
Abstract
Significant evidences have brought new insights on the mechanisms by which epigenetic machinery proteins regulate gene expression, leading to a redefinition of chromatin regulation in terms of modification of core histones, DNA methylation, RNA-mediated silencing pathways, action of methylation-dependent sensitive insulators and Polycomb/Trithorax group proteins. Consistent with these fundamental aspects, an increasing number of human pathologies have been found to be associated with aberrant epigenetics regulation, including cancer, mental retardation, neurodegenerative symptoms, imprinting disorders, syndromes involving chromosomal instabilities and a great number of human life-threatening diseases. The possibility of reversing epigenetic marks, in contrast to genetic code, may provide new pharmacological targets for emerging therapeutic intervention.
Similar content being viewed by others
Main
During the life cycle of an organism, epigenetic factors provide a heritable, reversible and dynamic manner to modulate gene expression without changing the DNA sequence. In this review, we discuss how the disruption of the principal epigenetic pathways can lead to silencing or inappropriate expression of genes, leading to a new category of pathologies, called epigenetic diseases. The control of eukaryotic gene transcription involves the dynamic positioning of histones, organizing the genome into open or condensed chromatin, governing transcriptional activity and the DNA availability for recombination, replication and repair. Histones N-terminal tails can suffer a variety of covalent, but dynamic post-translational modifications, including acetylation, phosphorylation and methylation, which determine their interactions with other proteins, regulating chromatin structure and remodeling.
Acetylation of conserved lysine residues present in histone tails has long been linked to transcriptional activity. Histone acetylation is a hallmark of transcriptionally active regions, whereas hypoacetylated histones tails are found in transcriptionally inactive euchromatin or heterochromatin regions. The equilibrium of histone acetylation/deacetylation is governed by the opposite actions of histone desacetylases (HDACs) and histone acetyl transferases. Aberrant acetylation or deacetylation leads to such diverse disorders as leukemia, epithelial cancers and Rubinstein–Taybi syndrome.1 Like histone acetylation, the addition of negatively charged phosphate groups to histone tails also neutralize their basic charge and reduce their affinity to DNA. In this context, phosphorylation of serine 10 in histone H3 has been correlated with gene activation in mammalian cells2 and phosphorylation of H2A seemed to occur after activation of DNA-damage signaling pathways, permitting chromatin exposure and, consequently, facilitating the DNA repair.3 Coffin–Lowry syndrome is a chromatin remodeling disorder caused presumably by insufficient H3 phosphorylation and acetylation owing to mutations in the RSK-2 gene (RPS6KA3), which inactivates its protein serine-threonine kinase activity and perturbs cell cycle regulation and cell proliferation.4 Histone methylation can be a mark for both active and inactive chromatin. Lysine residues can be mono-, di- or trimethylated at the ɛ amino group, thereby vastly expanding the possibility of such modification. Usually, methylation of lysine 9 on the N-terminus of histone H3 is associated to silent DNA and is globally distributed throughout heterochromatin regions such as centromeres and telomeres. It appears also at transcriptionally silenced promoters of inactive X chromosome.5, 6 Curiously, evidences indicate that different degrees of methylation could be correlated with distinct levels of gene silencing. For example, while trimethylation of lysine 9 of H3 is associated with constitutive heterochromatin, mono- and dimethylation of the same position were supposed to be associated with facultative heterochromatin.7 On the other hand, methylation of lysine 4 of histone H3 signals for transcriptional activity and is found at promoters of active genes. One explanation for this duality function of histone methylation is that the gene silencing of methylated lysine 9 of histone H3 tails is mediated by the specifically binding of the mammalian heterochromatin protein 1 (HP1) through its conserved chromo domain regions, whereas methylated lysine 4 of histone H3 is apparently not.8 A number of histone methyltransferases with distinct functions have been identified. Recently, Sun et al9proposed that a histone H3 lysine 36-specific HTMase activity could be seen for the huntingtin interacting protein B and could be implicated in the pathogenesis of Huntington disease. The relevance of chromatin remodeling is further emphasized by the action of ATP-dependent SWI/SNF chromatin remodeling complexes. These multisubunit complexes modify the nucleosomes topology in a ATP-dependent manner by disrupting DNA:histone interactions, allowing the access of transcription factors to their targets within the chromatin. Several members of the highly conserved SWI–SNF complex have been implicated in cancer10 and in ATRX syndrome.11 Owing to the multiplicity of these and others potentially histone modifications like ubiquination, poly-ADP ribosylation and sumoylation, we can consider that histones have their own epigenetic code, essential to efficient spatial and temporal gene expression control.
