamide riboside is uniquely and orally bioavailable in mice and humans. Nat. Commun. 7, 12948 (2016).
Tonnerre, P. et al. Evolution of the innate and adaptive immune response in women with acute Zika virus infection. Nat. Microbiol. 5, 76–83 (2020).
Ledur, P. F. et al. Zika virus infection leads to mitochondrial failure, oxidative stress and DNA damage in human iPSC-derived astrocytes. Sci. Rep. 10, 1218 (2020).
Lindenbach, B. D. & Rice, C. M. The ins and outs of hepatitis C virus entry and assembly. Nat. Rev. Microbiol. 11, 688–700 (2013).
Mackenzie, J. M., Khromykh, A. A. & Parton, R. G. Cholesterol manipulation by West Nile virus perturbs the cellular immune response. Cell Host Microbe 2, 229–239 (2007).
Martín-Acebes, M. A., Vázquez-Calvo, Á. & Saiz, J.-C. Lipids and flaviviruses, present and future perspectives for the control of dengue, Zika, and West Nile viruses. Prog. Lipid Res. 64, 123–137 (2016).
Wei, C. et al. HDL-scavenger receptor B type 1 facilitates SARS-CoV-2 entry. Nat. Metab. https://doi.org/10.1038/s42255-020-00324-0 (2020).
Li, C. et al. 25-Hydroxycholesterol protects host against Zika virus infection and its associated microcephaly in a mouse model. Immunity 46, 446–456 (2017).
Katsyuba, E., Romani, M., Hofer, D. & Auwerx, J. NAD+ homeostasis in health and disease. Nat. Metab. 2, 9–31 (2020).
Chiarugi, A., Dolle, C., Felici, R. & Ziegler, M. The NAD metabolome: a key determinant of cancer cell biology. Nat. Rev. Cancer 12, 741–752 (2012).
Verdin, E. NAD(+) in aging, metabolism, and neurodegeneration. Science 350, 1208–1213 (2015).
Araki, T., Sasaki, Y. & Milbrandt, J. Increased nuclear NAD biosynthesis and SIRT1 activation prevent axonal degeneration. Science 305, 1010–1013 (2004).
Cohen, M. S. Interplay between compartmentalized NAD(+) synthesis and consumption: a focus on the PARP family. Genes Dev. 34, 254–262 (2020).
Katsyuba, E. et al. De novo NAD(+) synthesis enhances mitochondrial function and improves health. Nature 563, 354–359 (2018).
Lautrup, S., Sinclair, D. A., Mattson, M. P. & Fang, E. F. NAD in brain aging and neurodegenerative disorders. Cell Metab. 30, 630–655 (2019).
Covarrubias, A. J., Perrone, R., Grozio, A. & Verdin, E. NAD(+) metabolism and its roles in cellular processes during ageing. Nat. Rev. Mol. Cell Biol. 22, 119–141 (2021).
Gardell, S. J. et al. Boosting NAD(+) with a small molecule that activates NAMPT. Nat. Commun. 10, 3241 (2019).
Ying, W. et al. Intranasal administration with NAD+ profoundly decreases brain injury in a rat model of transient focal ischemia. Front. Biosci. 12, 2728–2734 (2007).
Xie, L. et al. Nicotinamide adenine dinucleotide protects against spinal cord ischemia reperfusion injury-induced apoptosis by blocking autophagy. Oxid. Med. Cell Longev. 2017, 7063874 (2017).
Brown, G. C. & Neher, J. J. Microglial phagocytosis of live neurons. Nat. Rev. Neurosci. 15, 209–216 (2014).
Mitchell, S. J. et al. Nicotinamide improves aspects of healthspan, but not lifespan, in mice. Cell Metab. 27, 667–676 e664 (2018).
Scaturro, P. et al. An orthogonal proteomic survey uncovers novel Zika virus host factors. Nature 561, 253–257 (2018).
Bouhaddou, M. et al. The global phosphorylation landscape of SARS-CoV-2 infection. Cell 182, 685–712 e619 (2020).
Meineke, R., Rimmelzwaan, G. F. & Elbahesh, H. Influenza virus infections and cellular kinases. Viruses https://doi.org/10.3390/v11020171 (2019).
