
Abstract
It is important to characterise the amount of variation on the mammalian Y chromosome in order to assess its potential for use in evolutionary studies. We report very low levels of polymorphism on the Y chromosome of Saudi-Arabian hamadryas baboons, Papio hamadryas hamadryas. We found no segregating sites on the Y, despite sequence analysis of 3 kb noncontiguous intron sequence in 16 males with divergent autosomal microsatellite genotypes, and a further analysis of 1.1 kb intron sequence in 97 males from four populations by SSCP. In addition, we tested seven human-derived Y-linked microsatellites in baboons. Only four of these loci were male-specific and only one was polymorphic in our 97 male sample set. Polymorphism on the Y chromosome of Arabian hamadryas appears to be low compared to other primate species for which data are available (eg humans, chimpanzees and bonobos). Low effective population size (Ne) of paternal genes due to polygyny and female-biased adult sex ratio is a potential reason for low Y chromosome variation in this species. However, low Ne for the Y should be counterbalanced to some extent by the species' atypical pattern of male philopatry and female-biased dispersal. Allelic richness averaged over seven loci was not significantly different between an African and an Arabian population, suggesting that loss of variation during the colonisation of Arabia does not explain low Y variation. Finally, in the absence of nucleotide polymorphism, it is unclear to what extent selection could be responsible for low Y variation in this species.
Similar content being viewed by others

Introduction
There is substantial interest in using the mammalian Y chromosome in evolutionary genetics, particularly in combination with mitochondrial DNA (mtDNA), to give a more comprehensive perspective on a species' evolutionary history (Hurles and Jobling, 2001; Petit et al, 2002). This is primarily because a large portion of the Y does not recombine and is inherited from father to son. Haplotypic data are therefore readily accessible and patterns of variation should reflect the population history of males. Despite the success of Y chromosome analysis for the study of human population structure, evolutionary history and differences in behaviour of the sexes (reviewed in Hurles and Jobling, 2001; Stumpf and Goldstein, 2001; Jobling and Tyler-Smith, 2003), the Y has yet to realise its full potential in studies of non-human populations (exceptions include: Boissinot and Boursot, 1997; Tosi et al, 2000, 2002, 2003; Stone et al, 2002; van Hooft et al, 2002; Brändli et al, 2005).
One reason for the small number of studies employing the Y chromosome is that polymorphism is generally lower than in other regions of the genome (International SNP working Group, 2001; Hellborg and Ellegren, 2004). This may be explained by a greater influence of genetic drift on the Y because of its low effective population size (Ne, the copy number of the Y is one quarter that of the autosomes and variance in reproductive success is generally higher in males than in females), demographic history (such as recent bottlenecks), and directional selection (in the absence of recombination). Early studies of human Y chromosomes reported very low levels of polymorphism (eg Dorit et al, 1995; Hammer, 1995; Whitfield et al, 1995) and this has probably deterred researchers from using the Y, since a large sequencing effort is required to find variation. In human studies at least, the idea of the ‘uninformative Y’ has now been dismissed thanks to the development of a large panel of single nucleotide polymorphisms (SNPs) and microsatellite loci on the human Y (eg Underhill et al, 2000; Kayser et al, 2004). These developments have encouraged human population studies, but studies of other species still lag behind. Fortunately, thanks to recent genome sequencing efforts, it has been possible to develop conserved primers for the Y in a range of non-human mammals (eg Hellborg and Ellegren, 2003; Erler et al, 2004), and the use of the Y chromosome in population genetics of other species has become a real possibility. Characterisation of the amount of variation on the Y chromosome is the next step in realising the Y's potential.
In this study, we tested a panel of Y-linked markers (total ∼3.6 kb intron sequence and seven microsatellite loci) to quantify the amount of Y chromosome variation in samples of male hamadryas baboons (Papio hamadryas hamadryas) from four Saudi-Arabian populations. We interpret our results in terms of male effective population size, demographic history of our study populations and the possibility for selection on the Y, and consider the general implications of our findings for application of the Y chromosome in population genetics.