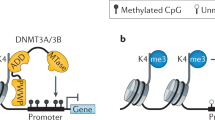
Besides modification of core histones, the genomes of all vertebrates are also modified by the post-synthetic addition of a methyl group in carbon 5 of the cytosine ring, predominantly in 5′-CpG-3′. Methylation of DNA plays a key role in several biological processes, including parental imprinting,12 development,13 X-chromosome inactivation,14 silencing of foreign DNA,15 proper centromeric segregation16 and T-cell function.17 Furthermore, methylation is a prominent factor in aging18 and has been suggested to be involved in the long-term memory process.19 The donation of carbon atoms for maintenance of methylation patterns is mainly realized through the folate cycle, which involves an integrated action of many gene products and other important micronutrients, like vitamin B12, vitamin B6, choline and methionine, obtained by dietary. All these elements are directly or indirectly required for the conversion of homocysteine to methionine, which is the immediate precursor of S-adenosyl methionine (SAM), the major intracellular methyl donor for DNA, protein and lipid methylation maintenance. After donating its methyl group, SAM is converted into S-adenosylhomocysteine (SAH), which then becomes homocysteine after losing its adenosine in a reversible manner. The presence of genetic polymorphisms that alter the functionality of key transport molecules and enzymes required for the folate/homocysteine cycle steps could predispose to genomic instability, altered recombination and abnormal segregation.20 It is clear, however, that the effects of these variants will be closely dependent on the individual folate nutritional status, as a high intake of folate and associated nutrients effectively could neutralize the negative biochemical consequence of the polymorphism(s). In this context, polymorphisms that diminish the bioavailability of folate/homocysteine in the mother around or during pregnancy associated with a low intake of these micronutrients have been proposed to contribute to abnormalities in the fetus, such as failure of neural tube closure,21 cleft palate,22 and non-disjunctions disorders as Down syndrome20 and trisomy 18.23 Furthermore, DNA hypomethylation and high levels of homocysteine associated with low folate intake have been described as important risk factors for cancer,24 including those of the colon, breast, pancreas, cervix, bronchus, as well as leukemia. It is also implicated in the etiology of cardiovascular25 and cerebrovascular diseases,26 diabetes27 and developmental/neurological, such as schizophrenia and depression abnormalities.28 The capacity of homocysteine induces oxidative stress and DNA damage suggests yet that increase of it may predispose the nervous system to aging and age-related neurodegenerative disorders like Parkinson and Alzheimer diseases.29 This aspect may represent one contribution factor to the association of Alzheimer disease (AD) and Down syndrome-related dementia, since by the fourth decade of life, individuals with Down syndrome display many of the same neuropathological features as do individuals with AD. Increasing levels of homocysteine also result in a greater amount of SAH, as well as free adenosine, which are potent inhibitors of transmethylation enzymes and can affect a variety of biochemical processes, including the decrease of hepatocellular detoxification.30 Some reports have yet demonstrated that low folate and vitamin B12 levels associated to demethylation patterns may facilitate the DNA integration of oncogenic viruses and may activate transcription of endogenous retroviruses.15 Corroborating these findings, the treatment of AIDS patients by using folic acid and vitamin B12 supplements has been successful.31 However, despite of the enormous proven benefit of folate nutrition, recent findings alert that the excessive supplemental levels of this vitamin during the periconceptional period could select embryos that carry the folate-associated mutant alleles and it could increase the prevalence of significant human life-threatening diseases associated with the presence of this variant allele along our evolution, like AD.32
Between the DNA methyltransferases, the first described was Dnmt1, which is indispensable for restoring full methylation from the hemi-methylated DNA sites that arises as a result of the semiconservative replication (maintenance activity). It is likely that Dnmt1 requires chromatin to be deacetylated before it can effectively methylate DNA.33 Four other DNA methyltransferases genes were described in mammals (Dnmt2, Dnmt3a, Dnmt3b and Dnmt3-like gene/Dnmt3L). Dnmt2 was recently shown to have a weak DNA methyltransferase activity and to act as a tRNA methyltransferase,34 whereas Dnmt3a and Dnmt3b are high expressed in embryonic cells and have de novo methyltransferase rather than maintenance methyltransferase activity.35 It implies that after a global pre implantation demethylation, Dnmt3a and Dnmt3b are responsible for initiating DNA methylation during early embryonic development. Li et al13 proposed that Dnmt3a and Dnmt3b may cooperate with Dnmt1 in stable maintenance of DNA methylation patterns and that inactivation of Dnmt3a and Dnmt3b in somatic cells leads to chromosomal instability. Recently, it was demonstrated that Dnmt3L physically associates with Dnmt3a and Dnmt3b and stimulates their catalytic activities both in cell culture assays and in vitro.36
Mutations in DNMTs genes, cis-acting sequences or their interacting partners could have profound limitations on specific DNA methylation patterns. All methyltransferases have been found to be overexpressed in human tumors, although to moderate levels only.37, 38 Thus, DNA methylation acts as a double-edged sword in cancer, as whereas oncogenesis is promoted by local hypermethylation of tumor suppressor genes, global genomic hypomethylation, on the other hand, affects oncogene expression and genomic instability.39 So, nowadays epigenetics silencing has been recognized as a third pathway satisfying Knudson's hypothesis. DNMT3B misexpression is also known to be the cause of the ICF syndrome, characterized by the lack of condensation of the pericentromeric heterochromatin regions of chromosomes 1, 9 and 16 owing to a marked hypomethylation of classical satellites II and III associated with centromere instability.40 It is intriguing, however, that although DNMT3B alterations lead to a widespread genomic DNA methylation defect, the ICF phenotype is restricted to facial abnormalities and immune deficiency.