Yang, J. et al. Pathological axonal death through a MAPK cascade that triggers a local energy deficit. Cell 160, 161–176 (2015).
Walker, L. J. et al. MAPK signaling promotes axonal degeneration by speeding the turnover of the axonal maintenance factor NMNAT2. eLife https://doi.org/10.7554/eLife.22540 (2017).
Yang, Y. et al. Development of a reverse transcription quantitative polymerase chain reaction-based assay for broad coverage detection of African and Asian Zika virus lineages. Virol. Sin. 32, 199–206 (2017).
Wang, S. et al. Sh3rf2 haploinsufficiency leads to unilateral neuronal development deficits and autistic-like behaviors in mice. Cell Rep. 25, 2963–2971 (2018).
Deng, Y. Q. et al. Isolation, identification and genomic characterization of the Asian lineage Zika virus imported to China. Sci. China Life Sci. 59, 428–430 (2016).
Kim, D. et al. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14, R36 (2013).
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA 102, 15545–15550 (2005).
Zhang, X. et al. CellMarker: a manually curated resource of cell markers in human and mouse. Nucleic Acids Res. 47, D721–D728 (2019).
Hanzelmann, S., Castelo, R. & Guinney, J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinf. 14, 7 (2013).
Ma, J. et al. iProX: an integrated proteome resource. Nucleic Acids Res. 47, D1211–D1217 (2019).
Luck, K. et al. A reference map of the human binary protein interactome. Nature 580, 402–408 (2020).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015).
Yu, G., Wang, L. G., Han, Y. & He, Q. Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16, 284–287 (2012).
Acknowledgements
We thank members of the Hu laboratory for critiquing the manuscript. We thank W. Zeng and G. Wang at Tsinghua for providing NMNAT2 and NAMPT antibodies. We thank Q. Ding at Tsinghua for providing advice on virology. We thank Y. Shi at Institute of Microbiology and C. Qin at Beijing Institute of Microbiology and Epidemiology for providing ZIKV stock. Some illustrations were created with BioRender.com. Z.H. is supported by grants from National Key R&D Program of China (2019YFA0802100-02), National Natural Science Foundation of China (92057209), National Science and Technology Major Project for ‘Significant New Drugs Development’ (2017ZX09304015), Tsinghua University (53332200517), Tsinghua-Peking Joint Center for Life Sciences and Beijing Frontier Research Center for Biological Structure. Z.X. is supported by grants from the National Natural Science Foundation of China (NSFC) (31730108, 31921002, 32061143026) and Chinese Academy of Science (QYZDJ-SSW-SMC007, XDB32020100, YJKYYQ20200052).
Author information
Authors and Affiliations
Contributions
H.P., Z.X. and Z.H. conceived the project and designed the study. H.P., M.N. and Z.H. wrote the paper. N.X., Y.J., J.L., Y.W., L.S., X.L. and Z.X. contributed to paper writing. H.P. and Z.H. designed and performed metabolomics, analyzed multi-omics data and interpreted results. Y.J. and H.P. designed and performed cell and animal experiments. J.L. and X.L. analyzed RNA-seq and multi-omics data. Y.W. and L.S. performed proteomics and phosphoproteomics and analyzed data. N.X. and K.Y. assisted in metabolomics data analysis. L.Y. assisted in cell and animal experiments. S.W. and Y.Z. assisted in animal experiments. Z.S. and F.J. assisted in RNA-seq data analysis. S.L. and P.L. assisted in multi-omics data analysis and interpretation. Y.C. and Z.X. provided the RNA-seq dataset. Z.H. supervised the project.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Metabolism thanks Patricia C. B. Beltrão-Braga, Charles Brenner and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editors: Pooja Jha; Isabella Samuelson.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Multi-omics workflow and quality assessments for transcriptomics, proteomics, phosphoproteomics, and metabolomics data.
a, General workflow of multi-omics experiments and data analysis. b-c, Number of detected proteins (b) and phosphorylated peptides (c) in mock- and ZIKV-infected brains. d, Abundance distributions of transcriptomics data in mock- and ZIKV-infected brains, with expression levels transformed to log10 (FPKM) (n = 3 per group). Data are shown as box plot, the bottom and top of the box are the first and third quartiles, and the band inside the box is the median of the log10 (FPKM). e-g, Pearson correlations of transcriptomics (e), proteomics (f), and phosphoproteomics (g) data between samples from mock- and ZIKV-infected brains. h, Overlap of detected proteins and phosphoproteins. 1,346 proteins were identified with 4,395 highly reliable phosphosites. 3,734 proteins were identified with non-phosphorylated forms (73.5% of all proteins). 581 proteins were identified with only their phosphorylated forms (30.1% of all phosphoproteins). i, Pearson correlations of metabolomics data between samples from mock- and ZIKV-infected brains.
