
Abstract
Changes in synaptic plasticity are involved in pathophysiology of depression and in the mechanism of antidepressants. Ca2+/calmodulin (CaM) kinase II, a protein kinase involved in synaptic plasticity, has been previously shown to be a target of antidepressants. We previously found that antidepressants activate the kinase in hippocampal neuronal cell bodies by increasing phosphorylation at Thr286, reduce the kinase phosphorylation in synaptic membranes, and in turn its phosphorylation-dependent interaction with syntaxin-1 and the release of glutamate from hippocampal synaptosomes. Here, we investigated the chronic effect of different antidepressants (fluoxetine, desipramine, and reboxetine) on the expression and function of the kinase in distinct subcellular compartments in order to dissect the different kinase pools affected. Acute treatments did not induce any change in the kinase. In total tissue extracts chronic drug treatments induced activation of the kinase; in hippocampus (HC), but not in prefrontal/frontal cortex, this was partially accounted for by increased Thr286 phosphorylation, suggesting the involvement of different mechanisms of activation. In synaptosomes, all drugs reduced the kinase phosphorylation, particularly in HC where, upon fractionation of the synaptosomal particulate into synaptic vesicles and membranes, we found that the drugs induced a redistribution and differential activation of the kinase between membranes and vesicles. Furthermore, a large decrease in the level and phosphorylation of synapsin I located at synaptic membranes was consistent with the observed decrease of CaM kinase II. Overall, antidepressants induce a complex pattern of modifications in distinct subcellular compartments; at presynaptic level, these changes are in line with a dampening of glutamate release.
Similar content being viewed by others


INTRODUCTION
It is well known that, although most antidepressants exert their initial effects by increasing the extraneuronal levels of serotonin and/or norepinephrine, their therapeutic effects occur only after chronic administration. Several different lines of evidence in recent years have suggested that a cascade of postreceptor downstream effects is ultimately responsible for their therapeutic efficacy. These effects involve selective modulation of intracellular signaling cascades, and in turn modifications in gene expression, dendritic remodeling, synaptic plasticity, and neurogenesis (Manji et al, 2001; Coyle and Duman, 2003; Zarate et al, 2003; Wong and Licinio, 2004; Spedding et al, 2005; Tardito et al, 2006). Several signaling pathways regulating cellular resilience and neuroplasticity have been implicated in the mechanism of action of antidepressants, including cyclic adenosine mono phosphate-protein kinase A (cAMP-PKA), mitogen-activated protein kinases, Ca2+/calmodulin (CaM)-dependent kinases, protein kinase C (PKC), glycogen synthase kinase-3, and phosphatidylinositol 3-kinase/Akt.
CaM kinase II is an abundant protein kinase in brain, regulating neuronal response to calcium fluxes with a pivotal role in neuroplasticity (Hudmon and Schulman, 2002; Lisman et al, 2002). The kinase is activated by the binding of Ca2+/CaM that generates Ca2+-dependent enzymatic activity. Upon activation, depending on the extent of calcium elevation and duration of stimulation, the kinase autophosphorylates at Thr286, thereby generating Ca2+-independent activity that outlasts the calcium signal and is involved in synaptic plasticity. CaM kinase II is a multifunctional and ubiquitous enzyme and it was shown recently that targeting of the kinase to distinct subcellular compartments is crucial to its specificity of action in terms of space and time (Griffith et al, 2003). Targeting of the kinase to the nucleus, cytoskeleton, sarcoplasmic reticulum, and postsynaptic density of dendritic spines is achieved by several mechanisms, including alternative splicing of sequences containing nuclear localization signal, change of α/β isoform ratio, alternatively splicing-generated anchoring protein (αCaM kinase II association protein, αKAP), activity-dependent-regulated dendritic translation, and binding to N-methyl-D-aspartate (NMDA) receptors (Srinivasan et al, 1994; Bayer et al, 1998; Ouyang et al, 1999; Shen et al, 1998, 2000; Fink et al, 2003).
A number of studies have addressed the role of CaM kinase II in antidepressant mechanisms, by exploring its expression and function in limbic and cortical areas (Popoli et al, 1995; Pilc et al, 1999; Celano et al, 2003; Tiraboschi et al, 2004a; Bonanno et al, 2005). Chronic, but not acute, treatment with different antidepressants upregulated the Thr286 autophosphorylation and the enzymatic activity in neuronal cell bodies of hippocampus (HC), without increasing the total expression of αCaM kinase II (Tiraboschi et al, 2004a). A consistent finding was that chronic antidepressants also upregulated the expression level, autophosphorylation and enzymatic activity of the kinase localized to synaptic vesicles in both HC and prefrontal/frontal cortex (P/FC) (Celano et al, 2003). Recently, by using Percoll gradient-purified synaptosomes, we found that chronic antidepressants downregulated, rather than upregulate, Thr286 phosphorylation of αCaM kinase II in purified synaptic terminals (synaptosomes) and synaptic membranes of HC. The decrease in the kinase phosphorylation reduced its interaction with the SNARE (soluble N-ethylmaleimide-sensitive factor attachment protein receptor) protein syntaxin-1, thereby changing protein–protein interactions at glutamatergic presynaptic terminals and reducing depolarization-evoked release of glutamate (Bonanno et al, 2005).
Taken together, these results suggested a complex regulation of CaM kinase II by these drugs, with different state of activation/inactivation in distinct subcellular compartments, likely related to different functions of the kinase. Therefore, in order to understand better the different cellular functions affected by antidepressants and identify nodal points in signaling that may be related to the therapeutic action of these drugs, in the present study we systematically investigated CaM kinase II in different cellular compartments. In particular, the main aim of the present work was to investigate the state of CaM kinase II at synaptic site as opposed to total tissue, and within different presynaptic compartments. By using purified synaptosomes and fractionating this pure preparation in different membrane compartments, we report here that after chronic antidepressants the kinase redistributed between the synaptic membranes and the total pool of synaptic vesicles, and that its activation (Thr286 phosphorylation) was complementary reduced in synaptic membranes and increased in total synaptic vesicles of drug-treated rats. Furthermore, we observed that likely as a consequence of the redistribution of αCaM kinase II, the level of synapsin I was also greatly reduced in synaptic membranes, a finding that may further explain the reduction of glutamate release induced by antidepressants in HC (Bonanno et al, 2005).