Methods
Samples
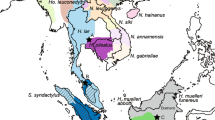
We analysed a total of 97 males sampled from four Saudi-Arabian populations (Abha n=69, Baha n=12, Taif n=13, Al-Akhal n=3), which are distributed along a north-south transect with about 200 km separating each population (Figure 1). Sampling methods and DNA extraction are described in Hammond et al (in revision). For sequence analysis, we first chose eight males (Abha n=5, Baha n=1, Taif n=1 and Al-Akhal n=1) from our total of 97 ensuring that there were animals from each population and avoiding males that were likely to be closely related given our knowledge of the social structure of the Abha population (Hammond et al, in revision). After finding no sequence variation in this sample set, we chose an additional eight individuals (Abha n=6, Baha n=1 and Taif n=1) by analysing published data from 19 autosomal loci (16 microsatellite and three protein loci; Hammond et al, in revision) using the programme STRUCTURE (Pritchard et al, 2000). We used STRUCTURE to identify the most genetically distinct eight groups of male baboons so as to maximise our chances of detecting variation in our sample of Y chromosomes. We specified eight genetic clusters (k=8) and used the admixture option, a burn-in of 50 000 iterations and 100 000 iterations of the Gibbs sampler. We then randomly selected one male from each of the eight clusters for sequencing.

Range and sampling. Geographic distribution of P. hamadryas hamadryas shown in grey, with sampling localities, represented by stars, separated by approximately 200 km each. ‘n’ is the number of males sampled and analysed at each locality.
Intron sequencing
Approximately 3 kb (2968 bp) from four Y-linked introns (UTY11, DBY1, DBY13 and DBY14, where the first three letters refer to the name of the gene and the numbers refer to the intron, Table 1, Hellborg and Ellegren, 2003) were amplified and sequenced in the subset of 16 males. PCR reactions contained ∼40 ng DNA, 0.2 μM each primer, 0.2 mM dNTPs, 1 × PCR buffer, 2.5 mM MgCl2 and 1.5 units Taq polymerase (Qiagen) in a 50 μl total volume, and PCRs were performed in a PE9700 (Perkin-Elmer) thermocycler. Cycling conditions, with a touchdown annealing step in the first 20 cycles, were as follows: initial denaturation at 95°C for 5 min, followed by 20 cycles of 95°C for 30 s, anneal (decreasing by 0.5°C/cycle from either 60–50°C or 55–45°C, Table 1) for 60 s, and extension at 72°C for 90 s, followed by a further 20 cycles using the lowest annealing temperature, and a final extension of 72°C for 10 min. Y chromosome specificity was verified by absence of PCR product in female controls. PCR products were purified using the QIAQuick PCR purification kit (Qiagen), and eluted in 30 μl dH2O. All loci were sequenced in both directions using the same primers used for PCR by a commercial sequencing service (Microsynth, Switzerland).
Intron SSCP
The loci DBY4, DBY8, DBY14 and ZFY4 (Hellborg and Ellegren, 2003) were analysed in all 97 males using single-strand conformational polymorphism (SSCP) of PCR products (note that DBY14 was analysed by both sequencing and by SSCP). PCR conditions were the same as described for sequence analysis except that a 60 s extension time was used. As for sequencing, Y chromosome specificity was verified by absence of PCR product in female controls. Post-PCR, 3 μl of PCR product was mixed with 7 μl of formamide (99%), 10 mM NaOH and bromophenol blue (0.005%) and denatured at 95°C for 10 min. SSCP analysis was carried out using GMA™ gels and SEA 2000™ Advanced submerged gel electrophoresis apparatus (Elchrom Scientific, Switzerland) for 12 h at 6 V/cm with the gel temperature maintained at 5±4°C in accordance with the manufacturer's guidelines for optimal separation of ssDNA. Gels were stained for 30 min in 1 × TAE containing 0.01% SYBR Gold (Molecular Probes, USA) and SSCP bands visualised by UV.