The repression of methylated CpG islands located at promoter sequences showed to be mediated by methyl-CpG binding proteins (MBPs). Upon specifically binding to methylated DNA, the MBPs recruit chromatin remodeling factors, like HDACs, acting as gene expression regulators. The founder member of the MBPs is the methyl-CpG binding protein 2 (MeCP2), which binds to symmetrically methylated cytosine throughout the genome, and is an important transcriptional silencer.41 Loss-of-function mutations in the MECP2 gene are associated to Rett syndrome in females and to a wide spectrum of neurological phenotypes in males.42 Interestingly, a mild overexpression of MeCP2 in neurons is also detrimental and leads to symptoms of severe motor dysfunction and seizures,43 suggesting that a proper dosage of MeCP2 is critical for neuronal function and that levels of MeCP2 in the CNS are tightly regulated. By searching for sequence homology to the conserved methyl-binding domain of MeCP2, other five similar proteins were found. Like MeCP2, the MBD1, MBD2, MBD3 and Kaiso proteins are all involved in transcription repression, whereas MBD4 acts in DNA mismatch repair. Unexpectedly, although MeCP2, MBD2 and MBD4 have been found to be downregulated in human cancers,44 no firm cancer connections have been established for any of the other MBP.45
Other DNA methylation-related epigenetic diseases originate from genetic alterations like tri-nucleotide expansions (as in Fragile X syndrome) and from parental imprinting defects (imprinting disorders). Fragile X syndrome is the best-characterized genetic alteration that modifies the epigenetic coding of a sequence, contributing to a disease phenotype. This syndrome elapses from an expansion of 5′CGG 3′ polymorphic repeats in the 5′UTR of the FMR1 gene and is associated to hypermethylation and changes in the chromatin structure, leading to gene silencing.46 It is conceivable that the expanded number of repeats is recognized as foreign and become methylated de novo like any integrated viral DNA.47 Concerning imprinting-based epigenetic diseases, it is vastly known that maternal and paternal genomes are both required for normal embryonic and postnatal development, although there is a functional asymmetry between them. CpG methylation is also closely associated with imprinting, having an important role in silencing certain genes as well in activating others. Initial parental-origin imprinting signals are established during gametogenesis. In the pre-implantation embryo, the methylation patterns of the gametes are erased with a dramatic decrease in the total level of methylated DNA. After implantation, a wave of de novo methylation establishes a new pattern, in which the majority of CpGs islands are methylated in a tissue-specific manner.48 Therefore, it is unlikely that all epigenetic information is erased in germ cells, as some heritable characteristics in humans may in fact have an inherited epigenetic basis through generations.49 Also, Moore and Haig50 postulated that during fetal growth there is a preferential expression of paternal genes, whereas maternal allele's expression should seek a protective performance of minimizing the absorption of mother resources for future pregnancies. Such details of the epigenetic reprogramming are responsible for the highest source of failure found in cloning approaches. The usual epigenetic alteration leading to imprinting syndromes is uniparental disomy. Angelman (AS) and PraderWilli (PWS) syndromes are the best-known imprinting syndromes, being caused by either deletion or epigenetic faults of the same cluster (15q11–q13). Evidences suggest that the AS cause resides in loss of expression of the E6-associated protein ubiquitin-protein ligase gene (UBE3A), maternal imprinted only in the brain. Otherwise, PWS main cause is attributed to the paternal imprinted SNRPN gene, which encodes the expressed SNURF and SmN spliceosomal (SNRPN – small nuclear ribonucleoprotein-associated polypeptide N) proteins.51 Imprinting alterations are frequently associated with many types of cancer, including acute myoblastic leukemia (paternal chromosome 7), Wilm's tumor (maternal chromosome 11p15.5), neuroblastoma (maternal chromosome 1p36 and paternal chromosome 2), rhabdomyosarcoma (maternal chromosome 11p15.5), glomus tumors (paternal chromosomes 11q13 and 11q22.3–q23.3) and sporadic osteosarcoma (maternal chromosome 13).
Besides modification of core histones and DNA methylation, three other types of epigenetic control have been recently described. The first consists in the activity of post-transcriptional gene silencing transcripts that can be expressed in the form of non-coding RNAs (eg, XIST), non-coding antisense RNAs (eg, TSIX) and RNA interference (RNAi). Non-coding RNAs regulate key processes, such as association of HP1 to chromatin52 and the role of XIST transcript in the initiation of X-chromosome inactivation.53 After fertilization, expression of the gene XIST from the inactive X is required for initiating X inactivation, by the ‘coating’ of a substantial portion of the future inactive X, leading to its DNA methylation and chromatin condensation. Small non-coding RNAs (microRNAs) are also involved in the silencing of repetitive DNA, such as retrotransposons, being important components for genome stability and integrity.54 A survey about the distribution of microRNAs along the genome evidenced that they are frequently located at fragile sites and loci involved in cancer.55 Consistent with this, the microRNA genes miR15 and miR16 are downregulated in two-thirds of chronic lymphocytic leukemia cases.56 Another class of non-coding RNAs, the antisense RNAs, is involved in the silencing of some imprinted genes57 and also in the repression of XIST on the selected active X chromosome by the antisense transcript TSIX and accumulation in cis of Xist.53 A case of α thalassemia was reported, in which a deletion-induced aberrant antisense transcript from a neighboring gene leads to DNA methylation and silencing of the intact HBA2 α-globin gene.58 It lifts primarily suspicion that double-strand RNAs (dsRNAs), formed by the complementary of sense and antisense transcripts, would recruit by their own other epigenetic repressors like those involved in DNA methylation and opened our mind to