MATERIALS AND METHODS
Animals and Drug Treatments
Experiments complied with guidelines for use of experimental animals of European Community Council Directive 86/609/EEC. Groups of 12 male Sprague–Dawley rats (170–200 g) were anesthetized and subcutaneously implanted with osmotic minipumps Alzet 2ML2 (release 5 μl/h, capacity 2 ml) (Charles River, Wilmington, MA), containing either vehicle (5% ethanol) or reboxetine (RBX), a selective norepinephrine reuptake inhibitor (NRI), fluoxetine (FLX), a selective serotonin reuptake inhibitor (SSRI) or desipramine (DMI), a tricyclic antidepressant mainly inhibiting norepinephrine release. Drug dosage was 10 mg/kg daily for each drug. Acute treatment was carried out by injecting rats (250–270 g) intraperitoneally. Animals were killed after 3 h for acute treatment and after 14 days for chronic treatment.
Preparation of Total Homogenate and Synaptosomes
Animals were killed and the HC was quickly dissected on ice. The whole frontal lobe was separated by a coronal section in correspondence to the optic chiasm (Glowinski and Iversen, 1966), and referred to as P/FC. The tissue was homogenized in 10 volumes of 0.28 M sucrose buffered at pH 7.4 with Tris, containing 20 mM NaF, 5 mM Na4P2O7, 1 mM Na3VO4 (protein phosphatase inhibitors), and 2 μl/ml of protease inhibitor cocktail (Sigma-Aldrich, St Louis, MO), using a glass–teflon tissue grinder (clearance 0.25 mm). Purified synaptosomes were prepared essentially according to Dunkley et al (1986), with minor modifications. The homogenate was centrifuged (5 min, 1000 g at 4°C) to remove nuclei and debris. The supernatant was centrifuged 5 min at 12 000 g; the resultant pellet was resuspended in homogenization buffer and gently stratified on a discontinuous Percoll gradient (2, 6, 10, and 20% v/v in Tris-buffered sucrose) and centrifuged at 33 500 g for 5 min. The layer between 10 and 20% Percoll (synaptosomal fraction) was collected and washed by centrifugation. To obtain cytosolic fraction (S3), the supernatant of 12 000 g centrifugation was ultracentrifuged at 135 000 g for 1 h. The synaptosomes were resuspended in lysis buffer: 120 mM NaCl, 20 mM HEPES pH 7.4, 0.1 mM EGTA, 0.1 mM DTT, containing 20 mM NaF, 5 mM Na2PO4, 1 mM Na3VO4, and 2 μl/ml of protease inhibitor cocktail (Sigma-Aldrich). Further fractionation of purified synaptosomes into synaptic membrane- (LP1) and synaptic vesicle-fraction (LP2) was carried out by differential centrifugation and ultracentrifugation, essentially according to Huttner et al (1983), as previously reported in Popoli and Paternò (1991) and Popoli et al (1995). The LP1 and LP2 fractions, previously characterized with regard to morphology and protein markers by several groups, were enriched with N-cadherin (a plasma membrane marker) and synaptophysin (a synaptic vesicle marker), respectively (Figure 1a).

Characterization of subcellular fractions and β-actin levels after drug treatments. (a) Subcellular distribution of protein markers. Proteins from HC subcellular fractions were fractionated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and subjected to Western blot with antibodies for NeuN, a soluble nuclear marker that is also found in cytosol (Mullen et al, 1992); N-cadherin, a marker for cell plasma membrane; synaptophysin, a marker for synaptic vesicles; H, total HC extract; P1, nuclear fraction; SPT, purified synaptosomes; LP1, synaptic membranes; LP2, synaptic vesicles; S3, high speed cytosol. (b) Immunoreactive bands for β-actin in total extract and subcellular fractions as above in rats treated with vehicle, FLX or RBX.
Assay of Ca2+-Dependent and -Independent Activity of CaM Kinase II
Ca2+-dependent activity of CaM kinase II was assayed by measuring initial rate of phosphate incorporation in the selective peptide substrate autocamtide-3 (Biosource, Camarillo, CA), as described previously (Celano et al, 2003). For assay of Ca2+-independent activity the samples contained, in the place of Ca2+/CaM, 2 mM EGTA, 2 μM heat-stable cAMP-dependent protein kinase inhibitor (New England, Beverly, MA), and 5 μM PKC (fragment 19–36) inhibitor (Sigma-Aldrich). Blanks were incubated in the absence of peptide.
Western Blot
Western-blot analysis was carried out as previously described (Verona et al, 2000; Tiraboschi et al, 2004b), by incubating poly-vinylidene difluoride membranes containing electrophoresed proteins with monoclonal or polyclonal antibodies in appropriate dilutions. Monoclonal antibodies used were: antibody for αCaM kinase II 1 : 3000, NeuN 1 : 500 (both from Chemicon International, Temecula, CA), βCaM kinase II 1 : 500, N-Cadherin 1 : 1000 (both from Zymed, South San Francisco, CA), synapsin I 1 : 2000, synaptophysin 1 : 350 (both from Synaptic Systems, Goettingen, Germany), and β-actin 1 : 5000 (Sigma-Aldrich). Polyclonal antibodies were: antibody for phospho-Thr286 αCaM kinase II 1 : 1000 (Promega, Milan, Italy) and phospho-Ser603 synapsin I 1 : 1000 (Chemicon). The membranes were blocked with 5% milk and incubated with primary antibody. Following incubation with peroxidase-coupled secondary antibodies, protein bands were detected by using ECL (Amersham Biosciences, Piscataway, NJ) or Super Signal Dura West (Pierce Biotechnology Inc., Rockford, IL). Standard curves were obtained by loading increasing amounts of samples on gels as described previously (Bonanno et al, 2005). All protein bands used were within linear range of standard curves, and normalized for actin level in the same membrane. Actin level did not change with drug treatment in any of the subcellular fractions analyzed (Figure 1b). Quantity One software (BioRad Laboratories, Hercules, CA) was used for standardization and quantitation, as previously reported (Tiraboschi et al, 2004b).