Microsatellite analysis
We tested seven published human-derived Y-specific microsatellite primer combinations. Six of these loci (DYS472, DYS557, DYS569, DYS576, DYS579, DYS645, Kayser et al, 1997, 2004) have been shown to be polymorphic, or are potentially so, in mandrills (Erler et al, 2004) but have never before been tested in baboons. The seventh locus (DYS391, Kayser et al, 1997, 2004) has been shown to be polymorphic in African hamadryas baboons (Woolley-Barker, 1999). Primer sequences for these loci are available from http://www.gdb.org/. All loci were tested for male-specific amplification in two males and one female, using a range of conditions. Optimised PCR reactions contained 1 μM of each primer (forward primer 5′ fluoro-labelled), 0.2 mM dNTPs, 1 × PCR buffer, 1.5–2.2 mM MgCl2 (Table 2), 0.5 units of Taq DNA polymerase (Qiagen) and approximately 20 ng DNA in a 25 μl total volume. For DYS557, DYS645 and DYS391 a touchdown programme identical to that described for the SSCP analysis was used (Table 2). For DYS472, DYS569, DYS576 and DYS579, cycling consisted of an initial denaturation for 10 min at 95°C followed by 32 cycles at 94°C for 45 s, annealing (see Table 2 for temperatures) for 45 s and 72°C for 45 s, followed by a final extension of 72°C for 5 min The loci that showed male-specific amplification were analysed in the total sample set of 97 males on an ABI377 automated sequencer (Applied Biosystems).
We tested whether the distribution of allele frequencies among the four Saudi-Arabian populations differed at the Y-linked DYS576 locus using an exact test, with the programme R × C (Miller, 1997). We also compared allelic richness, Rs, a measure of the number of alleles independent of sample size, at DYS576 among Abha, Baha and Taif populations (note that the Al-Akhal population was omitted from this analysis owing to very small sample size, n=3). Allelic richness was estimated with FSTAT v2.9.1 (Goudet, 2000) using the rarefaction method of El Mousadik and Petit (1996), in which genotype data are resampled to give sample sizes equal to the smallest population (in this case Baha, n=12).
Arabian hamadryas are thought to have African ancestry (Kummer et al, 1981; Wildman et al, 2004; Winney et al, 2004). If a founder event during colonisation has contributed to low Y chromosome variation in Saudi Arabia, we also expect lower variation at autosomal loci in Arabian compared to African populations. To test this hypothesis, we compared allelic richness at six autosomal loci (five microsatellites: D14S306, D3S1766, D4S243, D12S375, D7S817, and one allozyme locus: TF, transferrin) and a Y-linked microsatellite (DYS391), which have been typed in both a Saudi-Arabian population (Abha, Hammond et al, in revision, and the present study for DYS391) and in an African population (Filwoha, Ethiopia) of morphologically ‘pure’ hamadryas (data from Woolley-Barker, 1999). We calculated allelic richness (Rs) using rarefaction (see above for details) with the size of resampled data sets set to n=44, the size of the smallest sample (TF in Filwoha, Ethiopia). We tested the difference in mean allelic richness for the six autosomal loci between the two populations using a Mann–Whitney U score test.
Results
Intron sequencing
Sequences from all four introns (Genbank accession numbers: UTY11, DQ023315; DBY1, DQ023316; DBY13, DQ023317; DBY14, DQ023318) aligned with high sequence identities to Y-specific regions from the human genome database (UTY11>89% identity to Homo sapiens Y clone accession number AF265575; DBY1>90%, DBY13>86%, DBY14>91% identity to H. sapiens Y clone accession number AC004474) as determined using BLAST (http://www.ncbi.nlm.nih.gov/BLAST/). This confirms that our sequences were from the Y chromosome. We found no nucleotide variation among the 16 males sequenced for a total of 3 kb. The PCR product from DBY13 was about 300 bp larger in baboons than in humans and chimpanzees (Table 1, Hellborg and Ellegren, 2003) and aligning the sequence to the RepeatMasker database (http://www.repeatmasker.org/) revealed that the first 282 bp of the baboon DBY13 was an AluY insertion.
Intron SSCP
We also failed to detect any variation in the sample of 97 males that were analysed by SSCP for the shorter introns DBY4, DBY8, DBY14 and ZFY4 (comprising a total of 1.1 kb in each male). Importantly, the SSCP and sequencing results for DBY14, analysed using both methods, were consistent with one another.
Microsatellite analysis
Male-specific amplification of a single band per individual was achieved with DYS557, DYS569, DYS576, and DYS391. For DYS472 a single band of the same size was amplified in females as well as males, whereas DYS579 and DYS645 produced multiple bands in both sexes (conditions given in Table 2 for these loci are the best from a range of conditions tried). We genotyped DYS391, DYS569, and DYS576 in our sample of 97 males but not DYS557, as the amplification products for this locus were above the size range (>500 bp) routinely genotyped for microsatellites. Of the three genotyped loci, DYS391 and DYS569 were monomorphic in our sample but DYS576 had four alleles (Table 2 and 3). All four alleles were found in the southern-most population, Abha, whereas three alleles (292, 296 and 300) were found in Baha, two alleles (292 and 296) found in Taif and only one allele in Al-Akhal (292, Table 3). The distribution of allele frequencies was highly significantly different among populations (exact test P<0.0001).