a novel mechanism of human diseases. By this way, such kind of genomic rearrangement causing epigenetic silencing might elucidate several other unexplained but clinically well-defined disorders. RNAi is a highly conserved silencing pathway triggered by dsRNA, which regulates gene expression via targeted degradation of mRNAs and subsequently transcriptional repression of mRNAs. The hallmark of this pathway involves the cleavage of dsRNAs into ∼22 nucleotides double and small-interfering RNA molecules (siRNAs) by an Rnase-III-like enzyme called Dicer and the subsequent association of these siRNAs with a multiprotein RNAi effector complex called RISC to provide specificity for the degradation of complementary mRNAs. In some organisms, the RNAi machinery may also require an RNA-dependent RNA polymerase that is believed to amplify dsRNA.59 Recent data, from fission yeast, indicates that the RNAi machinery targets repressive histone modification to specific chromosomal loci destined for silencing.60 RNAi machinery is guided to heterochromatin through the formation of dsRNAs by the bidirectional transcription of repetitive DNA sequences.54, 61 In mammals, this RNAi machinery is conserved and recent evidences indicate that centromeric and pericentromeric heterochromatin repeats are transcribed and dsRNA can silence genes through de novo methylation in such organisms.62 In diseases associated with trinucleotide repeat instability and subsequent DNA methylation-mediated gene silencing, like Fragile X syndrome, one of the models for explaining how the large repeats are targeted for methylation includes the formation of hairpin RNA (similar to dsRNA) from the transcribed large repeats, which might be cleaved by Dicer and subsequently recruit the RNAi silencing machinery.63
The second additional class of epigenetic regulation involves the presence of methylation-dependent sensitive insulators, fundamental for the roles of insulin-like growth factor 2 (IGF2) and H19 genes. These are proximal and oppositely imprinted genes: expression is from the paternal copy of IGF2 and from maternal copy of H19. An insulator element has been found upstream of H19, between this gene and IGF2, and is identified as a differentially methylated domain (DMD), which interacts with enhancer elements downstream of H19 and acts as a chromatin boundary that regulates IGF2 imprinting. On the maternal chromosome, an enhancer-blocking protein binds to the DMD, preventing the enhancers from interacting with the promoter of IGF2, favoring only H19 expression. In contrast, on the paternal chromosome, DMD is methylated and, consequently, the blocking protein cannot bind and IGF2 can be expressed whereas H19 is not.64 Recently, the importance of these genes was thoroughly discussed by Kono et al65 who combined the chromosomal complements of a mature, fully grown mouse egg with a non-growing egg from a new-born mouse, in which the H19 gene and the DMD had been deleted for mimic parental imprint. Surprisingly, this manipulation was sufficient for providing normal results for the expression of 1000 between 11000 analyzed genes, including other apparently unconnected imprinted genes.
The latter form of epigenetic control refers to the action of Polycomb Group (PcG) and Trithorax Group (TrxG), which are an ancient group of chromatin modifiers proteins that constitute a cellular memory system responsible for controlling chromatin accessibility and maintaining transcription in the first stages of embryonic life, throughout development and in adulthood.66 For this purpose, these proteins usually have antagonistic effects on target genes, including developmentally and cell cycle regulated genes: PcG proteins act as stable repressors whereas TrxG proteins promote maintenance of gene activity. Evidences showed that components of the PcG complex and two polypeptides of the TrxG, TRX and ASH1, are histone methyltransferases, suggesting that the memory system uses a similar pathway to regulate the binding of both PcG and TrxG complexes to their sites.67, 68
The emerging role of PcG proteins in X-chromosome inactivation has calling attention for the involvement of PcG proteins with RNA machinery. Thus, X inactivation represents one of the great paradigms of epigenetics, as it involves among other mechanisms, the roles of antisense transcripts69 and other non-coding RNAs,70, 71 H3 K9 di-methylation,5 H3 K27 tri-methylation,6 H4 acetylation,72 DNA methylation,14 possible chromatin insulation73 and Eed and Enx1 PcG proteins.74 PcG EZH2 is overexpressed in many types of cancer, including those of prostate75 and breast.76 Moreover, the homolog of the Drosophila PcG PHO in humans, the YY1 transcriptional repressor, has been found to be part of a repressive complex bound to the D4Z4 3.3 kb repeat that regulates genes involved in the control of the facioscapulohumeral dystrophy.77 Dystrophic patients carry deletions of these repeats and show depression of neighboring and more distant transcription units. As 50% of the genome is composed by repetitive sequences, it will be interesting if and how repetitive sequences could be a major target of PcG proteins in mammalian genome.66
Elucidation of the precise way by which the human epigenome reprogramming occurs during gametogenesis, development and aging is the dream of returning differentiated cells into an undifferentiated stem cell state and it will certainly provide new therapeutic strategies for the treatment of epigenetic-based diseases. The investigation of the epigenome by high-throughput genomic technologies has been allowed the identification of differentially methylated sites/genes, which could be used as potential epigenetic markers for routine diagnosis and prognosis. Concerning cancer, many CpG islands that exhibit abnormal methylation have been identified and have been considered for identification of cancer classes, monitoring of tumor progression and evaluation of distinct drug intolerance between patients. In the future, it is expected that personalized medicine could be provided as a result of epigenotype profiling of critical genes. Although these approaches are not still available, an increasing number of agents that alter DNA methylation or histones positioning patterns are currently being tested in clinical trials.