RESULTS
Expression and Function of αCaM Kinase II in Total HC and P/FC after Chronic Antidepressant Treatments
We sought to investigate the outcome of long-term treatment with different representative antidepressants on expression and function of CaM kinase II in distinct cellular compartments. To this aim we first assayed the basal enzymatic activity of the kinase in total extracts prepared from HC and P/FC of rats treated with FLX, DMI, and RBX (Figure 2). Both basal Ca2+-dependent and Ca2+-independent activity of CaM kinase II were measured. Ca2+-dependent activity is induced by the binding of Ca2+/CaM to pseudosubstrate inhibitory domain and consequent activation of the kinase; Ca2+-independent activity is generated when the activated kinase autophosphorylates at Thr286 residue in the autoinhibitory domain, an event allowing the kinase to remain persistently activated when calcium concentration falls to basal levels (Lisman et al, 2002). Acute treatments did not induce any change in this or other mechanisms investigated here (not shown). After chronic treatment, basal Ca2+-dependent activity was slightly but significantly elevated by DMI in HC (32%) and P/FC (35%) and by FLX in HC (34%), with DMI inducing the highest increase in P/FC (Figure 2a). Basal Ca2+-independent activity showed greater changes after drug treatments, particularly in HC (FLX 140%, DMI 65%, and RBX 105%) (Figure 2b); in P/FC only DMI significantly increased the activity (60%). We further investigated the changes induced by drug treatments, measuring the expression level of αCaM kinase II (the major isoform in forebrain) and the basal phosphorylation level of Thr286 residue (Figure 2c and d). None of the drugs increased total expression level of the kinase in the two areas (except RBX in P/FC; 19%); rather, RBX (−13%) slightly decreased the total level of αCaM kinase II in HC. In both areas, Thr286 phosphorylation was not significantly changed by the three drugs. Overall, the results with Ca2+-independent activity are compatible with and extend our previous data showing that chronic antidepressants activate CaM kinase II in neuronal cell bodies of hippocampal neurons (Tiraboschi et al, 2004a). The same did not apply to P/FC in the present work, where the increase of Ca2+-independent activity observed was significant only for DMI and not paralleled by any increase of Thr286 phosphorylation by the same drug. This suggests that mechanisms different from phosphorylation are responsible for the kinase activation by antidepressants (see Discussion).

Enzymatic activity, expression level, and phosphorylation of CaM kinase II in total HC and P/FC. (a) Ca2+-dependent enzymatic activity of CaM kinase II in total HC or P/FC extract from rats chronically treated with vehicle (Control), FLX, DMI or RBX. HC, hippocampus; prefrontal/frontal cortex, P/FC. Activity expressed as pmoles of PO4 incorporated per minute per μg protein (mean±SEM). (b) Ca2+-independent enzymatic activity of CaM kinase II as in (a). (c) Western-blot analysis of total αCaM kinase II in HC and P/FC. The band intensities were normalized for β-actin. Data expressed as % immunoreactivity vs control (mean±SEM). (d) Western-blot analysis of phospho-Thr286 αCaM kinase II as in (c). Insets: representative immunoreactive bands from Western blots. Statistics: one-way analysis of variance followed by Newman–Keuls post hoc tests. p-values reported refer to drug treated vs control tests (*p<0.05, **p<0.01, and ***p<0.001; n=3 in triplicate or quadruplicate).
Expression and Function of αCaM Kinase II in Synaptosomes after Chronic Antidepressant Treatments
To investigate the action of antidepressants on expression and function of CaM kinase II at synaptic sites, we purified synaptic terminals (synaptosomes) from rats long-term treated with the three drugs as above, by centrifugation on Percoll gradients as reported previously (Bonanno et al, 2005). Basal Ca2+-dependent activity of the kinase in synaptosomes was significantly elevated by FLX (18%) and DMI (54%) in HC, and by DMI (78%) and RBX (37%) in P/FC (Figure 3a). FLX exerted a minimal effect on Ca2+-dependent activity in synaptosomes from both areas. Conversely, basal Ca2+-independent activity was robustly increased in both brain areas by all drugs, with the only exception of FLX in P/FC (Figure 3b). In P/FC, only pronoradrenergic drugs increased the kinase enzymatic activity (DMI, 130%; RBX, 162%). In HC, the results showed no selectivity of effect for drug classes (FLX, 280%; DMI, 110%; RBX, 365%). The changes in enzymatic activity were not justified by corresponding changes in the levels of synaptic kinase (Figure 3c). Interestingly, the effects of the drugs on Thr286 phosphorylation were different in the two brain areas (Figure 3d). Whereas in HC all drugs markedly and significantly decreased phosphorylation of the activator residue (FLX, −36%; DMI, −62%; RBX, −42%), in P/FC the reduction of phosphorylation was more modest and not significant (Figure 3d).

Enzymatic activity, expression level, and phosphorylation of CaM kinase II in synaptosomes from HC and P/FC. (a) Ca2+-dependent enzymatic activity of CaM kinase II in purified synaptosomes from HC or P/FC, from rats chronically treated as in Figure 1. (b) Ca2+-independent enzymatic activity of CaM kinase II in purified synaptosomes as in (a). (c) Western-blot analysis of total αCaM kinase II in purified synaptosomes. The band intensities were normalized for β-actin. Data expressed as % immunoreactivity vs control (mean±SEM). (d) Western-blot analysis of phospho-Thr286 αCaM kinase II in purified synaptosomes as in (c). Insets: representative immunoreactive bands from Western blots. Statistics as in Figure 2 (*p<0.05, **p<0.01, and ***p<0.001; n=3 in triplicate or quadruplicate).
Furthermore, the complete absence of upregulation of Thr286 phosphorylation in the synaptosomes from both areas suggested that the augmentation of Ca2+-independent activity induced by the drugs cannot be owing to this phosphorylation change. However, the present results confirm our previous data showing that antidepressants decrease Thr286 phosphorylation in HC synaptosomes (Bonanno et al, 2005).