Allelic richness was higher in Filwoha than in Abha for all loci except D12S375 (Table 4), but mean allelic richness (3.764 and 3.062, respectively) was not significantly different between the two populations (Mann–Whitney U Score test, two-tailed, U=31, P=0.406). Note that this difference remains nonsignificant when the Y-linked locus DYS391 is removed.
Discussion
Nucleotide variation on the Y chromosome of Saudi-Arabian hamadryas baboons is clearly low, with no differences found in approximately 3.6 kb noncontiguous intron sequence (sequencing and SSCP data combined) from animals sampled from a wide geographic area. In addition, out of seven human-derived microsatellite loci tested, we found only four to be male-specific and only one (DYS576) to be polymorphic in our sample of 97 males. The number of alleles found at DYS576 (four alleles) was similar to the average for autosomal loci (4.26 averaged over 16 microsatellite and three allozyme loci, Hammond et al, in revision) in these populations. These results have parallels in studies of humans, where microsatellite diversity and mutation rate is similar between the Y chromosome and autosomes (Roewer et al, 1992; Goldstein et al, 1996; Kayser et al, 2000; Hurles and Jobling, 2001), but nucleotide diversity on the Y is only 20% of that of the autosomes (International SNP Map Working Group, 2001).
Although there is a gathering consensus that Y chromosome polymorphism is lower than in the rest of the nuclear genome (eg International SNP Working group, 2001; Hellborg and Ellegren, 2004), there appears to be a large difference between species. Roughly one segregating site per 0.5–1 kb is found on the human Y depending on the sample set used (Shen et al, 2000; Hammer et al, 1997, 2003), and estimates of the same order have been found for other mammal species (bonobos and chimpanzees, Stone et al, 2002; wolves and field voles, Hellborg and Ellegren, 2004; shrews Brändli et al, 2005), whereas no variation has been detected in some species despite sampling of divergent populations and sequencing of several thousand base pairs of DNA (eg lynx, cattle, Hellborg and Ellegren, 2004; horses, Lindgren et al, 2004). Variation on the Y chromosome of Saudi-Arabian hamadryas baboons is clearly at the lower end of the mammalian spectrum.
As mentioned previously, there are several, nonmutually exclusive explanations for low variation on the Y chromosome, including low male effective population size, demographic history and selection. Firstly, random genetic drift is expected to reduce variation on the Y chromosome at a faster rate than the rest of the nuclear genome because the number of copies per breeding pair of paternal genes is one quarter that of the autosomes. In addition, hamadryas baboons are not only polygynous but the adult sex ratio is known to be biased about 2:1 in favour of females (Zinner et al, 2001), so male effective population size is probably much lower than for females. Consequently, Ne for the Y is likely to be low relative to maternal or biparentally inherited genes compared to a species with a less biased sex ratio. However, sex-biased dispersal patterns also influence effective population size of male and female-specific genes, with Ne being higher for the philopatric sex because substantial variation is maintained among groups (Chesser and Baker, 1996). In most mammals, females are philopatric and dispersal is male-biased (Greenwood, 1980; Dobson, 1982) so Ne of the Y should be lowered further, but hamadryas baboons are one of a few mammals with philopatric males and dispersing females (Pusey, 1987; Pusey and Packer, 1987; Stammbach, 1987; Hammond et al, 2006). Female-biased dispersal should therefore counteract, to some unknown degree, the low Ne of the Y caused by low copy number, polygynous mating and a female-biased adult sex ratio (Chesser and Baker, 1996).