Inhibitors of DNA methylation are able to reactivate the expression of previously silenced genes owing to an epigenetically pathological situation, like the case of tumor-suppressor genes. For this purpose, the most used hypomethylating agents are pyrimidine nucleosides analogs of cytidine (eg, 5-azacytidine and 5-aza-2′-deoxycytidine) that specifically inhibits DNA methylation by its incorporation into replicating DNA in place of cytosine, trapping DNA methyltransferases and resulting in heritable demethylated DNA. In addition to the fact that non-proliferating cells are relatively insensitive to azacytidine, the great disadvantage of these agents is their cytotoxicity, particularly when used in high doses. Therefore, the proved clinical efficiency of low-dose of 5-azacytidine in treating hemoglobinopathies,78 myeloid dysplastic syndrome and other leukemias79 prompted its regular approval by the US Food and Drug Administration for the treatment of patients with all subtypes of myelodysplastic syndrome as injectable suspension. Moreover, several in vitro/in vivo experiments of inhibitors of DNA methylation have showed positive results for autoimmune anomalies80 and Fragile X syndrome.81 Thus, epigenetic therapy might be also a good strategy to treat females bearing X-linked dominant disorders, like Rett syndrome. As X-inactivation is random, 50% of the females' cells harbor an unused but intact copy of the gene, which can be reactivated, ameliorating the clinical phenotype of the disorder. An alternative for preventing the cytotoxicity of the demethylating agents is the development of antisense oligonucleotides that specific target DNA methyltransferases.82
Pathological epigenetic silencing is almost associated with histone deacetylation. For therapy purpose, HDACs can be globally or specifically inhibited by a structurally diverse group of small molecules, including hydroxamates, cyclic peptides, aliphatic acids, benzamides and electrophilic ketones.83 The inhibitory effects occur owing to acetylated histones accumulation, leading to transcription activation of select genes whose previous expression had been epigenetically turned off. Therefore, as there are many types of HDACs, the success of the inhibition approach as a therapeutic role is directly linked to the selectiveness of each HDACs inhibitor and its cytotoxicity. Such therapy strategy has long being routinely present in the treatment of epilepsy and bipolar disorder with valproic acid.84 Experimentally, HDACs inhibition worked efficiently in cell lines of non-small lung cancer85 and several other types of solid and hematological tumors.86 Positive therapy results have been also found in animals models for cardiac hypertrophy,87 autoimmune encephalomyelitis88 and polyglutamine expansion-associated neurological disorders, such as Dentatorubral–pallidoluysian atrophy,89 Huntington disease90 and spinal and bulbar muscular atrophy.91
The closely link between DNA methylation and histone deacetylation stimulated the development of experimental combined and efficient therapies, in which demethylating agents are used simultaneously with HDACs inhibitors, like 5-aza-2′deoxycytidine and trichostatin or sodium phenylbutyrate.92 Moreover, cancer pre-clinical researchers have demonstrated that HDAC inhibitors and demethylating agents can be potentially used as sensitizing agents for subsequent therapies as chemotherapy,93 radiotherapy94 and immunotherapy.95 However, epigenetic therapy should be taken with care, as it could wake up dormant hypermethylated oncogenes, silenced alleles of imprinted genes, genes on the female inactive X chromosome or other intentionally spatial/temporal silenced genes.
The most intriguing and exciting feature concerning epigenetic diseases is that, except to cancer, neurocognitive disturbs (mainly mental disturbances or neurodegenerative disorders) are present in almost all of them. One possible explanation is that the complexity of brain depends on the precise spatial and temporal action of many genes, by the way that a minimal alteration is sufficient to disturb the balance of the integrated gene expression. Furthermore, complex cytoarchiteture and cellular signaling mechanisms of the nervous system are reflected at the molecular level with more genes being expressed in this tissue than in any other.29 Future eager research will certainly provide more mechanistic understanding into how epigenetics pathways can regulate or disturb the role of genetics code in neuronal differentiation and maturation and may therefore reveal novel approaches for preventing and treating a great number of epigenetic diseases.
References
Ausió J, Levin DB, de Amorim GV, Bakker S, Macleod PM : Syndromes of disordered chromatin remodeling. Clin Genet 2003; 64: 83–95.
Thomson S, Mahadevan LC, Clayton AL : MAP kinase-mediated signaling to nucleosomes and immediate-early gene induction. Semin Cell Dev Biol 1999; 10: 205–214.
Downs JA, Lowndens NF, Jackson SP : A role for Saccharomyces cerevisiae histone H2A in DNA repair. Nature 2000; 408: 1001–1004.
Jacquot S, Zeniou M, Touraine R, Hanauer A : X-linked Coffin–Lowry syndrome (CLS, MIM 303600, RPS6KA3 gene, protein product known under various names: pp. 90 (rsk2), RSK2, ISPK, MAPKAP1). Eur J Hum Genet 2002; 10: 2–5.
Heard E, Rougeulle C, Arnaud D, Avner P, Allis CD, Spector DL : Methylation of histone H3 at Lys-9 is an early mark on the X chromosome during X inactivation. Cell 2001; 107: 727–738.
Plath K, Fang J, Mlynarczyk-Evans SK et al: Role of histone H3 lysine 27 methylation in X inactivation. Science 2003; 300: 131–135.
Rice JC, Briggs SD, Ueberheide B et al: Histone methyltransferases direct different degrees of methylation to define distinct chromatin domains. Mol Cell 2003; 12: 1591–1598.
Bannister AJ, Zegerman P, Partridge JF et al: Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature 2001; 410: 120–124.
Sun XJ, Wei J, Wu XY et al: Identification and characterization of a novel human histone H3 lysine 36-specific methyltransferase. J Biol Chem 2005; 280: 35261–35271.
Roberts CW, Orkin SH : The SWI/SNF complex – chromatin and cancer. Nat Rev Cancer 2004; 4: 133–142.
Picketts DJ, Higgs DR, Bachoo S, Blake DJ, Quarrell OW, Gibbons RJ : ATRX encodes a novel member of the SNF2 family of proteins: mutations point to a common mechanism underlying the ATR-X syndrome. Hum Mol Genet 1996; 5: 1899–1907.
Bartolomei MS, Tilghman SM : Genomic imprinting in mammals. Annu Rev Genet 1997; 31: 493–525.
Li E, Chen T, Dodge J, Hata K, Okano M, Ueda Y : DNA methylation in development and diseases. Human Genome Meeting Symposium Abstracts: 2003, Presentation 13. 2003, http://hgm2003.hgu.mrc.ac.uk/Abstracts/Publish/Symposia/Symposium04/hgm13.html.