Differential Expression Level and Activation of αCaM Kinase II in Presynaptic Compartments after Chronic Antidepressant Treatments
To further investigate the changes induced in the regulation of CaM kinase II at presynaptic level, we fractionated purified synaptosomes by using differential centrifugation and ultracentrifugation in the two particulate compartments containing the kinase: synaptic vesicles and synaptic membranes. The remaining part of this work was conducted on HC, because in this area the reduction of kinase phosphorylation was more marked and significant (see Figure 3d). For further characterization of subsynaptic compartments, we used rats treated with two (FLX and RBX) of the three drugs used in the first part of this work, representative of different primary mechanisms (SSRI and NRI, respectively). Preparation of synaptic vesicles and membranes must be carried out using freshly prepared synaptosomes, and requires further centrifugation and ultracentrifugation. Using additional samples from DMI-treated rats would have slowed down the procedure; for this reason we chose to limit the second set of experiments to controls, FLX- and RBX-treated rats. Overall, by this way the second part of this study compares the effects of two drugs with different and complementary mechanisms on synaptic effectors.
Surprisingly, we found that long-term treatment with both drugs induced opposite changes in local expression level and Thr286 phosphorylation of αCaM kinase II in the two presynaptic compartments (Figure 4). In synaptic vesicles, both expression level and phosphorylation of the kinase were markedly and significantly increased by drug treatments, whereas in synaptic membranes protein level and, particularly, Thr286 phosphorylation were reduced. Although part of the effect seems to be owing to actual changes in protein level, in both cases phosphorylation changes largely exceeded protein level changes. We recently showed that the down-regulation of kinase autophosphorylation in synaptic membranes induces a redistribution of the binding of the SNARE protein syntaxin-1 between αCaM kinase II and Munc-18, a molecular event with a physiological counterpart represented by a decrease of depolarization-evoked release of glutamate (Bonanno et al, 2005). Our present results suggest that two antidepressants with different primary mechanisms (eg, SSRI and NRI) may alter the function of the presynaptic machinery by inducing redistribution of αCaM kinase II and of its basal phosphorylation level between synaptic membranes and synaptic vesicles. The lack of major changes in total expression level (Figure 2c) and synaptosomal expression level (Figure 3c) of the kinase supports this conclusion.

Expression level and phosphorylation of αCaM kinase II in synaptic vesicles and membranes from HC. Western-blot analysis of αCaM kinase II in synaptic vesicles and synaptic membranes isolated from hippocampal synaptosomes of rats chronically treated with vehicle (Control), FLX, or RBX. (a) Western-blot analysis of total αCaM kinase II in synaptic vesicles. The band intensities were normalized for β-actin. Data expressed as % immunoreactivity vs control (mean±SEM). (b) Western-blot analysis of total αCaM kinase II in synaptic membranes. (c) Western-blot analysis of phospho-Thr286 αCaM kinase II in synaptic vesicles. (d) Western-blot analysis of phospho-Thr286 αCaM kinase II in synaptic membranes. Insets: representative immunoreactive bands from Western blots. Statistics as in previous figures (***p<0.001; n=3 in triplicate or quadruplicate).
Regulation of Synapsin I and βCaM Kinase II in Presynaptic Compartments after Chronic Antidepressant Treatments
We speculated that the redistribution of total and phosphorylated αCaM kinase II between synaptic membranes and synaptic vesicles induced by antidepressants could be due to a number of reasons, including: (1) translocation or different targeting of the kinase (change in the binding to other membrane-associated proteins or to the hydrophobic core of the membrane); (2) changes in the α- vs β-subunit composition of the kinase holigomer that was shown to be sensitive to changes in neuronal activity and NMDA/α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors activation (Thiagarajan et al, 2002); (3) changes in local αCaM kinase II synthesis (possibly involving kinase associated with portions of postsynaptic densities that copurify with presynaptic terminals).
-
1)
While it was shown that both α- and βCaM kinase II copurifying with synaptic vesicles are tightly associated with the vesicle membrane, suggesting that this is due to either covalent modification or high-affinity association with other membrane-associated components (Benfenati et al, 1996), no data are available regarding the association of the kinase with the presynaptic membrane. However, because in presynaptic terminals the particulate kinase is tightly bound to the vesicles, it can be speculated that the association with presynaptic membranes is mainly due to the presence of vesicles docked to the plasma membrane (see Discussion). It has been demonstrated that αCaM kinase II is an anchoring protein for synapsin I, a presynaptic protein regulating the vesicle cycle and neurotransmitter release, and a major substrate for αCaM kinase II (Benfenati et al, 1992). We sought further evidence for changes in the membrane targeting of the kinase by measuring the level of synapsin I and its phosphorylation at Ser603 (the consensus site for CaM kinase II) in the rats chronically treated with FLX and RBX. Although the drugs did not reduce synapsin I in the total HC (Figure 5a,d), both the level and CaM kinase II-dependent phosphorylation of synapsin I (likely as a consequence of protein level decrease) were reduced in synaptosomes and synaptic membranes after treatment (Figure 5b,e and c,f). In particular, the level of synapsin I in synaptic membranes of FLX- and RBX-treated rats was reduced by 70–80%. First, these results suggest that the reduction of synapsin I associated with synaptic membranes is a likely consequence of the downregulation of the anchoring site for this protein (eg, CaM kinase II) in this compartment after drug treatments. Second, the drugs also reduced the localization of synapsin I to synaptic terminals in toto, in the absence of reduction in total expression.