Second, it has been suggested that the Arabian populations of hamadryas baboons were founded from north-east African ancestors fairly recently (<5–10 kya), with introduction by humans a possibility (Kummer et al, 1981), although mtDNA divergence between Africa and Saudi Arabia indicates an older colonisation event (Wildman et al, 2004), perhaps via the land bridge which formed across the Strait of Bab el Mandeb, 85–119 kya (Winney et al, 2004). We found only slightly lower values of allelic richness at five out of six autosomal loci and at one Y-linked locus in an Arabian (Abha, Saudi Arabia) compared to an African population (Filwoha, Ethiopia; Table 4), and no difference between the two populations in mean allelic richness. Furthermore, the slightly higher number of alleles in Filwoha may be the result of gene flow or dispersal from the hybrid zone between Papio hamadryas hamadryas and P. h. anubis and, although Filwoha is considered morphologically ‘pure’ hamadryas a male P. h. anubis was recorded from this population (see page 10 in: Woolley-Barker, 1999). These findings suggest that a bottleneck during the foundation of the Arabian range of hamadryas is unlikely to be the cause of low Y chromosome variation in Arabian hamadryas. A survey of African diversity at the polymorphic Y-linked DYS576 microsatellite would help to establish whether low Y chromosome variation is a feature of the species rather than just the Arabian populations.
Finally, if selection is acting on the Y, this will tend to eliminate polymorphism and reduce the time to the most recent Y chromosome ancestor. While the efficacy of selection is reduced by the low Ne of Y chromosomes, the absence of recombination along most of the length of the Y means that it behaves as a single linkage group and so selective sweeps (whereby a favourable mutation is driven to fixation, dragging along with it linked neutral variants, Rice, 1987), and/or background selection (in which chromosomes containing deleterious mutations are eliminated from the population, Charlesworth and Charlesworth, 2000) are potentially important. Evidence for positive selection in coding regions of the UTY gene has recently been described in the primate lineage (Gerrard and Filatov, 2005). Unfortunately, in the absence of nucleotide polymorphism we are unable to test for selection directly, and it is therefore unclear to what extent selection is responsible for our results.
In summary, nucleotide variation on the Y chromosome of Saudi-Arabian hamadryas baboons appears to be low relative to other species. High variance in male reproductive success and female-biased adult sex ratios are probably important in reducing Ne of paternal genes, although this effect is slightly counterbalanced in hamadryas by male philopatry and female-biased dispersal. We found no difference in allelic richness between an Arabian and an African population and so it is unlikely that the low level of variation on the Y chromosome in Arabia was caused by a population bottleneck during the colonisation of Arabia by African hamadryas baboons. Whether low nucleotide diversity is a feature of the hamadryas nuclear genome, needs to be addressed by comparison with sequence data from the autosomes and/or X chromosome. Finally, this study indicates that extensive efforts may be required to find sufficient Y chromosome variation in wild populations, particularly in those species with high variance in male reproductive success and a history of small population size. Our finding of variation at a microsatellite but no nucleotide variation in introns suggests that it would be wise for researchers to focus on finding Y-linked microsatellite variation rather than SNPs for population genetic studies.
References
Boissinot S, Boursot P (1997). Discordant phylogeographic patterns between the Y chromosome and mitochondrial DNA in the house mouse: selection on the Y chromosome? Genetics 146: 1019–1034.
Brändli L, Lawson Handley LJ, Vogel P, Perrin N (2005). Evolutionary history of the greater white-toothed shrew (Crocidura russula) inferred from analysis of mtDNA, Y, and X chromosome markers. Mol Phylogenet Evol 37: 832–844.
Charlesworth B, Charlesworth D (2000). The degeneration of Y chromosomes. Phil Trans Roy Soc B 355: 1563–1572.
Chesser RK, Baker RJ (1996). Effective sizes and dynamics of uniparentally and diparentally inherited genes. Genetics 144: 1225–1235.
Dobson FS (1982). Competition for mates and predominant juvenile male dispersal in mammals. Anim Behav 30: 1183–1192.
Dorit RL, Akashi H, Gilbert W (1995). Absence of polymorphism at the ZFY locus on the human Y chromosome. Science 268: 1183–1185.
El Mousadik A, Petit RJ (1996). High level of genetic differentiation for allelic richness among populations of the argan tree Argania spinosa (L.) Skeels endemic to Morocco. Theor Appl Genet 92: 832–839.
Erler A, Stoneking A, Kayser M (2004). Development of Y-chromosomal microsatellite markers for non-human primates. Mol Ecol 13: 2921–2930.
Gerrard DT, Filatov DA (2005). Positive and negative selection on mammalian Y chromosomes. Mol Biol Evol 22: 1423–1432.