Hornstra I K, Yang TP : High resolution methylation analysis of the human HPRT gene 5′ region on the active and inactive X chromosomes: correlation with binding sites for transcriptional factors. Mol Cell Biol 1994; 14: 1419–1430.
Jones PA : DNA methylation errors and cancer. Cancer Res 1996; 56: 2463–2467.
Cleveland DW, Mao Y, Sullivan KF : Centromeres and kinetochores: from epigenetics to mitotic checkpoint signaling. Cell 2003; 112: 407–421.
Richardson B : DNA methylation and autoimmune disease. Clin Immunol 2003; 109: 72–79.
Issa JP : CpG-island methylation in aging and cancer. Curr Top Microbiol Immunol 2000; 249: 101–118.
Levenson JM, Roth TL, Lubin FD et al: Evidence that DNA (cytosine-5) methyltransferase regulates synaptic plasticity in the hippocampus. J Biol Chem 2006; 281: 15763–15773.
James SJ, Pogribna M, Pogribny IP et al: Abnormal folate metabolism and mutation in the methylenetetrahydrofolate reductase gene may be maternal risk factors for Down syndrome. Am J Clin Nutr 1999; 70: 495–501.
van der Put NM, Thomas CM, Eskes TK et al: Altered folate and vitamin B12 metabolism in families with spina bifida offspring. Q J Med 1997; 90: 505–510.
Murray JC : Gene/environment causes of cleft lip and/or palate. Clin Genet 2002; 61: 248–256.
Hassold TJ, Burrage LC, Chan ER et al: Maternal folate polymorphisms and the etiology of human nondisjunction. Am J Hum Genet 2001; 69: 434–439.
Skibola CF, Smith MT, Kane E et al: Polymorphisms in the methylenetetrahydrofolate reductase gene are associated with susceptibility to acute leukemia in adults. Proc Natl Acad Sci USA 1999; 96: 12810–12815.
Boushey CJ, Beresford SA, Omenn GS, Motulsky AG : A quantitative assessment of plasma homocysteine as a risk factor for vascular disease. Probable benefits of increasing folic acid intakes. JAMA 1995; 274: 1049–1057.
Hankey GJ, Eikelboom JW : Homocysteine and stroke. Curr Opin Neurol 2001; 14: 95–102.
Sun J, Xu Y, Zhu Y, Lu H : Genetic polymorphism of methylenetetrahydrofolate reductase as a risk factor for diabetic nephropathy in Chinese type 2 diabetic patients. Diabetes Res Clin Pract 2004; 64: 185–190.
Herran A, Garcia-Unzueta MT, Amado JA, Lopez-Cordovilla JJ, Diez-Manrique JF, Vazquez-Barquero JL : Folate levels in psychiatric outpatients. Psychiatry Clin Neurosci 1999; 53: 531–533.
Mattson MP : Methylation and acetylation in nervous system development and neurodegenerative disorders. Ageing Res Rev 2003; 2: 329–342.
Miller A : The methionine-homocysteine cycle and its effect on cognitive diseases. Alt Med Rev 2003; 8: 7–19.
Mathe G : Why have ten or so nontoxic, retrovirus integrase inhibitors not been made available for AIDS treatment? A ten-year experience [correction of experiment] must liberate them. Biomed Pharmacother 1999; 53: 484–486.
Lucock M, Yates Z : Folic acid – vitamin and panacea or genetic time bomb? Nat Rev Genet 2005; 6: 235–240.
Fuks F, Burgers WA, Brehm A, Hughes-Davies L, Kouzarides T : DNA methyltransferase Dnmt1 associates with histone deacetylase activity. Nat Genet 2000; 24: 88–91.
Jeltsch A, Nellen W, Lyko F : Two substrates are better than one: dual specificities for Dnmt2 methyltransferases. Trends Biochem Sci 2006; 31: 306–308.
Okano M, Bell DW, Haber DA, Li E : DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 1999; 99: 247–257.
Kareta MS, Botello ZM, Ennis JJ, Chou C, Chedin F : Reconstitution and mechanism of the stimulation of DE NOVO methylation by human DNMT3L. J Biol Chem 2006; July 7; [Epub ahead of print].
Robertson KD, Uzvolgyi E, Liang G et al: The human DNA methyltransferases (DNMTs) 1, 3a and 3b: coordinate mRNA expression in normal tissues and overexpression in tumors. Nucleic Acids Res 1999; 27: 2291–2298.
Chen CL, Yan X, Gao YN, Liao QP : Expression of DNA methyltransferase 1, 3A and 3B mRNA in the epithelial ovarian carcinoma. Zhonghua Fu Chan Ke Za Zhi 2005; 40: 770–774.
Karpf AR, Matsui S : Genetic disruption of cytosine DNA methyltransferase enzymes induces chromosomal instability in human cancer cells. Cancer Res 2005; 65: 8635–8639.
Xu GL, Bestor TH, Bourc'his D et al: Chromosome instability and immunodeficiency syndrome caused by mutations in a DNA methyltransferase gene. Nature 1999; 402: 187–191.
Jones PL, Veenstra GJ, Wade PA et al: Methylated DNA and MECP2 recruit histone deacetylase to repress transcription. Nat Genet 1998; 19: 187–191.
Moog U, Smeets EE, Roozendaal KEV et al: Neurodevelopmental disorders in males related to the gene causing Rett syndrome in females (MECP2). Eur J Paediatr Neurol 2003; 7: 5–12.