Figure 5 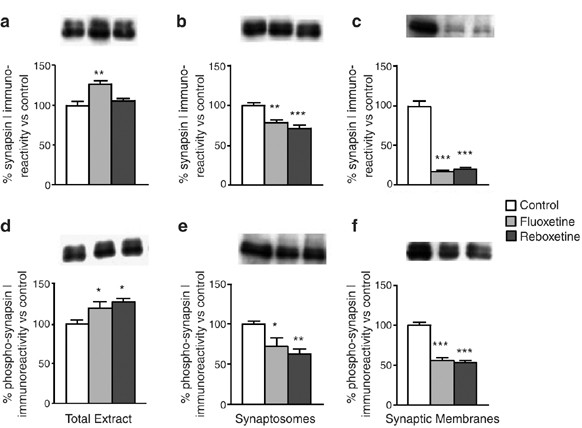
Expression level and phosphorylation of synapsin I in total extract, synaptosomes, and synaptic membranes from HC. Western-blot analysis of total synapsin I in: (a) total extract; (b) synaptosomes; and (c) synaptic membranes from HC of rats chronically treated with vehicle (Control), FLX, or RBX. Western-blot analysis of phospho-Ser603 synapsin I in: (d) total extract; (e) synaptosomes; and (f) synaptic membranes from HC of rats as above. Insets: representative immunoreactive bands from Western blots. Statistics as in previous figures (*p<0.05, **p<0.01, and ***p<0.001; n=3 in triplicate or quadruplicate).
-
2)
Interestingly, the expression of βCaM kinase II in total HC was robustly increased by the drug treatments (Figure 6a), showing a regulation different from the α-isoform (compare with Figure 2c). Furthermore, no major changes in the level of βCaM kinase II were found in synaptosomes and synaptic membranes, with the exception of a small downregulation by FLX in the latter (Figure 6b, c). Therefore, both at total and synaptosomal levels, βCaM kinase II was differently regulated compared to αCaM kinase II.
Figure 6 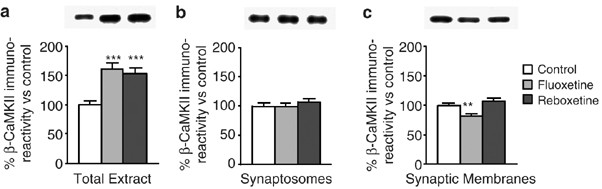
Expression level of βCaM kinase II in total extract, synaptosomes, and synaptic membranes from HC. Western-blot analysis of total βCaM kinase II in: (a) total extract; (b) synaptosomes; and (c) synaptic membranes from HC of rats chronically treated with vehicle (Control), FLX, or RBX. Insets: representative immunoreactive bands from Western blots. Statistics as in previous figures (**p<0.01 and ***p<0.001; n=3 in triplicate or quadruplicate).
-
3)
Portions of the dendritic postsynaptic membrane, containing postsynaptic densities enriched with αCaM kinase II, are a typical contaminant of synaptosomes. It has been shown that neuronal activation induces local dendritic synthesis of αCaM kinase II (Ouyang et al, 1999; Bagni et al, 2000); therefore, changes in local synthesis of the kinase might contribute to the changes observed here following drug treatments. In a parallel study, we subjected purified synaptosomes from RBX-treated rats as above to the analysis of mRNA expression and of mRNA translation for several different mRNAs known to be present and locally translated at synapses. Although we found significant changes for some mRNAs, the efficiency of translation for αCaM kinase II transcript was unchanged in the RBX-treated rats (Veneri et al, manuscript in preparation), ruling out an involvement of dendritic αCaM kinase II synthesis in the modifications observed here.


DISCUSSION
Chronic Antidepressant Treatments Activate CaM Kinase II in HC and P/FC by Canonical and Non-Canonical Mechanisms
The main aim of this work was a complete description of the changes in enzymatic activity, expression level, and Thr286 phosphorylation of CaM kinase II in distinct subcellular compartments after chronic antidepressant treatments. We found that drugs with different primary mechanisms (serotonin and/or norepinephrine reuptake inhibition) upregulated CaM kinase II enzymatic activity in total extracts from HC and P/FC; the increase was more remarkable for Ca2+-independent activity in HC (see Figure 2a and b). In this area, the kinase activation was accompanied by a slight but not significant increase of autophosphorylation at Thr286 (for FLX and RBX), which represents the canonical mechanism for activity-induced activation of this kinase (Hudmon and Schulman, 2002; Lisman et al, 2002). We reported previously upregulation of CaM kinase II activity by pronoradrenergic antidepressants in cell bodies of pyramidal and granular neurons of HC (Tiraboschi et al, 2004a); our previous and present results strongly suggest that the modifications observed in total HC extract are mainly related to cell bodies and that this mechanism seems to be common to pronoadrenergic (DMI and RBX) and proserotonergic (FLX) antidepressants. However, in total HC, the increase of kinase autophosphorylation did not reach significance; and in P/FC, the kinase activation (significant only for DMI) was not accompanied by any increase of Thr286 phosphorylation. These findings suggest that other mechanisms of kinase activation are involved in both brain areas. CaM kinase II can be persistently activated by interacting with other proteins, including the NMDA receptor and ether-a-go-go (Eag) K+ channel (Strack et al, 2000; Bayer et al, 2001; Sun et al, 2004). These non-canonical mechanisms of activation of CaM kinase II could be a preferential target of antidepressants, because they also seem to be involved at synaptic level (see next section).
Chronic Antidepressant Treatments Induce a Redistribution of CaM Kinase II between Synaptic Membranes and Synaptic Vesicles in HC
First, antidepressants upregulated CaM kinase II enzymatic activity in synaptosomes from HC and P/FC. The increase was particularly remarkable for Ca2+-independent activity (in P/FC restricted to noradrenergic drugs). However, in both areas again the kinase activation was neither related to an increase of expression level nor of Thr286 phosphorylation. By contrast, the kinase autophosphorylation was reduced in synaptosomes from both areas, particularly in HC where up-regulation of Ca2+-independent activity was maximal. This suggests that the modulation of CaM kinase II activity by antidepressants at synaptic terminals is independent from autophosphorylation and may involve protein–protein interactions (Strack et al, 2000; Bayer et al, 2001; Sun et al, 2004). Indeed, it has been shown that interaction of CaM kinase II with subunits of the NMDA receptor or the Eag potassium channel activates the kinase independent on autophosphorylation (Bayer et al, 2001; Sun et al, 2004). A future study of CaM kinase II interaction with NR2A/B and NR1 subunits of NMDA receptor, or with Eag potassium channel, in synaptosomes of antidepressant-treated rats will verify this hypothesis. Therefore, the present results showed that activation of HC CaM kinase II by antidepressants in synaptosomes is obtained by a mechanism independent on Thr286 phosphorylation.