Goldstein D, Zhivotovsky LA, Nayar K, Linares AR, Cavalli-Sforza LL, Feldman MW (1996). Statistical properties of the variation at linked microsatellite loci: implications for the history of human Y chromosomes. Mol Biol Evol 13: 1213–1218.
Goudet J (2000). FSTAT, a programme to estimate and test gene diversities and fixation indices (version 2.9.1). Available from http://www.unil.ch/izea/softwares/fstat.html.
Greenwood PJ (1980). Mating systems, philopatry and dispersal in birds and mammals. Anim Behav 28: 1140–1162.
Hammer MF (1995). A recent common ancestry for human Y chromosomes. Nature 378: 376–380.
Hammer MF, Blackmer F, Garrigan D, Nachman MW, Wilder JA (2003). Human population structure and its effects on sampling Y chromosome sequence variation. Genetics 164: 1495–1509.
Hammer MF, Spurdle AB, Karafet T, Bonner MR, Wood ET, Novelletto A et al (1997). The geographic distribution of human Y chromosome variation. Genetics 145: 787–805.
Hammond RL, Lawson Handley LJ, Winney BJ, Bruford MW, Perrin N (2006). Genetic evidence for female-biased dispersal and gene-flow in a polygynous primate. Proc Roy Soc B 273: 479–484.
Hammond RL, Winney BJ, Macasero W, Flores B, Boug A, Scheffrahn W et al (in revision). Kin structure in Arabian hamadryas baboons, Papio hamadryas hamadryas. Behav Ecol Sociobiol.
Hellborg L, Ellegren H (2003). Y chromosome conserved anchor tagged sequences (YCATS) for the analysis of mammalian male-specific DNA. Mol Ecol 12: 283–291.
Hellborg L, Ellegren H (2004). Low levels of nucleotide diversity in mammalian Y chromosomes. Mol Biol Evol 21: 158–163.
Hurles ME, Jobling MA (2001). Haploid chromosomes in molecular ecology: lessons from the human Y. Mol Ecol 10: 1599–1613.
International SNP Map Working Group (2001). A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms. Nature 409: 928–933.
Jobling M, Tyler-Smith C (2003). The human Y chromosome: an evolutionary marker comes of age. Nat Rev Genet 4: 598–612.
Kayser M, Kittler R, Erler A, Hedman M, Lee AC, Mohyuddin A et al (2004). A comprehensive survey of human Y-chromosomal microsatellites. Am J Hum Genet 74: 1183–1197.
Kayser M, Roewer L, Hedman M, Henke L, Henke J, Brauer S et al (2000). Characteristics and frequency of germline mutations at microsatellite loci from the human Y chromosome, as revealed by direct observation in father/son pairs. Am J Hum Genet 66: 1580–1588.
Kayser MA, Caglia D, Corach N, Fretwell C, Gehrig G, Graziosi F et al (1997). Evaluation of Y-chromosomal STRs: a multicenter study. Int J Legal Med 110: 125.
Kummer H, Banaja AA, Abo-Khatwa AN, Ghandour AM (1981). Mammals of Saudi Arabia: Primates: a survey of hamadryas baboons in Saudi Arabia. Fauna Saudi Arabia 3: 441–471.
Lindgren G, Backstrom N, Swinburne J, Hellborg L, Einarsson A, Sandberg K et al (2004). Limited number of patrilines in horse domestication. Nat Genet 36: 335–336.
Miller MP (1997). RxC: A Program for the Analysis of Contingency Tables. Department of Biological Sciences, North Arizona University: Flagstaff.
Petit E, Balloux F, Excoffier L (2002). Mammalian population genetics: why not Y? Trends Ecol Evol 17: 28–33.
Pritchard JK, Stephens M, Donnelly P (2000). Inference of population structure using multilocus genotype data. Genetics 155: 945–959.
Pusey A (1987). Sex-biased dispersal and inbreeding avoidance in mammals. Trends Ecol Evol 2: 295–299.
Pusey A, Packer C (1987). Dispersal and philopatry. In: Smuts BB, Cheney DL, Seyfarth RM, Wrangham RW, Struhsaker TT (eds) Primate Societies. University of Chicago Press: Chicago. pp 250–266.
Rice WR (1987). Genetic hitchhiking and the evolution of reduced genetic activity of the Y sex chromosome. Genetics 116: 161–167.