Collins AL, Levenson JM, Vilaythong AP et al: Mild overexpression of MeCP2 causes a progressive neurological disorder in mice. Hum Mol Genet 2004; 13: 2679–2689.
Zhu Y, Spitz MR, Zhang H, Grossman HB, Frazier ML, Wu X : Methyl-CpG-binding domain 2: a protective role in bladder carcinoma. Cancer 2004; 100: 1853–1858.
Bader S, Walker M, McQueen HA et al: MBD1, MBD2 and CGBP genes at chromosome 18q21 are infrequently mutated in human colon and lung cancers. Oncogene 2003; 22: 3506–3510.
O'Donnell WT, Warren ST : A decade of molecular studies of fragile X syndrome. Annu Rev Neurosci 2002; 25: 315–338.
Doerfler W : Patterns of DNA methylation – evolutionary vestiges of foreign DNA inactivation as a host defense mechanism. A proposal. Biol Chem Hoppe Seyler 1991; 372: 557–564.
Kafri T, Ariel M, Brandeis M et al: Developmental pattern of gene-specific DNA methylation in the mouse embryo and germ line. Genes Dev 1992; 6: 705–714.
Rakyan V, Whitelaw E : Transgenerational epigenetic inheritance. Curr Biol 2003; 13: R6.
Moore T, Haig D : Genomic imprinting in mammalian development: a parental tug-of-war. Trends Genet 1991; 7: 45–49.
Nicholls RD, Knepper JL : Genome organization, function, and imprinting in Prader–Willi and Angelman syndromes. Annu Rev Genomics Hum Genet 2001; 2: 153–175.
Maison C, Bailly D, Peters AH et al: High-order structure in pericentric heterochromatin involves a distinct pattern of histone modification and an RNA component. Nat Genet 2002; 30: 329–334.
Avner P, Heard E : X-chromosome inactivation: counting, choice and initiation. Nat Rev Genet 2001; 2: 59–67.
Schramke V, Allshire R : Hairpin RNAs and retrotransposon LTRs effect RNAi and chromatin-based gene silencing. Science 2003; 301: 1069–1073.
Calin GA, Sevignani C, Dumitru CD et al: Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancer. Proc Natl Acad Sci 2004; 101: 2999–3004.
Calin GA, Dumitru CD, Shimizu M et al: Frequent deletions and down-regulation of micro-RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci 2002; 99: 15524–15529.
Sleutels F, Zwart R, Barlow DP : The non-coding Air RNA is required for silencing autosomal imprinted genes. Nature 2002; 415: 810–813.
Tufarelli C, Stanley JA, Garrick D et al: Transcription of antisense RNA leading to gene silencing and methylation as a novel cause of human genetic disease. Nat Genet 2003; 34: 157–165.
Huang L, Gledhill J, Cameron CE : RNA-dependent RNA polymerase in gene silencing; In Hannon G (ed): RNAi: a guide for gene silencing. Cold Spring Habor Laboratory Press: NY, 2003, pp 175–203.
Verdel A, Jia S, Gerber S et al: RNAi-mediated targeting of heterochromatin by the RITS complex. Science 2004; 303: 672–676.
Noma K, Sugiyama T, Cam H et al: RITS acts in cis to promote RNA interference-mediated transcriptional and posttranscriptional silencing. Nat Genet 2004; 36: 1174–1180.
Kawasaki H, Taira K : Induction of DNA methylation and gene silencing by short interfering RNAs in human cells. Nature 2004; 431: 211–217.
Robertson KD : DNA methylation and human disease. Nat Rev Genet 2005; 6: 597–610.
Arney KL : H19 and Igf2 – enhancing the confusion? Trends Genet 2003; 19: 17–23.
Kono T, Obata Y, Wu Q et al: Birth of parthenogenetic mice that can develop to adulthood. Nature 2004; 428: 860–864.
Orlando V : Polycomb, epigenomes and control of cell identity. Cell 2003; 112: 599–606.
Kuzmichev A, Nishioka K, Erdjument-Bromage H, Tempst P, Reinberg D : Histone methyltransferase activity associated with a human multiprotein complex containing the Enhancer of Zeste protein. Genes Dev 2002; 16: 2893–2905.
Nakamura T, Mori T, Tada S et al: ALL-1 is a histone methyltransferase that assembles a supercomplex of proteins involved in transcriptional regulation. Mol Cell 2002; 10: 1119–1128.
Plath K, Mlynarczyk-Evans S, Nusinow DA, Panning B : Xist RNA and the mechanism of X chromosome inactivation. Annu Rev Genet 2002; 36: 233–278.
Johnston CM, Newall AE, Brockdorff N, Nesterova TB : Enox, a novel gene that maps 10 kb upstream of Xist and partially escapes X inactivation. Genomics 2002; 80: 236–244.
Ogawa Y, Lee JT : Xite, X-inactivation intergenic transcription elements that regulate the probability of choice. Mol Cell 2003; 11: 731–743.
O'Neill LP, Keohane AM, Lavender JS et al: A developmental switch in H4 acetylation upstream of Xist plays a role in X chromosome inactivation. EMBO J 1999; 18: 2897–2907.
Chao W, Huynh KD, Spencer RJ, Davidow LS, Lee JT : CTCF, a candidate trans-acting factor for X-inactivation choice. Science 2002; 295: 345–347.
Silva J, Mak W, Zvetkova I et al: Establishment of histone h3 methylation on the inactive X chromosome requires transient recruitment of Eed-Enx1 polycomb group complexes. Dev Cell 2003; 4: 481–495.