Second, antidepressants induced a redistribution of αCaM kinase II between synaptic membranes and synaptic vesicles of HC. Based on previous studies, it can be argued that the presence of CaM kinase II in synaptic membranes is mainly due to the presence of a pool of vesicles docked to the plasma membrane (Mehta et al, 1996). This morphologically defined pool of vesicles has been correlated with the functionally defined readily releasable pool (RRP), containing the vesicles that fuse with the membrane and release glutamate upon stimulation (Schikorski and Stevens, 2001). Overall, our results would suggest that the redistribution is due to a translocation or a change in the targeting of the kinase during the synaptic vesicle cycle. This is supported by the following results: (a) the total level of αCaM kinase II in synaptosomes was little or not changed after drug treatments; (b) the level of synapsin I, which is anchored to CaM kinase II in synaptic vesicles (Benfenati et al, 1992), was dramatically reduced in synaptic membranes; (c) the level of βCaM kinase II in synaptic membranes was not increased by the drug treatments, ruling out a replacement of α- with β-isoform (but see next paragraph); and (d) local protein synthesis of αCaM kinase II was unchanged (Veneri et al, manuscript in preparation). Similar to our previous results showing a downregulation of the interaction αCaM kinase II/syntaxin-1 in synaptic membranes (Bonanno et al, 2005), the extent of kinase redistribution we observed is large enough to involve glutamatergic synaptic terminals (estimated to represent up to 60% of hippocampal terminals).
With regard to βCaM kinase II, it is interesting that, although not replacing the αCaM kinase II missing at synaptic membranes, the changes observed in total expression of β after drug treatments will alter the normal ratio between α and β isoform in HC (normally ≈3 : 1; Benfenati et al, 1996). In particular, the large increase of β expression in total HC will likely affect the kinase function in neuronal cell bodies, whereas the lack of expression changes in β in synaptic membranes in the face of a large reduction of α will still considerably change the ratio between subunits. It has been shown that in HC neurons, the α/β ratio increases during periods of higher electrical activity and decreases with reduced activity. The expression level of α-isoform was found to be regulated by the activity of NMDA receptor (Thiagarajan et al, 2002). The reduction induced by antidepressants in the α/β ratio at both total and synaptic levels could be related to a drug-induced reduction of glutamate neurotransmission. It is worth mentioning that these same drug treatments also induced a robust reduction in the expression level of NR1 subunit of NMDA receptor in synaptic membranes, a finding that could be functionally coupled to the reduction of αCaM kinase II in the same compartment (not shown).
Functional Changes Induced by Antidepressants in the Presynaptic Machinery of HC: Implications for Synaptic Plasticity
Preclinical and clinical studies have shown that molecular/cellular mechanisms underlying synaptic plasticity are involved in both stress-related disorders and in the action of antidepressants (McEwen, 1999; Sapolsky, 2000; Popoli et al, 2002; Shakesby et al, 2002; Zarate et al, 2002). With regard to presynaptic mechanisms, chronic stress has been shown to induce in HC terminals a redistribution of synaptic vesicles suggesting increased glutamate release, and microdialysis in vivo studies reported increased extracellular glutamate following stress paradigms (Bagley and Moghaddam, 1997; Magarinos et al, 1997). Conversely, we showed that different antidepressants reduce depolarization-evoked release of glutamate from hippocampal synaptosomes, by reducing syntaxin-1/CaM kinase II interaction and increasing syntaxin-1/Munc-18 interaction (Bonanno et al, 2005). Therefore, although independent evidence suggests that stress induces an increase of glutamate release in HC and cortical areas, antidepressants seem to exert their restorative action partly by dampening glutamate release. The combination of these presynaptic effects with the documented drug-induced postsynaptic effects on different types of glutamate receptors (Zarate et al, 2003; Popoli et al, 2002) could explain the impact of antidepressants on synaptic plasticity (Popoli et al, 2002; Shakesby et al, 2002).
Overall, our present and previous (Bonanno et al, 2005) data suggest that the functional presynaptic compartments involved here are the RRP of vesicles (associated with the synaptic membrane) and the reserve pool of synaptic vesicles (largely coincident with the vesicle fraction). We previously showed that the decreased kinase phosphorylation in the RRP brings about consistent changes in the presynaptic machinery, leading to a decrease of depolarization-evoked glutamate release (Bonanno et al, 2005). Therefore, the antidepressant-induced change in αCaM kinase II phosphorylation seems to be a crucial event in the molecular modifications leading to modulation of glutamate release. Indeed, αCaM kinase II has been proposed to regulate probability of release in central synapses during high-frequency presynaptic stimulation (Hinds et al, 2003); the drug-induced molecular and functional changes we found related to αCaM kinase II are in line with this role for the enzyme. The mechanism whereby antidepressants may induce the adaptive changes we observed in αCaM kinase II is at present unknown. A possibility is that these drugs affect calcium fluxes, because in preliminary experiments with Fura-2 employing synaptosomes from chronic FLX-treated rats we found consistent upregulation of basal and depolarization-induced calcium fluxes (not shown).
However, our results also suggest an additional functional change that may be a consequence of the large decrease of synapsin I in synaptic membranes, likely due to the decrease of its anchoring protein (αCaM kinase II). It has been shown that synapsin I is required for stimulation of glutamate release by brain-derived neurotrophic factor (BDNF) (Jovanovic et al, 2000). BDNF expression is increased and indirect evidence showed that more BDNF is released after chronic antidepressant treatments (Saarelainen et al, 2003). Now, if antidepressants increase the synaptic concentration of BDNF one would expect an upregulation of glutamate release rather than a downregulation, as we observed. However, if synapsin I is nearly absent from the RRP of vesicles after drug treatment (≈80% of synapsin I was gone, see Figure 5), elevated BDNF will not increase glutamate release, but at the same time will be able to induce different neurotrophic effects. This mechanism may represent an additional explanation for the antidepressant-induced dampening of glutamate release.