Roewer L, Arnemann J, Spurr NK, Grzeschik KH, Epplen JT (1992). Simple repeat sequences on the human Y-Chromosome are equally polymorphic as their autosomal counterparts. Hum Genet 89: 389–394.
Shen P, Wang F, Underhill PA, Franco C, Yang WH, Roxas A et al (2000). Population genetic implications from sequence variation in four Y chromosome genes. Proc Natl Acad Sci USA 97: 7354–7359.
Stammbach E (1987). Desert, Forest and Montane Baboons: Multilevel societies. In: Smuts BB, Cheney DL, Seyfarth RM, Wrangham RW, and Struhsaker TT (eds) Primate Societies. University of Chicago Press: Chicago. pp 112–120.
Stone AC, Griffiths RC, Zegura SL, Hammer MF (2002). High levels of Y-chromosome nucleotide diversity in the genus Pan. Proc Natl Acad Sci USA 99: 43–48.
Stumpf MPH, Goldstein DB (2001). Genealogical and evolutionary inference with the human Y chromosome. Science 291: 1738–1747.
Tosi AJ, Morales JC, Melnick DJ (2000). Comparison of Y chromosome and mtDNA phylogenies leads to unique inferences of macaque evolutionary history. Mol Phylogenet Evol 17: 133–144.
Tosi AJ, Morales JC, Melnick DJ (2002). Y-chromosome and mitochondrial markers in Macaca fascicularis indicate introgression with Indochinese M. mulatta and a biogeographic barrier in the Isthmus of Kra. Int J Primatol 23: 161–178.
Tosi AJ, Morales JC, Melnick DJ (2003). Paternal, maternal, and biparental molecular markers provide unique windows onto the evolutionary history of macaque monkeys. Evolution 57: 1419–1435.
Underhill PA, Shen P, Lin AA, Jin L, Passarino G, Yang WH et al (2000). Y chromosome sequence variation and the history of human populations. Nat Genet 26: 358–361.
van Hooft WF, Groen AF, Prins HHT (2002). Phylogeography of the African Buffalo based on mitochondrial and Y-chromosomal loci: Pleistocene origin and population expansion of the Cape buffalo subspecies. Mol Ecol 11: 267–279.
Whitfield LS, Sulston JE, Goodfellow PN (1995). Sequence variation of the human Y chromosome. Nature 378: 379–380.
Wildman DE, Bergman TJ, al-Aghbari A, Sterner KN, Newman TK, Phillips-Conroy JE et al (2004). Mitochondrial evidence for the origin of hamadryas baboons. Mol Phylogenet Evol 32: 287–296.
Winney BJ, Hammond RL, Macasero W, Flores B, Boug A, Biquand V et al (2004). Crossing the Red Sea: phylogeography of the hamadryas baboon, Papio hamadryas hamadryas. Mol Ecol 13: 2819–2827.
Woolley-Barker T (1999). Social Organization and Genetic Structure in a Baboon Hybrid Zone. PhD Thesis, New York University.
Zinner D, Pelaez F, Torkler F (2001). Group composition and adult sex-ratio of hamadryas baboons (Papio hamadryas hamadryas) in central Eritrea. Int J Primatol 22: 415–430.
Acknowledgements
We thank Sylvain Biquand, Veronique Biquand and Ahmed Boug for collecting the samples used in this study, and two anonymous reviewers for their helpful comments. Financial support for this project was obtained from the University of Lausanne 2ème cycle funds for teaching.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Lawson Handley, L., Hammond, R., Emaresi, G. et al. Low Y chromosome variation in Saudi-Arabian hamadryas baboons (Papio hamadryas hamadryas). Heredity 96, 298–303 (2006). https://doi.org/10.1038/sj.hdy.6800803
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.hdy.6800803
Keywords
This article is cited by
-
Assessing the recovery of Y chromosome microsatellites with population genomic data using Papio and Theropithecus genomes
Scientific Reports (2023)
-
Genomic variation in baboons from central Mozambique unveils complex evolutionary relationships with other Papio species
BMC Ecology and Evolution (2022)
-
Sex-linked and autosomal microsatellites provide new insights into island populations of the tammar wallaby
Heredity (2014)
-
Genetics of the Shimokita macaque population suggest an ancient bottleneck
Primates (2008)