Extended Data Fig. 2 Transcriptomics and proteomics analyses of mock- and ZIKV-infected brains.
a, Principal component analysis for the detected proteins in mock- and ZIKV-infected brains. b-c, Top 10 most significant GO terms enriched by significantly upregulated (red) and downregulated (blue) proteins (FDR < 0.05 and fold change > 2) in ZIKV-infected brains, respectively. d-e, Heatmap of significantly altered proteins (FDR < 0.05 and fold change > 2) related to cytokine response (d) and axon development (e). f, Inflammatory cytokine alterations induced by ZIKV infection at mRNA level (data from our transcriptomics datasets, n = 3 mice per group). Data are shown as mean ± s.e.m. Two-tailed unpaired t-test was used for statistical analysis. Exact P values are indicated. g, Cell type enrichment scores (ES) calculated by GSVA using proteomics data of ZIKV-infected brains and mock-infected controls. Enrichment scores demonstrating the relative abundance of distinct cell types were shown in orange (increased) and green (decreased).
Extended Data Fig. 3 Protein-protein interaction in distinct cell types and proteomic changes in ZIKV-infected brains on E18.5 and P3.
a, Correlations between cell type marker proteins and metabolic related proteins. Cell type specific KEGG metabolism pathways were enriched based on protein pairs with spearman correlation coefficient > 0.8 (P < 0.05). Cell type and related marker proteins were listed in the figure. b, Proteomic changes of OXPHOS and TCA cycle in E18.5 and P3 models upon ZIKV infection. Ratios of relative protein levels between ZIKV-infected (ZIKV) and mock-infected (Ctrl) mouse brains in E18.5 model (infected on E13.5 and analyzed on E18.5, left) and P3 model (infected on E15.5 and analyzed on P3, right) were shown. Altered proteins between mock- and ZIKV-infected mouse brains in either model were shown in the figure (p < 0.05).
Extended Data Fig. 4 Metabolic differences between mock- and ZIKV-infected brains on P3.
a, Hierarchical cluster analysis of metabolite abundance in ZIKV-infected brains (n = 8) and mock-infected controls (n = 8). b-c, Transcriptomic, proteomic, and metabolic analyses of purine (b) and pyrimidine metabolism (c). Protein and mRNA changes of metabolic enzymes in mock- and ZIKV-infected mouse brains were indicated. Metabolites significantly upregulated and downregulated in ZIKV-infected mouse brains were marked in red or blue color, respectively (FDR < 0.05).
Extended Data Fig. 5 Metabolic changes of mock- and ZIKV-infected brains on E18.5 and P3.
a, Hierarchical cluster analysis of metabolite abundance in ZIKV-infected brains (n = 7) and mock-infected controls (n = 8) on E18.5. b, Altered KEGG metabolic pathways in ZIKV-infected brains on E18.5 compared with mock-infected brains enriched by significantly altered metabolites (FDR < 0.05). c, Changes of NAD+ metabolism, tryptophan metabolism, and TCA cycle between E18.5 and P3 models upon ZIKV infection. Ratios of relative metabolite levels between ZIKV-infected (ZIKV) and mock-infected (Ctrl) mouse brains in E18.5 (left) and ratios in P3 model (right) were shown. Altered metabolites between mock- and ZIKV-infected mouse brains in either model were shown (P < 0.05).