In summary, CaM kinase II is a target of antidepressants, which induce a complex pattern of modifications in the kinase at different subcellular compartments. At presynaptic level, these changes are consistent with the dampening of glutamate release chronically induced by these drugs. It will be crucial in order to understand if these mechanisms can be targeted by novel pharmacological strategies, to identify the anchoring protein for the kinase in synaptic vesicles, and the factors regulating phosphorylation changes in the presynapse.
References
Bagley J, Moghaddam B (1997). Temporal dynamics of glutamate efflux in the prefrontal cortex and in the hippocampus following repeated stress: effects of pretreatment with saline or diazepam. Neuroscience 77: 65–73.
Bagni C, Mannucci L, Dotti CG, Amaldi F (2000). Chemical stimulation of synaptosomes modulates alpha -Ca2+/calmodulin-dependent protein kinase II mRNA association to polysomes. J Neurosci 20: RC76.
Bayer KU, De Koninck P, Leonard AS, Hell JW, Schulman H (2001). Interaction with the NMDA receptor locks CaMKII in an active conformation. Nature 411: 801–805.
Bayer KU, Harbers K, Schulman H (1998). alphaKAP is an anchoring protein for a novel CaM kinase II isoform in skeletal muscle. EMBO J 17: 5598–5605.
Benfenati F, Onofri F, Czernik AJ, Valtorta F (1996). Biochemical and functional characterization of the synaptic vesicle-associated form of CA2+/calmodulin-dependent protein kinase II. Brain Res Mol Brain Res 40: 297–309.
Benfenati F, Valtorta F, Rubenstein JL, Gorelick FS, Greengard P, Czernik AJ (1992). Synaptic vesicle-associated Ca2+/calmodulin-dependent protein kinase II is a binding protein for synapsin I. Nature 359: 417–420.
Bonanno G, Giambelli R, Raiteri L, Tiraboschi E, Zappettini S, Musazzi L et al (2005). Chronic antidepressants reduce depolarization-evoked glutamate release and protein interactions favoring formation of SNARE complex in hippocampus. J Neurosci 25: 3270–3279.
Celano E, Tiraboschi E, Consogno E, D'Urso G, Mbakop MP, Gennarelli M et al (2003). Selective regulation of presynaptic calcium/calmodulin-dependent protein kinase II by psychotropic drugs. Biol Psychiatry 53: 442–449.
Coyle JT, Duman RS (2003). Finding the intracellular signaling path-ways affected by mood disorder treatments. Neuron 38: 157–160.
Dunkley PR, Jarvie PE, Heath JW, Kidd GJ, Rostas JA (1986). A rapid method for isolation of synaptosomes on Percoll gradients. Brain Res 372: 115–129.
Fink CC, Bayer KU, Myers JW, Ferrell Jr JE, Schulman H, Meyer T (2003). Selective regulation of neurite extension and synapse formation by the beta but not the alpha isoform of CaMKII. Neuron 39: 283–297.
Glowinski J, Iversen LL (1966). Regional studies of catecholamines in the rat brain. I. The disposition of [3H]norepinephrine, [3H]dopamine and [3H]dopa in various regions of the brain. J Neurochem 13: 655–669.
Griffith LC, Lu CS, Sun XX (2003). CaMKII, an enzyme on the move: regulation of temporospatial localization. Mol Interv 3: 386–403.
Hinds HL, Goussakov I, Nakazawa K, Tonegawa S, Bolshakov VY (2003). Essential function of alpha-calcium/calmodulin-dependent protein kinase II in neurotransmitter release at a glutamatergic central synapse. Proc Natl Acad Sci USA 100: 4275–4280.
Hudmon A, Schulman H (2002). Neuronal CA2+/calmodulin-dependent protein kinase II: the role of structure and autoregulation in cellular function. Annu Rev Biochem 71: 473–510.
Huttner WB, Schiebler W, Greengard P, De Camilli P (1983). Synapsin I (protein I), a nerve terminal-specific phosphoprotein. III. Its association with synaptic vesicles studied in a highly purified synaptic vesicle preparation. J Cell Biol 96: 1374–1388.
Jovanovic JN, Czernik AJ, Fienberg AA, Greengard P, Sihra TS (2000). Synapsins as mediators of BDNF-enhanced neurotransmitter release. Nat Neurosci 3: 323–329.
Lisman J, Schulman H, Cline H (2002). The molecular basis of CaMKII function in synaptic and behavioural memory. Nat Rev Neurosci 3: 175–190.
Magarinos AM, Verdugo JM, McEwen BS (1997). Chronic stress alters synaptic terminal structure in hippocampus. Proc Natl Acad Sci USA 94: 14002–14008.
Manji HK, Drevets WC, Charney DS (2001). The cellular neurobiology of depression. Nat Med 7: 541–547.
McEwen BS (1999). Stress and hippocampal plasticity. Annu Rev Neurosci 22: 105–122.
Mehta PP, Battenberg E, Wilson MC (1996). SNAP-25 and synaptotagmin involvement in the final Ca(2+)-dependent triggering of neurotransmitter exocytosis. Proc Natl Acad Sci USA 93: 10471–10476.
Mullen RJ, Buck CR, Smith AM (1992). NeuN, a neuronal specific nuclear protein in vertebrates. Development 116: 201–211.
Ouyang Y, Rosenstein A, Kreiman G, Schuman EM, Kennedy MB (1999). Tetanic stimulation leads to increased accumulation of Ca(2+)/calmodulin-dependent protein kinase II via dendritic protein synthesis in hippocampal neurons. J Neurosci 19: 7823–7833.
Pilc A, Branski P, Palucha A, Aronowski J (1999). The effect of prolonged imipramine and electroconvulsive shock treatment on calcium/calmodulin-dependent protein kinase II in the hippocampus of rat brain. Neuropharmacology 38: 597–603.