Varambally S, Dhanasekaran SM, Zhou M et al: The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature 2002; 419: 624–629.
Raaphorst FM, Meijer CJ, Fieret E et al: Poorly differentiated breast carcinoma is associated with increased expression of the human polycomb group EZH2 gene. Neoplasia 2003; 5: 481–488.
Gabellini D, Green MR, Tupler R : Inappropriate gene activation in FSHD: a repressor complex bnds a chromosomal repeat deleted in dystrophic muscle. Cell 2002; 110: 339–348.
Saunthararajah Y, Hillery CA, Lavelle D et al: Effects of 5-aza-2′-deoxycytidine on fetal hemoglobin levels, red cell adhesion, and hematopoietic differentiation in patients with sickle cell disease. Blood 2003; 102: 3865–3870.
Issa JP, Garcia-Manero G, Giles FJ et al: Phase I study of low-dose prolonged exposure schedules of the hypomethylating agent 5-aza-2′-deoxycytidine (decitabine) in hematopoietic malignancies. Blood 2004; 103: 1635–1640.
Mizugaki M, Yamaguchi T, Ishiwata S et al: Alteration of DNA methylation levels in MRL lupus mice. Clin Exp Immunol 1997; 110: 265–269.
Tabolacci E, Pietrobono R, Moscato U, Oostra BA, Chiurazzi P, Neri G : Differential epigenetic modifications in the FMR1 gene of the fragile X syndrome after reactivating pharmacological treatments. Eur J Hum Genet 2005; 13: 641–648.
Yan L, Nass SJ, Smith D, Nelson WG, Herman JG, Davidson NE : Specific inhibition of DNMT1 by antisense oligonucleotides induces re-expression of estrogen receptor-alpha (ER) in ER-negative human breast cancer cell lines. Cancer Biol Ther 2003; 2: 552–556.
Marks PA, Miller T, Richon VM : Histone deacetylases. Curr Opin Pharmacol 2003; 3: 344–351.
Phiel CJ, Zhang F, Huang EY, Guenther MG, Lazar MA, Klein PS : Histone deacetylase is a direct target of valproic acid, a potent anticonvulsant, mood stabilizer, and teratogen. J Biol Chem 2001; 276: 36734–36741.
Komatsu N, Kawamata N, Takeuchi S et al: SAHA, a HDAC inhibitor, has profound anti-growth activity against non-small cell lung cancer cells. Oncol Rep 2006; 15: 187–191.
Marks PA, Dokmanovic M : Histone deacetylase inhibitors: discovery and development as anticancer agents. Expert Opin Investig Drugs 2005; 14: 1497–1511.
Kee HJ, Sohn IS, Nam KI et al: Inhibition of histone deacetylation blocks cardiac hypertrophy induced by angiotensin II infusion and aortic banding. Circulation 2006; 113: 51–59.
Camelo S, Iglesias AH, Hwang D et al: Transcriptional therapy with the histone deacetylase inhibitor trichostatin A ameliorates experimental autoimmune encephalomyelitis. J Neuroimmunol 2005; 164: 10–21.
McCampbell A, Taye AA, Whitty L, Penney E, Steffan JS, Fischbeck KH : Histone deacetylase inhibitors reduce polyglutamine toxicity. Proc Natl Acad Sci USA 2001; 98: 15179–15184.
Hockly E, Richon VM, Woodman B et al: Suberoylanilidehydroxamic acid, a histone deacetylase inhibitor, ameliorates motor deficits in a mouse model of Huntington's disease. Proc Natl Acad Sci USA 2003; 100: 2041–2046.
Minamiyama M, Katsuno M, Adachi H et al: Sodium butyrate ameliorates phenotypic expression in a transgenic mouse model of spinal and bulbar muscular atrophy. Hum Mol Genet 2004; 13: 1183–1192.
Belinsky SA, Klinge DM, Stidley CA et al: Inhibition of DNA methylation and histone deacetylation prevents murine lung cancer. Cancer Res 2003; 63: 7089–7093.
Plumb JA, Strathdee G, Sludden J, Kaye SB, Brown R : Reversal of drug resistance in human tumor xenografts by 2′-deoxy-5-azacytidine-induced demethylation of the hMLH1 gene promoter. Cancer Res 2000; 60: 6039–6044.
Chinnaiyan P, Vallabhaneni G, Armstrong E, Huang SM, Harari PM : Modulation of radiation response by histone deacetylase inhibition. Int J Radiat Oncol Biol Phys 2005; 62: 223–229.
Karpf AR, Jones DA : Reactivating the expression of methylation silenced genes in human cancer. Oncogene 2002; 21: 5496–5503.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Santos-Rebouças, C., Pimentel, M. Implication of abnormal epigenetic patterns for human diseases. Eur J Hum Genet 15, 10–17 (2007). https://doi.org/10.1038/sj.ejhg.5201727
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5201727
Keywords
This article is cited by
-
Cognitive Dysfunction and Exercise: From Epigenetic to Genetic Molecular Mechanisms
Molecular Neurobiology (2024)
-
Particulate matter exposure shapes DNA methylation through the lifespan
Clinical Epigenetics (2019)
-
Investigating the genetic and epigenetic basis of big biological questions with the parthenogenetic marbled crayfish: A review and perspectives
Journal of Biosciences (2018)
-
Physical activity in the prevention of human diseases: role of epigenetic modifications
BMC Genomics (2017)
-
A Comprehensive View of the Epigenetic Landscape Part I: DNA Methylation, Passive and Active DNA Demethylation Pathways and Histone Variants
Neurotoxicity Research (2015)