Extended Data Fig. 6 Nominating potential druggable phosphoproteins for ZIKV-induced microcephaly.
a, iFOT values of altered phosphopeptides and corresponding proteins in MAPK pathway in ZIKV-infected (ZIKV) and mock-infected (Ctrl) brain samples (p < 0.05 and fold change > 2); linear regression model is used to fit scatter between ZIKV and Ctrl groups; shadow area represents 95% confidence interval of iFOT values in individual group. b, Expression of phosphorylated p38 and JNK in mock- and ZIKV-infected mouse brains (n = 3 per group). Data are representative of three independent experiments. Data are shown as mean ± s.e.m. Two-tailed unpaired t-test was used for statistical analysis. Exact P values are indicated. c, Major upregulated phosphoproteomic pathways and FDA-approved drugs targeting these signaling pathways. Significantly altered phosphoproteins (P < 0.05 and fold change > 2) in either E18.5 or P3 model involved in MAPK, calcium, cGMP-PKG, and cAMP signaling pathways were shown. d, Significantly altered kinases detected in phosphoproteomics data (P < 0.05 and fold change > 2). e, Kinase specific predictions in phosphoproteomics data. Sequence logos, credible motif and predictive kinases for significantly upregulated (left) or downregulated (right) phosphopeptides (FDR < 0.05 and fold change > 2) were shown. f, Expression levels of predicted kinases in phosphoproteomics data (P < 0.05 and fold change > 2).
Extended Data Fig. 7 Effects of NAD+ and NAM supplementation on ZIKV-induced microcephaly.
a-b, Brain weight (a) and body weight (b) of mock-infected and ZIKV-infected mice with or without NAD+ supplementation (n = 10 - 12 mice, each dot represents one mouse). c, Immunostaining images of ZIKV (green), DAPI (blue) and Cleaved caspase-3 (Cleaved Cas3, red) on ZIKV-infected brains with (ZIKV + NAM) or without (ZIKV + Veh.) NAM supplementation. Veh., Vehicle. Scale bar: 200 μm. Data are representative of two independent experiments. d-e, ZIKV intensity (d) and cell death (e) in cortex (left in each panel) and hippocampus (right in each panel) of ZIKV-infected brains with (ZIKV + NAM, n = 4) or without (ZIKV + Veh., n = 3) NAM supplementation, respectively. Three slices for each brain. f-g. Brain weight (f) and body weight (g) of mock- and ZIKV-infected mice with or without NAM supplementation (n = 4 - 6 mice, each dot represents one mouse). All data are shown as mean ± s.e.m. Two-tailed unpaired t-test (a-b, d-g) was used for statistical analysis. Exact P values are indicated in the figure.
Extended Data Fig. 8 Absolute quantification of NAD+ and its precursors (NAM, NR and NMN) in the brains, primary neurons, and culture media after NAD+ or NAM supplementation.
a, NAD+ concentrations in the homogenous extract of cortex and hippocampus of ZIKV-infected mouse at indicated time points post vehicle (RPMI medium 1640 basic + 2% FBS) or NAD+ (500 μM, 2 μL) injection into the λ point (n = 4 mice for 0 h, 1 h, 2 h, 12 h time points, n = 3 mice for 6 h time point). b, NAD+ and NAM concentrations in the homogenous extract of cortex and hippocampus of ZIKV-infected mouse at indicated time points post vehicle (RPMI medium 1640 basic + 2% FBS) or NAM (500 μM, 2 μL) injection into the λ point (n = 5 mice for 0 h, 2 h, 6 h, 12 h time points; n = 4 mice for 1 h time point). c, Concentrations of NAD+ and its precursors in primary neurons before (zero time point) and after being cultured with 1 mM NAD+ for 0.5 h to 12 h (n = 5 biological replicates per time point). d, Concentrations of NAD+ and its precursors in culture media at indicated time points (n = 5 biological replicates per time point). Fresh media at zero time point contains 1 mM NAD+. Data are shown as mean ± s.e.m. (a-d). Statistical analysis was performed using One-way ANOVA followed by Benjamini and Hochberg multiple comparisons (a-d). Each time point was compared with zero time point. Exact P values are indicated.