Popoli M, Gennarelli M, Racagni G (2002). Modulation of synaptic plasticity by stress and antidepressants. Bipolar Disord 4: 166–182.
Popoli M, Paternò R (1991). Properties of a synaptic vesicle protein binding plasma membranes. Neuroreport 2: 93–95.
Popoli M, Vocaturo C, Perez J, Smeraldi E, Racagni G (1995). Presynaptic Ca2+/calmodulin-dependent protein kinase II: autophosphorylation and activity increase in the hippocampus after long-term blockade of serotonin reuptake. Mol Pharmacol 48: 623–629.
Saarelainen T, Hendolin P, Lucas G, Koponen E, Sairanen M, MacDonald E et al (2003). Activation of the TrkB neurotrophin receptor is induced by antidepressant drugs and is required for anti-depressant-induced behavioral effects. J Neurosci 23: 349–357.
Sapolsky RM (2000). The possibility of neurotoxicity in the hippocampus in major depression: a primer on neuron death. Biol Psychiatry 48: 755–765.
Schikorski T, Stevens CF (2001). Morphological correlates of functionally defined synaptic vesicle populations. Nat Neurosci 4: 391–395.
Shakesby AC, Anwyl R, Rowan MJ (2002). Overcoming the effects of stress on synaptic plasticity in the intact hippocampus: rapid actions of serotonergic and antidepressant agents. J Neurosci 22: 3638–3644.
Shen K, Teruel MN, Connor JH, Shenolikar S, Meyer T (2000). Molecular memory by reversible translocation of calcium/calmodulin-dependent protein kinase II. Nat Neurosci 3: 881–886.
Shen K, Teruel MN, Subramanian K, Meyer T (1998). CaMKIIbeta functions as an F-actin targeting module that localizes CaMKIIalpha/beta heterooligomers to dendritic spines. Neuron 21: 593–606.
Spedding M, Jay T, Costa e Silva J, Perret L (2005). A pathophysiological paradigm for the therapy of psychiatric disease. Nat Rev Drug Discov 4: 467–476.
Srinivasan M, Edman CF, Schulman H (1994). Alternative splicing introduces a nuclear localization signal that targets multifunctional CaM kinase to the nucleus. J Cell Biol 126: 839–852.
Strack S, Robison AJ, Bass MA, Colbran RJ (2000). Association of calcium/calmodulin-dependent kinase II with developmentally regulated splice variants of the postsynaptic density protein densin-180. J Biol Chem 275: 25061–25064.
Sun XX, Hodge JJ, Zhou Y, Nguyen M, Griffith LC (2004). The eag potassium channel binds and locally activates calcium/calmodulin-dependent protein kinase II. J Biol Chem 279: 10206–10214.
Tardito D, Perez J, Tiraboschi E, Musazzi L, Racagni G, Popoli M (2006). Signaling pathways regulating gene expression, neuroplasticity and neurothrophic mechanisms in the action of antidepressants. A critical overview. Pharmacol Rev 58: 115–134.
Thiagarajan TC, Piedras-Renteria ES, Tsien RW (2002). Alpha- and betaCaMKII. Inverse regulation by neuronal activity and opposing effects on synaptic strength. Neuron 36: 1103–1114.
Tiraboschi E, Giambelli R, D'Urso G, Galietta A, Barbon A, de Bartolomeis A et al (2004a). Antidepressants activate CaMKII in neuron cell body by Thr286 phosphorylation. Neuroreport 15: 2393–2396.
Tiraboschi E, Tardito D, Kasahara J, Moraschi S, Pruneri P, Gennarelli M et al (2004b). Selective phosphorylation of nuclear CREB by fluoxetine is linked to activation of CaM kinase IV and MAP kinase cascades. Neuropsychopharmacology 29: 1831–1840.
Verona M, Zanotti S, Schafer T, Racagni G, Popoli M (2000). Changes of synaptotagmin interaction with t-SNARE proteins in vitro after calcium/calmodulin-dependent phosphorylation. J Neurochem 74: 209–221.
Wong ML, Licinio J (2004). From monoamines to genomic targets: a paradigm shift for drug discovery in depression. Nat Rev Drug Discov 3: 136–151.
Zarate Jr CA, Du J, Quiroz J, Gray NA, Denicoff KD, Singh J et al (2003). Regulation of cellular plasticity cascades in the pathophysiology and treatment of mood disorders: role of the glutamatergic system. Ann NY Acad Sci 1003: 273–291.
Zarate CA, Quiroz J, Payne J, Manji HK (2002). Modulators of the glutamatergic system: implications for the development of improved therapeutics in mood disorders. Psychopharmacol Bull 36: 35–83.
Acknowledgements
This study was supported by Grants from NARSAD (USA) to MP and from Ministry of University, PRIN # 2001054224 and 2003053993 to GR and MP (Italy). RG was founded by the PhD Program in Neuropharmacology, University of Catania Medical School.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Barbiero, V., Giambelli, R., Musazzi, L. et al. Chronic Antidepressants Induce Redistribution and Differential Activation of αCaM Kinase II between Presynaptic Compartments. Neuropsychopharmacol 32, 2511–2519 (2007). https://doi.org/10.1038/sj.npp.1301378
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.npp.1301378
Keywords
This article is cited by
-
Time-dependent activation of MAPK/Erk1/2 and Akt/GSK3 cascades: modulation by agomelatine
BMC Neuroscience (2014)
-
Stress and corticosterone increase the readily releasable pool of glutamate vesicles in synaptic terminals of prefrontal and frontal cortex
Molecular Psychiatry (2014)
-
Antidepressants that inhibit both serotonin and norepinephrine reuptake impair long-term potentiation in hippocampus
Psychopharmacology (2014)
-
Chronic treatment with agomelatine or venlafaxine reduces depolarization-evoked glutamate release from hippocampal synaptosomes
BMC Neuroscience (2013)
-
Serotonin–glutamate and serotonin–dopamine reciprocal interactions as putative molecular targets for novel antipsychotic treatments: from receptor heterodimers to postsynaptic scaffolding and effector proteins
Psychopharmacology (2013)