Extended Data Fig. 9 NR supplementation alleviates metabolic disturbances in the ZIKV-infected mice.
a, Heatmap of metabolites detected in mock- and ZIKV-infected mouse brains after vehicle or NR supplementation (n = 4 – 5 mice, each square represents a mouse). b, Partial least squares discriminant analysis (PLS-DA) of ZIKV-infected mouse brains after vehicle (n = 5) or NR (n = 4) supplementation. c, Altered metabolic pathways in ZIKV-infected mouse brains after NR supplementation enriched by differential metabolites between vehicle and NR supplementation groups (Variable importance in the projection, VIP > 1). d-g, Absolute concentrations of NAD+ and its precursors in nicotinamide and nicotinamide metabolism – NAD+ (d), NR (e), NMN (f), and NAM (g) – in mock- and ZIKV-infected mouse brains. h-i, Relative levels of selected metabolites in mock- and ZIKV-infected mouse brains. Only metabolites that were both significantly altered by ZIKV-infection (Ctrl + Veh. group vs ZIKV + Veh group) and NR treatment (ZIKV + Veh. group vs ZIKV + NR group) were selected and shown. The average levels of each metabolite in control group (Ctrl + Veh.) were considered as 1, a.u. (arbitrary unit). n = 5 mice for Ctrl + NR and ZIKV + Veh. groups, n = 4 mice for Ctrl + Veh. and ZIKV + NR groups (d-i). j, Relative expression levels of transcripts, Nmrk and Nampt, in mock- and ZIKV-infected mouse brains from transcriptomics dataset (infected at E18.5 and inspected at P3, n = 3). Expression levels of Nmrk and Nampt in mock-infected brains were considered as 1, respectively. Data are shown as mean ± s.e.m. (d-j). Two-tailed unpaired t-test (d-j) was used for statistical analysis. Exact P values are indicated in the figure (d-g, j) or in the source data (h and i), *P < 0.05.
Supplementary information
Supplementary Data 1
Raw metabolomics data
Source data
Source Data Fig. 1
Statistical Source Data Fig. 1.
Source Data Fig. 2
Statistical Source Data Fig. 2.
Source Data Fig. 3
Statistical Source Data Fig. 3.
Source Data Fig. 4
Statistical Source Data Fig. 4.
Source Data Fig. 4
Unprocessed western blots.
Source Data Fig. 5
Statistical Source Data Fig. 5.
Source Data Fig. 5
Unprocessed western blots.
Source Data Fig. 6
Statistical Source Data Fig. 6.
Source Data Fig. 7
Statistical Source Data Fig. 7.
Source Data Extended Data Fig. 1
Statistical Source Data Extended Fig. 1.
Source Data Extended Data Fig. 2
Statistical Source Data Extended Fig. 2
Source Data Extended Data Fig. 3
Statistical Source Data Extended Fig. 3.
Source Data Extended Data Fig. 4
Statistical Source Data Extended Fig. 4.
Source Data Extended Data Fig. 5
Statistical Source Data Extended Fig. 5.
Source Data Extended Data Fig. 6
Statistical Source Data Extended Fig. 6.
Source Data Extended Data Fig. 6
Unprocessed western blots.
Source Data Extended Data Fig. 7
Statistical Source Data Extended Fig. 7.
Source Data Extended Data Fig. 8
Statistical Source Data Extended Fig. 8.
Source Data Extended Data Fig. 9
Statistical Source Data Extended Fig. 9.
Rights and permissions
About this article

Cite this article
Pang, H., Jiang, Y., Li, J. et al. Aberrant NAD+ metabolism underlies Zika virus–induced microcephaly. Nat Metab 3, 1109–1124 (2021). https://doi.org/10.1038/s42255-021-00437-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s42255-021-00437-0
This article is cited by
-
Tryptophan catabolism via the kynurenine pathway regulates infection and inflammation: from mechanisms to biomarkers and therapies
Inflammation Research (2024)
-
Molecular Mechanisms Associated with Neurodegeneration of Neurotropic Viral Infection
Molecular Neurobiology (2024)
-
Stress-Activated Protein Kinase JNK Modulates Depression-like Behaviors in Mice
Molecular Neurobiology (2023)
-
Zika virus vertical transmission in interferon receptor1-antagonized Rag1−/− mice results in postnatal brain abnormalities and clinical disease
Acta Neuropathologica Communications (2022)
-
Treatment of SARS-CoV-2-induced pneumonia with NAD+ and NMN in two mouse models
Cell Discovery (2022)
